
Fe-Catalyzed reductive NO-bond cleavage – a route to the diastereoselective 1,4-aminohydroxylation of 1,3-dienes†
S.
Scholz
and
B.
Plietker
*
Institut für Organische Chemie, Universität Stuttgart, Pfaffenwaldring 55, DE-70569 Stuttgart, Germany. E-mail: bernd.plietker@oc.uni-stuttgart.de
First published on 3rd August 2016
Abstract
The low-valent Fe-complex Bu4N[Fe(CO)3(NO)] (TBA[Fe]) catalyzes the reductive cleavage of N–O-bonds in oxazines under salt-free conditions using malononitrile as reductant. The mild reduction conditions allow the development of a sequence of [4 + 2]-cycloaddition/reductive cleavage, a process that can be considered as an efficient way for the diastereoselective 1,4-aminohydroxylation of 1,3-dienes.
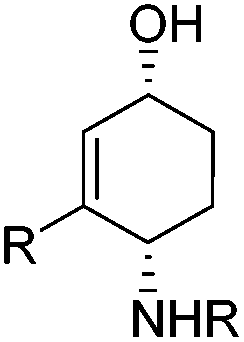
1,4-Aminoalkenols are ubiquitous structural motives in various natural products and useful building blocks in organic synthesis (Fig. 1).1 Several ways to generate this assembly of functional groups are reported with the oxidative diastereoselective Pd-catalyzed 1,4-aminooxygenation of 1,3-dienes as the most versatile process.2 However, acidic conditions and strong oxidizing agents are required and hence limit the application range of these methods to substrates possessing no oxidation labile and acid stable functional groups.
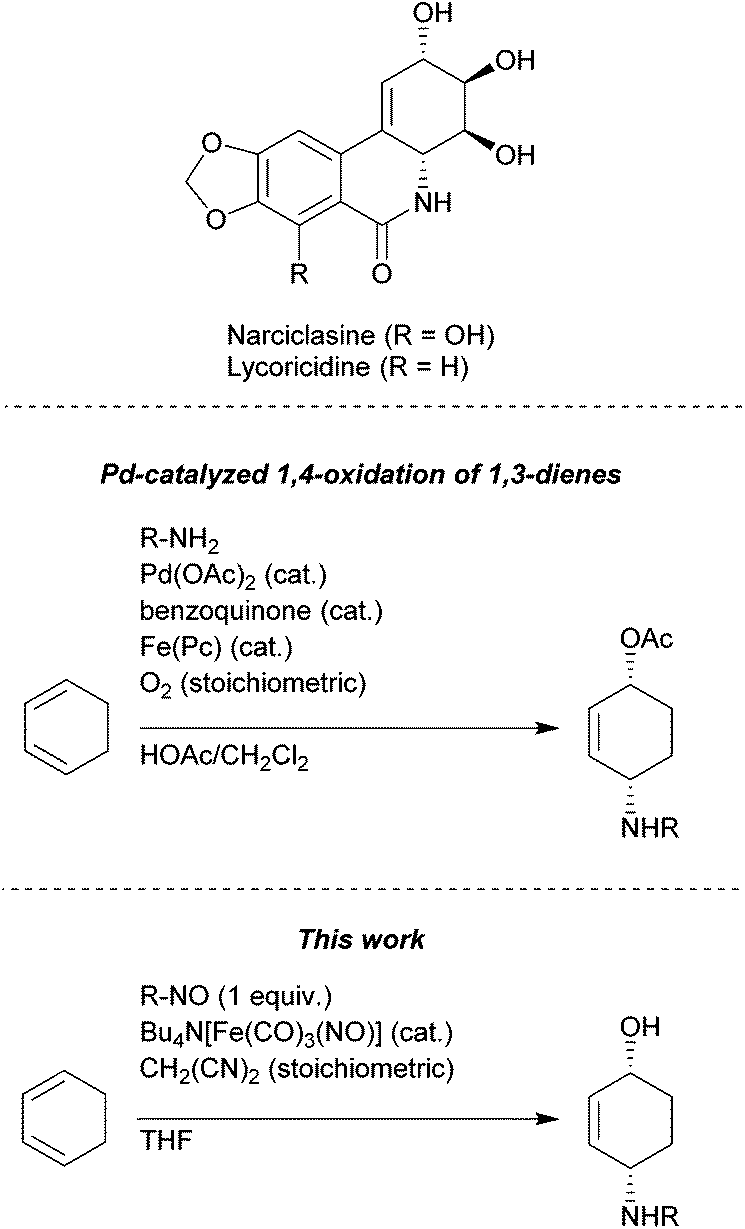 | ||
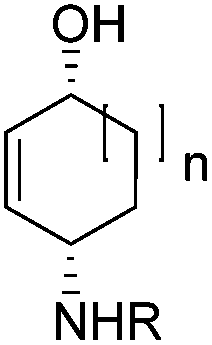
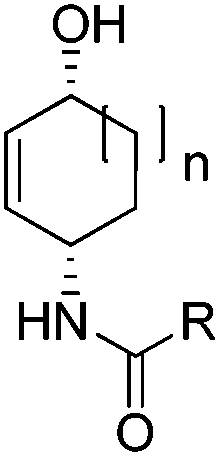
| Fig. 1 Non-oxidative Fe-catalyzed 1,4-aminohydroxylation. | ||
The two-step [4 + 2]-cycloaddition of organic nitroso compounds to 1,3-dienes/reductive N–O-bond cleavage represents a classical alternative strategy for a non-oxidative 1,4-aminooxygenation. However up to date mostly stoichiometric reagents like Zn, SmI2, Mo(CO)6etc. are required for the reductive cleavage of the N–O-bond in oxazines obtained in the cycloaddition event.3–6 The catalytic N–O-bond reduction using RANEY®-Ni and H2-gas circumvents the production of undesired salt byproducts, however it is limited to substrates possessing no functional group that could react under these conditions (e.g. C–S-bonds, carbonyl groups, etc.). Another catalytic method described by Miller et al. uses Cp2TiCl as catalyst (0.2 equiv.) and manganese (8 equiv.) as stoichiometric reductant.7 Since the yields are moderate with this catalytic protocol, Miller uses a system where Cp2TiCl is used in excess (2.5 equiv.) together with zinc (5 equiv.) as reductant.7 Despite the disadvantage of having two consecutive transformations we considered the [4 + 2]-cycloaddition/reductive N–O-bond cleavage to be a powerful addition to the existing oxidative transition metal catalyzed one-pot procedures since the relative configuration of the p-bonds in the starting material is directly transferred into the relative configuration of the resulting products.
In order to streamline the overall synthetic efficiency, a catalytic method for the highly chemoselective N–O-bond cleavage of the primary formed oxazines is required. Herein we describe an operationally simple Fe-catalyzed N–O-bond reduction that allows the efficient synthesis of diastereomerically pure 1,4-aminoalcohols.
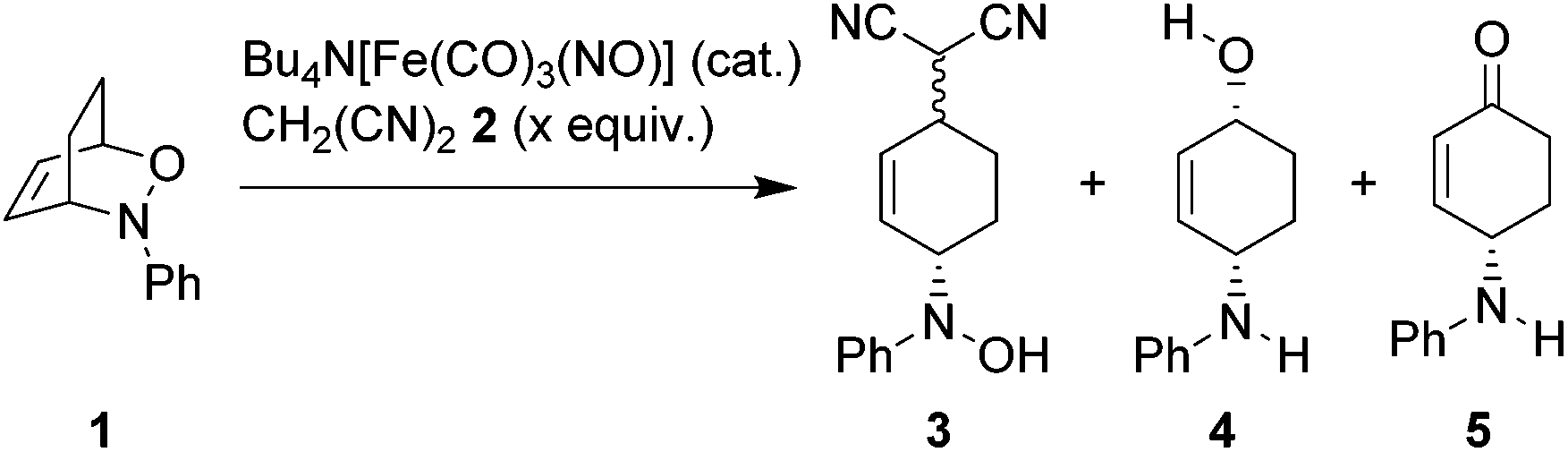
During our investigations on Bu4N[Fe(CO)3(NO)] (TBA[Fe])-catalyzed allylic substitutions we were wondering whether the 1,2-oxazine 1 derived from the Diels–Alder reaction between 1,3-cyclohexadiene and nitrosobenzene would react with malononitrile 2 in an allylic substitution to give the corresponding oxime 3, however unexpectedly we observed a clean reductive cleavage of the N–O-bond to give the corresponding aminoalcohol 4 alongside with traces of ketone 5. This unexpected reaction course caught our attention and we set out to systematically screen the influence of various reaction parameters (Table 1).
|
|
||||||
|---|---|---|---|---|---|---|
| Entrya | CH2(CN)2 (equiv.) | Solvent | Reaction time | T (°C) | TBA[Fe] (mol%) | Yield 4b (%) |
| a Reactions were carried out on a 0.2 mmol scale. b Isolated yield. | ||||||
| 1 | 1.5 | THF | 16 | 40 | 10 (16) | 37 |
| 2 | 1.5 | THF | 16 | 60 | 10 (16) | 80 |
| 3 | 1.5 | THF | 16 | 80 | 10 (16) | 80 |
| 4 | 1.5 | THF | 16 | 100 | 10 (16) | 77 |
| 5 | 1.5 | THF | 16 | 80 | 5 (16) | 75 |
| 6 | 1.5 | THF | 16 | 80 | 20 (16) | 90 |
| 7 | 2.5 | THF | 16 | 80 | 10 (16) | 90 |
| 8 | 1.5 | DME | 16 | 80 | 10 (16) | 70 |
| 9 | 1.5 | Dioxane | 16 | 80 | 10 (16) | 68 |
| 10 | 1.5 | THF | 6 | 80 | 10 (6) | 80 |
| 11 | 1.5 | THF | 1 | 80 | 10 (1) | 54 |
From these results it became obvious that stoichiometric amounts of malonontrile are required in order to get good yields of the cis-1,4-aminoalcohol 4. Best yields are obtained at a temperature of 60–80 °C (entries 1–4, Table 1). A reasonable amount of catalyst is 10 mol%, although 5 mol% also gave a good yield of 75% of aminalcohol 4 (entries 3, 5 and 6, Table 1). A change in solvent to DME or dioxane gave lower yield, compared to THF (entries 3, 8 and 9, Table 1) and reduction of the reaction time indicated the reaction to be fast leading to about 54% (isolated yield) product formation after only 1 h and 80% after 6 h (entries 10 and 11, Table 1). When used on a 1 mmol scale, the standard substrate 1 gave 85% isolated yield of the corresponding alcohol 4 under conditions given in Table 1, entry 3.
Recently, we were able to develop a TBA[Fe]-catalyzed conjugate addition in which a protonation of the ferrate anion appears to be the initial step of the catalytic reaction.8 We assume that malononitrile might also act as a weak acid that is protonating the catalyst within the initial step. In order to shed some light into the role of the malononitrile (pKa ∼ 11.1) more compounds of the general structure EWG1CH2EWG2 were tested under otherwise identical reaction conditions. Ethylcyanoacetate (pKa ∼ 13.1) and chloroacetonitrile gave 30% and 24% of alcohol 4, while diiodomethane and dichloromethane gave 19% and 11%, respectively. Without the addition of any of these agents, no conversion was detected.
Although more detailed mechanistic investigations on the mode of action are certainly necessary, the following mechanistic hypothesis might serve as a preliminary working model (Fig. 2). Accordingly, we propose a protonation of the ferrate catalyst by malondinitrile to be the initial step in this transformation. The corresponding Fe–H-complex acts as a Brønstedt-acid thus opening the oxazine through hydrometallation of the N–O-bond. Release of the catalyst through nucleophilc substitution and hydrolysis of the alkoxymalodinitrile leads to the formation of the observed product.
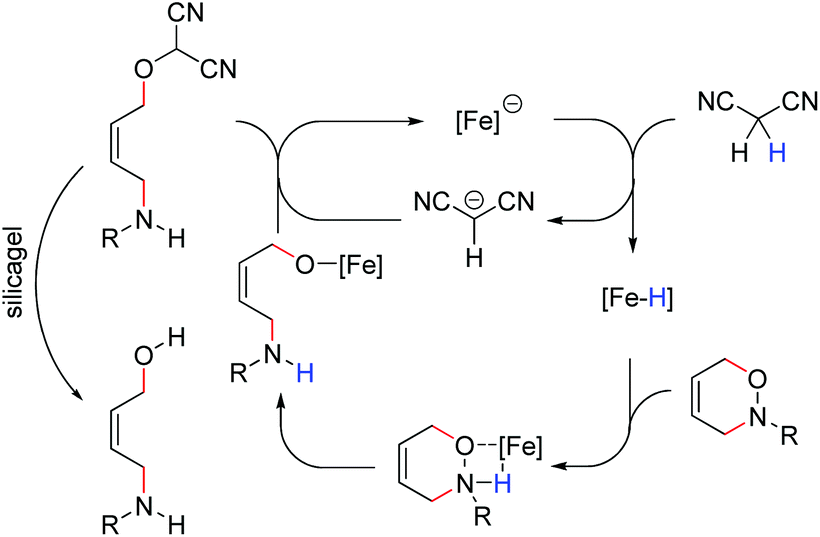 | ||
| Fig. 2 Mechanistic proposal. | ||
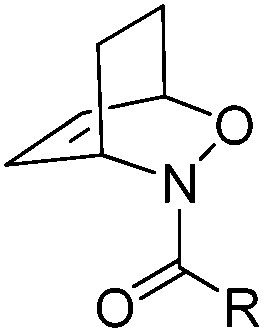
To show the applicability of this catalytic system, various cyclic and acyclic substituted 1,3-dienes were transferred into the corresponding oxazines via [4 + 2]-cycloaddition using stable nitroso arenes or in situ prepared acyl nitroso compounds (Table 2). The respective oxazines were obtained in moderate to excellent yields and excellent regio- and stereoselectivities (entries 1–20, middle column, Table 2). Treatment of these oxazines with catalytic amounts of Bu4N[Fe(CO)3(NO)] and malononitrile at 80 °C for 16 h led to the formation of the desired diastereomerically pure 1,4-aminoalcohols in moderate to excellent yields (entries 1–20, right column, Table 2). From these data it is obvious that the ringsize has a significant influence on the yield. Both cyclopentadiene and cylcooctadiene derived oxazines proved less reactive as compared to the substrates derived from cyclohexadiene or cycloheptadiene (entries 1–4, right column, Table 2), while the cyclopentadiene derived oxazine 6 could not be isolated and was therefore immediately reduced in a one-pot reaction sequence, as will be shown later. The reaction is tolerant toward different functional N-aryl substituents (entries 5–7, right column, Table 2). However, clear limitations for N-acyl groups were observed. Whereas a N-benzoyl, -cinnamoyl and -piperonoyl substituted oxazine was cleaved in a clean way (entries 8, 10 and 13, right column, Table 2), the corresponding carbamate, pivalate and phenylacetic acid derived oxazines did not give any conversion to the desired product (entries 9, 11 and 12, right column, Table 2).
|
|
|||
|---|---|---|---|
| Entry | Diene | Oxazinea | Aminoalcoholb |
| a Reactions were carried out on a 5 mmol scale using literature known procedures (see ESI). Isolated yields are given in parenthesis. b Reactions were performed on a 0.2 mmol scale in THF (0.5 mL) using TBA[Fe] (10 mol%) and malononitrile (1.5 equiv.) at 80 °C. Isolated yields are given in parenthesis. c Yield over two steps. | |||

|
|
|
|
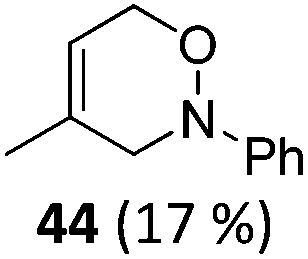
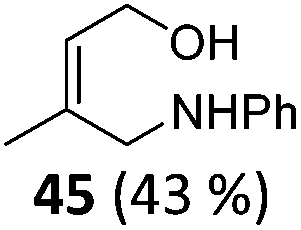
| 1 | 6 Not isolated (n = 0, R = Ph) | 7 (43%)c | |
| 2 | 1 (98%) (n = 1, R = Ph) | 4 (80%) | |
| 3 | 8 (97%) (n = 2, R = Ph) | 9 (70%) | |
| 4 | 10 (39%) (n = 3, R = Ph) | 11 (21%) | |
| 5 | 12 (27%) (n = 1, R = p-Cl–C6H4) | 13 (77%) | |
| 6 | 14 (39%) (n = 1, R = o-Cl–C6H4) | 15 (74%) | |
| 7 | 16 (13%) (n = 1, R = m-(CH3)2–C6H4) | 17 (74%) | |
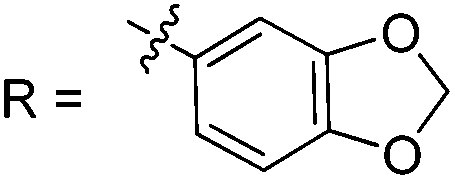
| 8 |
18 (70%) (n = 1, R = C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)Ph) O)Ph) |
19 (58%) | |
|
|
|
|
| 9 | 20 (72%) (R = OC2H5) | 21 (—) | |
| 10 | 22 (67%) (R = CHCHPh) | 23 (58%) | |
| 11 | 24 (55%) (R = C(CH3)3) | 25 (—) | |
| 12 | 26 (99%) (R = CH2Ph) | 27 (—) | |
| 13 | 28 (82%) | 29 (79%) | |
|
|||
|
|
|
|
| 14 | 30 (78%) (R = C4H9) | 31 (67%) | |
| 15 | 32 (99%) (R = Ph) | 33 (56%) | |
| 16 | 34 (93%) (R = (CH2)2CO2Et) | 35 (79%) | |
| 17 | 36 (88%) (R = (CH2)3OH) | 37 (68%) | |
| 18 | 38 (73%) (R = (CH2)3NBn2) | 39 (55%) | |
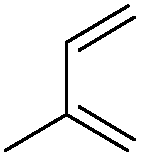
| 19 |
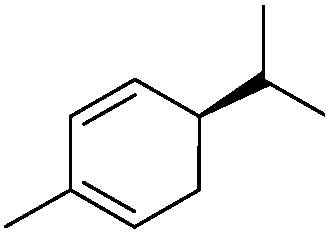
|
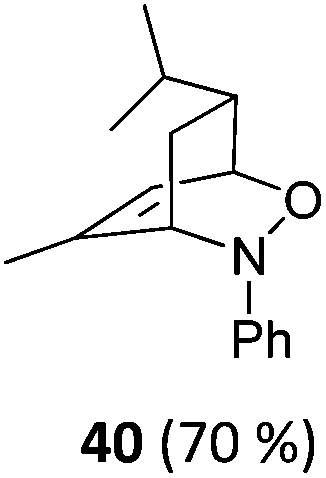
|

|
| 20 |
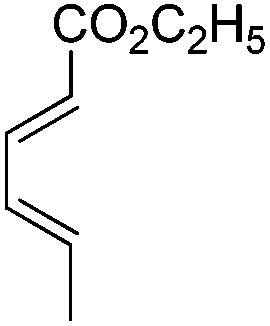
|
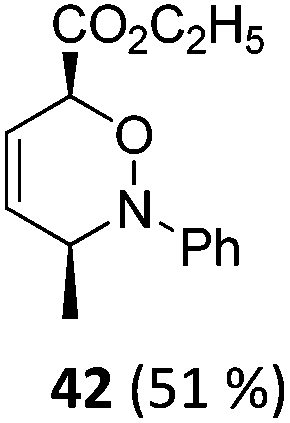
|

|
| 21 |
|
|
|
The high degree of regio- and diastereoselectivity obtained in the [4 + 2]-cycloaddition of substituted 1,3-cyclohexadienes was directly transferred into the formation of isomerically pure 1,4-aminoalcohols (entries 14–19, right column, Table 2). Even acyclic substrates were transferred into the corresponding diastereomerically pure aminoalcohols with high regioselectivities (entries 20 and 21, right column, Table 2).
Finally, we became interested in combining both processes into a one-pot protocol. Indeed, treating 1,3-dienes with nitrosobenzene in THF for 2 h at room temperature and subsequent addition of the TBA[Fe]-catalyst plus malononitrile resulted in the clean formation of the corresponding syn-1,4-aminoacohols in moderate to good yields (Table 3).
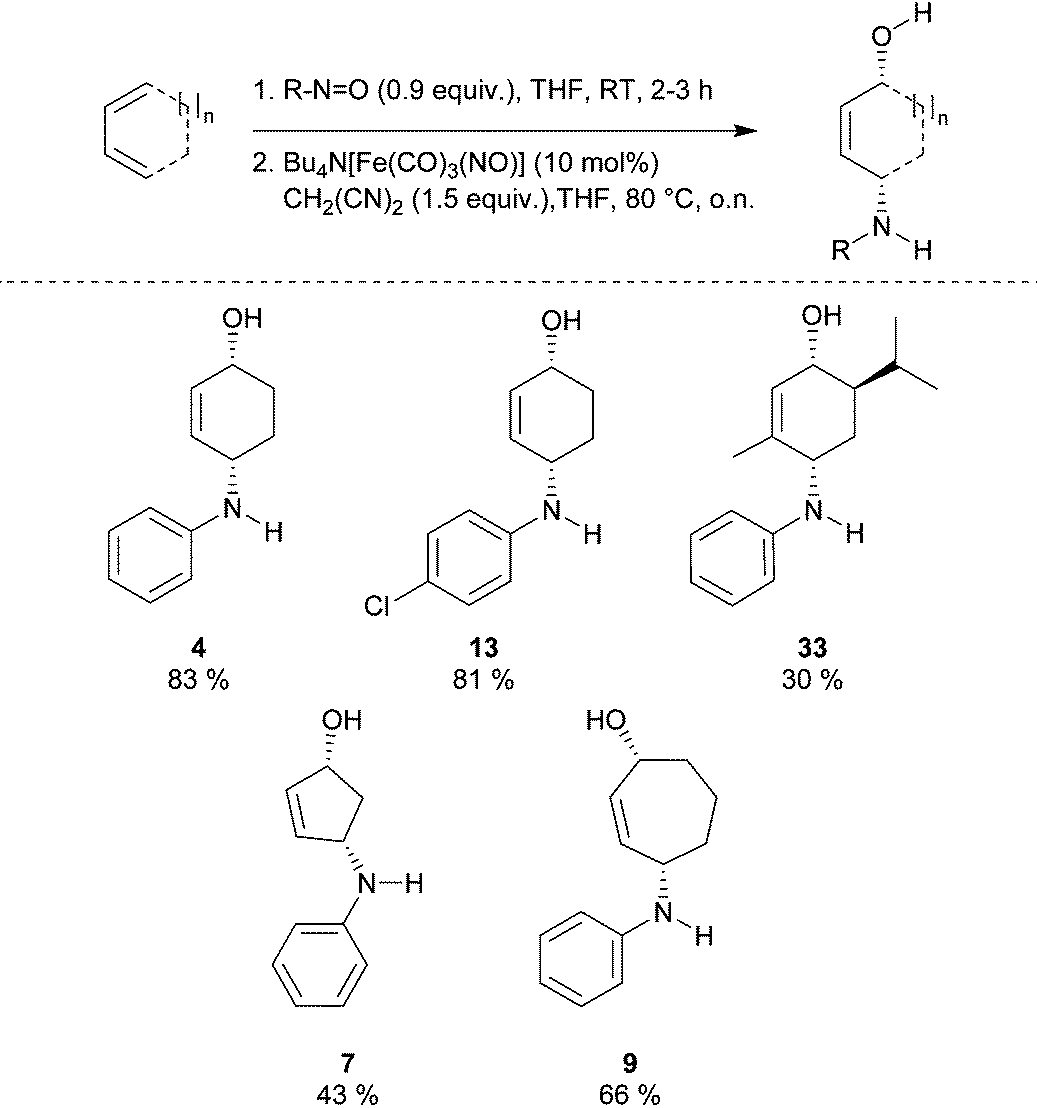
Conclusions
Herein we describe a Fe-catalyzed reductive N–O-bond cleavage of 1,2-oxazines that accepts a variety of different functional groups, like halides, carbonyls, alcohols and esters, as well as different ring sizes in bicyclic substrates together with open-chained substrates. Furthermore the relative configuration of the [4 + 2]-cycloaddition could be transferred into the product to obtain diastereomerically pure aminoalcohols. Additionally we could perform a consecutive [4 + 2]-cycloaddition/N–O-bond cleavage that can be regarded as a 1,4 aminohydroxylation of 1,3 dienes with nitroso compounds. This displays one of very few catalytic N–O-bond cleaving reactions and broadens the scope of iron catalyzed reactions.Notes and references
- (a) For a review on amaryllidacae alkaloids see: J. R. Lewis, Nat. Prod. Rep., 1997, 14, 303–308 RSC; (b) For a total synthesis of Lyco-ricidine see: T. Hudlicky and H. F. Olivo, J. Am. Chem. Soc., 1992, 114, 9694–9696 CrossRef CAS; (c) For a total synthesis of narciclasine and pancratista-tine see: J. H. Rigby, U. S. M. Maharoof and M. E. Mateo, J. Am. Chem. Soc., 2000, 122, 6624–6628 CrossRef CAS.
- (a) J.-E. Bäckvall, J.-E. Nystrom and R. E. Nordberg, J. Am. Chem. Soc., 1985, 107, 3676–3686 CrossRef; (b) J.-E. Bäckvall and P. G. Andersson, J. Am. Chem. Soc., 1990, 112, 3683–3685 CrossRef.
- (a) NO-bond cleavage with Zn/HCOOH: Y. A. Arbuzov and T. A. Mastryukova, Bull. Acad. Sci. USSR Div. Chem. Sci., 1952, 1, 613–616 CrossRef; (b) G. Kresze and G. Schulz, Tetrahedron, 1961, 12, 7–12 CrossRef CAS.
- (a) NO-bond cleavage with SmI2: G. E. Keck, T. T. Wager and S. F. McHardy, Tetrahedron, 1999, 55, 11755–11772 CrossRef CAS; (b) G. E. Keck, S. F. McHardy and T. T. Wager, Tetrahedron Lett., 1995, 41, 7419–7422 CrossRef.
- (a) NO-bond cleavage with Mo(CO)6: S. Cicchi, A. Goti, A. Brandi, A. Guarna and F. De Sarlo, Tetrahedron Lett., 1990, 31, 3351–3354 CrossRef CAS; (b) M. Nitta and T. Kobayashi, J. Chem. Soc., Perkin Trans. 1, 1985, 1401–1406 RSC.
- (a) Other NO-bond cleaving reactions: K. Klier, G. Kresze, O. Werbitzky and H. Simon, Tetrahedron Lett., 1987, 28, 2677–2680 CrossRef CAS; (b) Y. H. Lee and D. J. Choo, Bull. Korean Chem. Soc., 1993, 4, 423–424 Search PubMed; (c) G. Galvani, G. Calvet, N. Blanchard and C. Kouklovsky, Org. Biomol. Chem., 2008, 6, 1063–1070 RSC; (d) B. Yang and M. J. Miller, Org. Lett., 2010, 12, 392–395 CrossRef CAS PubMed; (e) I. Ramakrishna, H. Sahoo and M. Baidya, Chem. Commun., 2016, 52, 3215–3218 RSC.
- C. Cesario, L. P. Tardibono and M. J. Miller, J. Org. Chem., 2009, 74, 448–451 CrossRef CAS PubMed.
- D. Zhang, J. Knelles and B. Plietker, Adv. Synth. Catal., 2016 DOI:10.1002/adsc.201600278 , in press.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details, characterization, as well as NMR-spectra. See DOI: 10.1039/c6qo00255b |
| This journal is © the Partner Organisations 2016 |