
Learning lessons from directed evolution of stereoselective enzymes†
Guangyue
Li
ab and
Manfred T.
Reetz
*ab
aMax-Planck-Institut für Kohlenforschung, Kaiser-Wilhelm-Platz 1, 45470, Mülheim an der Ruhr, Germany. E-mail: reetz@mpi-muelheim.mpg.de
bFachbereich Chemie, Philipps-Universität, Hans-Meerwein-Strasse, 35032 Marburg, Germany
First published on 27th June 2016
Abstract
With the advent of directed evolution of stereoselective enzymes almost 20 years ago and the rapid development of this exciting area of research, the traditional limitations of biocatalysts in organic chemistry have been eliminated. It is now possible to enhance or invert enantioselectivity, broaden the substrate scope and increase the activity of many different types of enzymes. In addition to providing a prolific source of catalysts for asymmetric transformations, many lessons can be learned from directed evolution on the molecular level, because stereoselectivity is a sensitive probe. This review focuses on two types of lessons arising from studies focusing on (1) uncovering the source of altered stereoselectivity, and (2) constructing fitness landscapes which reveal additive and non-additive mutational effects as well as ways to escape from local minima. Case studies featuring enzymes of the type epoxide hydrolase, lipase and Baeyer–Villiger monooxygenase are presented.
 Guangyue Li | Guangyue Li studied protein engineering at the Institute of Plant Protection, Chinese Academy of Agricultural Sciences (Laboratory of Prof. Dewen Qiu), obtaining his master degree in 2010. He then joined the group of Prof. Dunming Zhu at the Tianjin Institute of Industrial Biotechnology, Chinese Academy of Sciences, obtaining a doctoral degree in 2014 in the area of enzyme engineering. Currently he is a postdoctoral fellow in the group of M. T. Reetz focusing on directed evolution of epoxide hydrolase, alcohol dehydrogenase and monoamine oxidase. |
 Manfred T. Reetz | Manfred T. Reetz obtained his doctoral degree in 1969 with Ulrich Schöllkopf at Göttingen University and then performed his postdoctoral work in the group of Reinhard W. Hoffmann at Marburg University. Following an appointment at Bonn University, he became a Full Professor of organic chemistry back at Marburg University (1980–1991) before joining the Max-Planck-Institut fur Kohlenforschung in Mülheim/Germany, where he served as Managing Director from 1993 to 2002. Following formal retirement as Director in 2011, he accepted the offer to become the first Hans-Meerwein-Research-Professor in the Chemistry Department of Marburg University while also being an external (emeritus) group leader of the Mülheim Max-Planck-Institute. |
Introduction
Directed evolution1 of stereoselective enzymes is a protein engineering technique which constitutes a prolific source of biocatalysts for asymmetric transformations in organic chemistry and biotechnology.1,2 The underlying principle involves recursive cycles of gene mutagenesis, protein expression and screening (Scheme 1). The genetically modified gene of the best mutant displaying the highest stereoselectivity in the first round is subjected to another cycle, thereby exerting “evolutionary pressure”. The process is continued until the desired degree of stereoselectivity has been achieved. The experimenter simulates Darwinian evolution as it occurs in nature (“evolution in the test tube”). Thus, this approach is quite different from creating chiral transition metal catalysts or organocatalysts, which requires knowledge of the mechanism, experience, intuition and often trial and error.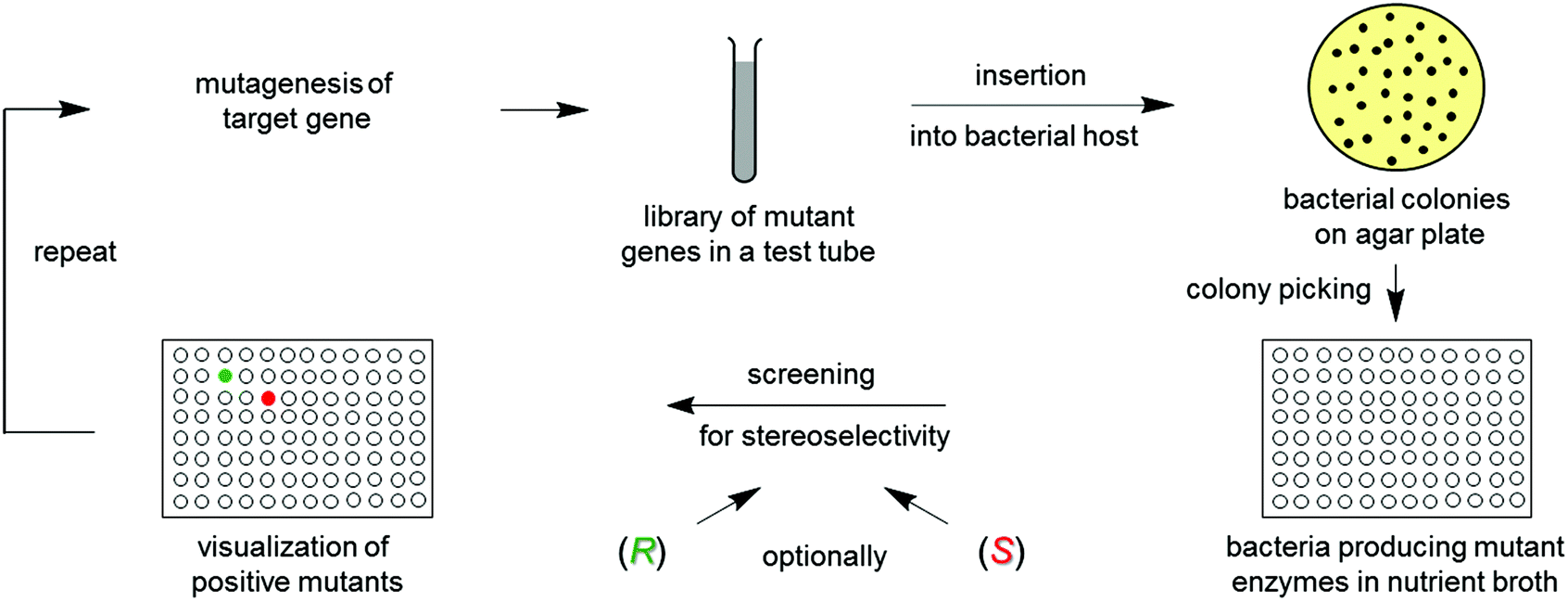 | ||
| Scheme 1 The concept of directed evolution of stereoselective enzymes, allowing the researcher to achieve R- and/or S-selectivity on an optional basis. | ||
Following the proof-of-concept in 1997 using a lipase as a catalyst in the hydrolytic kinetic resolution of a racemic ester,3 we and others have applied the concept to many different enzyme types including lipases, esterases, nitrilases, amidases, epoxide hydrolases, glycosidases, alcohol dehydrogenases, enoate-reductases, P450-monooxygenases, Baeyer–Villiger monooxygenases, oxynitrilases, aldolases and pyruvate decarboxylases.1,2 The bottleneck in the overall process is the labor-intensive screening step.4 In the early days of this fascinating research area, efficiency played no role, and indeed, very large mutant libraries were usually generated which had to be screened for enantiomeric excess (ee).5 In many cases this required extensive assaying, sometimes using expensive instrumentation such as multiplexing mass spectrometry.6
Around that time it became clear that methodology development with the aim of creating small but “smart” libraries is essential for true progress.5,7 This means that strategies are required which provide highest-quality mutant libraries with a high frequency of notably improved biocatalysts, the real challenge in current directed evolution. Our contribution is iterative saturation mutagenesis (ISM) at sites lining the binding pocket of an enzyme.8 In the first step, residues (amino acids) lining the binding pocket of an enzyme are identified on the basis of X-ray data or a homology model, which are then grouped into randomization sites dubbed A, B, C, etc., each site comprising one, two, three or more amino acid positions as part of the Combinatorial Active-site Saturation Test (CAST)9 (Scheme 2a). The gene of the best hit in one library is subsequently used as the starting point (template) for saturation mutagenesis at another site, and the process is continued accordingly. The situation for the A–B, A–B–C and A–B–C–D systems is illustrated in Scheme 2b, which reveals that 2, 6 and 24 ISM pathways are involved, respectively.
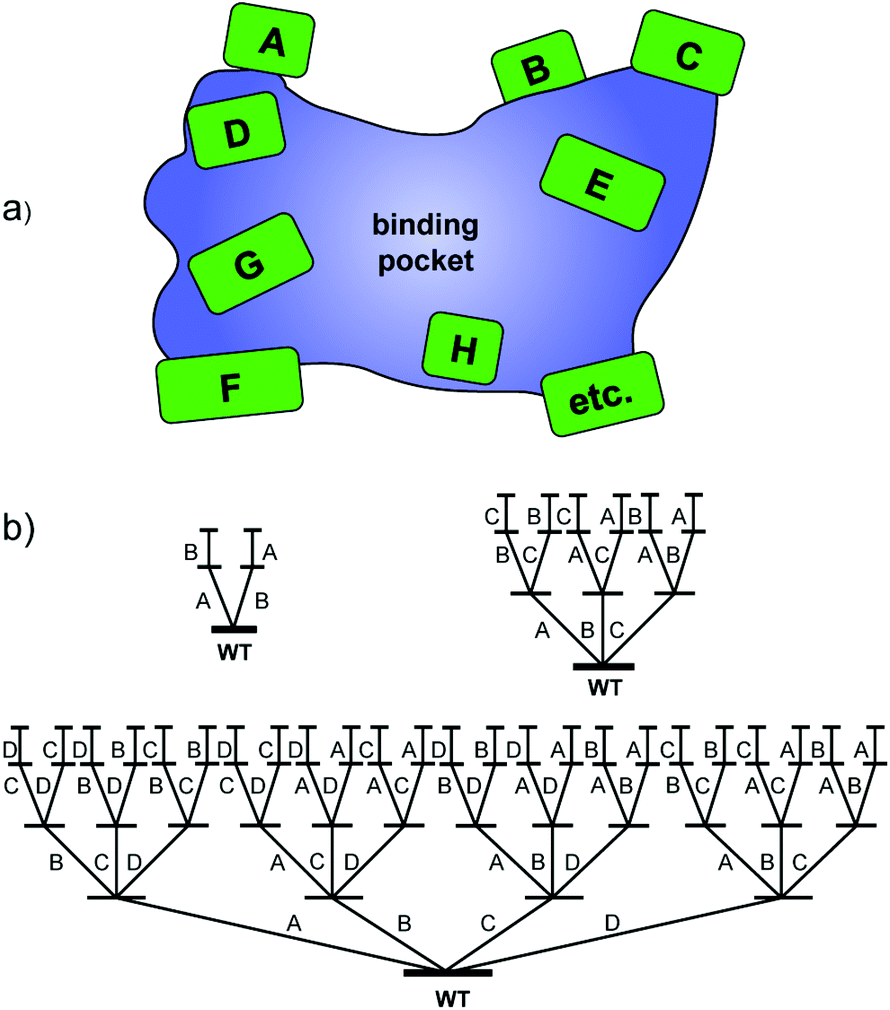 | ||
| Scheme 2 (a) CAST sites lining the binding pocket of an enzyme; (b) ISM schemes for A–B, A–B–C and A–B–C–D systems involving 2, 6 and 24 evolutionary pathways, respectively. | ||
ISM is a stochastic process which needs to be analyzed mathematically. The purpose is to provide the experimenter with the degree of oversampling necessary to ensure a given degree of library coverage, e.g., 95%, as a function of the size of a randomization site.8b,c This has been implemented with the development of the computer aid CASTER,8b which is based on the Patrick/Firth algorithm.10 The Nov metric is an alternative which focuses on the nth best mutant.11 When employing the so-called NNK codon degeneracy encoding all 20 canonical amino acids as building blocks in the combinatorial randomization of sites larger than three amino acid positions, an astronomically high number of transformants (bacterial colonies as shown in Scheme 1) need to be screened for 95% library coverage.2a,7b,8b,c Therefore, reduced amino acid alphabets were introduced in the CASTing process,7b beginning with the NDT codon degeneracy encoding 12 amino acids. Recently the extreme case of a single amino acid (valine) as the building block in a 10-residue randomization site of an epoxide hydrolase was described, resulting in the introduction of 3–4 valines lining the binding pocket and the generation of several R- and S-selective mutants as catalysts in the hydrolytic desymmetrization of cyclohexene oxide.12 In further methodology development, it was subsequently demonstrated that triple code saturation mutagenesis (TCSM) based on the use of three amino acids as building blocks is even more efficient, requiring minimal screening (often less than 1000 transformants).13
In addition to the practical results of directed evolution projects, different types of lessons can be learned, depending upon the degree of extra work that the experimenter is willing to invest. The following aspects are illustrated in this essay:
• Drawing sound mechanistic conclusions regarding the source of enhanced or inverted enantioselectivity on the molecular level when flanked by MD/docking computations and X-ray data.
• Studying the interaction of two or more point mutations with regard to additive or non-additive effects.
• Constructing evolutionary fitness landscapes which reveal the existence or absence of local minima.
Exploring the source of altered stereoselectivity
When aiming to define an enzyme's mechanism in greatest detail, stereoselectivity data are crucial. In the case of high enantioselectivity of the wild type (WT) in a model reaction involving the natural or unnatural substrate, sound mechanistic interpretations can be postulated if X-ray structural data are available. Mechanistic intricacies are also uncovered if a pronounced increase or reversal of stereo- and/or regioselectivity of an unselective enzyme has been achieved by directed evolution. Then a detailed analysis requires MD/docking computations, ideally flanked by X-ray structure determination(s), kinetic characterization and inhibition experiments. The majority of the current and past directed evolution studies aimed at manipulating stereo- and/or regioselectivity, activity or substrate scope does not include X-ray structures of the mutants, and QM calculations are rarely reported.An informative study which includes the crystal structure of a stereoselective variant generated earlier by directed evolution concerns the epoxide hydrolase from Aspergillus niger (ANEH) as the catalyst in the hydrolytic kinetic resolution of rac-1 with preferential formation of (S)-2 (Scheme 3).14 QM computations were not reported, but the study nevertheless provides several novel mechanistic insights. WT ANEH shows poor (S)-selectivity (E = 4.6). In the initial investigation,8a six CAST sites were chosen for ISM: A (comprising amino acid positions 193/195/196), B (215/217/219), C (329/330), D (349/350), E (317/318), and F (244/245/249). Pathway WT → B → C → D → F → E was arbitrarily chosen, leading to the best variant LW202 at the time with a selectivity factor of E = 115 in favor of (S)-1. A further upward climb by visiting site A was not attempted. The mutant has 9 point mutations L215F/A217N/R219S/L249Y/T317W/T318V/M329P/L330Y/C350V, which accumulated along the ISM pathway WT → B (variant LW081) → C(LW086) → D(LW123) → F(LW44) → E(LW202).8a
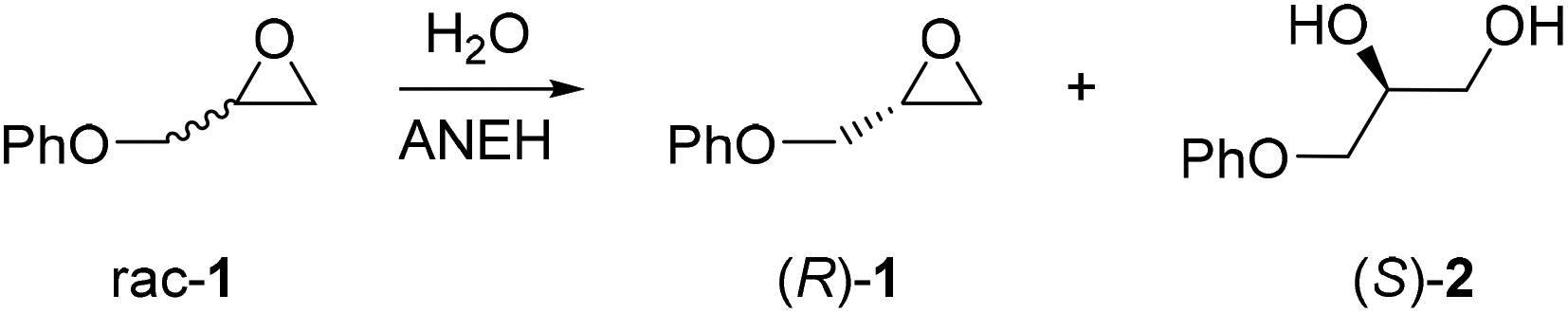 | ||
| Scheme 3 Hydrolytic kinetic resolution of rac-1 catalyzed by the epoxide hydrolase ANEH. | ||
The crystal structure of WT ANEH had already been determined, followed by a proposal of the gross features of the mechanism.15 Accordingly, the substrate is bound and activated by H-bonds to the epoxide O-atom originating from Tyr251 and Tyr314. Then catalytically active Asp192 induces an SN2 reaction in the rate determining initial step followed by fast hydrolysis of the short-lived acyl-enzyme intermediate (Scheme 4).15
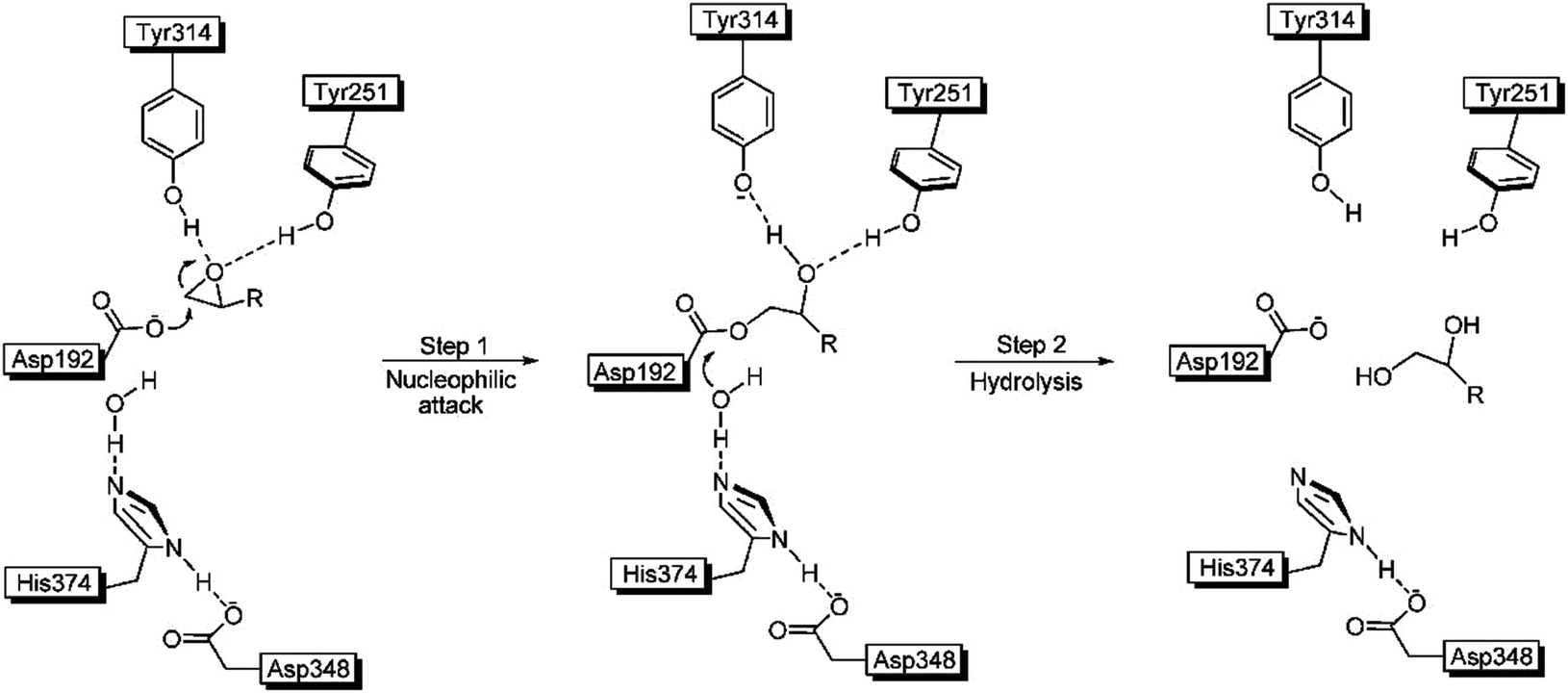 | ||
| Scheme 4 Mechanism of ANEH as the catalyst in hydrolytic ring-opening of epoxides.15 | ||
In an attempt to unravel the source of enhanced stereoselectivity of the best variant LW202, Michaelis–Menten kinetics were determined using in separate experiments enantiomerically pure (R)- and (S)-1, respectively.14 A nearly ideal behavior of a kinetic resolution was uncovered, since the reaction of the disfavored (R)-enantiomer is essentially shut down by the mutational influence (Fig. 1). Moreover, the data allowed a more exact determination of the selectivity factor, which is even higher (E = 195) than the original estimation based on the standard Sih-equation. The relative values of kcat/Km for the two enantiomers also reflect pronounced efficiency in (S)-selectivity.14 In order to identify the factors which lead to enhanced (S)-enantioselectivity at every stage of the 5-step ISM process in the directed evolution study, WT → LW081 → LW086 → LW123 → LW44 → LW202, extensive molecular dynamics (MD) simulations were carried out using (R)- and (S)-1 as substrates separately.14 The distance, d, between the attacking O-atom of Asp192 and the epoxide C-atom undergoing SN2 reaction was chosen as the crucial parameter (Fig. 2). It was postulated that a sufficiently small d-value would correspond fairly well to a near-attack pose,14 an assumption proposed earlier for many enzyme-catalyzed reactions.16 This means that productive binding can be expected if d is in the range of ≈3.5 Å. Large values should characterize the reaction of the disfavored enantiomer (R)-1. For the two enantiomeric substrates a close correlation (R2 = 0.86) was observed between the experimental E-values and the differences in the computed distance, ΔdR–S (Table 1).14 Strikingly, this difference increases as the evolutionary process proceeds. In the final variant LW202, dR amounts to 5.4 Å, too long for the disfavored substrate (R)-1 to undergo a smooth reaction, in full agreement with the kinetics (Fig. 2).14 It means that LW202 binds (R)-1 in an unproductive mode which essentially shuts down the reaction, very different from activated and perfectly positioned (S)-1. In contrast, both enantiomers take on productive poses in WT ANEH, leading to the observed poor enantioselectivity. The reasons for the different binding modes in LW202 were identified by MD and docking computations. The disfavored substrate (R)-1 is bound in a pose in which the C-atom of the epoxide undergoing SN2 reaction is pointing away from the nucleophilic Asp192. Moreover, differences in the dynamic properties of side-chain conformers (flexibility) contribute to differences in binding modes, as shown by the MD calculations.
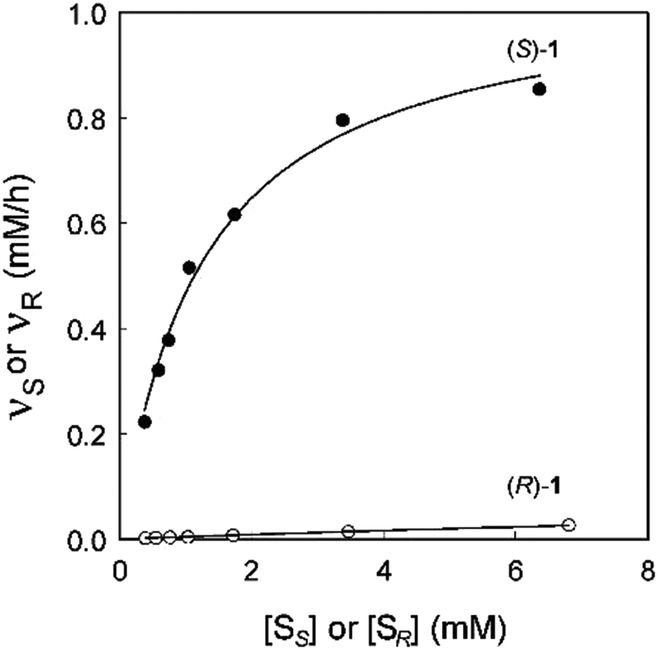 | ||
| Fig. 1 Michaelis–Menten kinetics of mutant LW202 as the catalyst in separate reactions of (R)- and (S)-1, where vR and vS are the initial rates of hydrolysis of (R)- and (S)-1 at different substrate concentrations [SR] or [SS].14 | ||
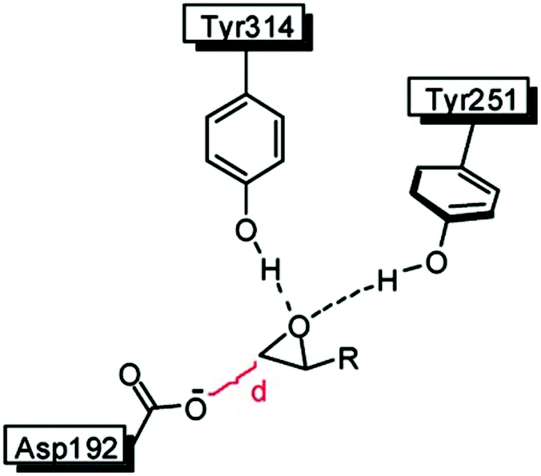 | ||
| Fig. 2 Definition of the distance d in the rate- and stereoselectivity-determining step of the ANEH-catalyzed reaction of rac-1.14 | ||
| Mutant | d R | d S | ΔdR–S | E (expl) |
|---|---|---|---|---|
| WT | 4.3 | 3.5 | 0.8 | 4.6 |
| LW081 | 4.8 | 4.0 | 0.8 | 14 |
| LW086 | 4.9 | 4.0 | 0.9 | 21 |
| LW123 | 5.1 | 4.0 | 1.1 | 24 |
| LW44 | 5.1 | 3.9 | 1.2 | 35 |
| LW202 | 5.4 | 3.8 | 1.6 | 115 |
This study includes the determination of two crystal structures: WT ANEH harboring the inhibitor valpromide (2-propyl-pentanoic acid amide) and apo (unbound) variant LW2002.14 Apo WT ANEH was also compared. The gross features of all structures are almost identical (essentially the same fold). Moreover, the positions of the amino acids participating in the catalytic machinery have not been perturbed. However, dramatic differences in the shape of the binding pocket of mutant LW202 relative to apo or bound WT became visible. The structures were employed in the manual docking of the favored compound (S)-1 and disfavored (R)-1 in the respective binding pockets so that from a purely geometric viewpoint smooth attack by nucleophilic Asp192 should occur (Fig. 3).14 It was found that both enantiomers, preferred (S)-1 and disfavored (R)-1, fit well into the WT ANEH in a way that avoids any steric clashes while maintaining activation by Tyr251/Tyr314 as well as optimal positioning for nucleophilic attack by Asp192 (Fig. 3a and b). This explains the low enantioselectivity as well as the kinetic results. The same applies to the favored (S)-substrate bound in the (S)-selective mutant LW202 (Fig. 3c). In sharp contrast, the disfavored (R)-enantiomer does not fit optimally into mutant LW202 because in this “forced” pose severe steric clashes occur between the substrate and the sidechains of mutated residues at sites B and E (Fig. 3d). Consequently, productive binding is hardly possible. A sterically less demanding pose is possible, but then the H-bond based activating effect of Tyr251/Tyr314 is prevented. Finally, inhibition experiments proved to be in accord with this model.14
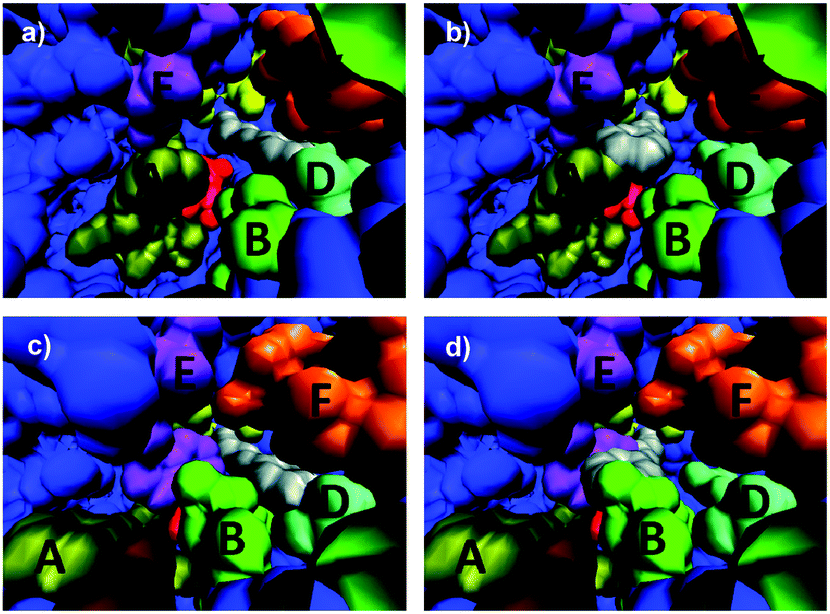 | ||
| Fig. 3 Interpretation of crystal structures of WT ANEH and of evolved (S)-selective mutant LW202 by manually docking (R)- and (S)-1 into the respective binding pockets.14 A, B, C, D, E and F represent the originally designed randomization sites in the ISM process. (a) Favored (S)-1 in the WT ANEH binding pocket; (b) disfavored (R)-1 in the binding pocket of WT ANEH; (c) favored (S)-1 in variant LW202; (d) disfavored (R)-1 in mutant LW202. | ||
Upon close inspection of the docked substrates in the binding pockets (Fig. 3), it becomes clear that the angle of attack is not likely to be 180° as in traditional trajectories Nu–C–X (Nu = nucleophile; X = leaving group). Rather, it should be smaller. It has been shown that in (non-enzymatic) SN2 reactions of epoxides the situation is different from reactions such as CH3I undergoing nucleophilic substitution.17a Several QM calculations for certain epoxides and nucleophiles suggest trajectories of 105–114°.17b,c This computational result is in accord with a QM/MM study of limonene epoxide hydrolase (LEH) in which activated water functions as the nucleophile.18 It is also in accord with MD computations of an evolved LEH mutant.
In a more recent directed evolution study of another epoxide hydrolase, crystal structures of several mutants provided valuable structural and mechanistic information for interpreting enhanced and reversed enantioselectivity.12a It concerns limonene epoxide hydrolase (LEH), which reacts by a different mechanism. In this case the epoxide is bound also by H-bonds, but activated water is the nucleophile, this machinery also requiring perfect substrate positioning for smooth reactions. Hydrolytic desymmetrization of cyclohexene oxide was used as the model reaction, and saturation mutagenesis using a single amino acid as a building block at a 10-residue CAST site served as the directed evolution technique. Both (R,R)- and (S,S)-selective mutants were evolved. Several X-ray structures of the respective apo and product-bound forms supported by MD/docking computations led to insightful models for explaining the origin of enhanced and inverted enantioselectivity.12a
In conclusion, studies of this type flanked by crystal structures of evolved mutants not only explain the reasons for enhanced enantioselectivity on the molecular level but also contribute to a deeper understanding of the mechanistic intricacies of enzymes. Several other case studies have been published which include X-ray structures of mutants.12 QM investigations in such cases would provide even more insight.
Additive versus non-additive mutational effects
Long before the advent of directed evolution,1 enzymologists exploited site-specific mutagenesis in order to study the mechanism of enzymes.19 In some studies two mutations, rationally chosen for mechanistic purposes, were combined with the formation of the respective double mutant. In many early cases it was found that the effect on activity was additive, but exceptions were noted.19 A commonly practiced technique in directed evolution is to combine separately evolved point mutations in order to optimize catalytic properties such as stereoselectivity.1 Unfortunately, this strategy does not always work for reasons that are not well understood.20 However, upon going the reverse way by deconvoluting multi-mutational mutants, important insight has been gained.21Using enantioselectivity as the catalytic parameter, additive and non-additive mutational effects as revealed by deconvolution experiments can be systematized as illustrated in Scheme 5. Non-additive mutational effects may be cooperative (more than conventional additivity), or they can prove to be deleterious (less than additive). Scheme 5 shows the case of an initial set of mutations A followed by a second set of mutations B. Since this is an accumulation process in a hypothetical directed evolution experiment, the catalytic action of B alone is not known at this point. Deconvolution of the mutant with generation of B alone provides this information. In the simplest case a mutational set comprises a single point mutation, but in practice several point mutations may also be involved. Classical additivity may be unveiled by deconvolution, in which case mutational set A does not interact with mutational set B (Scheme 5a). This means that both mutational sets favor the same direction of enantioselectivity, e.g., (R), but are independent of each other. Several types of non-additivity are theoretically possible. For example, deconvolution may reveal that the contribution of B is less than expected, but the sense of enantioselectivity is the same as exerted by A (Scheme 5b). This indicates a cooperative effect caused by interaction (more than additivity), and is a highly desirable feature. Another type of non-additivity results whenever mutational set B favors the opposite sense of enantioselectivity relative to A (Scheme 5c). This is a deleterious effect, unless complete reversal of enantioselectivity is strived for.21
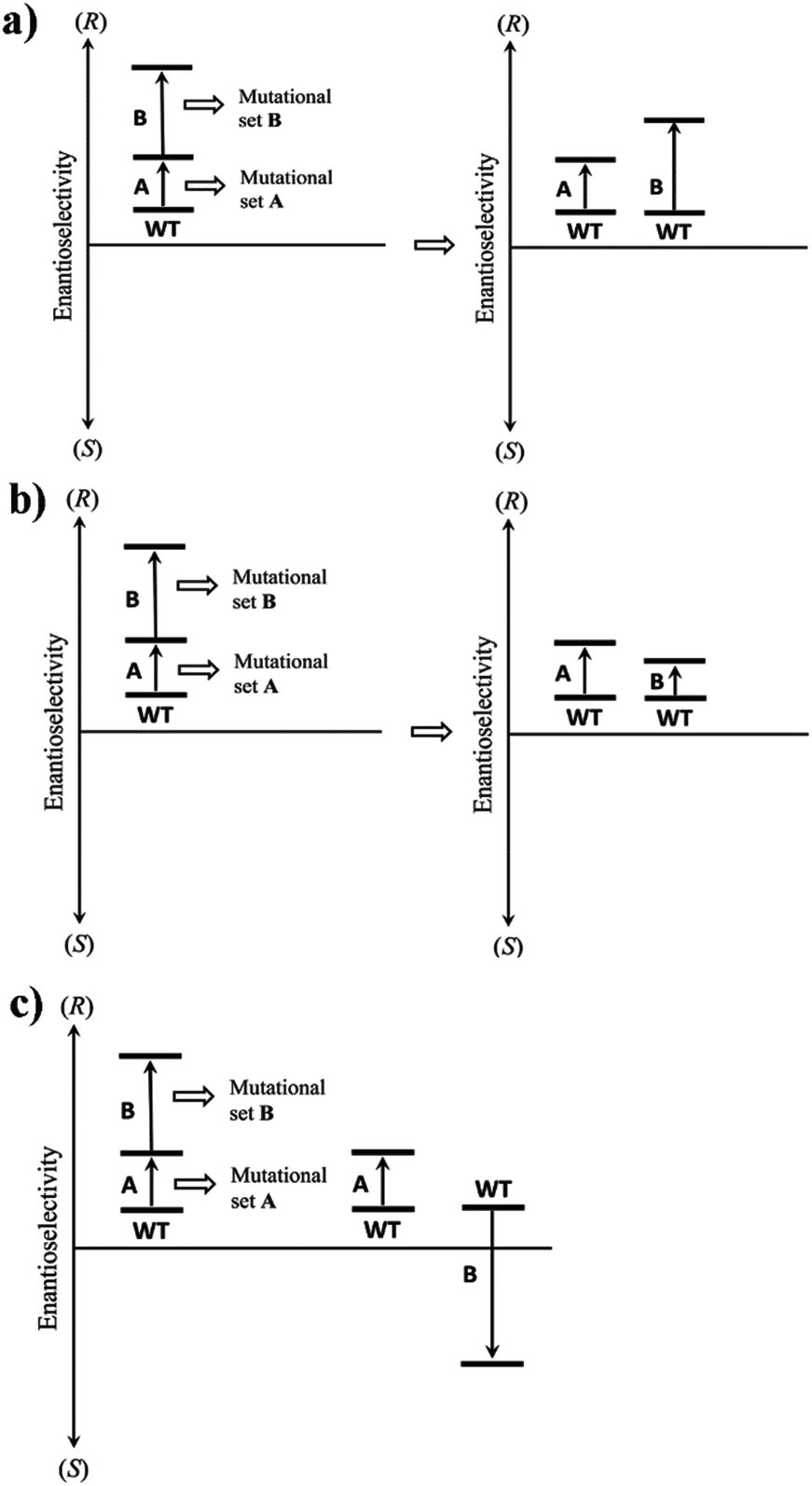 | ||
| Scheme 5 Systematization of additive and non-additive mutational effects in directed evolution with enantioselectivity being the catalytic parameter, as illustrated here using two sets of mutations A and B. (a) Conventional additive mutational effect; (b) non-additive mutational effect with B showing lower than expected enantioselectivity in the same direction; (c) non-additive effect in which mutational set B alone induces reversed enantioselectivity. | ||
The situation becomes more complex when a given mutational set comprises more than one point mutation. In such cases the interaction of the sets A and B (or more sets C, D, E, etc.) can be ascertained by a limited number of deconvolution experiments. However, additivity versus non-additivity also pertains to the point mutations within a set. Thus, complete deconvolution provides maximal information, but may also require formidable lab work, which is the reason why such studies are rare.21–23 The lessons learned from a number of these investigations include the realization that when applying CAST-based iterative saturation mutagenesis (ISM), all three types of effects as shown in Scheme 5 may result. Thus far the highly desirable cooperative case occurs most often, which sheds light on the efficacy of ISM.
A case in point concerns the hydrolytic kinetic resolution of rac-3 catalyzed by the lipase from Pseudomonas aeruginosa (PAL) as showed in Scheme 6.24 As already pointed out, this transformation served as the model reaction in the first case of directed evolution of stereoselectivity,3 and has been used in numerous follow-up studies with the aim of developing more efficient, reliable and fast directed evolution strategies.5,24
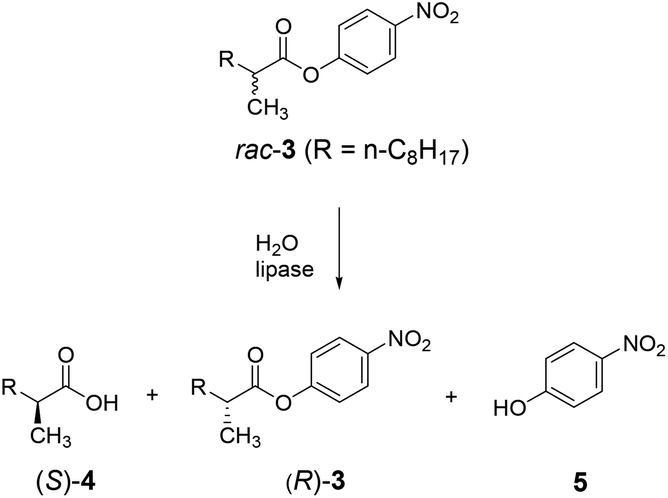 | ||
| Scheme 6 Hydrolytic kinetic resolution of rac-3 with preferential formation of (S)-4, catalyzed by mutants of the lipase from Pseudomonas aeruginosa (PAL) which were evolved by iterative saturation mutagenesis (ISM). | ||
The 3-site ISM scheme composed of CAST sites A (Met16/Leu17), B (Leu159/Leu162) and C (Leu231/Val232) was designed.24 It was discovered early in the project with minimal screening that pathway WT → B → A leads to a triple mutant 1B2 (Leu162Asn/Met16Ala/Leu17Phe) showing a selectivity factor of E = 594 in favor of (S)-4 (Scheme 7). Since this dramatic degree of catalyst improvement was unprecedented, further genetic optimization by visiting site C was not necessary. Moreover, the reaction rate of the preferred enantiomer (S)-3 and therefore of the overall kinetic resolution was increased significantly: WT PAL (kcat = 37 × 10−3 s−1; kcat/Km = 43.5 s−1 M−1) versus variant 1B2 (kcat = 1374 × 10−3 s−1; kcat/Km = 4041 s−1 M−1). Higher activity clearly correlates with higher enantioselectivity. Thus, the ISM strategy is clearly more efficient and productive than the previous best approach based on a combination of error-prone PCR, DNA shuffling and limited saturation mutagenesis (E = 51),25 which required the screening of 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 transformants.
000 transformants.
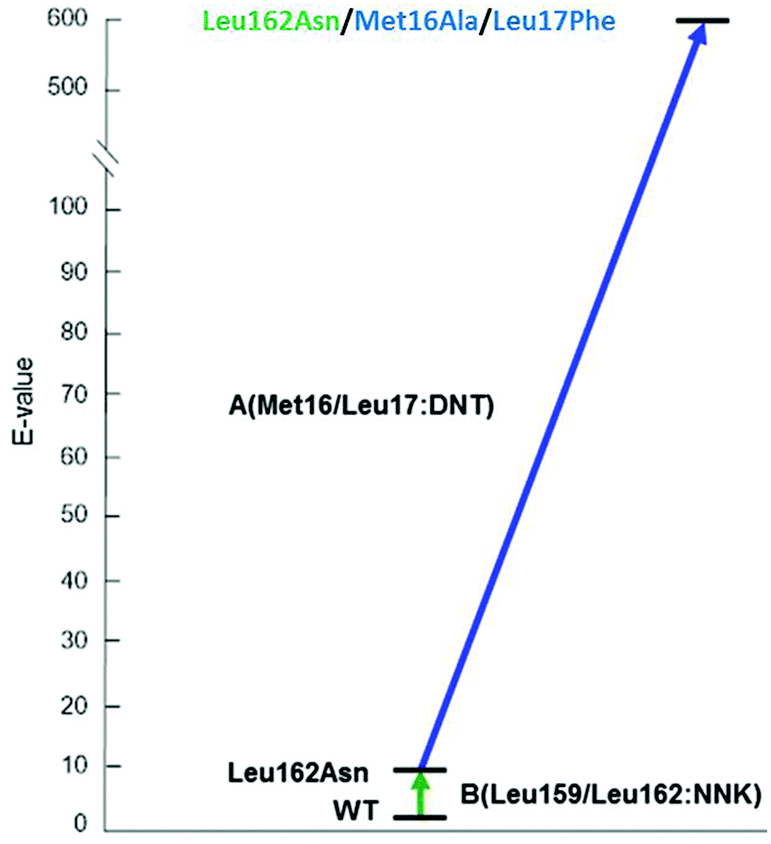 | ||
| Scheme 7 Optimal ISM pathway WT → B → A providing the triple mutant 1B2 (Leu162Asn/Met16Ala/Leu17Phe) with a selectivity factor of E = 594 (S) in the hydrolytic kinetic resolution of rac-3.24 | ||
A superficial glance at Scheme 7 might suggest that the second set of mutations that accumulated upon randomizing site A, namely Met16Ala/Leu17Phe, is mainly responsible for the high enantioselectivity. However, such a conclusion assumes additivity.21 In order to shed light on this issue, deconvolution was performed by preparing and testing the double mutant Met16Ala/Leu17Phe.24 It alone leads to a selectivity factor of only E = 2.6 (S), which corresponds to very low stereoselectivity. Assuming additivity, the selectivity factor should be a mere E = ≈22, not E = 594. Therefore, a significant cooperative non-additive effect is involved. This synergy correlates with an energy contribution of ≈2 kcal mol−1. Relative to WT PAL, the difference in stabilization energy of the two enantiomeric reactions amounts to about 3 kcal mol−1, which is formidable in an asymmetric transformation. Further deconvolution by generating separately the two single mutants Met16Ala and Leu17Phe was not reported.
The deconvolution study throws light on the reason why ISM is so effective, certainly in this particular case. A second lesson that was learned concerns the challenging question why the huge cooperative effect is occurring. In order to understand the effect on a molecular level, the known mechanism of PAL-catalyzed hydrolysis must be considered.26 It is characterized by the catalytic triad Asp229/His251/Ser82, which ensures rate- and stereoselectivity-determining nucleophilic addition of activated Ser82 to the carbonyl function of esters with the formation of short-lived oxyanion intermediates, followed by rapid product formation, a typical lipase mechanism (Scheme 8).
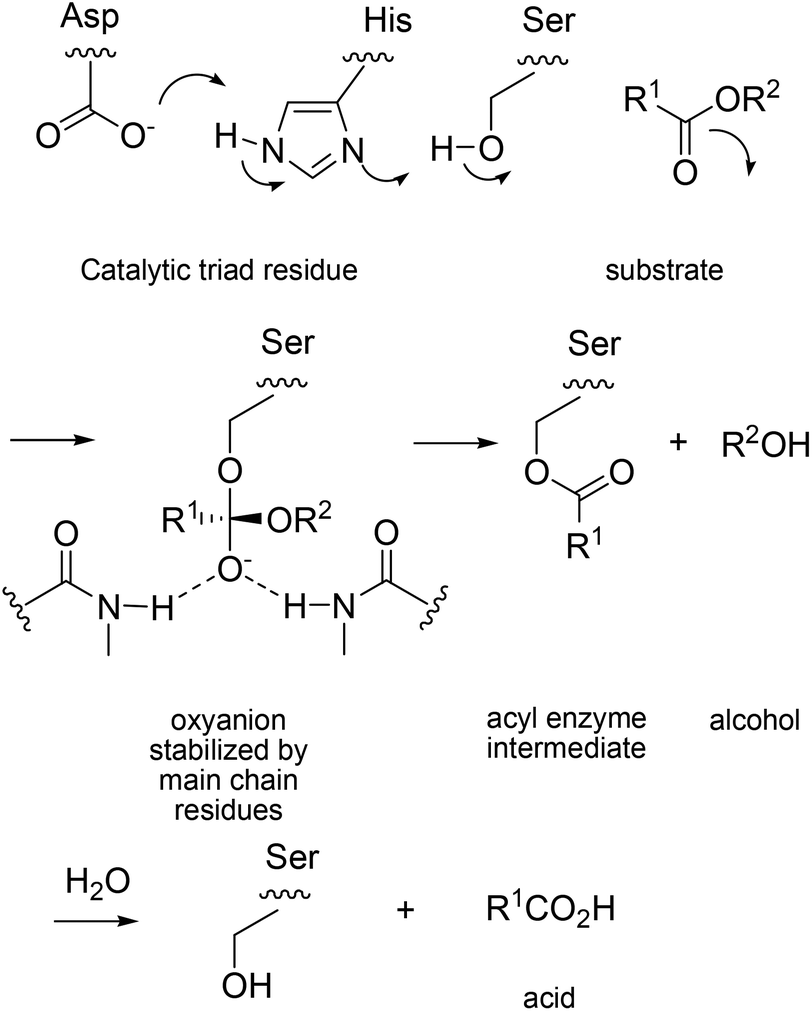 | ||
| Scheme 8 Mechanism of PAL-catalyzed hydrolysis.26 | ||
Using the crystal structure of WT PAL as the starting point,27 MD and induced fit docking computations were performed with the three point mutations of the best mutant 1B2 then being introduced by a docking program.24 Thereafter substrates (R)- and (S)-3 were introduced separately in the PAL binding pocket as the respective oxyanions covalently bound to Ser82. Fig. 4 shows the case of the favored (S)-substrate bound in WT PAL and in mutant 1B2. It can be seen that in the case of WT PAL, the n-octyl residue of (S)-3 clashes sterically with Leu162, whereas in mutant 1B2 the Leu162Asn exchange provides space for easy accommodation of this part of the substrate, while mutation Met16Ala provides space for His83 to result in additional stabilization of the oxyanion by H-bond formation (the normal stabilizing interactions have been deleted in Fig. 4 for clarity). Finally, the Leu17Phe mutation results in stabilizing π-stacking between phenylalanine and the p-nitrophenyl moiety of the ester substrate. Notice the extended H-bond network involving Asn162, formerly innocent Ser158, His83 and the oxyanion. Thus, it was postulated that the residues undergo a kind of communication in the case of disfavored (R)-3, because the methyl group at the stereogenic center would prevent stabilization by His83 due to steric clashes.
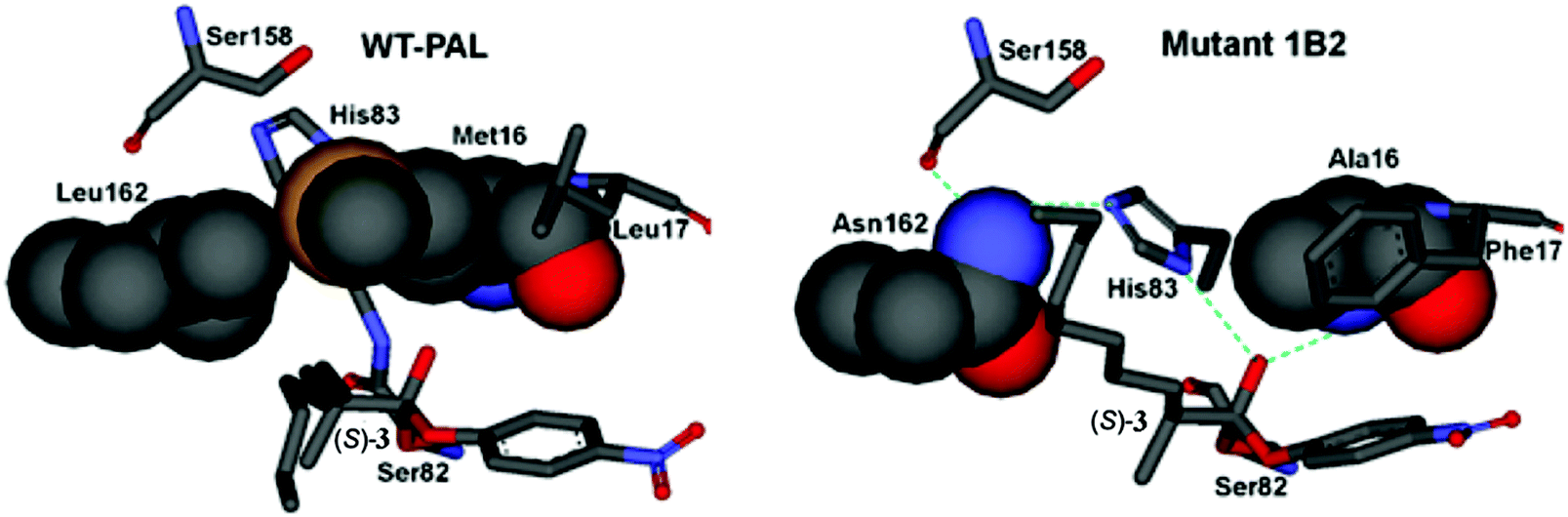 | ||
| Fig. 4 Comparison of the oxyanions with bound (S)-3 at the catalytically active Ser82 of WT PAL (left) and the best variant 1B2 (right).2 | ||
Constructing evolutionary fitness landscapes
Fitness landscapes stand at the heart of directed evolution, yet only a limited number have been constructed experimentally, which results in the cooperative effect as reflected in the rate and enantioselectivity enhancement.24 This effect is not possible in a systematic manner using empirically derived free energy values.1 When focusing on ISM, two different approaches are possible: (1) an unconstrained system in which all pathways of a designed ISM scheme are explored experimentally;28 (2) a constrained system in which all theoretically possible pathways leading from WT to a given mutant evolved earlier.22,29 In the former case, only one example has been reported thus far, in which all 24 pathways of a 4-site ISM system using an epoxide hydrolase were transversed28 (see Scheme 2b). In contrast, several cases of constrained systems have been published which focus on enantioselectivity as the crucial parameter.21,22,29 It should be mentioned that constrained systems have also been employed in the area of evolutionary biology by exploring the number of pathways leading from WT to fitter β-lactamases in antibiotic resistance.30An interesting case of a constrained system involves the directed evolution of the Baeyer–Villiger monooxygenase PAMO as the catalyst in the asymmetric sulfoxidation of methyl tolyl thio-ether (6) (Scheme 9).22 WT PAMO favors the formation of (S)-7 with 90% ee. Therefore, the goal was complete reversal of stereoselectivity with evolution of an R-selective PAMO mutant. This was achieved by ISM using a 2-site system comprising sites A and B, each composed of two residues. In two ISM steps a highly R-selective mutant ZGZ-2 (I67Q/P440F/A442N/L443I) was evolved displaying 95% ee.
 | ||
| Scheme 9 Asymmetric sulfoxidation catalyzed by mutants of the Baeyer–Villiger monooxygenase PAMO.22 | ||
Using these data, a constrained fitness landscape was constructed. Complete deconvolution dissects a multi-mutational mutant into the respective single mutants, which can be tested in the reaction of interest individually. Moreover, the generation of all theoretically possible combinations of point mutations (double, triple mutants, etc.) was implemented. In the present case these were likewise prepared by site-specific mutagenesis and used as catalysts in the asymmetric sulfoxidation of 6. Consequently, such data allow the construction of a complete fitness pathway landscape which features the mapping of all theoretically possible pathways (4! = 24) leading from WT to the final best mutant ZGZ-2 (I67Q/P440F/A442N/L443I) (Fig. 5).22
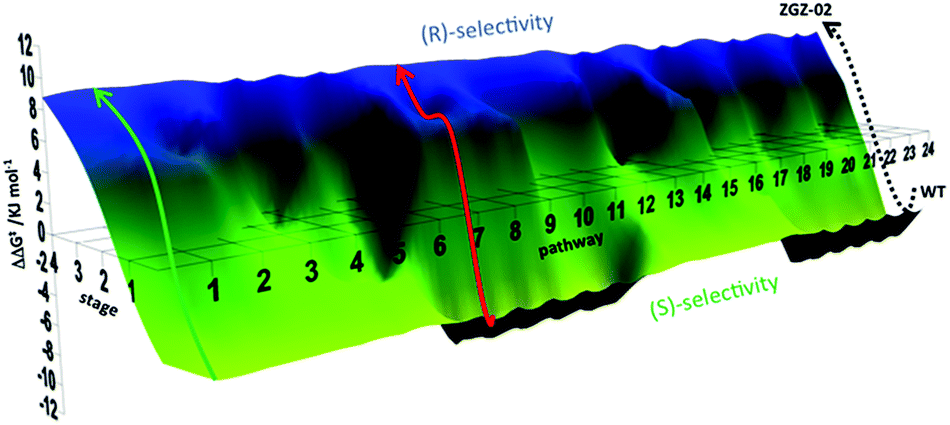 | ||
| Fig. 5 Fitness landscape featuring the 24 pathways leading from WT PAMO (bottom) to the best (R)-selective variant ZGZ-02 characterized by four point mutations I67Q/P440F/A442N/L443I. A typical trajectory lacking local minima is the green pathway, and one having a local minimum is the red pathway.22 | ||
By necessity all 24 pathways end up with the evolved quadruple mutant showing reversed enantioselectivity, but the topologies of the upward climbs are all different. Out of the 24 pathways, 18 have local minima, and 6 go smoothly to the endpoint. The data allow the analysis of all steps along each of the 24 trajectories. For illustrative purposes one of them is featured here, namely one of the “favored” pathways lacking local minima (Fig. 6). Here as in the other 23 pathways extreme cooperative non-additive effects occur. For example, each of the four single mutants corresponding to the evolved R-selective mutant I67Q/P440F/A442N/L443I is actually S-selective!22 If these four single mutants had been generated by some other means, traditional thinking would not lead to the conclusion that combining them would induce complete reversal of enantioselectivity, an important lesson.
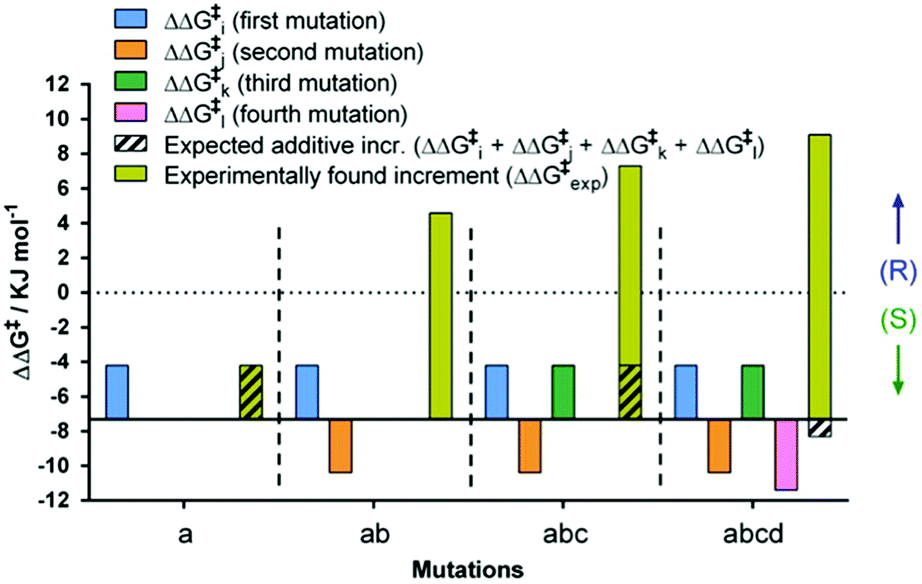 | ||
| Fig. 6 Mutational deconvolution along a favored pathway from WT to the final variant ZGZ-2 having reversed enantioselectivity in the sulfoxidation of 6. | ||
Conclusions and perspectives
This short overview featuring lessons that can be learned from directed evolution is not meant to be comprehensive. Rather, on the basis of selected examples it outlines the different types of insights that can be generated by investing additional experimental work when performing this type of protein engineering. Intricacies of enzyme mechanisms can be uncovered, while also exploring the reasons for enhanced efficacy of a given mutagenesis strategy. Finally, complete deconvolution of multi-mutational mutants allow the construction of fitness landscapes which reveal additive and non-additive mutational effects as well as ways to escape from local minima. It is hoped that future research in directed evolution along these lines will reveal further novel insights.Acknowledgements
Support from the Max-Planck-Society and the LOEWE Research Cluster SynChemBio is gratefully acknowledged.Notes and references
- Recent reviews on directed evolution: (a) A. S. Bommarius, Annu. Rev. Chem. Biomol. Eng., 2015, 6, 319–345 CrossRef CAS PubMed; (b) A. Currin, N. Swainston, P. J. Day and D. B. Kell, Chem. Soc. Rev., 2015, 44, 1172–1239 RSC; (c) C. A. Denard, H. Ren and H. Zhao, Curr. Opin. Chem. Biol., 2015, 25, 55–64 CrossRef CAS PubMed; (d) E. M. J. Gillam, J. N. Copp and D. F. Ackerley, in Methods in Molecular Biology, Humana Press, Totowa, NJ, 2014, vol. 1179 Search PubMed; (e) M. Goldsmith and D. S. Tawfik, Methods Enzymol., 2013, 523, 257–283 CAS; (f) E. M. Brustad and F. H. Arnold, Curr. Opin. Chem. Biol., 2011, 15, 201–210 CrossRef CAS PubMed; (g) M. T. Reetz, Angew. Chem., Int. Ed., 2011, 50, 138–174 CrossRef CAS PubMed; (h) C. Jäckel and D. Hilvert, Curr. Opin. Biotechnol., 2010, 21, 753–759 CrossRef PubMed; (i) N. J. Turner, Nat. Chem. Biol., 2009, 5, 568–574 CrossRef PubMed; (j) S. Lutz and U. T. Bornscheuer, Protein Engineering Handbook, Wiley-VCH, Weinheim, 2009 Search PubMed.
- Reviews on directed evolution of stereoselective enzymes: (a) M. T. Reetz, Angew. Chem., Int. Ed., 2011, 50, 138–174 CrossRef CAS PubMed; (b) M. T. Reetz, J. Am. Chem. Soc., 2013, 135, 12480–12496 CrossRef CAS PubMed.
- M. T. Reetz, A. Zonta, K. Schimossek, K. E. Jaeger and K. Liebeton, Angew. Chem., Int. Ed. Engl., 1997, 36, 2830–2832 CrossRef CAS.
- (a) M. T. Reetz, Select Protocols of High-Throughput ee-Screening Systems for Assaying Enantioselective Enzymes, in Methods in Molecular Biology, Humana Press, Totowa, New Jersey, 2003, vol. 230, pp. 283–290 Search PubMed; (b) J.-L. Reymond, Enzyme assays, John Wiley & Sons, 2006 Search PubMed.
- M. T. Reetz, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5716–5722 CrossRef CAS PubMed.
- M. T. Reetz, M. H. Becker, H. W. Klein and D. Stöckigt, Angew. Chem., Int. Ed., 1999, 38, 1758–1761 CrossRef CAS.
- (a) S. Lutz and W. M. Patrick, Curr. Opin. Biotechnol., 2004, 15, 291–297 CrossRef CAS PubMed; (b) M. T. Reetz, Adv. Catal., 2006, 49, 1–69 CAS.
- (a) M. T. Reetz, L. W. Wang and M. Bocola, Angew. Chem., Int. Ed., 2006, 45, 1236–1241 CrossRef CAS PubMed; (b) M. T. Reetz and J. D. Carballeira, Nat. Protocols, 2007, 2, 891–903 CrossRef CAS PubMed; (c) C. G. Acevedo-Rocha, S. Hoebenreich and M. T. Reetz, Directed Evolution Library Creation: Methods and Protocols, 2014, pp. 103–128 Search PubMed.
- M. T. Reetz, M. Bocola, J. D. Carballeira, D. Zha and A. Vogel, Angew. Chem., Int. Ed., 2005, 44, 4192–4196 CrossRef CAS PubMed.
- W. M. Patrick and A. E. Firth, Biomol. Eng., 2005, 22, 105–112 CrossRef CAS PubMed.
- Y. Nov, Appl. Environ. Microbiol., 2012, 78, 258–262 CrossRef CAS PubMed.
- (a) Z. Sun, R. Lonsdale, X. D. Kong, J. H. Xu, J. Zhou and M. T. Reetz, Angew. Chem., Int. Ed., 2015, 54, 12410–12415 CrossRef CAS PubMed; (b) G. Li, H. Zhang, Z. Sun, X. Liu and M. T. Reetz, ACS Catal., 2016, 6, 3679–3687 CrossRef CAS.
- (a) Z. Sun, R. Lonsdale, L. Wu, G. Li, A. Li, J. Wang, J. Zhou and M. T. Reetz, ACS Catal., 2016, 6, 1590–1597 CrossRef CAS; (b) Z. Sun, R. Lonsdale, A. Ilie, G. Li, J. Zhou and M. T. Reetz, ACS Catal., 2016, 6, 1598–1605 CrossRef CAS.
- M. T. Reetz, M. Bocola, L.-W. Wang, J. Sanchis, A. Cronin, M. Arand, J. Zou, A. Archelas, A.-L. Bottalla and A. Naworyta, J. Am. Chem. Soc., 2009, 131, 7334–7343 CrossRef CAS PubMed.
- J. Zou, B. M. Hallberg, T. Bergfors, F. Oesch, M. Arand, S. L. Mowbray and T. A. Jones, Structure, 2000, 8, 111–122 CrossRef CAS PubMed.
- T. C. Bruice, Acc. Chem. Res., 2002, 35, 139–148 CrossRef CAS PubMed.
- (a) G. Stork, L. D. Cama and D. Coulson, J. Am. Chem. Soc., 1974, 96, 5268–5270 CrossRef CAS; (b) J. Na, K. Houk, C. G. Shevlin, K. D. Janda and R. A. Lerner, J. Am. Chem. Soc., 1993, 115, 8453–8454 CrossRef CAS; (c) T. Laitinen, J. Rouvinen and M. Peräkylä, J. Org. Chem., 1998, 63, 8157–8162 CrossRef CAS.
- (a) M. E. Lind and F. Himo, Angew. Chem., Int. Ed., 2013, 52, 4563–4567 CrossRef CAS PubMed; (b) Q. Hou, X. Sheng, J. Wang, Y. Liu and C. Liu, Biochim. Biophys. Acta, Proteins Proteomics, 2012, 1824, 263–268 CrossRef CAS PubMed.
- (a) P. J. Carter, G. Winter, A. J. Wilkinson and A. R. Fersht, Cell, 1984, 38, 835–840 CrossRef CAS PubMed; (b) J. J. A. Wells, D. B. Powers, R. R. Bott, T. P. Graycar and D. A. Estell, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 1219–1223 CrossRef CAS PubMed; (c) J. A. Wells, Biochemistry, 1990, 29, 8509–8517 CrossRef CAS PubMed; (d) Z. Huang, C. R. Wagner and S. J. Benkovic, Biochemistry, 1994, 33, 11576–11585 CrossRef CAS PubMed.
- M. T. Reetz, J. D. Carballeira, J. Peyralans, H. Höbenreich, A. Maichele and A. Vogel, Chem. – Eur. J., 2006, 12, 6031–6038 CrossRef CAS PubMed.
- M. T. Reetz, Angew. Chem., Int. Ed., 2013, 52, 2658–2666 CrossRef CAS PubMed.
- Z.-G. Zhang, R. Lonsdale, J. Sanchis and M. T. Reetz, J. Am. Chem. Soc., 2014, 136, 17262–17272 CrossRef CAS PubMed.
- S. Bartsch, R. Kourist and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2008, 47, 1508–1511 CrossRef PubMed.
- M. T. Reetz, S. Prasad, J. D. Carballeira, Y. Gumulya and M. Bocola, J. Am. Chem. Soc., 2010, 132, 9144–9152 CrossRef CAS PubMed.
- M. T. Reetz, S. Wilensek, D. Zha and K. E. Jaeger, Angew. Chem., Int. Ed., 2001, 40, 3589–3591 CrossRef CAS.
- M. Bocola, N. Otte, K. E. Jaeger, M. T. Reetz and W. Thiel, ChemBioChem, 2004, 5, 214–223 CrossRef CAS PubMed.
- M. Nardini, D. A. Lang, K. Liebeton, K.-E. Jaeger and B. W. Dijkstra, J. Biol. Chem., 2000, 275, 31219–31225 CrossRef CAS PubMed.
- Y. Gumulya, J. Sanchis and M. T. Reetz, ChemBioChem, 2012, 13, 1060–1066 CrossRef CAS PubMed.
- M. T. Reetz and J. Sanchis, ChemBioChem, 2008, 9, 2260–2267 CrossRef CAS PubMed.
- D. M. Weinreich, N. F. Delaney, M. A. DePristo and D. L. Hartl, Science, 2006, 312, 111–114 CrossRef CAS PubMed.
Footnote |
| † Dedicated to Barry M. Trost on the occasion of his 75th birthday with great admiration for his seminal contributions to synthetic organic chemistry. |
| This journal is © the Partner Organisations 2016 |