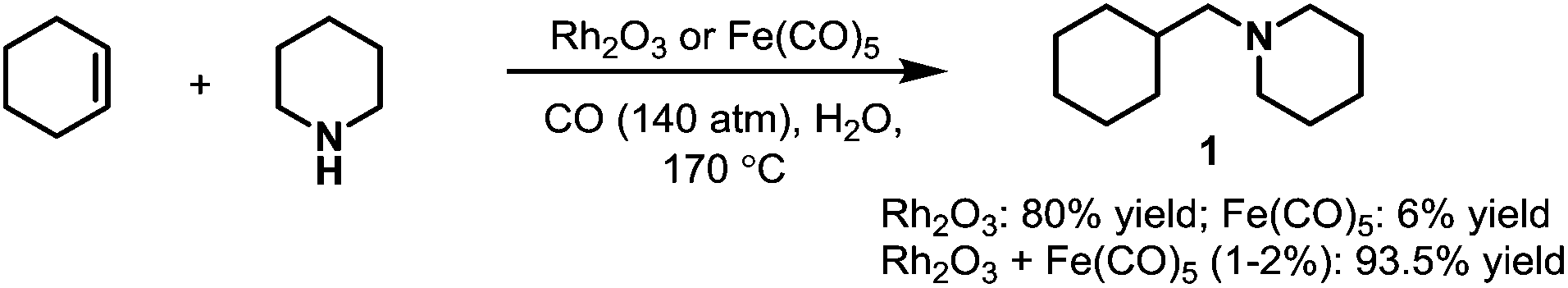DOI:
10.1039/C6QO00233A
(Review Article)
Org. Chem. Front., 2016,
3, 1359-1370
Recent progress in rhodium-catalyzed hydroaminomethylation†
Received
30th May 2016
, Accepted 5th July 2016
First published on 5th July 2016
Abstract
Hydroaminomethylation is a perfect reaction for converting alkenes into valuable amines with high atom economy in the presence of the syngas and amines. Significant progress has been made in the past decades; however, there still remain challenges for the control of chemo- and regioselectivity concurrently. Rhodium has proved to be a better metal in hydroaminomethylation for higher activity in hydroformylation and hydrogenation steps. Although promising results were shown by unmodified rhodium catalysts, phosphine ligand modified rhodium complexes generally displayed better activity and regioselectivity. Among the phosphorus ligands developed, tetraphosphorus ligands exhibited much better regioselectivity due to their stronger chelating ability. Apart from the phosphorus ligands, carbene and nitrogen-containing ligands have also been developed which showed good activity due to the promotion of the hydrogenation step. Although non-enantioselective hydroaminomethylation reactions have been intensively studied, reports on asymmetric hydroaminomethylation are rare. Direct asymmetric hydroaminomethylation is very challenging and only reaction systems with two different catalysts showed promising results.
 Caiyou Chen | Caiyou Chen was born in Hubei Province, China in 1989. He obtained his B.S. degree (2012) from Wuhan University. He joined Prof. Xumu Zhang's group during his fourth year of undergraduate study and started his Ph.D. study in September 2012. His research work is focused on hydroformylation, hydroaminomethylation, asymmetric hydrogenation and nickel-catalyzed copolymerization reactions. |
 Xiu-Qin Dong | Xiu-Qin Dong was born in 1985. She received her B.S. degree from Huanggang Normal College in 2007, and a Ph.D. degree from the Wuhan University under the guidance of Professor Chun-Jiang Wang in 2012. After postdoctoral research (2012–2014) at Hong Kong University of Science and Technology in Prof. Jianwei Sun's group, she joined Wuhan University as an Associate Professor in 2015. Her research interests is mainly focused on asymmetric metal-organo catalysis, hydroformylation, hydrogenation and the development of new synthetic methods for biologically active compounds. |
 Xumu Zhang | Xumu Zhang received his Ph.D. degree from Stanford University under the guidance of Professor James P. Collman in 1992. After two-year postdoctoral work at Stanford University, he joined the department of chemistry at The Penn State University as an Assistant Professor. In 1999, he was promoted to an Associate Professor. 2003–2007, he was appointed as a full professor. In 2007, he moved to The State University of New Jersey as a distinguished Professor. 2011-present, he was appointed as Professor (Thousand Talent Program) in Wuhan University. His research interests include the development of chiral phosphine ligands for asymmetric reactions. |
1. Introduction
Among the most important bulk and fine chemicals in the agrochemical and pharmaceutical industry, amines have been used to give access to biologically active molecules, dyes, solvents and functional materials,1 with a production on a million-ton scale annually.2 Although there exist available methods for the preparation of amines such as hydrocyanation of alkenes followed by reduction, nucleophilic substitution of alkyl halides, reductive amination, hydroamination, etc., amine preparation often suffers from low generality, expensive starting materials, side reactions and the requirement for protecting groups.3 In this regard, a general and selective method for the preparation of amines is of vital importance. From both economical and environmental points of view, hydroaminomethylation meets the requirements of the “perfect reaction” for the preparation of amines, which gives amines from the readily available and cheap olefins in the presence of syngas in a one-pot and environmental benign and atom-efficient way.
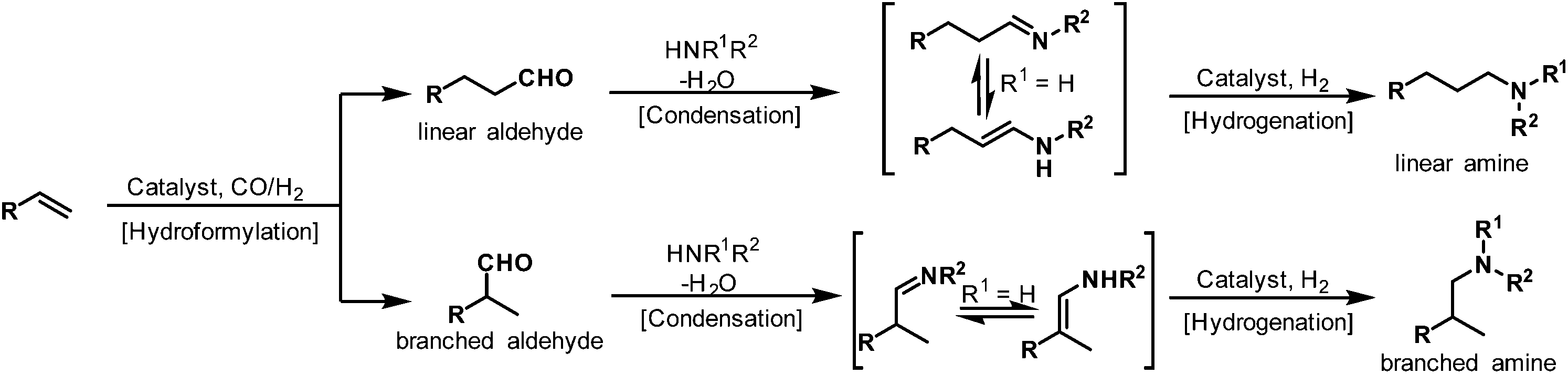
Some terms like cascade, domino, and tandem reaction are often used to describe the hydroaminomethylation reaction.4 The one-pot hydroaminomethylation reaction involves the succession of three steps. In the first step, the alkenes are converted into the linear or branched aldehydes (hydroformylation), which react with the primary or secondary amines present in the reaction medium to give the enamines or imines in the second step (condensation). The last step involves the hydrogenation of the enamine or imine intermediates to give the final amine products (hydrogenation) (Scheme 1).
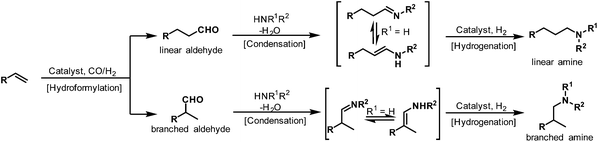 |
| | Scheme 1 Three successive steps of hydroaminomethylation. | |
Although hydroaminomethylation is a perfect reaction for the preparation of amines, there still remain challenges. The main difficulty is that chemoselectivity and regioselectivity should be controlled concurrently to give amines efficiently. As a matter of fact, side reactions are often observed which can occur with the reaction intermediates. For example, the aldehyde intermediate formed in the reaction system can undergo the aldol reaction to give non-desired products. Furthermore, byproducts like alkanes and alcohols can be observed due to the hydrogenation of the alkene substrates and the aldehyde intermediates. Although hydroaminomethylation is a challenging issue, recent years have seen considerable progress.
Hydroaminomethylation was first discovered by Reppe et al. at BASF, who found that acetylenic compounds reacted with carbon monoxide in the presence of ammonia and water, and the amine products were obtained.5 However, this reaction required a stoichiometric amount of Fe(CO)5 as the catalyst and the reaction conditions were harsh (T > 300 °C, pressure up to 150 bar). Owing to the low efficiency of iron catalysts for hydroaminomethylation, efforts have been made to develop new cobalt catalysts and the subsequent studies have proved that cobalt catalysts are superior and that hydroaminomethylation can proceed under catalytic conditions. The first cobalt-catalyzed hydroaminomethylation employed ethylene and ammonia under high pressure and temperature (40–75 MPa, 170–262 °C) to afford propylamine and dipropylamine, and dipropylamine was shown to be the major product.6 It was further found that the reaction can be conducted under relatively lower pressure by using the cobalt precursor Co2(CO)8 modified with phosphine ligands.7 However, low selectivity was observed in the hydroaminomethylation of terminal olefins with ammonia in the presence of syngas. In addition to cobalt catalysts, manganese and nickel carbonyl complexes were also shown to enable the hydroaminomethylation to proceed in a catalytic way under harsh conditions.8 Nowadays, rhodium, ruthenium and iridium precursors modified with certain ligands have been shown to be much better catalysts for hydroaminomethylation under mild reaction conditions, which makes hydroaminomethylation an ideal way for the preparation of amines. Among the metals used in hydroaminomethylation, rhodium was found to be more active and selective in catalyzing both the hydroformylation and the hydrogenation steps. The development of hydroaminomethylation has already been summarized in several reviews.8,9 The main emphasis of this review is to cover the recent studies on the rhodium-catalyzed hydroaminomethylation. Catalysts based on other metals will not be discussed herein. Moreover, the current review will emphasize ligand effects and the development of ligands for hydroaminomethylation will be discussed in detail.
2. Unmodified rhodium catalysts for hydroaminomethylation
The first rhodium-catalyzed hydroaminomethylation was reported by Iqbal in 1971.10 Rh2O3 was utilized as the main catalyst in the hydroaminomethylation of cyclohexene. It was found that rhodium was a much better catalyst compared with the iron catalyst used by Reppe et al.5 A much higher yield (up to 80%) was obtained with Rh2O3 as the only catalyst in the presence of CO and water compared with Fe(CO)5 (up to 6%). It was further found that the yield can be improved (up to 93.5%) by the addition of Fe(CO)5 as a co-catalyst with the byproducts highly suppressed (Scheme 2). Since the work by Iqbal et al., rhodium catalysts have attracted great attention in hydroaminomethylation.
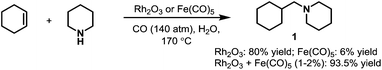 |
| | Scheme 2 The first rhodium-catalyzed hydroaminomethylation. | |
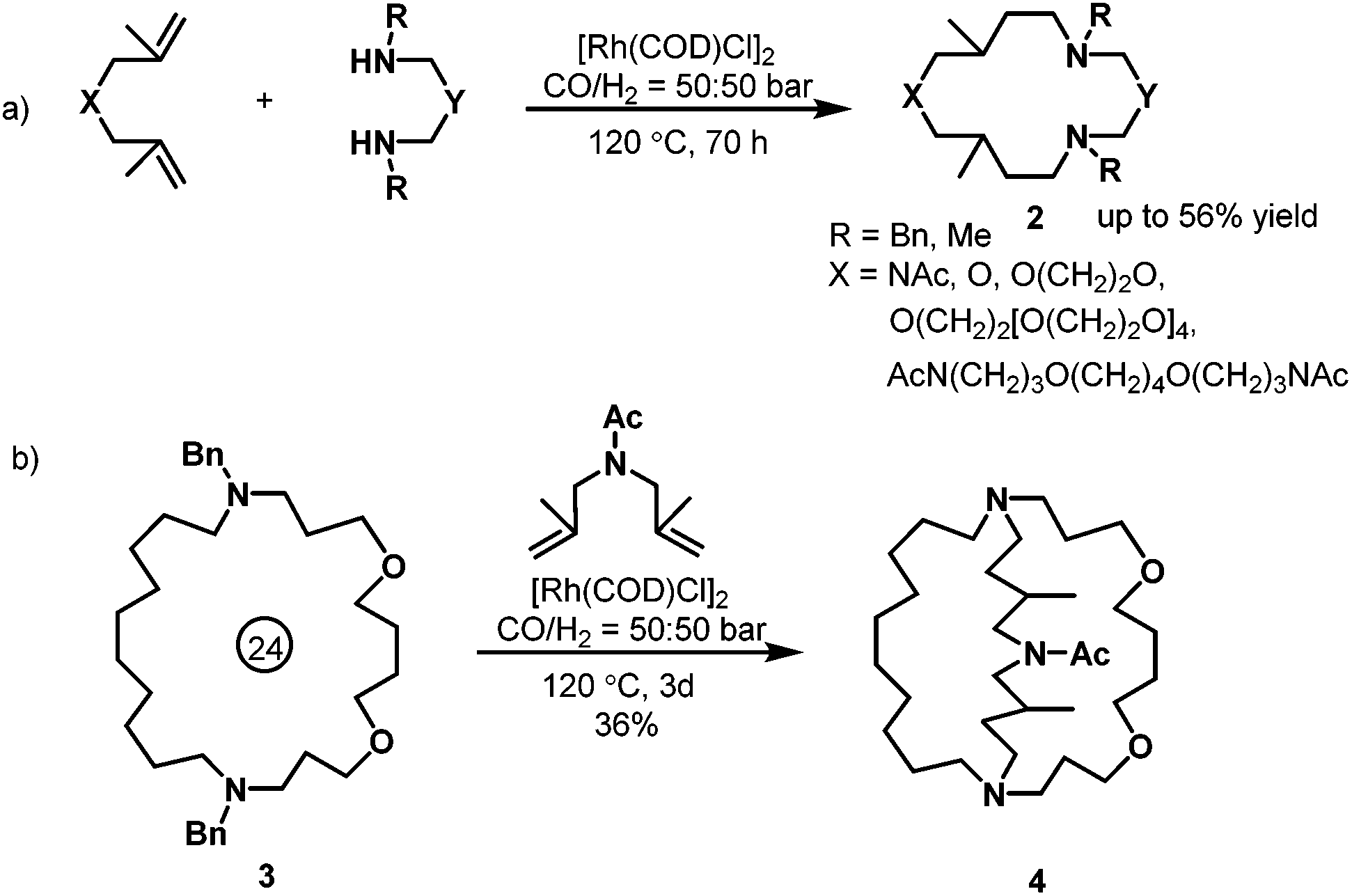
With regard to the rhodium-catalyzed hydroaminomethylation, Eilbracht et al. contributed many efforts in the last decade. In 2000, Eilbracht et al. conducted an interesting hydroaminomethylation reaction with unmodified [Rh(COD)Cl]2 as the catalyst.11 The Rh(I)-catalyzed hydroaminomethylation of dienes in the presence of primary amines or secondary α,ω-diamines was applied to the synthesis of a variety of macroheterocyclic rings (2). As shown in Scheme 3, starting from (hetero)diallylic systems, 12- to 36-membered polyheterocycles were obtained in up to 56% yield. In addition, it was found that the macrocyclic systems 3 thus obtained can be debenzylated and that the resulting macrocyclic diamines undergo a second ring-closing bis(hydroaminomethylation) to give cryptand systems 4.
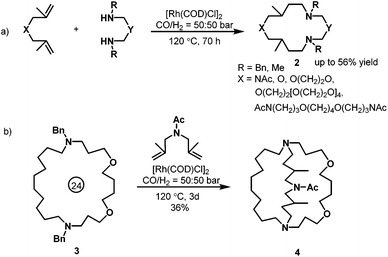 |
| | Scheme 3 Synthesis of macroheterocyclic rings via hydroaminomethylation. | |
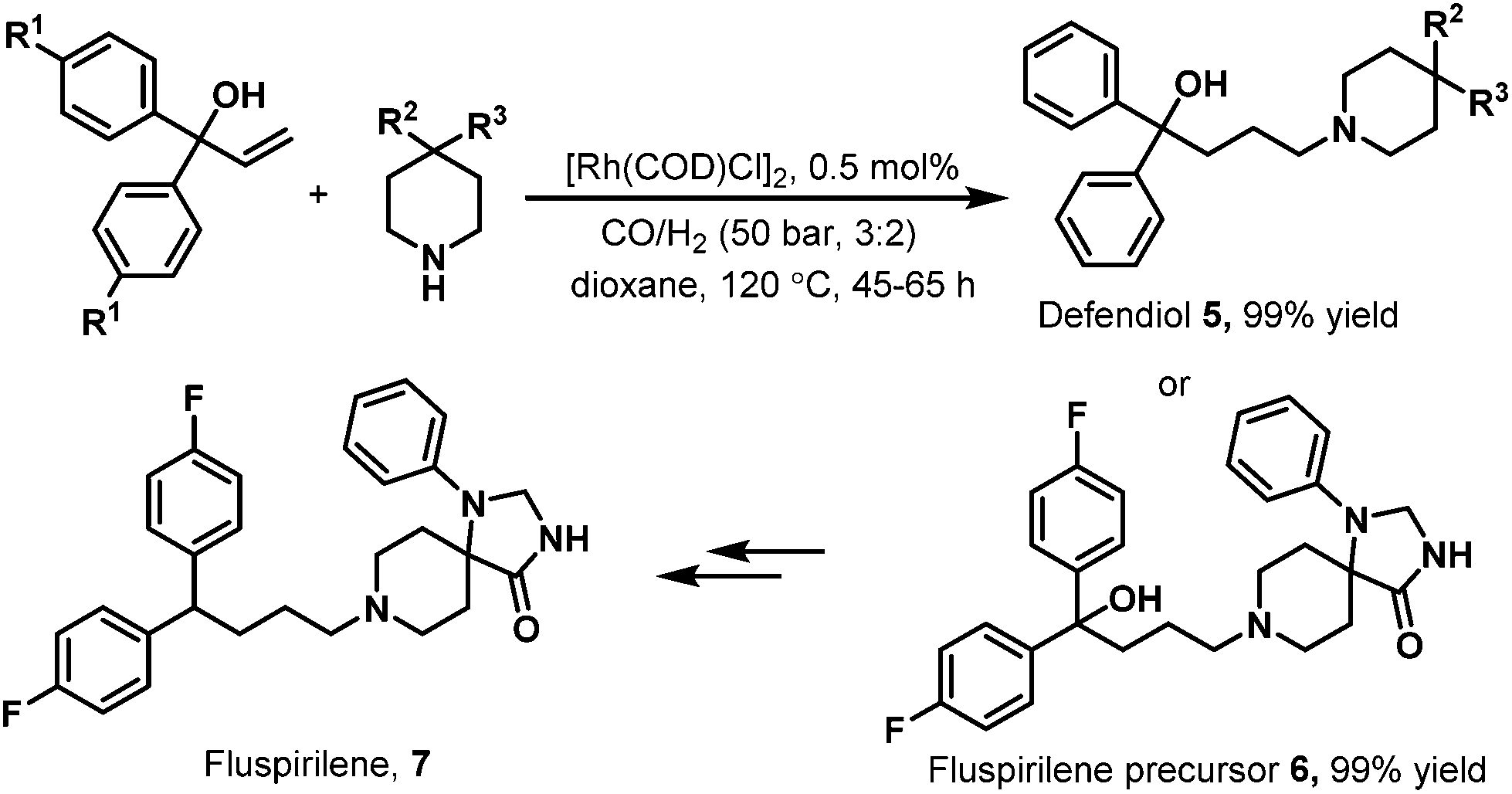
In 2004, Eilbracht et al. conducted the hydroaminomethylation of 1,1-diaryl-allyl-alcohols with unmodified [Rh(COD)Cl]2 as the catalyst.12 Hydroaminomethylation of 1,1-diaryl-allyl-alcohols provides a new access to 4,4-diarylbutylamines, which possess therapeutic activity and are commercially available therapeutic agents as exemplified by the antihistaminic agent difenidol 5 and fluspirilene 7 (Scheme 4).12 It was found that the hydroaminomethylation of 1,1-diaryl-allyl-alcohols proceeded smoothly with [Rh(COD)Cl]2 as the catalyst. As shown in Scheme 4, difenidol (5) and the fluspirilene precursor 6 were obtained in almost quantitative yield. Starting from 6, fluspirilene (7) can be easily obtained.
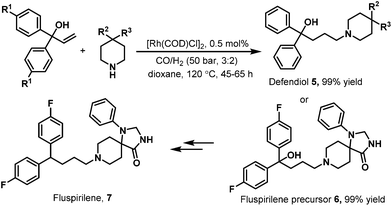 |
| | Scheme 4 Hydroaminomethylation of 1,1-diaryl-allyl-alcohols. | |
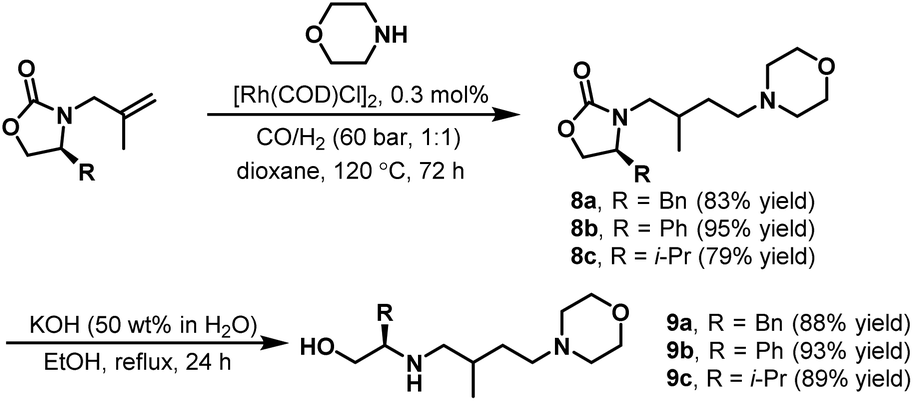
Later, in 2009, Eilbracht et al. continued work on hydroaminomethylation with unmodified [Rh(COD)Cl]2 as the catalyst.13 An efficient preparation of chiral polyamino alcohols (PAA) via the hydroaminomethylation of chiral N-olefinic oxazolidinones with different amines followed by hydrolysis was reported. As shown in Scheme 5, the hydroaminomethylation of chiral N-olefinic oxazolidinones proceeded smoothly in the presence of the unmodified catalyst [Rh(COD)Cl]2 to give the amine products 8a–c in high yields. The subsequent hydrolysis gave the desired chiral amino alcohols 9a–c readily. Based on these results, the dendritic chiral PAAs (Mw = 1300–1400 g mol−1) were also synthesized efficiently through a multifold hydroaminomethylation/hydrolysis procedure. Interestingly, the obtained chiral PAAs were investigated as ligands in the ruthenium-catalyzed asymmetric transfer hydrogenation of acetophenone to 1-phenyl alcohol.
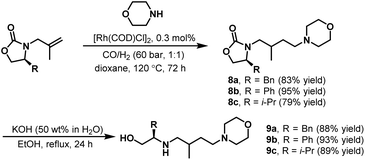 |
| | Scheme 5 Synthesis of chiral amino alcohols via hydroaminomethylation/hydrolysis. | |
3. Monophosphorus ligands for hydroaminomethylation
Although good results were obtained in the hydroaminomethylation of alkenes with amines in the presence of syngas with the unmodified rhodium catalysts, efforts have been made to develop more efficient catalytic systems with rhodium catalysts modified by ligands to further address the control of chemoselectivity and regioselectivity in the hydroaminomethylation reaction.
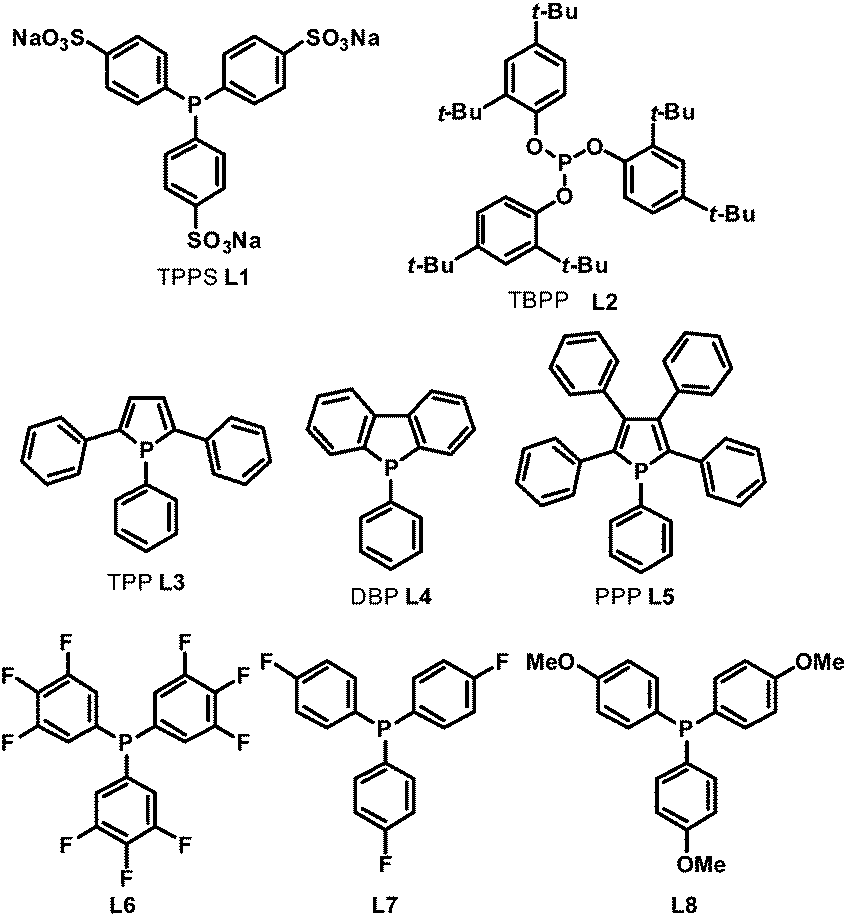
In 1999, Beller et al. described a selective hydroaminomethylation of olefins with ammonia to form linear primary and secondary aliphatic amines in a two phase system.14L1 (Scheme 6) was utilized as the ligand for its high water solubility. A high yield with moderate regioselectivity was observed in the hydroaminomethylation of terminal olefins with ammonia. Later, in 2005, Whiteker et al. used the phosphite ligand L2 (Scheme 6) in the hydroaminomethylation of 1-pentene with piperidine.15 Excellent amine selectivity was observed (100%); however, the regioselectivity was low.
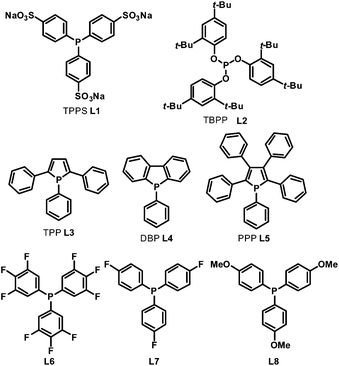 |
| | Scheme 6 Monophosphorus ligands for hydroaminomethylation. | |
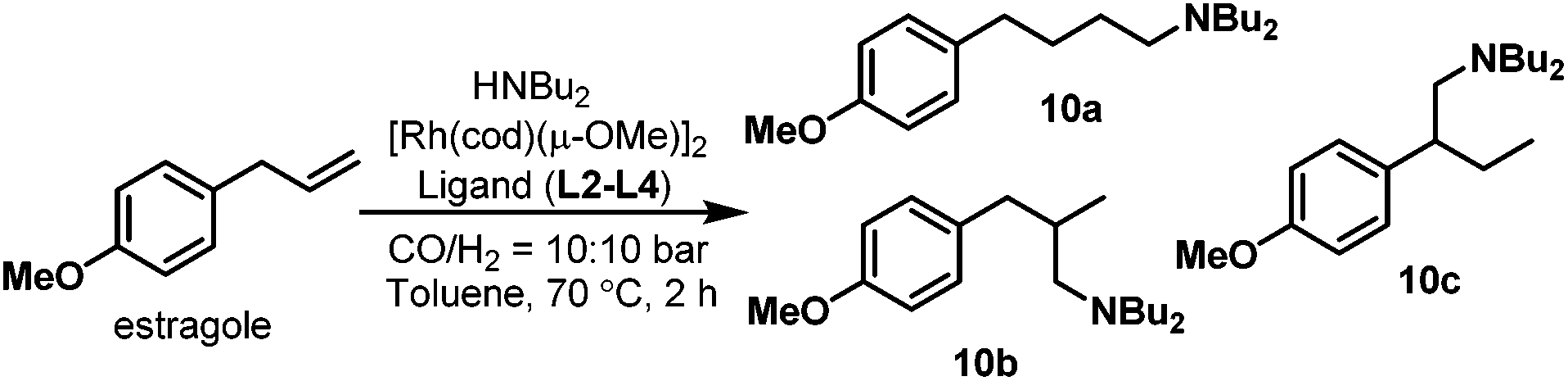
In the hydroaminomethylation reaction, fine tuning of the electronic and steric properties of the ligand is necessary to combine higher activity and selectivity. Recently, efforts have been made into the systematic investigation of monophosphorus ligands for hydroaminomethylation. Urrutigoïty et al. compared three class of phosphorus(III) ligands in hydroaminomethylation: phosphines, phosphites and phospholes (Scheme 6).16 The hydroaminomethylation (HAM) of estragole, a biorenewable starting material available from essential oils of various plants, was reported for the first time. Di-n-butylamine was used as the amine counterpart and the corresponding amines (10a–c) were obtained in high yields (Scheme 7). The monophospholes TPP (L3), DBP (L4) and PPP (L5) were employed for the first time as ancillaries for hydroaminomethylation and proved to be promising options for a more efficient manner of promoting the reductive amination than the classic PPh3 ligand and resulted in less side products than the systems with phosphite (L2).
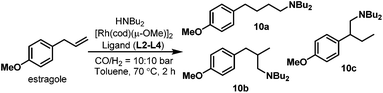 |
| | Scheme 7 Hydroaminomethylation of estragole with ligands L2–L4. | |
In 2011, Clarke et al. investigated ligand effects in the rhodium-catalyzed hydroaminomethylation of styrene with the ligands L6–L8.17 In the course of their studies, it was found that fluorinated mono-phosphines L6 and L7 were more active than their more electron-donating counterpart L8 in the enamine hydrogenation step of the reaction, which is in contrast with the widely held view that alkene hydrogenation activity increases with ligand donor strength. The subsequent DFT calculations comparing the reaction pathways for a simple alkene and a representative enamine showed that the rate-determining step changes from the first insertion into the Rh–H bond for but-2-ene to the final reductive elimination step from the Rh–hydride–alkyl species in the enamine hydrogenations.
4. Bisphosphorus ligands for hydroaminomethylation
4.1. Bisphosphine ligands
Owing to the much better chelating ability and steric environment, bisphosphine ligands generally give higher chemoselectivities and regioselectivities compared with monophosphorus ligands in alkene hydroaminomethylation. As a result, great efforts have been made in the development of efficient bisphosphorus ligands. In this regard, Beller et al. contributed great efforts to the development of tailor-made rhodium catalytic systems to transform both terminal and internal alkenes into linear amines.
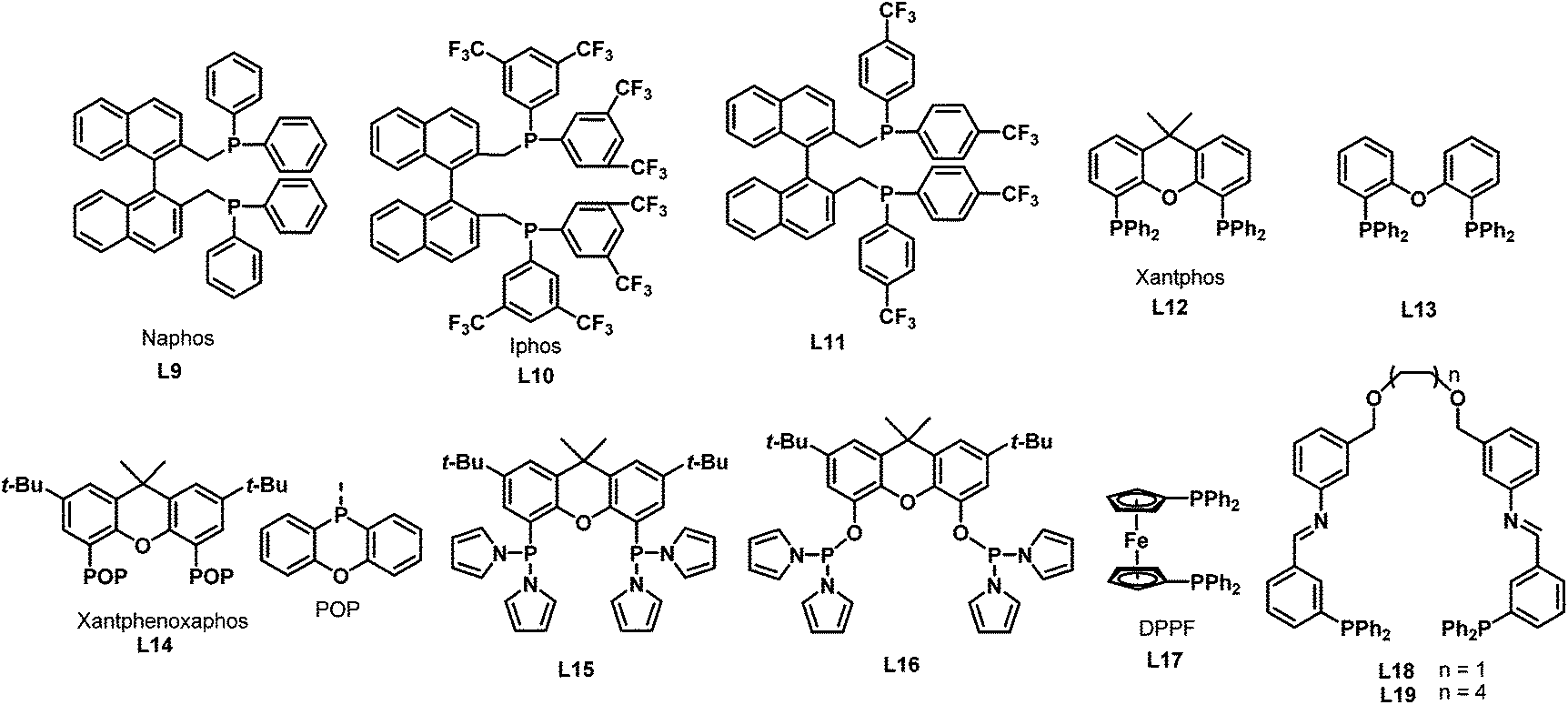
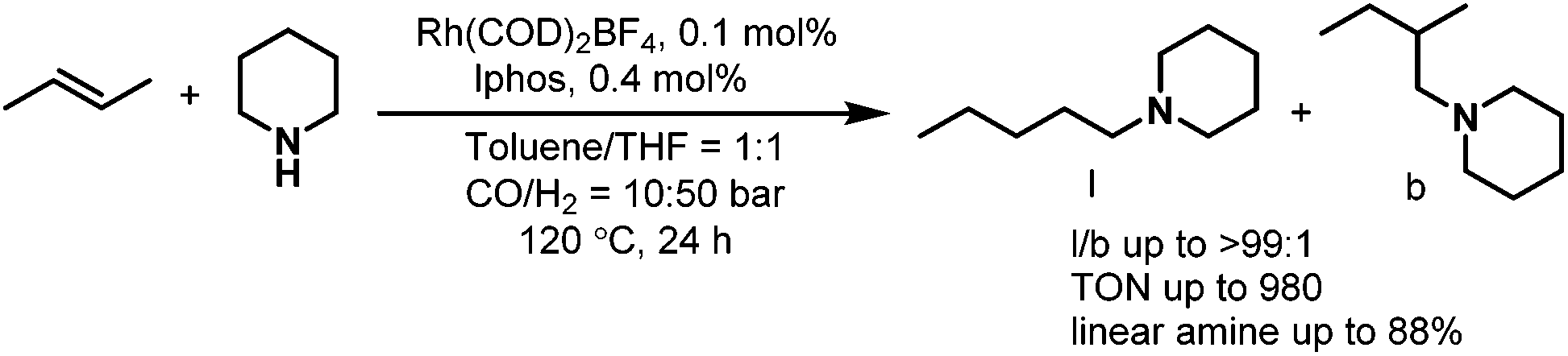
In 2002, Beller et al. reported the selective synthesis of linear amines from internal olefins or olefin mixtures via a catalytic one-pot reaction consisting of an initial olefin isomerization followed by hydroaminomethylation.18 This reaction constitutes an economically attractive and environmentally favorable synthesis of linear aliphatic amines. The use of the cationic rhodium precursor [Rh(COD)2]BF4 with the sterically crowded diphosphine ligands naphos (L9), iphos (L10) and L11 (Scheme 8) were reported for the first time. Among all the ligands tested, it was found that iphos (L10) gave the best result and excellent chemoselectivity and regioselectivity were obtained in the isomerization–hydroaminomethylation of 2-butene (Scheme 9). Later in 2003, the same group found that xantphos 12 was also an excellent ligand for the hydroaminomethylation of a variety of terminal alkenes with different amines.19 Investigation into xantphos type ligands such as L13 and L14 showed that the natural bite angles and steric hindrance of ligands have significant influence on the hydroaminomethylation results.20 It was also found that the regioselectivity for the linear product follows a similar trend to that observed in the hydroformylation of internal alkenes with the use of xantphos ligands. Furthermore, each of the individual steps in hydroaminomethylation was monitored by high-pressure infrared spectroscopy. The results suggest that hydroaminomethylations take place by a sequential isomerization/hydroformylation/amination/hydrogenation pathway.
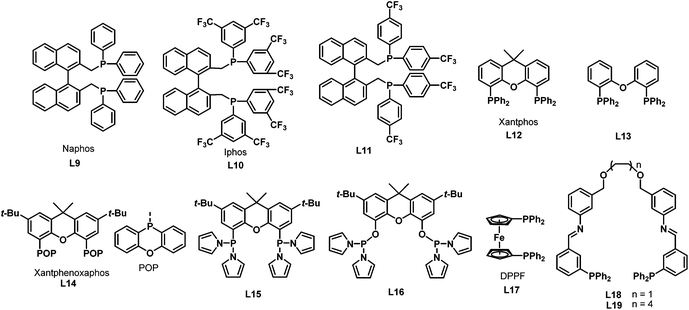 |
| | Scheme 8 Bisphosphine ligands for hydroaminomethylation. | |
 |
| | Scheme 9 Isomerization–hydroaminomethylation of 2-butene with piperidine utilizing L10. | |
Apart from the natural bite angles and steric hindrance, the electronic effects of ligands also have a crucial influence on the activity and selectivity of hydroaminomethylation. With the π-acidic ligands coordinated to the metal center, CO dissociation will be facilitated via the trans-effect and the reaction rate will be improved as a consequence in the hydroformylation step.21 Based on this concept, Vogt et al. developed ligands L15 and L16 with an outstanding π-acceptor character by modifying the xanthene backbone with dipyrrolylphosphine,22 which has χ values approaching those of phosphites.23 Accordingly, the bis-[(dipyrrolyl)phosphino]xanthene ligand L15 showed outstanding activities and selectivities with turnover frequencies of 6200 h−1 as well as very high n/i ratios exceeding 200 in the hydroaminomethylation of terminal alkenes. In addition, it was found that the pKa value of the alcohol used in the solvent mixture has a profound effect on the catalytic performance. By using acidic media, the activity was enhanced; on the contrary, less acidic media led to increased regio- and chemoselectivity, as well as an increased degree of double-bond isomerization.
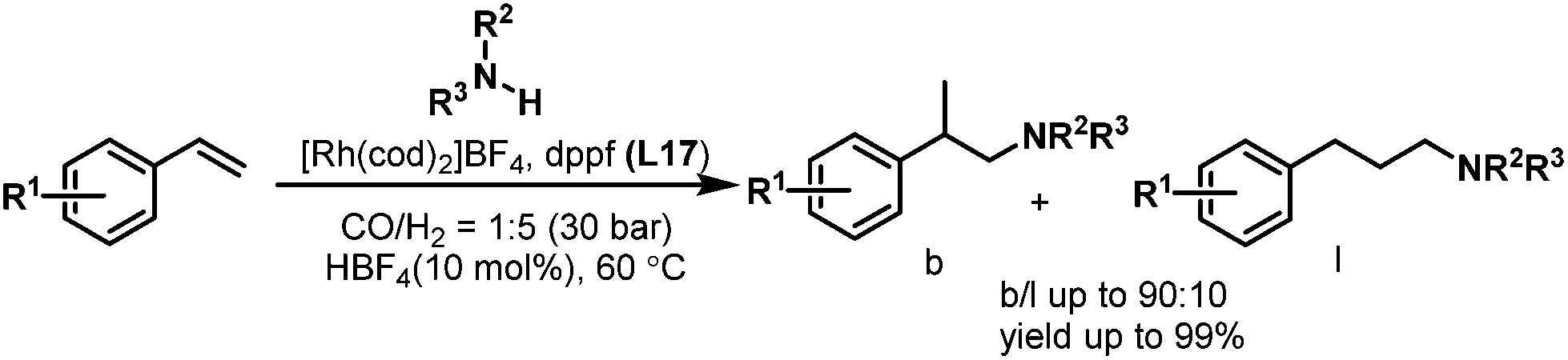
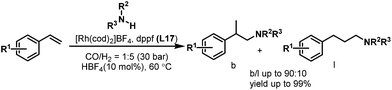
The hydroaminomethylation of styrene provides easy access to arylethylamines, which are a class of pharmaceutically important compounds.24 However, it is a great challenge to obtain the branched amines. In 2005, Beller et al. developed the [Rh(cod)2BF4]/dppf- (L17) catalyzed hydroaminomethylation of aromatic olefins with different amines in the presence of HBF4 with good regioselectivity towards the branched products (Scheme 10).25 The described catalytic system allows the hydroaminomethylation of styrenes under mild conditions.
 |
| | Scheme 10 Hydroaminomethylation of styrene with ligand L17. | |
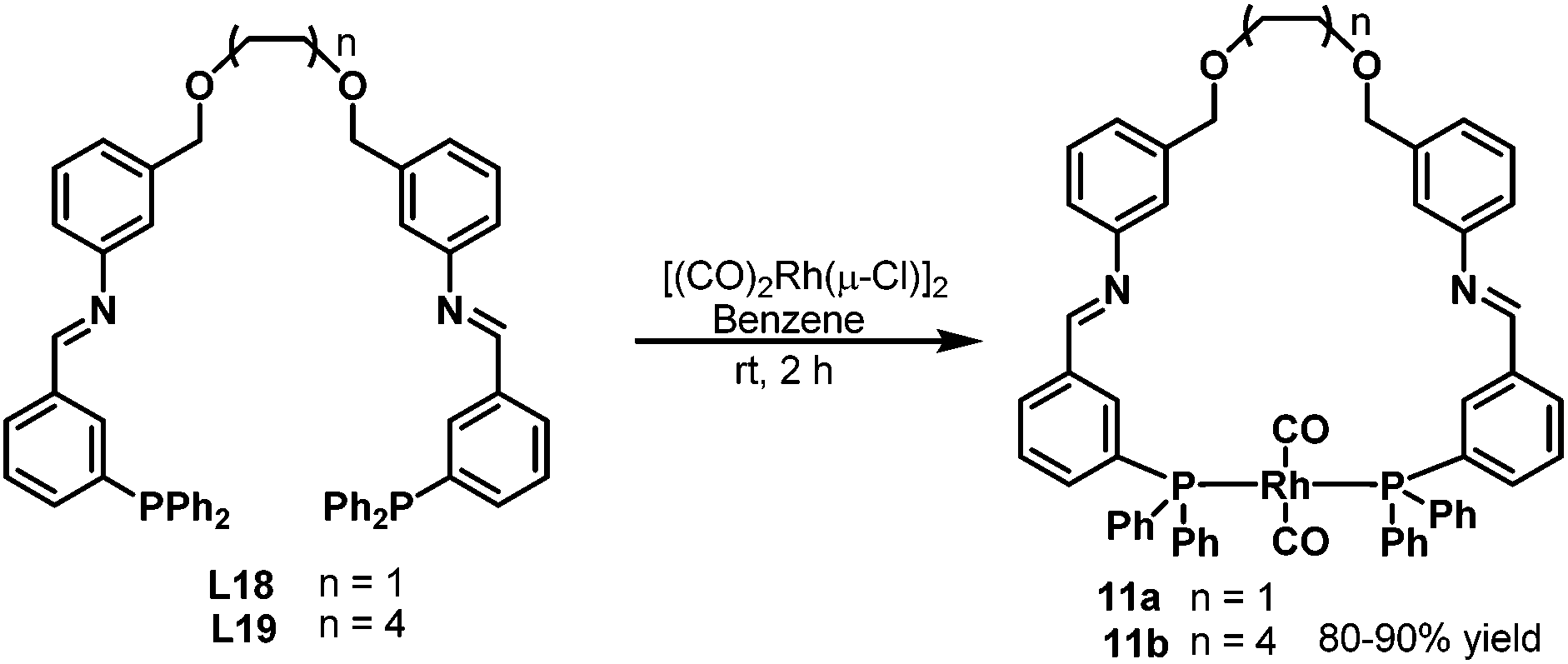
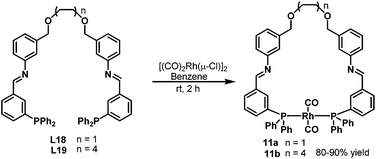
In 2004, Thiel et al. developed the new bisphosphine ligands L18–L19 (Scheme 8) containing the imine donor functionality in the backbone.26 The new bisphosphine ligands were coordinated to the [(CO)2Rh(μ-Cl)]2 precursor to give the rhodium complexes 11a–b in high yields (Scheme 11). The resulting complexes were characterized by means of 31P NMR, 13C NMR and infrared spectroscopy, which revealed that the two phosphine atoms in rhodium complexes 11a–b are in the trans configuration (Scheme 11). Significant changes were observed in the 31P NMR spectra when the Lewis acid KPF6 or Zn(OTf)2 was added to the solution of the rhodium complexes 11a–b, which proves the existence of secondary interactions between the ligands and the Lewis acids. The rhodium complexes 11a–b were subsequently tested in the hydroaminomethylation of 1-pentene or styrene with piperidine. It was found that the amine selectivity increased evidently with the addition of co-catalyst, due to the easier hydrogenation of the enamine.
 |
| | Scheme 11 Preparation of the rhodium complexes for hydroaminomethylation. | |
4.2. Bisphosphite ligands
Most of the current rhodium-catalyzed hydroaminomethylation reactions have predominantly employed phosphine ligands. In a sharp contrast, the use of phosphite ligands in hydroaminomethylation has been rare. In studies with phosphine ligands, it was concluded that phosphite ligands are unsuitable for hydroaminomethylation and give low yields of amine products.27 The inability of phosphite ligands to promote hydroaminomethylation was attributed to the hydrolytic instability of the ligands under the reaction conditions. Regardless of the hydrolytic instability, bisphosphite ligands are very reactive and afford reaction rates significantly higher compared with those obtained with the bisphosphine ligands, due to their good π-acidic character.28 Owing to these advantages, it is very attractive to employ phosphite ligands in hydroaminomethylation.
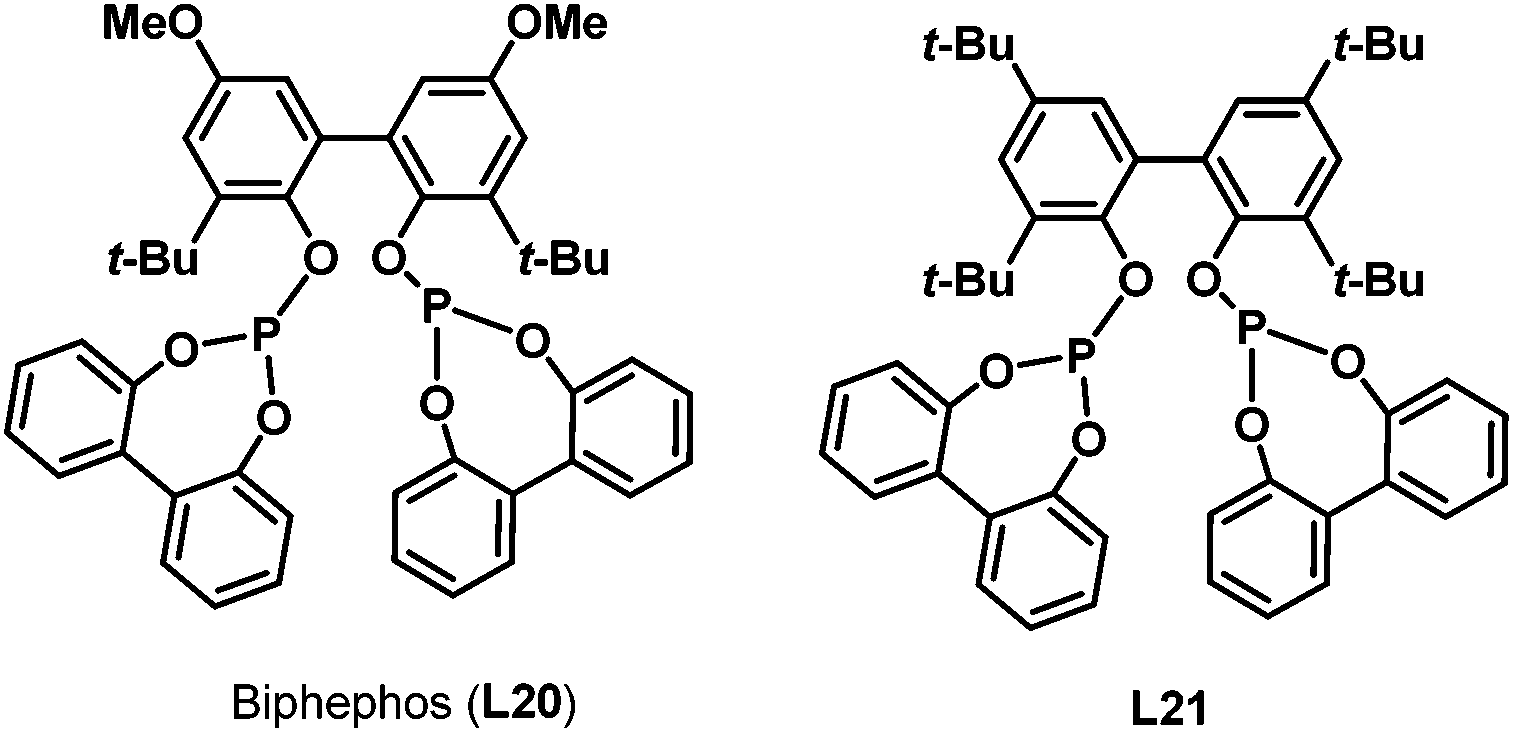
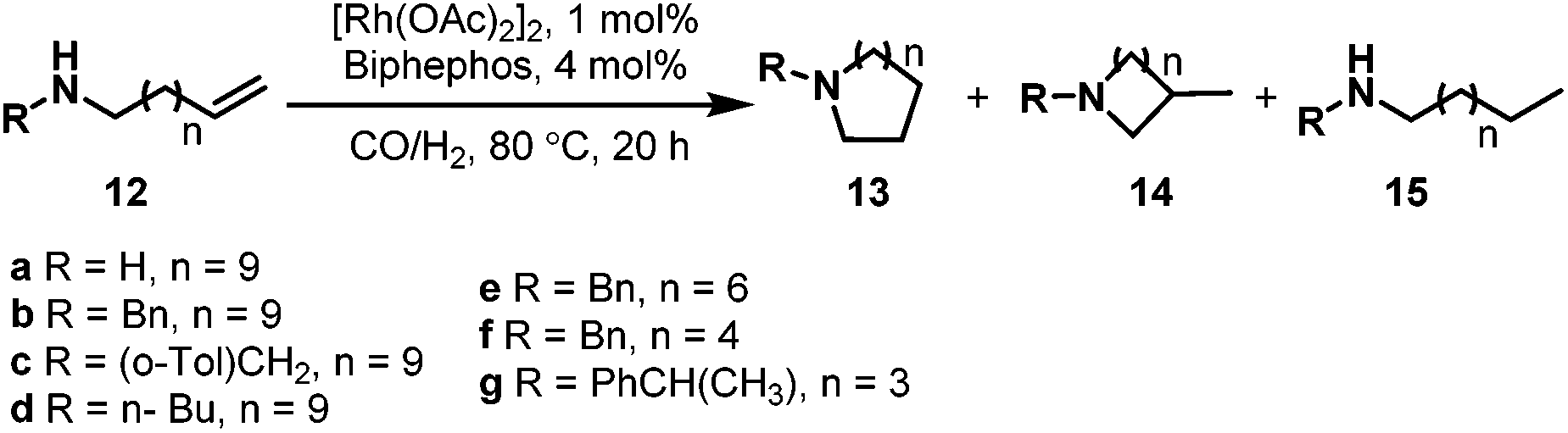
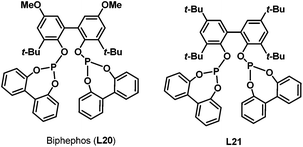
In 1999, Saylik et al. first employed the bisphosphite ligand biphephos (L20) (Scheme 12) in intramolecular hydroaminomethylation.29 A range of aminoalkenes (12) were converted to give cyclic amines with a range of medium and large ring sizes in yields up to 85% in the presence of biphephos as a ligand. High regioselectivity was observed for the non-branched products 13 when biphephos was used as the ligand. However, the hydrogenation products (15) of the substrates were also observed (Scheme 13).
 |
| | Scheme 12 Bisphosphite ligands for hydroaminomethylation. | |
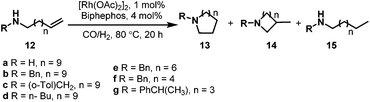 |
| | Scheme 13 Intramolecular hydroaminomethylation with biphephos (L20) as the ligand. | |
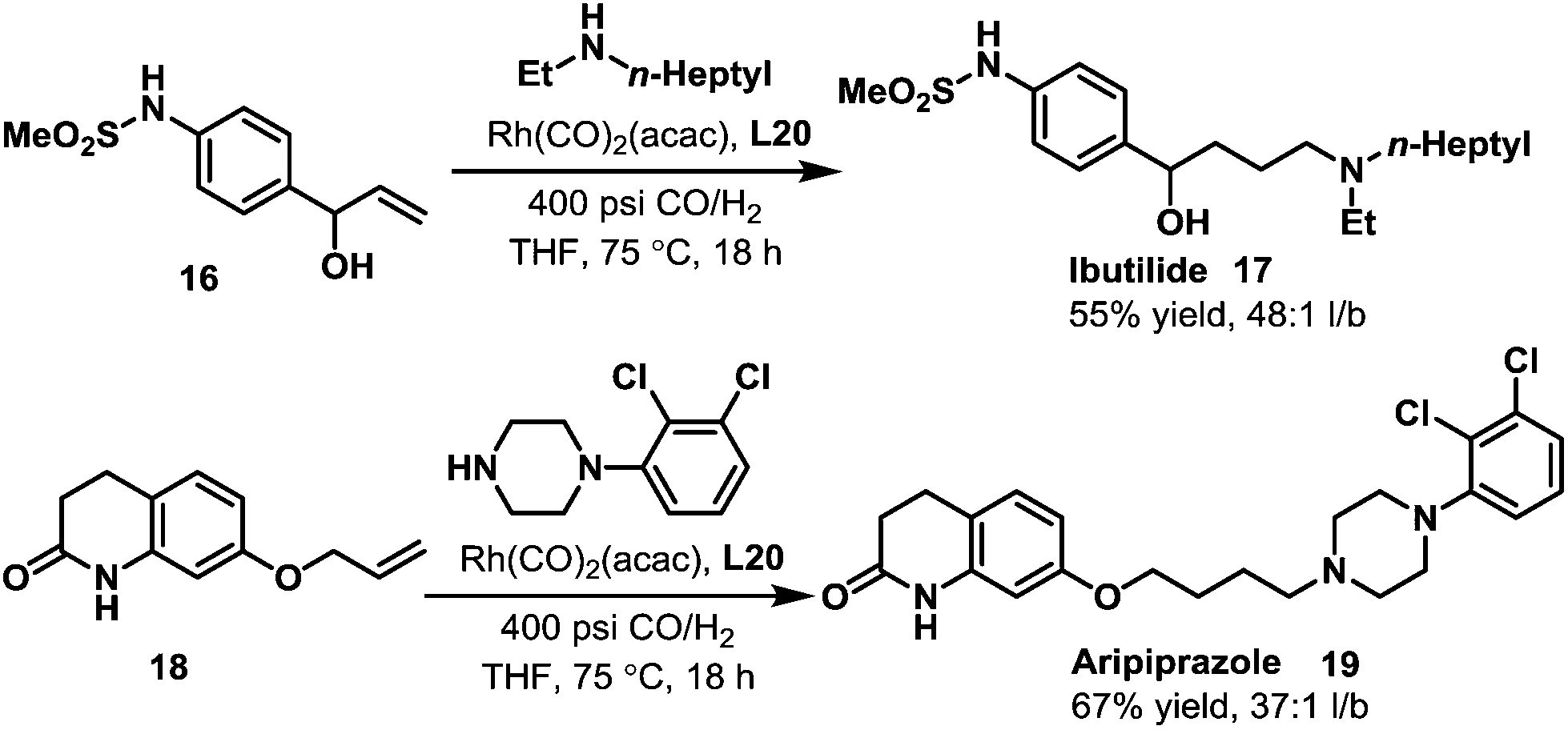
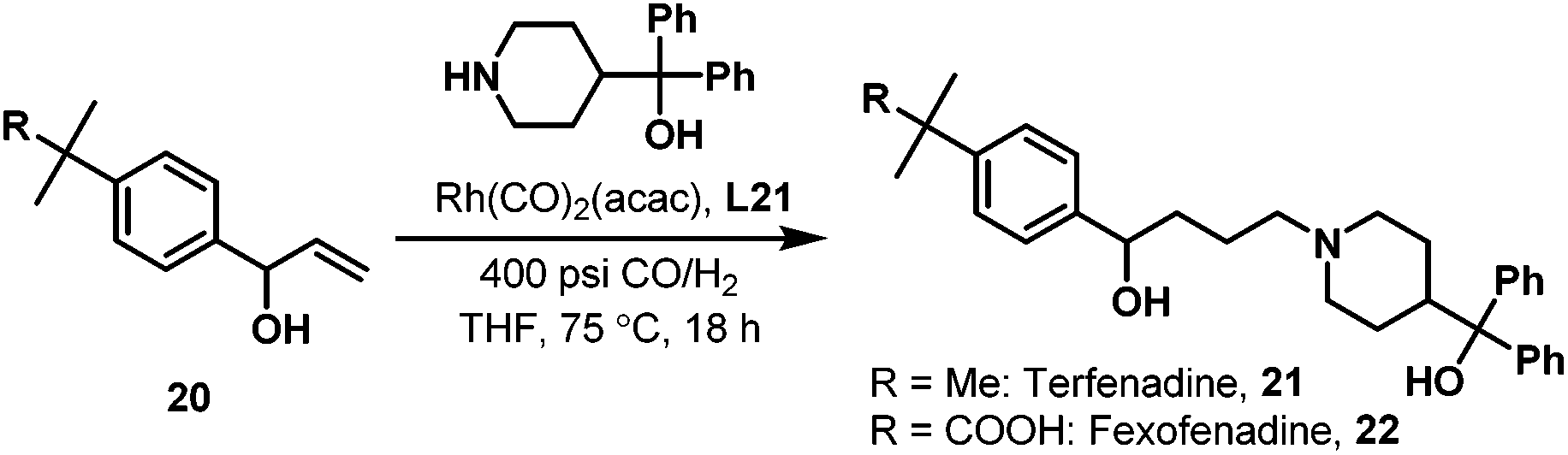
Later in 2005, Whiteker et al. developed the highly regioselective synthesis of two biologically active targets, the anti-arrhythmia compound ibutilide (17)30 and the antidepressant aripiprazole (19)31via rhodium-catalyzed hydroaminomethylation with biphephos (L20) as the ligand.32 Under very mild reaction conditions (75 °C, CO/H2 = 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :14 bar), ibutilide (17) and aripiprazole (19) were obtained in moderate yields (Scheme 14). Importantly, the regioselectivity (up to 48:1) was very high when biphephos (L20) was used as the ligand. In 2010, the same group also published the synthesis of the other structurally related antihistamine drugs, terfenadine (21)and fexofenadine (22) (Scheme 15).33 Under similar reaction conditions, terfenadine (21) and fexofenadine (22)were synthesized with high regioselectivities using the bisphosphite ligand L21 (Scheme 15).
:14 bar), ibutilide (17) and aripiprazole (19) were obtained in moderate yields (Scheme 14). Importantly, the regioselectivity (up to 48:1) was very high when biphephos (L20) was used as the ligand. In 2010, the same group also published the synthesis of the other structurally related antihistamine drugs, terfenadine (21)and fexofenadine (22) (Scheme 15).33 Under similar reaction conditions, terfenadine (21) and fexofenadine (22)were synthesized with high regioselectivities using the bisphosphite ligand L21 (Scheme 15).
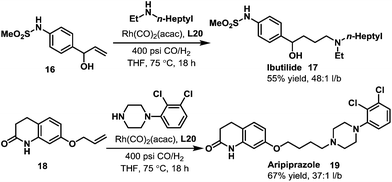 |
| | Scheme 14 Highly selective synthesis of ibutilide (17) and aripiprazole (19) via the rhodium-catalyzed hydroaminomethylation. | |
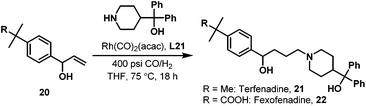 |
| | Scheme 15 Highly selective synthesis of terfenadine (21) and fexofenadine (22) via the rhodium-catalyzed hydroaminomethylation. | |
5. Tetraphosphorus ligands for hydroaminomethylation
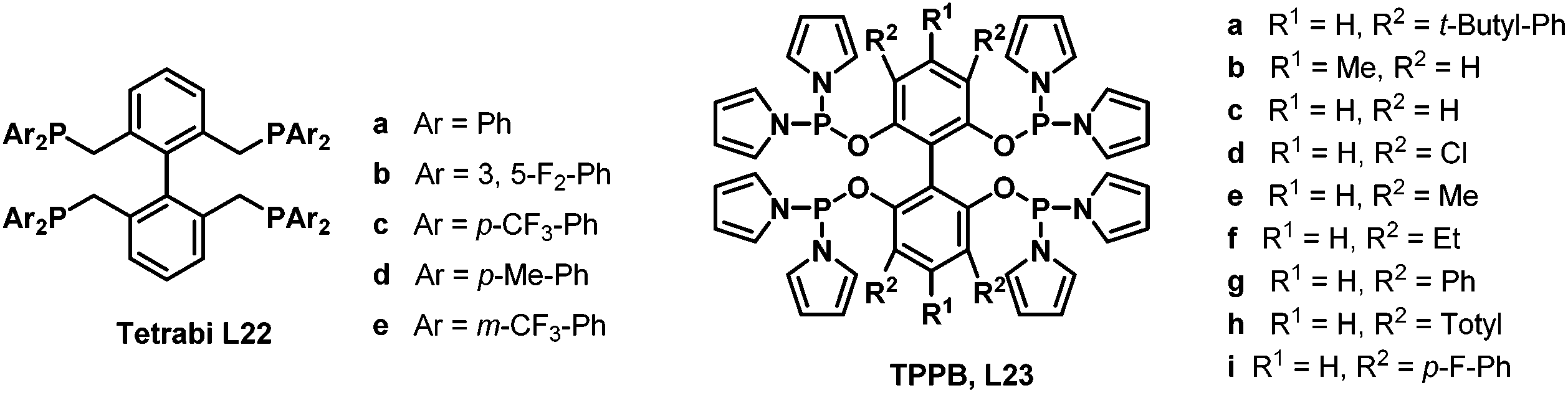
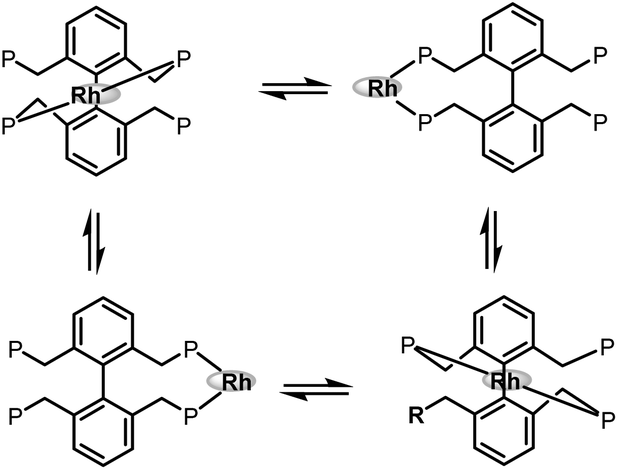
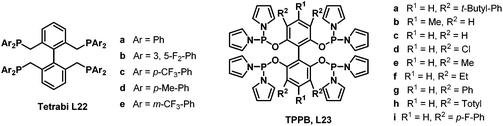
In 2006 and 2007, our group developed two systems of tetraphosphorous ligands,34 which are based on a biphenyl backbone and can be successfully applied in hydroformylation (Scheme 16). Unprecedented regioselectivities have been achieved in the hydroformylation of both terminal and internal olefins. It is believed that there are four identical coordination modes of the tetraphosphorus ligands with rhodium (Scheme 17). The phosphine concentration around the metal center is much increased compared with the corresponding bisphosphorus ligands, which ensured much stronger chelating ability. As a consequence, better regioselectivity was observed.
 |
| | Scheme 16 Tetraphosphorus ligands for hydroaminomethylation. | |
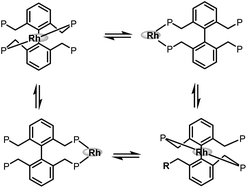 |
| | Scheme 17 Four identical coordination modes of tetraphosphorus ligands with rhodium. | |
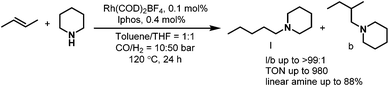
As the regioselectivity in the hydroaminomethylation was determined by the hydroformylation step, we envisioned the tetraphosphorus ligands tetrabi (L22) and TPPB (L23) will also afford excellent regioselectivities in the hydroaminomethylation. The tetrabi L22a (Scheme 16) was then tested in the hydroaminomethylation of a variety of terminal alkenes and amines. Corresponding to our hypothesis, the tetrabi L22a showed excellent regioselectivities and amine selectivities. Regioselectivity as high as >525:1 (n:i) was obtained in the hydroaminomethylation of 1-hexene with piperidine, which represents the highest among all the reported results. Amine selectivity as high as 99.7% was also observed. Moreover, the rhodium–L22a catalytic system exhibited excellent activities, affording TONs as high as 6930.35
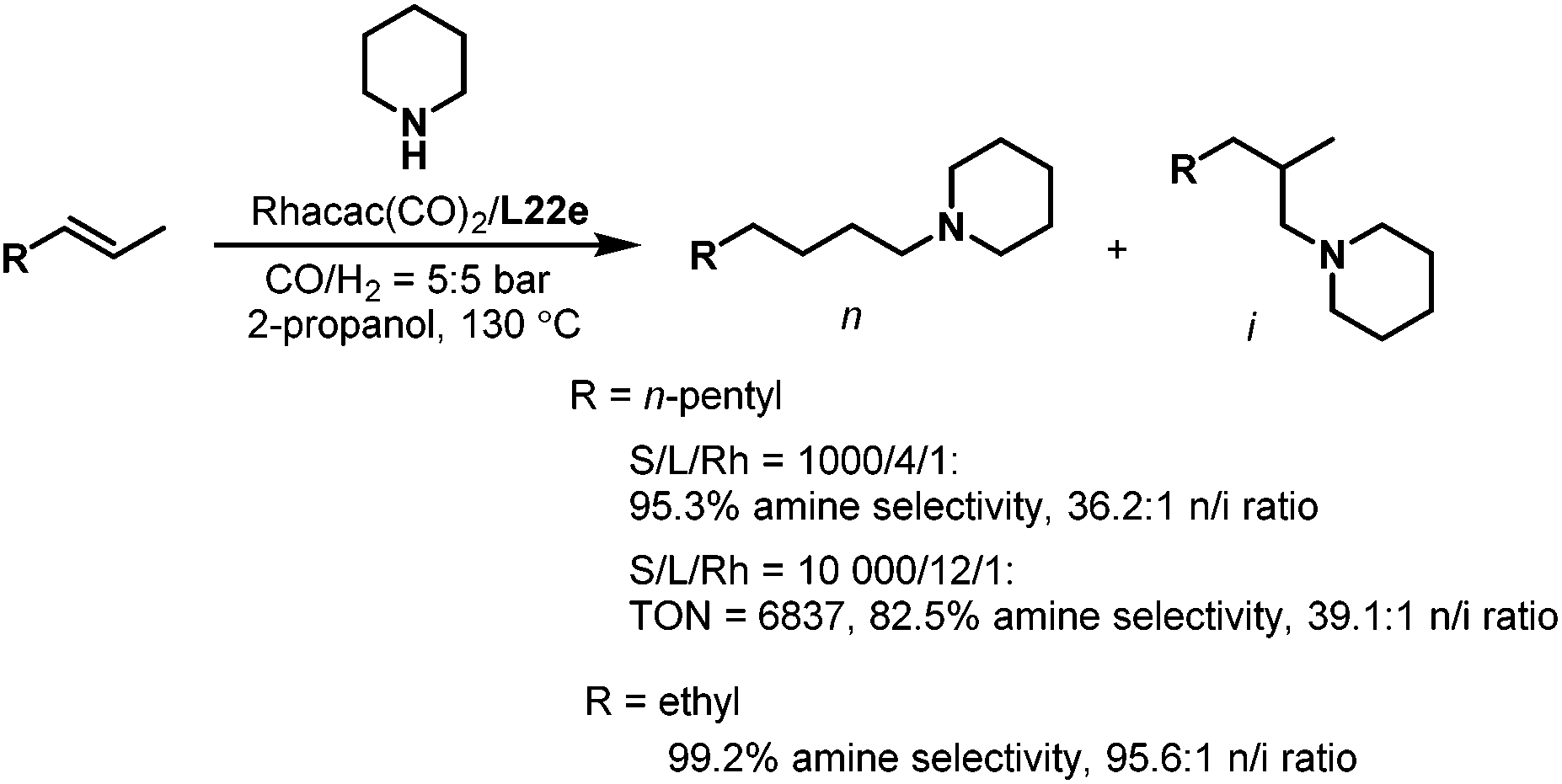
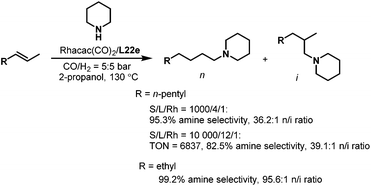
Subsequently, the tetrabi ligands L22 were tested in the isomerization–hydroaminomethylation of internal alkenes.36 Remarkably, a 95.3% amine selectivity and a 36.2 n/i ratio were obtained in the isomerization–hydroaminomethylation of 2-octene with piperidine using the tetrabi ligand at an S/L/Rh ratio of 1000/4/1, and the TON could reach 6837 with an 82.5% amine selectivity and a 39.1 n/i ratio at an S/L/Rh ratio of 10000/12/1. The meta-CF3-Ph substituted ligand L22e (Scheme 16) was found to be the best ligand with up to a 99.2% amine selectivity and 95.6 n/i ratio for 2-pentene (Scheme 18). The tetrabi ligands L22 afforded much better regioselectivity than the other ligands applied in the isomerization–hydroaminomethylation of internal olefins, and these results were also the best among all the reported results.
 |
| | Scheme 18 Isomerization–hydroaminomethylation of internal alkenes using tetrabi ligand L22e. | |
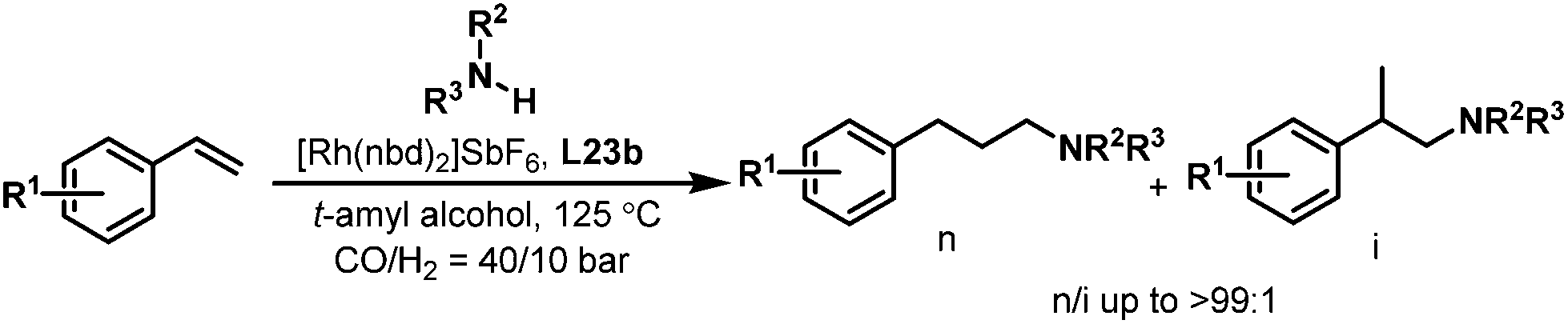
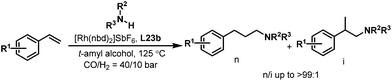
Encouraged by the excellent results achieved with the tetrabi ligands L22, the tetraphosphoramidite ligands L23 were also employed in the rhodium-catalyzed hydroaminomethylation reactions. It is well-known that the linear-selective hydroaminomethylation of styrenes is very challenging among the different alkenes due to the intrinsic trend to form branched amines. There is no general report on the highly linear selective hydroaminomethylation of styrenes to produce 3-arylpropylamines. Recently, we developed an efficient and unprecedented linear-selective (n/i up to >99:1) rhodium-catalyzed hydroaminomethylation of styrenes with the tetraphosphoramidite ligands L23 as the ligands.37 A very high n:i ratio (up to >99:1) was achieved using Rh(nbd)2SbF6 modified with L23b, which is the highest linear selectivity for the hydroaminomethylation of styrene and its derivatives (Scheme 19). The performance of the present system can be explained by the electron-withdrawing property of the pyrrole moiety and the steric interactions between the more bulky tetraphosphorus ligands and the substrates.
 |
| | Scheme 19 Highly linear selective hydroaminomethylation of styrenes using the tetraphosphoramidite ligand L23b. | |
The ligand effects of the tetraphosphoramidite ligands L23 were investigated in the rhodium-catalyzed regioselective hydroaminomethylation of 1-hexene and 1-pentene with piperidine.38 It was found that the substituents of the diphenylphosphane moiety of the ligands greatly affected the amine selectivity and regioselectivity. The 3,3′,5,5′-tetramethylsubstituted pyrrole-based tetraphosphorus ligand L23e was found to be the best ligand, with up to a 70.9 n/i ratio and 99.5% amine selectivity for 1-pentene and a 31.3 n/i ratio and 97.9% amine selectivity for 1-hexene. Moreover, the 4,4′-dimethyl-substituted ligand L23b also showed excellent reactivity, 99.9% amine selectivity and a 27.7 n/i ratio for the regioselective hydroaminomethylation of 1-pentene.
6. Other ligands for hydroaminomethylation
6.1. Carbene ligands
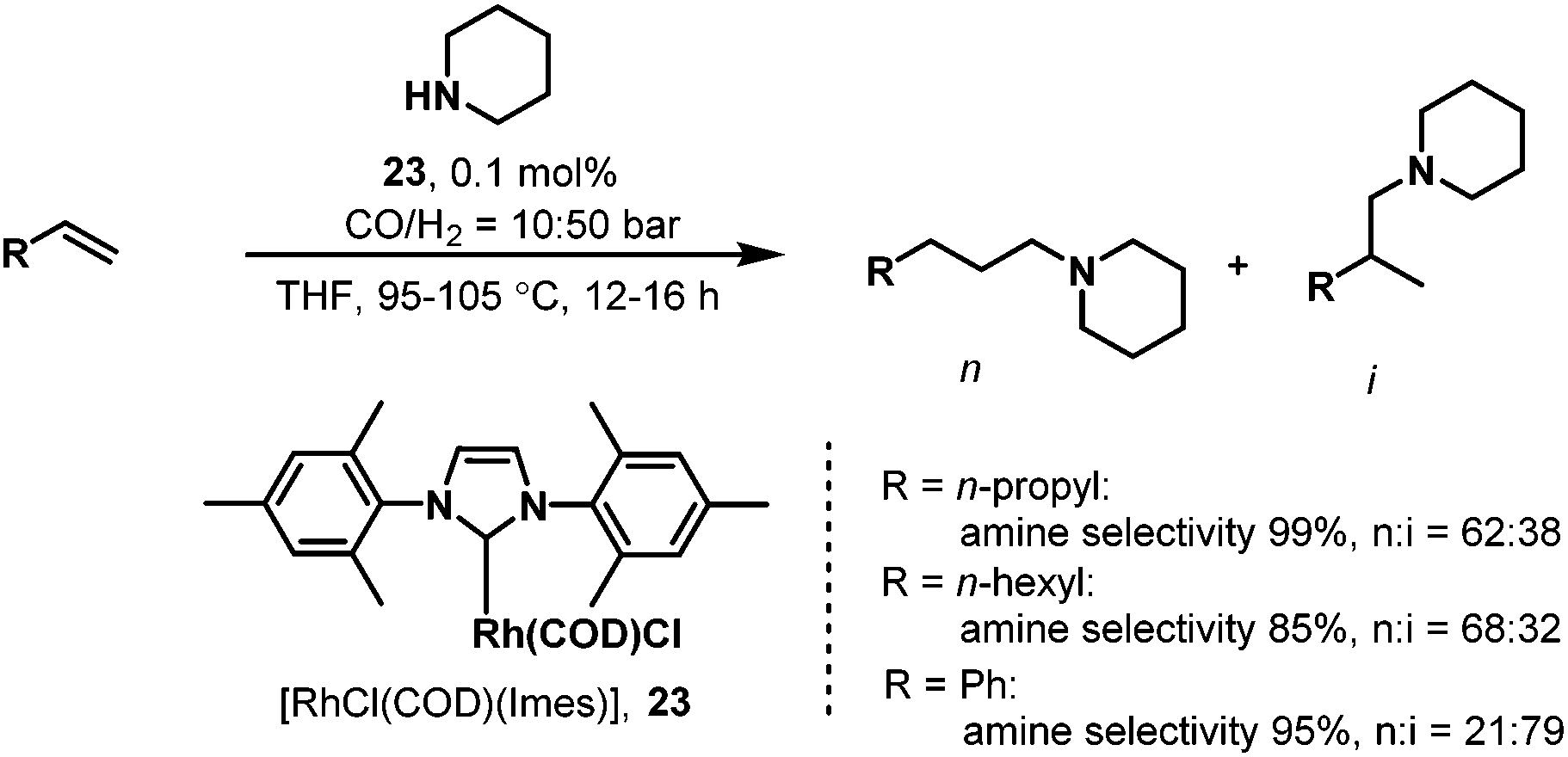
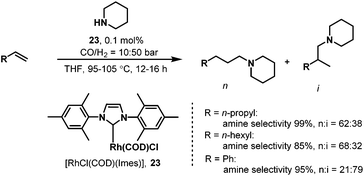
While carbene ligands have been widely used in palladium-catalyzed coupling reactions,39 only very few applications of rhodium–carbene complexes have been reported.40 The first example of carbene ligands for hydroaminomethylation was reported by Beller et al. in 2003.41 It was proposed that the use of a defined rhodium monocarbene complex might solve the problem of the low hydrogenation activity of rhodium–phosphine catalytic systems in which an excess of strongly coordinating phosphine ligands slows down the hydrogenation of the imine or enamine. The new rhodium complex Rh(cod)(Imes)Cl (Imes = 1,3-dimesitylimidazol-2-ylidene, cod = cyclooctadiene) was prepared from [Rh(cod)Cl]2. The newly prepared rhodium–carbene catalyst was then tested in the hydroaminomethylation of 1-pentene with piperidine. Good to excellent activities and chemoselectivities to amines were achieved under comparatively mild reaction conditions. Notably, a high degree of regioselectivity (n/i ratio) was obtained in the case of substituted styrenes. However, terminal aliphatic olefins gave mixtures of linear and branched products (Scheme 20).
 |
| | Scheme 20 Rhodium–carbene complex catalyzed hydroaminomethylation. | |
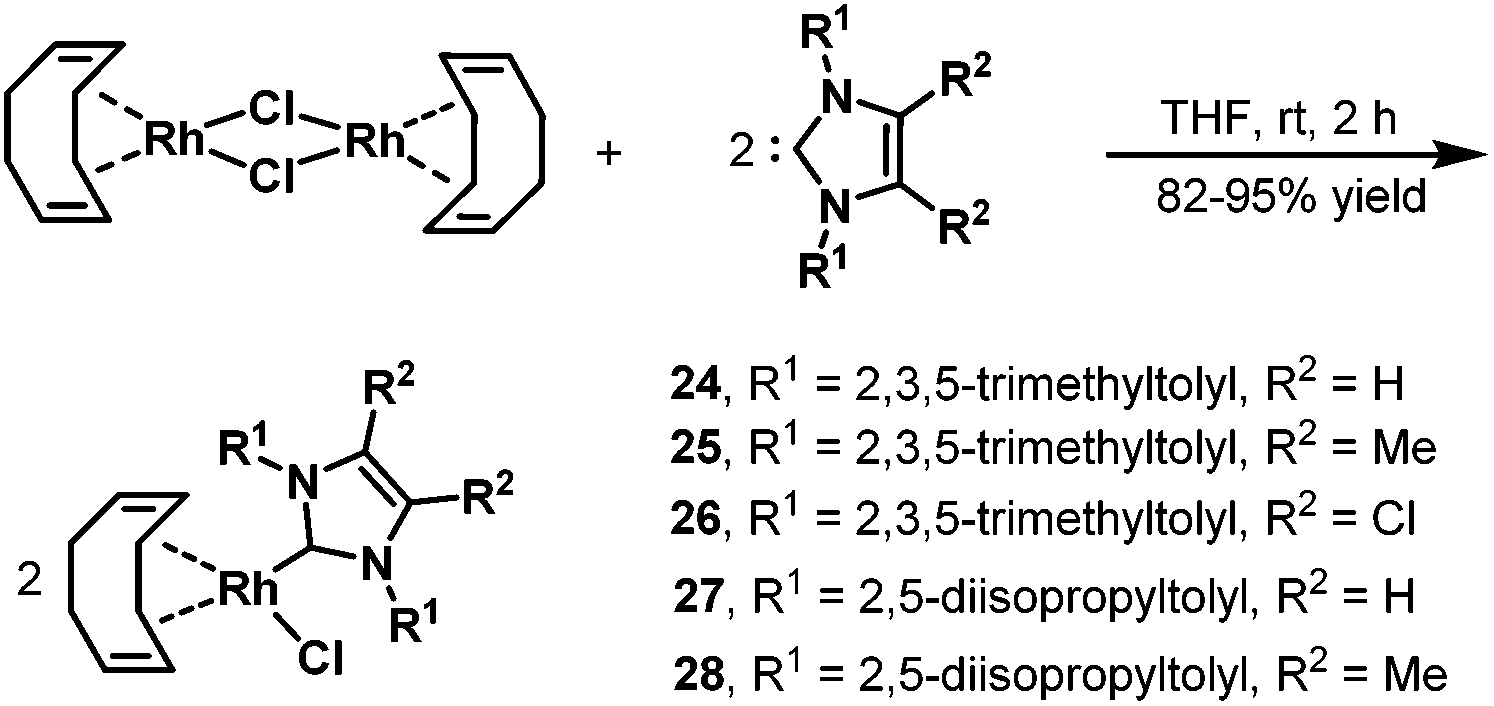
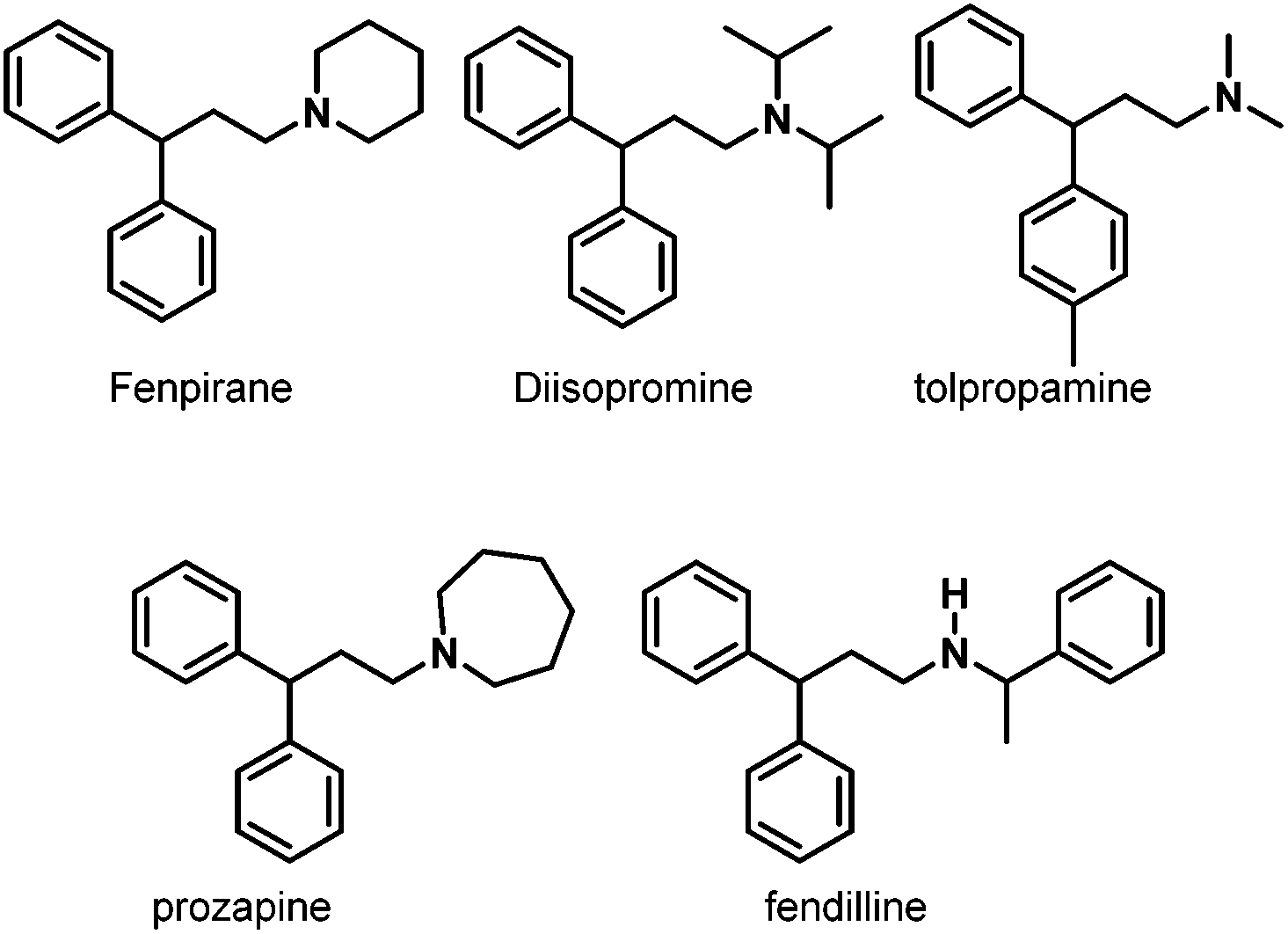
Later, in 2006, the same group prepared a variety of novel rhodium–carbene complexes for hydroaminomethylation.42 Starting from [Rh(cod)Cl]2 and 1,3-dimesitylimidazole-2-ylidenes, the corresponding rhodium–carbene complexes 24–28 were prepared in 82–95% yields (Scheme 21). The rhodium–carbene complexes 24–28 were then tested in the hydroaminomethylation of 1,1-diphenylethylenes. Hydroaminomethylation of 1,1-diphenylethylenes will give 3,3-diarylpropylamines (pheniramines), which represent the well-known first-generation family of H1 antihistaminic agents. The biological activity of 3,3-diarylpropylamines can be tuned from antiallergic to choleretic, antipyretic, coronardilatic, and antispasmodic by varying the amine core (Scheme 22).43 Synthesis of 3,3-diarylpropylamines has attracted immense interest, and a variety of methods have been reported via rhodium-catalyzed hydroaminomethylation.44 However, the reported methods often suffered from long reaction time, high catalyst loading, and low catalyst activity. The novel rhodium–carbene complexes showed promising results for the synthesis of the bioactive 3,3-diarylpropylamines via the hydroaminomethylation reaction. Better activity (TOF up to 288 h−1) was achieved compared with any previously reported procedure for the hydroaminomethylation of 1,1-diphenylethylenes. In the presence of 0.1 mol% of the catalyst, the corresponding 3,3-diarylpropylamines were obtained in high yield and selectivity.42
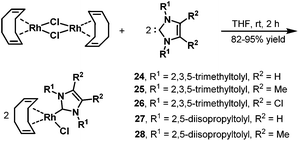 |
| | Scheme 21 Preparation of the rhodium–carbene complexes 24–28. | |
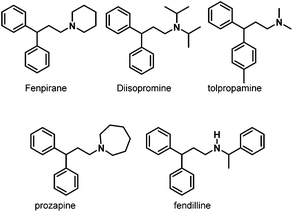 |
| | Scheme 22 Pharmacologically active secondary and tertiary 1-(3,3-diarylpropyl)amines. | |
6.2. Nitrogen-containing ligands
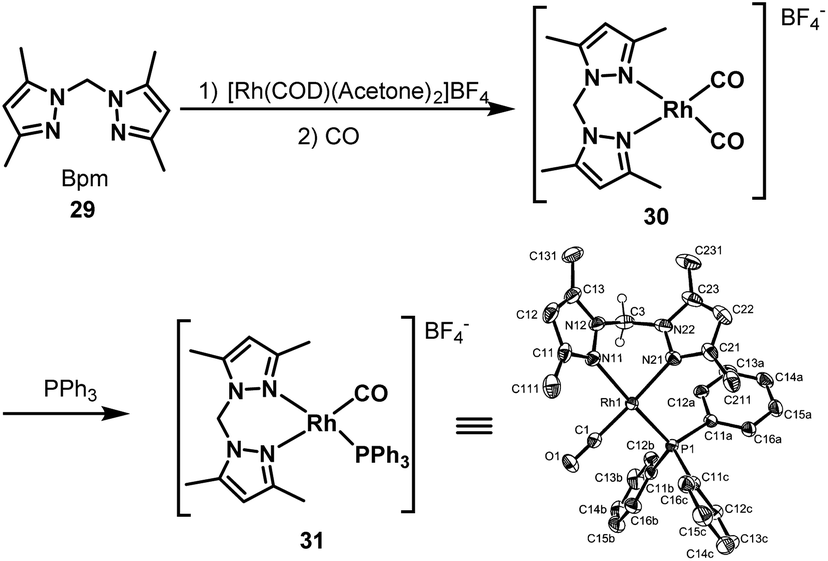
In 2003, Kalck et al. developed the [Bpm*Rh(CO)2]BF4 complex 30 by the addition of H2C(3,5-Me2pz)2 (Bpm*) (29) to [(η4-1,5-COD)Rh(acetone)2]BF4 followed by carbonylation (Scheme 23).45 The new nitrogen-containing ligand modified rhodium complex displayed good activity in the hydroaminomethylation of 1-octene with diethylamine. The hydroaminomethylation reaction was conducted under mild reaction conditions (12 bar CO/H2 (1:1), 80 °C, 6 h) with an 85% amine selectivity and a 1.6 n:i ratio. Notably, the only byproduct in this reaction system was the internal alkenes. The other rhodium complex [Rh(BpmMe2)(CO)(PPh3)]BF4 (31) was also prepared by substituting one CO ligand in the [Bpm*Rh(CO)2]BF4 complex with the addition of PPh3 and the structure was determined by X-ray spectrum analysis (Scheme 23). The rhodium complex [Rh(BpmMe2)(CO)(PPh3)]BF4 (31) increased the n:i ratio to 2.6 and only a small amount of the internal alkene byproduct was observed. The reason that the introduction of the triphenylphosphine ligand gives rise to the increase of the n/i ratio is probably due to the greater hydridic character of the Rh–H bond, and the formation of the linear alkyl moiety is favored. However, the system remains very slow for both the formation of enamines and their hydrogenation.
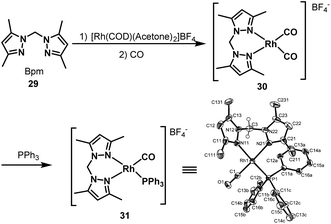 |
| | Scheme 23 Preparation of the [Bpm*Rh(CO)2]BF4 and [Rh(BpmMe2)(CO)(PPh3)]BF4 complexes. | |
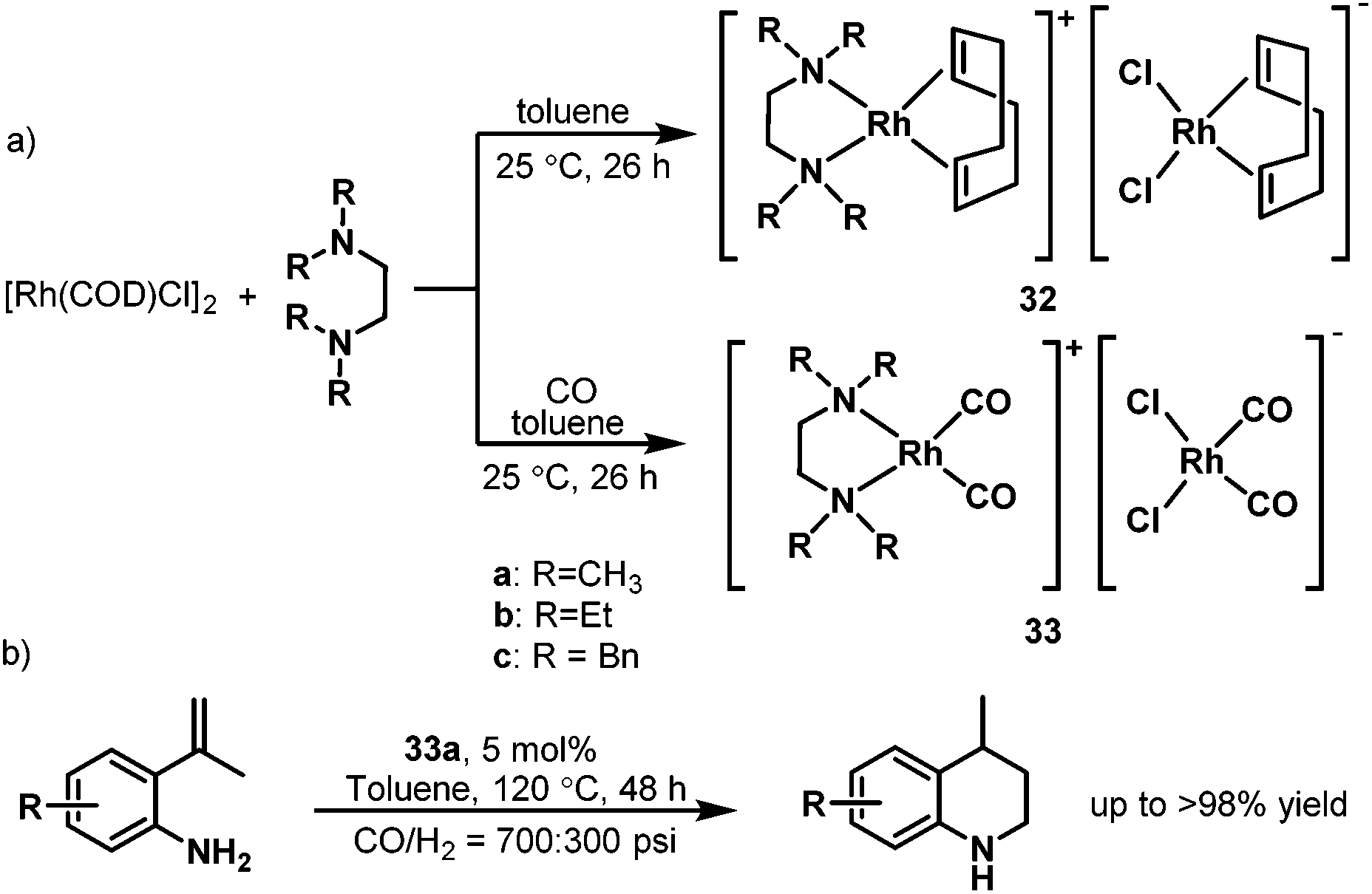
In 2007, Alper et al. developed the hydroaminomethylation of 2-isopropenylanilines and isopropenylamines to produce 1,2,3,4-tetrahydroquinolines catalyzed by the cationic rhodium complex 33a coordinated by N,N,N′,N′-tetramethylethylendiamine (TMEDA).46 This intramolecular hydroaminomethylation is highly regioselective and gives the 1,2,3,4-tetrahydroquinolines in up to over 98% yield (Scheme 24b) albeit with a relatively high catalyst loading. The catalytic precursor was synthesized from [{RhCl(COD)}2] and the bidentate nitrogen-containing ligands, which afforded the cationic [Rh(COD)(TMEDA)]+ rhodium species associated to the anionic [RhCl2(COD)] moiety (32). The carbonyl rhodium species [Rh(CO)2(TMEDA)]-[RhCl2(CO)2] 33 were obtained under a CO atmosphere (Scheme 24a).47 This rhodium carbonyl complex also showed high activity and regioselectivity in the hydroformylation of various substituted styrenes under mild conditions.47
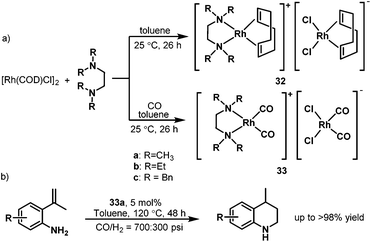 |
| | Scheme 24 Preparation of the rhodium complexes 32–32 and the intramolecular hydroaminomethylation. | |
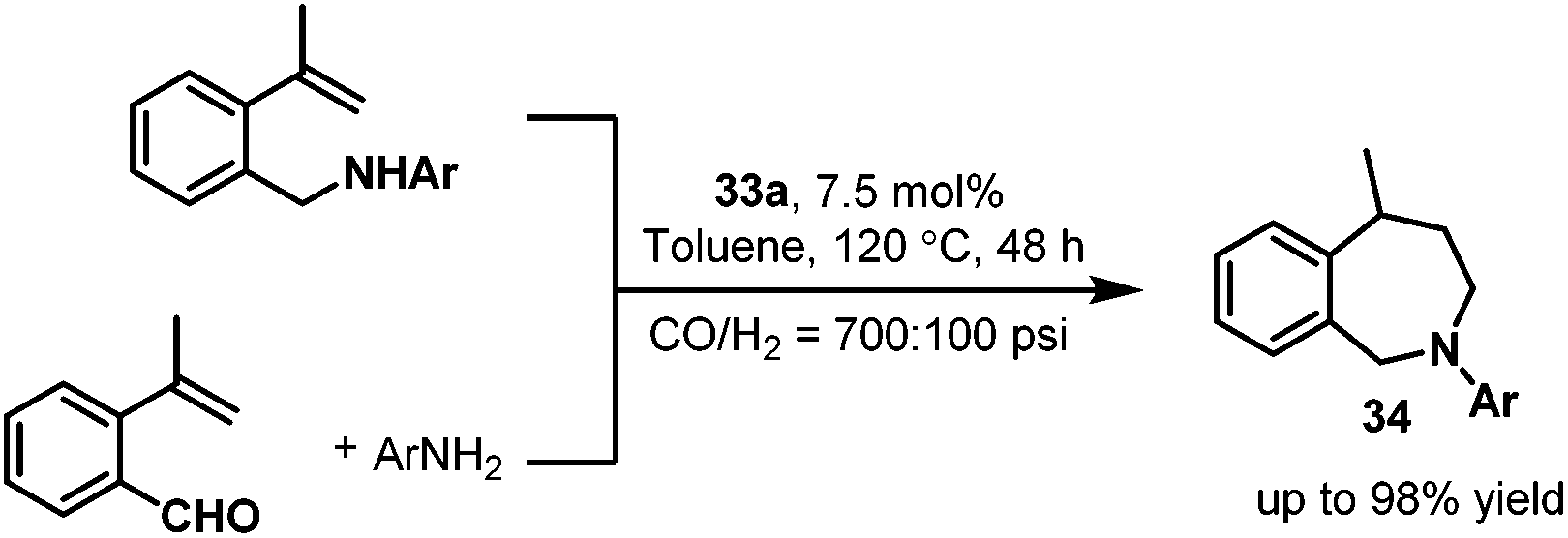
Later, in 2008 and 2010, the nitrogen-containing ligands modified rhodium carbonyl species 33 were applied by the same group to the synthesis of the seven-member-ring 2-benzazepines 34,48 which have proven to be interesting for their biological activity.49 Two synthetic routes for the synthesis of the seven-member-ring 2-benzazepines 34 were described (Scheme 25). The first route is the intramolecular hydroaminomethylation starting from isopropenylamines or allylanilines. The second route is the intermolecular hydroaminomethylation starting from 2-isopropenylbenzaldehyde and aniline derivatives. The two synthetic routes both gave the desired biologically active seven-member-ring 2-benzazepines 34 in similar high yields. Interestingly, it was found that the solvent had a significant influence on the regioselectivity.
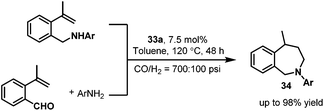 |
| | Scheme 25 Two synthetic routes for the synthesis of seven-membered-ring 2-benzazepines via hydroaminomethylation. | |
7. Asymmetric hydroaminomethylation
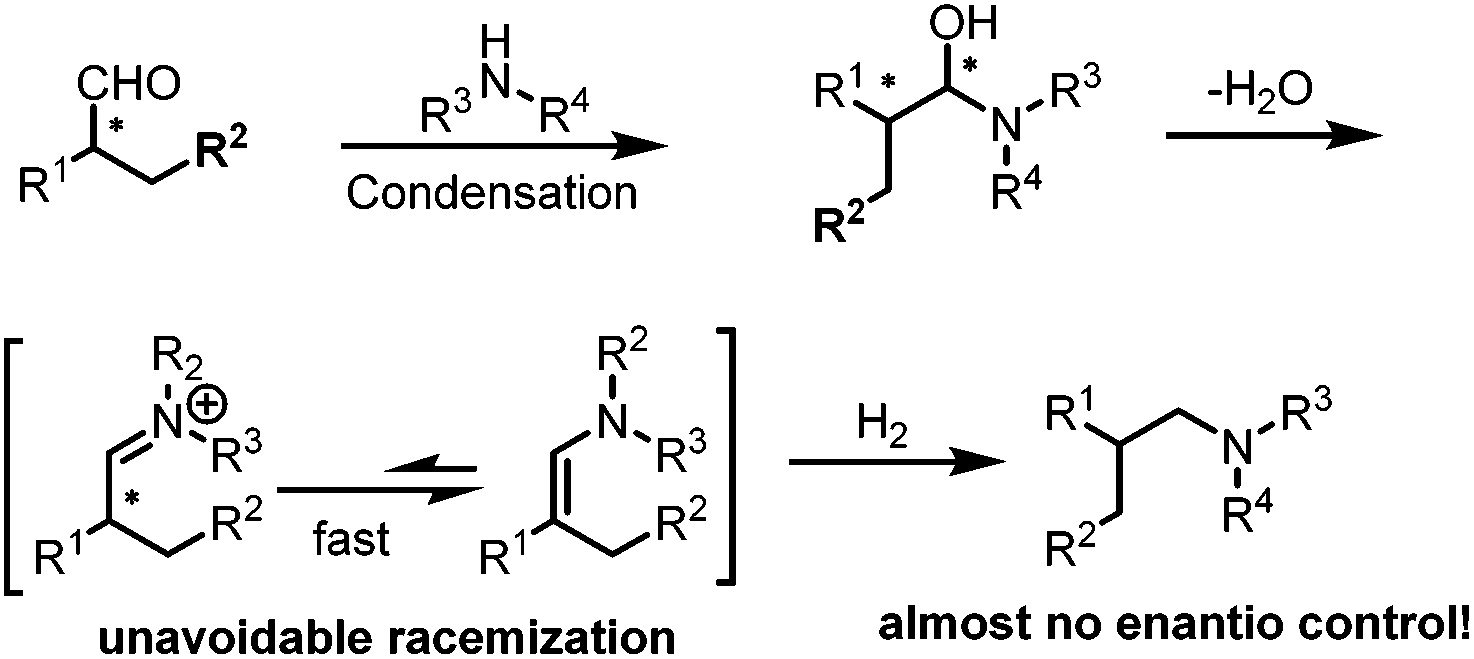
While non-enantioselective hydroaminomethylation has been intensively studied and many efficient catalytic systems have been reported with high activities and regioselectivities, in a sharp contrast, very little work has been reported in terms of asymmetric hydroaminomethylation. One of the major reasons is probably due to the unavoidable racemization of the chiral iminium intermediates (Scheme 26). Recently, Kalck et al. tried the rhodium complex modified by chiral diphosphine ligands to catalyze asymmetric HAM.50 Various chiral diphosphine ligands were introduced in the coordination sphere of neutral or cationic rhodium complexes, and the generated species efficiently catalyzed the hydroaminomethylation reaction of styrene with piperidine with good chemo- and regioselectivity. Disappointingly, only racemic amine was obtained. The results were explained by a detailed computational study which revealed that the hydrogenation of the imine intermediates follows a similar energetic pathway regardless of the chiral phosphine ligands used.
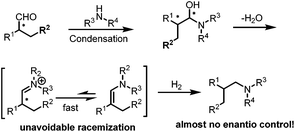 |
| | Scheme 26 Racemization of the chiral iminium intermediate. | |
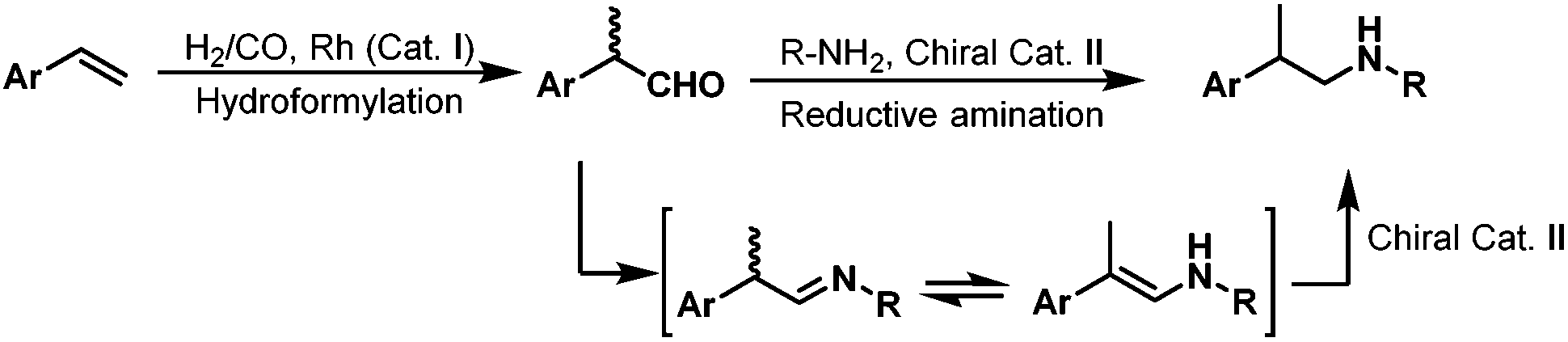
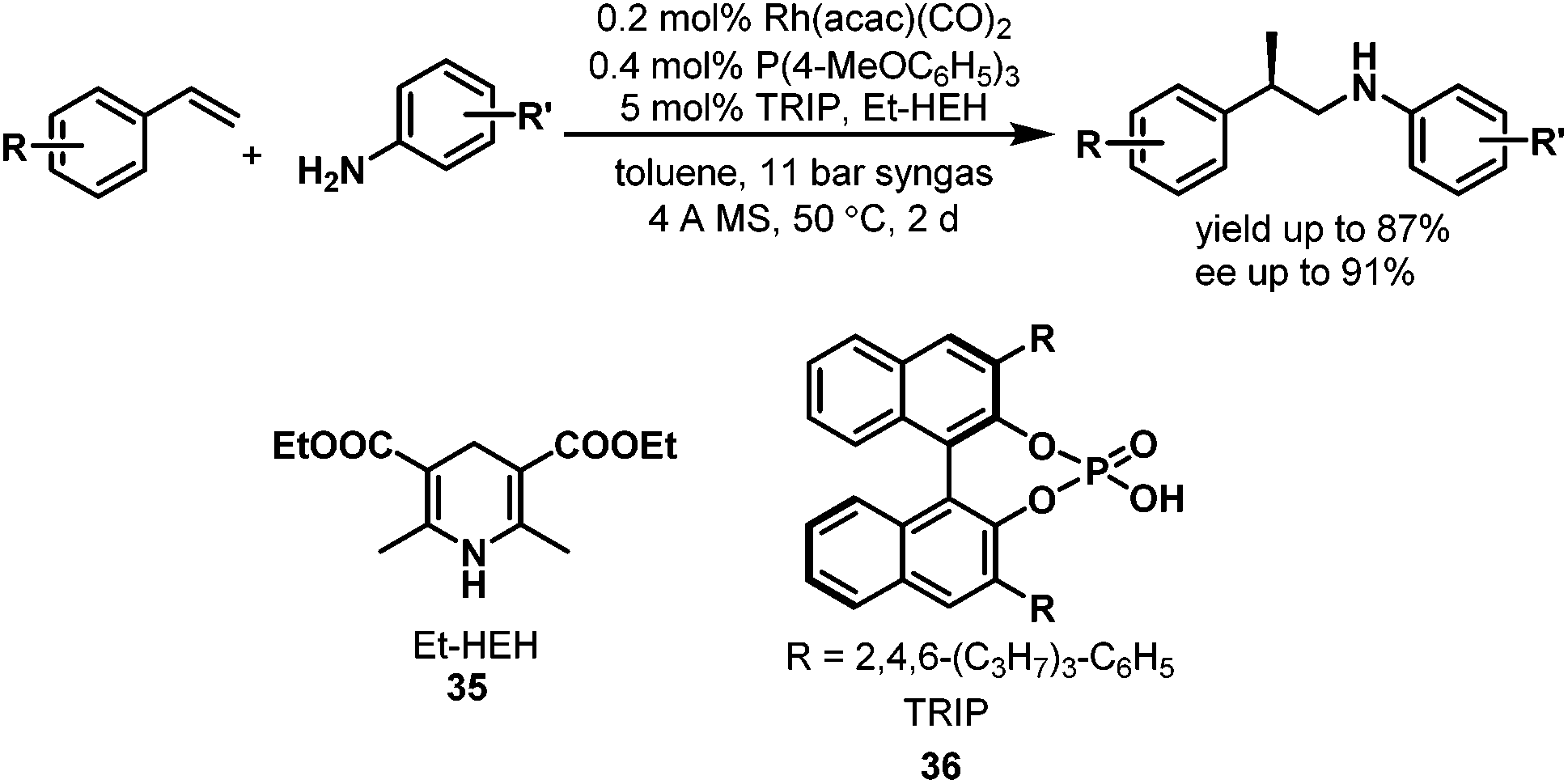
Although direct asymmetric hydroaminomethylation is extremely challenging due to the unavoidable racemization of the chiral iminium or imine intermediates, it is still possible to get the chiral amine products if the racemic iminium or imine intermediates can be hydrogenated in an enantioselective way with an appropriate chiral catalyst (Scheme 27).51 Recently, Xiao et al. reported the asymmetric hydroaminomethylation of styrenes using dual metal and organo-catalysts.51 In this dual metal and organo-catalytic system, the non-chiral rhodium catalyst modified with P(4-MeOC6H5)3 is responsible for the non-enantioselective hydroformylation giving the branched aldehyde with high regioselectivity, and the chiral organo-catalyst is responsible for the enantioselective hydrogenation of the iminium or the imine intermediates. Based on the studies of List et al.,52 a chiral phosphoric acid TRIP (36) was selected as the second catalyst and the Hantzsch ester (HEH) 35 was selected as the hydrogen source (Scheme 28), due to the excellent performance of this catalytic system in asymmetric reductive amination reactions.52 A variety of styrene derivatives were converted to the chiral amines via asymmetric hydroaminomethylation with the dual metal and organo-catalyst. Good to excellent yields (up to 87%) and ee (up to 91%) were obtained (Scheme 28). Although this hydroaminomethylation was non-direct and needed an equivalent amount of Hantzsch ester as the hydrogen source for the hydrogenation of the iminium or imine intermediates, this method is attractive and represents the first asymmetric hydroaminomethylation.
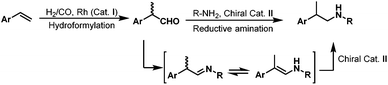 |
| | Scheme 27 Asymmetric hydroaminomethylation with two catalysts. | |
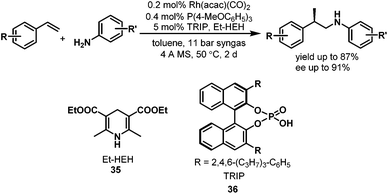 |
| | Scheme 28 Asymmetric hydroaminomethylation of styrene derivatives with two different catalysts. | |
8. Conclusion
Since the first discovery of hydroaminomethylation by Reppe et al. at BASF, considerable progress has been made in the past decades. Among the metals used in hydroaminomethylation, rhodium was found to be more active and selective to catalyze both the hydroformylation and the hydrogenation steps. Significant efforts have been made in the unmodified rhodium-catalyzed hydroaminomethylations. In order to achieve better activity and selectivity, phosphine ligands have been introduced into the rhodium catalytic system. Monophosphorus, bisphosphorus and tetraphosphorus ligands have been developed for highly selective hydroaminomethylations. Compared with the corresponding mono- and bisphosphorus ligands, the tetraphosphorus ligands exhibited better regioselectivities due to the multiple chelating modes leading to stronger chelating ability. Apart from the phosphine ligands, some carbene and nitrogen-containing ligands were also reported for rhodium-catalyzed hydroaminomethylations. The carbene and nitrogen-containing ligands seemed to facilitate the hydrogenation of the iminium or the imine intermediates, affording good activities. Although the hydroaminomethylation reaction has been intensively studied in the past decades, the generality and selectivity of this method still need to be improved. Furthermore, few reports have been reported in terms of the asymmetric hydroaminomethylation, probably due to the racemization of the chiral iminium or imine intermediates. Direct asymmetric hydroaminomethylation with chiral phosphines gives only racemic products. Good results were reported with two different catalysts, which is promising for asymmetric hydroaminomethylation.
Acknowledgements
We are grateful for financial support by the grant from Wuhan University (203273463, 203410100064), “111” Project of the Ministry of the Education of China and the National Natural Science Foundation of China (Grant No. 21372179, 21432007, 21502145).
Notes and references
-
(a)
J. March, Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, Wiley, New York, 4th edn, 1992, p. 768 Search PubMed;
(b)
J. E. McMurry, Organic Chemistry, Wadsworth, Belmont, 3rd edn, 1992 Search PubMed;
(c)
K. Eller, E. Henkes, R. Rossbacher and H. Hoke, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2005 Search PubMed;
(d) T. E. Muller and M. Beller, Chem. Rev., 1998, 98, 675–703 CrossRef PubMed;
(e) Y. Yamamoto and U. Radhakrishnan, Chem. Soc. Rev., 1999, 28, 199–207 RSC.
-
K. Eller, E. Henkes, R. Rossbacher and H. Hoke, Amines, Aliphatic, in Ullman's Encycl. Ind. Chem, Wiley-VCH, Weinheim, 2012, vol. 2, pp. 2–14 Search PubMed.
- M. Ahmed, A. M. Seayad, R. Jackstell and M. Beller, J. Am. Chem. Soc., 2003, 125, 10311–10318 CrossRef CAS PubMed.
- J.-C. Wasilke, S. J. Obrey, R. T. Baker and G. C. Bazan, Chem. Rev., 2005, 105, 1001–1020 CrossRef CAS PubMed.
- W. Reppe and H. Vetter, Liebigs Ann. Chem., 1953, 582, 133–163 CrossRef CAS.
-
A. T. Larson, US2497310, 1950 Search PubMed;
Chem. Abstr.
1950
44
4489
Search PubMed.
- K. Matrata, A. Matsuda and T. Matsuda, J. Mol. Catal., 1984, 23, 121 CrossRef.
- D. Crozet, M. Urrutigoity and P. Kalck, ChemCatChem, 2011, 3, 1102–1118 CrossRef CAS.
-
(a) P. Eilbracht, L. Barfacker, C. Buss, C. Hollmann, B. E. Kitsos-Rzychon, C. L. Kranemann, T. Rische, R. Roggenbuck and A. Schmidt, Chem. Rev., 1999, 99, 3329–3365 CrossRef CAS PubMed;
(b)
P. Eilbracht and A. M. Schmidt, in Transition Metals for Oganic Synthesis, ed. M. Beller and C. Bolm, Wiley-VCH, Weinheim, 2nd edn, 2004, vol. 1, pp. 57–85 Search PubMed;
(c) P. Eilbracht and A. M. Schmidt, Top. Organomet. Chem., 2006, 18, 65–95 CrossRef CAS;
(d) F. Ungvary, Coord. Chem. Rev., 1996, 147, 547–570 CrossRef CAS.
- A. F. M. Iqbal, Helv. Chim. Acta, 1971, 54, 1440 CrossRef CAS.
- C. L. Kranemann and P. Eilbracht, Eur. J. Org. Chem., 2000, 2367–2377 CrossRef CAS.
- A. Schmidt, M. Marchetti and P. Eilbracht, Tetrahedron, 2004, 60, 11487–11492 CrossRef CAS.
- M. A. Subhani, K.-S. Mller and P. Eilbracht, Adv. Synth. Catal., 2009, 351, 2113–2123 CrossRef CAS.
- B. Zimmermann, J. Herwig and M. Beller, Angew. Chem., Int. Ed., 1999, 38, 2732–2735 CrossRef.
- J. R. Briggs, J. Klosin and G. T. Whiteker, Org. Lett., 2005, 7, 4795–4798 CrossRef CAS PubMed.
- K. C. B. Oliveira, S. N. Carvalho, M. F. Duarte, E. V. Gusevskaya, E. N. dos Santos, J. El Karroumi, M. Gouygou and M. Urrutigoïty, Appl. Catal., A, 2015, 497, 10–16 CrossRef CAS.
- J. A. Fuentes, P. Wawrzyniak, G. J. Roff, M. Buhl and M. L. Clarke, Catal. Sci. Technol., 2011, 1, 431–436 CAS.
-
(a) A. Seayad, M. Ahmed, H. Klein, R. Jackstell, T. Gross and M. Beller, Science, 2002, 297, 1676–1678 CrossRef CAS PubMed;
(b) J. F. Hartwig, Science, 2002, 297, 1653–1654 CrossRef CAS PubMed.
- M. Ahmed, A. Seayad, R. Jackstell and M. Beller, J. Am. Chem. Soc., 2003, 125, 10311–10318 CrossRef CAS PubMed.
- M. Ahmed, R. P. J. Bronger, R. Jackstell, P. C. J. Kamer, P. W. N. M. van Leeuwen and M. Beller, Chem. – Eur. J., 2006, 12, 8979–8988 CrossRef CAS PubMed.
-
P. C. J. Kamer, J. N. H. Reek and P. W. N. M. van Leeuwen, in Rhodium Catalyzed Hydroformylation, ed. P. W. N. M. van Leeuwen and C. Claver, Kluwer Academic Publisher, 2000, pp. 35–62 Search PubMed.
-
W. Ahlers, R. Paciello, D. Vogt and P. Hofmann, WO02/083695A1, BASF AG, 2002 Search PubMed.
-
(a) C. A. Tolman, J. Am. Chem. Soc., 1970, 92, 2953–2956 CrossRef CAS;
(b)
Rhodium Catalyzed Hydroformylation, ed. P. W. N. M. van Leeuwen and C. Claver, Kluwer Acad. Pub., 2000 Search PubMed.
-
(a) C. Botteghi, L. Cazzolato, M. Marchetti and S. Paganelli, J. Org. Chem., 1995, 60, 6612–6615 CrossRef CAS;
(b) T. Rische and P. Eilbracht, Tetrahedron, 1999, 55, 1915–1920 CrossRef CAS.
- L. Routaboul, C. Buch, H. Klein, R. Jackstell and M. Beller, Tetrahedron Lett., 2005, 46, 7401–7405 CrossRef CAS.
- Y. Sun, M. Ahmed, R. Jackstell, M. Beller and W. R. Thiel, Organometallics, 2004, 23, 5260–5267 CrossRef CAS.
- G. Angelovski and P. Eilbracht, Tetrahedron, 2003, 59, 8265–8274 CrossRef CAS.
-
(a)
E. Billig, A. G. Abatjoglou and D. R. Bryant, US Pat, 4769498, 1988 Search PubMed;
(b) G. D. Cuny and S. L. Buchwald, J. Am. Chem. Soc., 1993, 115, 2066–2068 CrossRef CAS.
- D. J. Bergmann, E. M. Campi, W. R. Jackson, A. F. Patti and D. Saylik, Tetrahedron Lett., 1999, 40, 5597–5600 CrossRef CAS.
- K. S. Lee, J. Pharm. Exp. Theor., 1992, 262, 99–108 CAS.
- A. Inoue, S. Miki, M. Seto, T. Kikuchi, S. Morita, H. Hueda, Y. Misu and Y. Nakata, Eur. J. Pharmacol., 1997, 321, 105–111 CrossRef PubMed.
-
(a) J. R. Briggs, J. Klosin and G. T. Whiteker, Org. Lett., 2005, 7, 4795–4798 CrossRef CAS PubMed;
(b)
J. R. Briggs, G. T. Whiteker and J. Klosin, Union Carbide chemicals & Plastics Technology Corporation, Patent WO2005/077884A2, 2005 Search PubMed.
-
(a) G. T. Whiteker, Top. Catal., 2010, 53, 1025–1030 CrossRef CAS;
(b)
J. R. Briggs, G. T. Whiteker and J. Klosin, Dow Global Technnologies, Inc., Patent US7220884B2, May, 2007 Search PubMed.
-
(a) Y. Yan, X. Zhang and X. Zhang, J. Am. Chem. Soc., 2006, 128, 16058–16061 CrossRef CAS PubMed;
(b) Y. Yan, X. Zhang and X. Zhang, Adv. Synth. Catal., 2007, 349, 1582–1586 CrossRef CAS;
(c) S. Yu, Y. Chie, Z. Guan and X. Zhang, Org. Lett., 2008, 10, 3469–3472 CrossRef CAS PubMed;
(d) S. Yu, X. Zhang, Y. Yan, C. Cai, L. Dai and X. Zhang, Chem. – Eur. J., 2010, 16, 4938–4943 CrossRef CAS PubMed.
- G. Liu, K. Huang, C. Cai, B. Cao, M. Chang, W. Wu and X. Zhang, Chem. – Eur. J., 2011, 17, 14559–14563 CrossRef CAS PubMed.
- G. Liu, K. Huang, B. Cao, M. Chang, S. Li, S. Yu, L. Zhou, W. Wu and X. Zhang, Org. Lett., 2012, 14, 102–105 CrossRef CAS PubMed.
- S. Li, K. Huang, J. Zhang, W. Wu and X. Zhang, Org. Lett., 2013, 15, 3078–3081 CrossRef CAS PubMed.
- G. Liu, Z. Li, H. Geng and X. Zhang, Catal. Sci. Technol., 2014, 4, 917–921 CAS.
-
(a) L. Jafarpour and S. P. Nolan, Adv. Organomet. Chem., 2001, 46, 181–222 CrossRef;
(b) W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290–1309 CrossRef CAS;
(c) A. C. Hillier and S. P. Nolan, Platinum Met. Rev., 2002, 46, 50–64 CAS.
-
(a) A. C. Chen, L. Ren, A. Decken and C. M. Crudden, Organometallics, 2000, 19, 3459–3461 CrossRef CAS;
(b) C. Kocher and W. A. Herrmann, J. Organomet. Chem., 1997, 532, 261–265 CrossRef;
(c)
W. A. Herrmann, M. Elison, J. Fischer and C. Kocher, US5663451, 1997 Search PubMed.
- A. Seayad, K. Selvakumar, M. Ahmed and M. Beller, Tetrahedron Lett., 2003, 44, 1679–1683 CrossRef CAS.
- M. Ahmed, C. Buch, L. Routaboul, R. Jackstell, H. Klein, A. Spannenberg and M. Beller, Chem. – Eur. J., 2007, 13, 1594–1601 CrossRef CAS PubMed.
-
(a)
A. Kleemann and J. Engel, Pharmazeutische Wirkstoffe, Thieme, Stuttgart, 1987 Search PubMed;
(b) H. Certa and G. Blaschke, Arch. Pharm., 1990, 323, 744–747 Search PubMed;
(c) H. M. Piper, J. F. Hutler and P. G. Spieckermann, Arzneim.-Forsch., 1985, 35, 1495–1498 CAS.
-
(a) T. Rische and P. Eilbracht, Tetrahedron, 1999, 55, 1915–1920 CrossRef CAS;
(b) C. Botteghi, L. Cazzolato, M. Marchetti and S. Paganelli, J. Org. Chem., 1995, 60, 6612–6615 CrossRef CAS;
(c) M. Ahmed, A. M. Seayad, R. Jackstell and M. Beller, J. Am. Chem. Soc., 2003, 125, 10311–10318 CrossRef CAS PubMed;
(d) S. Li, K. Huang, J. Zhang, W. Wu and X. Zhang, Org. Lett., 2013, 15, 1036–1039 CrossRef CAS PubMed.
- E. Teuma, M. Loy, C. Le Berre, M. Etienne, J.-C. Daran and P. Kalck, Organometallics, 2003, 22, 5261–5267 CrossRef CAS.
- T. O. Vieira and H. Alper, Chem. Commun., 2007, 2710–2711 RSC.
- J. J. Kim and H. Alper, Chem. Commun., 2005, 3059–3061 RSC.
-
(a)
J. R. Briggs, G. T. Whiteker and J. Klosin, Dow Global Technnologies, Inc., Patent US7220884B2, May, 2007 Search PubMed;
(b) H. Alper and T. O. Vieira, Org. Lett., 2008, 10, 485–487 CrossRef PubMed;
(c) K. Okuro and H. Alper, Tetrahedron Lett., 2010, 51, 4959–4961 CrossRef CAS.
- V. Kouznetsov, A. Palma and C. Ewert, Curr. Org. Chem., 2001, 5, 519–551 CrossRef CAS.
- D. Crozet, C. E. Kefalidis, M. Urrutigoïty, L. Maron and P. Kalck, ACS Catal., 2014, 4, 435–447 CrossRef CAS.
- B. Villa-Marcos and J. Xiao, Chin. J. Catal., 2015, 36, 106–112 CrossRef CAS.
- S. Hoffmann, M. Nicoletti and B. List, J. Am. Chem. Soc., 2006, 128, 13074–13075 CrossRef CAS PubMed.
Footnote |
| † This review is for celebrating the 75th birthday of Prof. Barry Trost. |
|
| This journal is © the Partner Organisations 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.