
Copper(II)-catalyzed methoxylation of unactivated (hetero)aryl C–H bonds using a removable bidentate auxiliary†
Xue-Song
Yin
a,
Yi-Chen
Li
a,
Jun
Yuan
a,
Wen-Jia
Gu
a and
Bing-Feng
Shi
*ab
aDepartment of Chemistry, Zhejiang University, Hangzhou 310027, China. E-mail: bfshi@zju.edu.cn
bState Key Laboratory of Bioorganic & Natural Products Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, China
First published on 17th December 2014
Abstract
A copper(II)-catalyzed methoxylation of unactivated (hetero)aryl C–H bonds directed by our newly developed PIP directing group with methanol has been achieved. This protocol occurs efficiently with a catalytic amount of copper (5 mol%), is a simple and environmentally benign reaction system, has a broad substrate scope and high functional group tolerance.
Aryl ethers are important structural motifs because of their ubiquity in biologically active molecules, such as natural products, pharmaceuticals and pesticides.1 Among the methods for access to aryl ethers, transition-metal-catalyzed C(sp2)–O bond formation reactions, such as the Buchwald–Hartwig coupling of aryl halides with alcohols,2 and the Chan–Evans–Lam coupling of boronic acids with alcohols, have been studied extensively.3 These methods allow the incorporation of ether functionality into functionalized molecules under mild conditions. However, these methods still need the use of prefunctionalized starting materials, such as aryl halides or boronic acids, and generate stoichiometric wastes.
Recently, ether formation via transition-metal-catalyzed direct alkoxylation of unactivated C–H bonds with alcohols has received tremendous interest in terms of atom and step economy.4 A number of research groups have made considerable progress in palladium-catalyzed alkoxylation of arenes employing pyridyl,5a oxime,5bN-methoxybenzamide,6a cyano,7 anilide,6bN-(quinolin-8-yl)benzamide,8N-tosylcarboxamide,9N-(2-(pyridin-2-yl)isopropyl)benzamide,10 and triazole11 as directing groups. Despite significant success in this area, the direct alkoxylation of arenes catalyzed/mediated by cheap, abundant copper catalysts remains largely undeveloped.12–18 In 2006, the pioneering work by Yu demonstrated the first aryloxylation of 2-phenylpyridine with 4-cyanophenol.13 Zhu's group reported the intramolecular aryloxylation of electron-deficient phenols.14 Song and Hao reported a copper-mediated mono- and diaryloxylation of benzamides using a N,O-bidentate directing group.15 However, these protocols were restricted to the formation of biaryl ethers. In 2013, Goossen and coworkers reported the synthesis of aryl ethers via copper-catalyzed ortho-alkoxylation of benzoic acids followed by protodecarboxylation.16a Shortly after, the same group reported the copper/silver-mediated alkoxylation of 2-arylpyridine with alcohols.16b However, the requirement of stoichiometric amounts of expensive silver salts and the use of an unremovable directing group hampered the synthetic application of this protocol. Stahl reported a mechanistic study of the methoxylation of N-(8-quinolinyl)benzamides with 2 equivalents of Cu(OAc)2.17 More recently, Daugulis and coworkers reported the alkoxylation and aryloxylation of benzamides directed by 8-aminoquinoline with catalytic amounts of copper catalyst using O2 as the oxidant.18 However, the alkoxylation only worked well for 3-trifluoromethylbenzamide. Thus, it is highly desirable to develop an efficient C–H alkoxylation reaction with a broad substrate scope, and a simple and environmentally benign reaction system using low catalyst loading.
As a part of our continuous interest towards using the newly developed bidentate directing group derived from (pyridin-2-yl)isopropyl amine (PIP-amine),10,19 we have recently realized the copper-mediated PIP-directed C–H hydroxylation19d and sulfenylation.19e We speculated that the PIP directing group may facilitate the copper-catalyzed alkoxylation of a broad range of inert aryl C–H bonds, because the nitrogen atom on the PIP directing group is more electron-rich and sterically bulky than Daugulis's 8-aminoquinoline auxiliary.20,21 We found that it is indeed the case, and we report herein a highly efficient protocol that enables the methoxylation of benzamides bearing various functional groups and a wide range of aromatic heterocycles by applying a low loading of copper catalyst under aerobic conditions (eqn (1)).

We initiated our investigation with N-(2-(pyridin-2-yl)propan-2-yl) benzamide (1a) as a model substrate (Table 1). To our delight, the reaction was found to proceed efficiently in the presence of KOCN as a base with 10 mol% Cu(OAc)2 under O2 at 100 °C (entry 1, 75%).22 The screening of copper salts revealed that the reaction with (CuOH)2CO3 gave the best yield (entries 2–4). By increasing the reaction temperature to 120 °C, the yield was improved to 86% (entry 5). However, no obvious improvement could be observed when the reaction was conducted at 140 °C (entry 6). Further investigations showed that the reaction proceeded efficiently under an air atmosphere and the amount of (CuOH)2CO3 could be reduced to 5 mol% (entry 8, 86% isolated yield). Thus, this is a simple and environmentally benign catalyst system for the synthesis of methoxyarenes.
|
|
||||
|---|---|---|---|---|
| Entry | [Cu] (X mol%) | KOCN (Y equiv.) | T (°C)/atmosphere | Yieldb (%) |
| a Reaction conditions: 1a (0.15 mmol), [Cu] (X mol%), KOCN (Y equiv.), in MeOH (2 mL). b 1H NMR yield using CH2Br2 as the internal standard. c Isolated yields. | ||||
| 1 | Cu(OAc)2 (10) | 1.5 | 100 °C/O2 | 75 |
| 2 | CuBr2 (10) | 1.5 | 100 °C/O2 | 69 |
| 3 | Cu(OTf)2 (10) | 1.5 | 100 °C/O2 | 70 |
| 4 | (CuOH)2CO3 (10) | 1.5 | 100 °C/O2 | 78 |
| 5 | (CuOH)2CO3 (10) | 1.5 | 120 °C/O2 | 86 |
| 6 | (CuOH)2CO3 (10) | 3.0 | 140 °C/O2 | 72 |
| 7 | (CuOH)2CO3 (10) | 3.0 | 120 °C/Air | 84 |
| 8 | (CuOH)2CO3 (5) | 1.5 | 120 °C/Air | 88 (86) |

We then investigated the efficiency of other directing groups under our optimized reaction conditions (Scheme 1). No reaction occurred when 3a was used as the substrate, indicating that the gem-dimethyl substitution is essential for the reaction to proceed.10 8-Aminoquinoline (3b) was found to be less efficient under current reaction conditions (47% yield). Other directing groups, such as 2-thiomethylaniline (3c),20dN-methoxyamide (3d) and N-arylamide (3e), which were widely used in Pd-catalyzed C–H functionalization reactions, failed to give the desired products either.
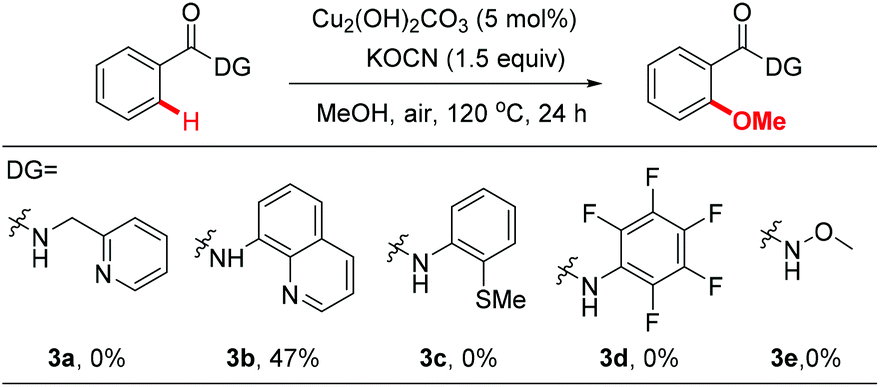 | ||
| Scheme 1 Directing groups for methoxylation of benzamide. | ||
The reaction scope with respect to benzamide substrates under the optimized conditions is presented in Table 2. A variety of functional groups, such as OMe (1c and 1q), F (1d, 1i and 1n), Cl (1e and 1o), Br (1f), CF3 (1g, 1j and 1p) and NO2 (1h), were tolerated under the optimized conditions and the reaction proceeded with moderate to high yields. The reaction protocol was sensitive to steric hindrance, since ortho-substituted substrates generally gave lower yields (1i–1m). Due to the strong electron-withdrawing nature of the nitro functional group, 2h was obtained in moderate yield and no monomethoxylated product was detected. Methoxylation of meta-substituted substrates proceeded exclusively at the less hindered C–H bonds, irrespective of the electronic nature of the substituents (2n–2r). However, 2,3-dihydrobenzo[1,4]dioxine-6-carboxamides 1t led to a mixture of methoxylation at C2 and C6 positions, indicating the coordinating effect of the dioxin group in stabilizing the copper intermediate.19d The methoxylation structure was confirmed unambiguously by X-ray analysis of products 2p and 2s (Fig. 1).23 Finally, the reaction of 1a with ethanol gave the ethoxylated product 2u in 50% yield; however, alkoxylation with other alcohols gave no encouraging results.
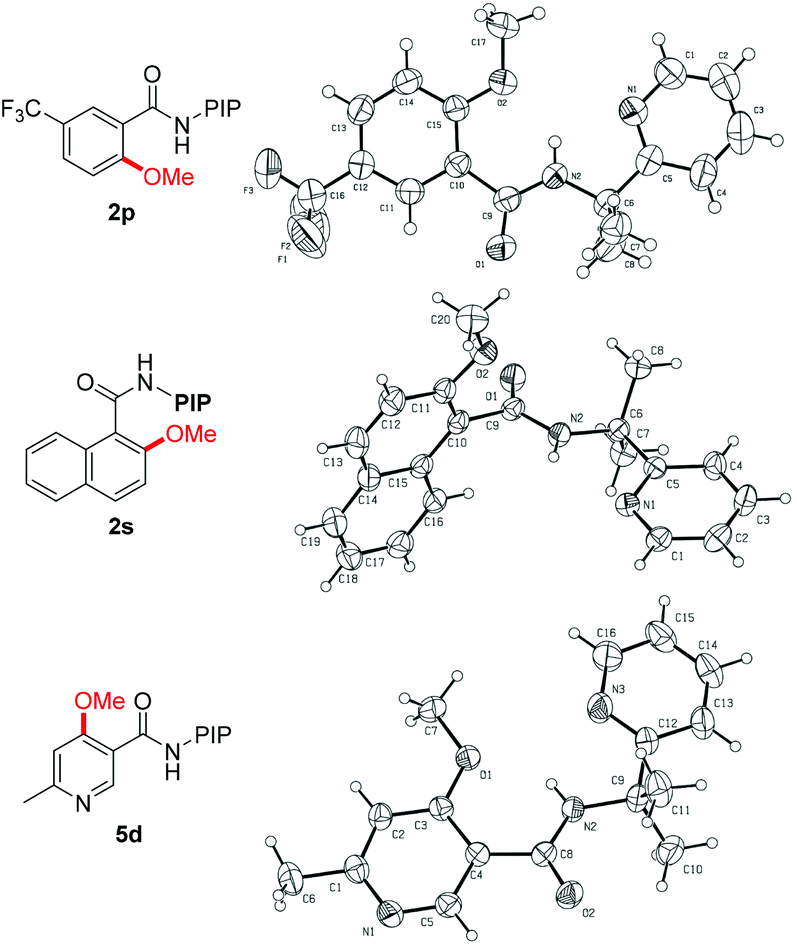 | ||
| Fig. 1 X-ray analysis of products 2p, 2s and 5d. | ||

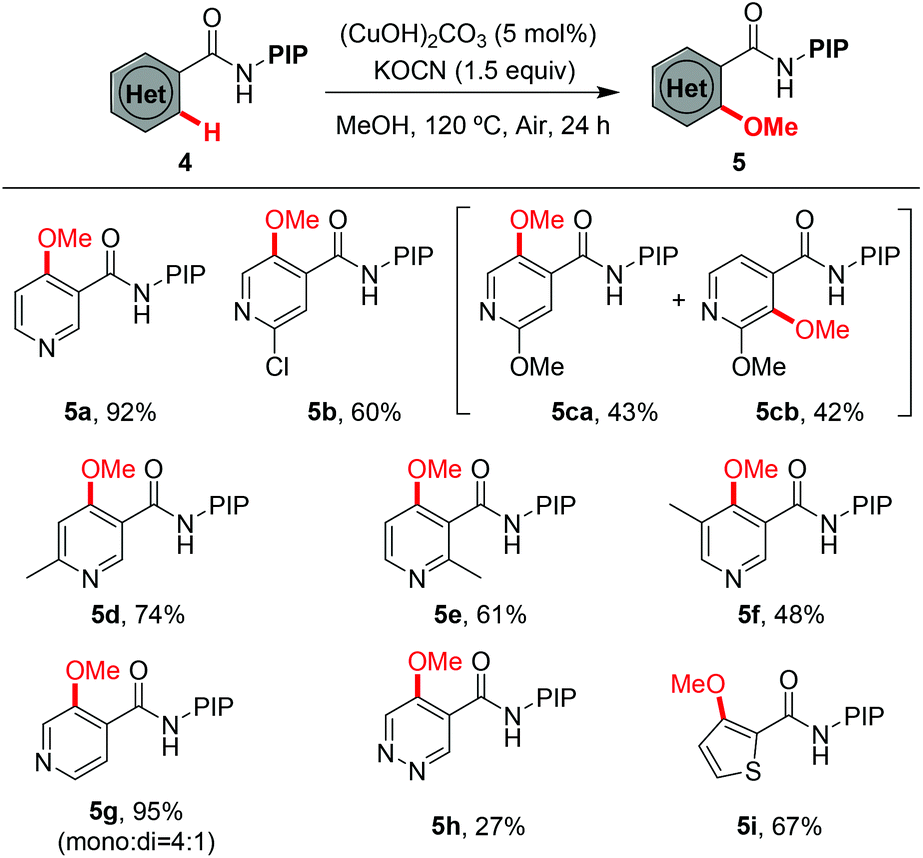
Encouraged by the excellent results from the functionalized benzamides, we then tested the scope of carboxamides with heteroarenes, in view of the importance of heterocycles in pharmaceuticals (Table 3). We were delighted to find that the optimized conditions were compatible with a wide range of heteroarenes. Consistent with copper-mediated hydroxylation that we have reported recently,19d we also found that the methoxylation of nicotinamides bearing various substituents reacted exclusively at the C-4 position, even with the sterically bulky group such as methyl (5d–5f). The regioselectivity of the product was unambiguously confirmed by X-ray analysis of product 5d (Fig. 1).23 These observations revealed that the C(4)–H bond is more reactive than the C(2)–H ones in the methoxylation protocol. Therefore, a mechanistic pathway identical to the copper-mediated hydroxylation reaction may be involved. Isonicotinamides (4b–4c and 4g), pyridazine (4h) and 2-thiophenecarboxamide (4i) were also tolerated under the reaction conditions.
Finally, the PIP directing group can be removed under mild conditions,10 indicating the synthetic versatility of this reaction. As shown in Scheme 2, a mild nitrosylation/hydrolysis sequence was conducted and gave 2-methoxybenzoic acid 6 in 77% yield.
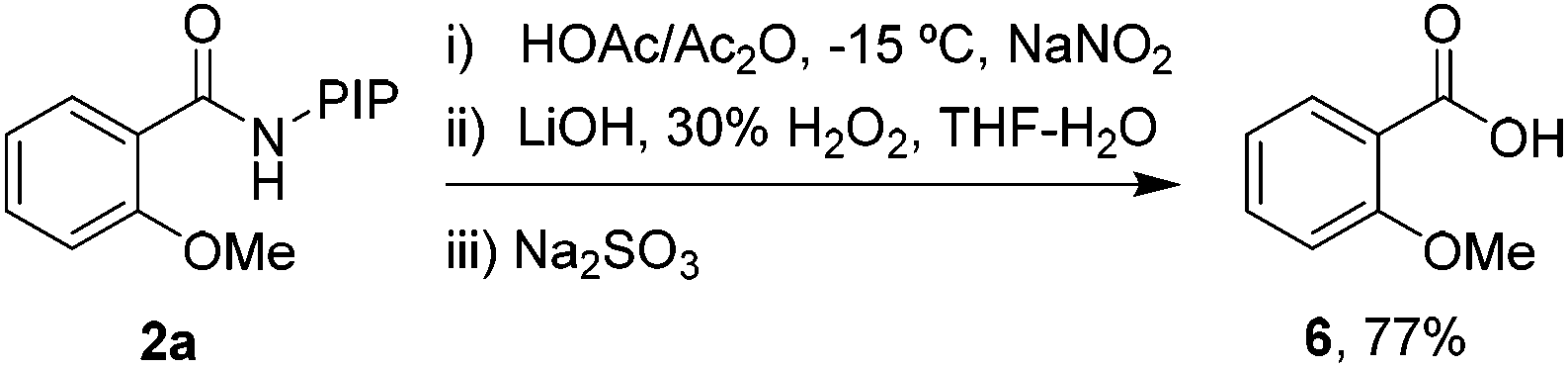 | ||
| Scheme 2 Removal of the directing group. | ||
A series of experiments were conducted to gain further insight into the reaction mechanism (Scheme 3). Addition of 0.5 equivalent of radical scavengers, such as 1,4-dinitrobenzene, 1,1-diphenylethylene and 2,6-di-tert-butyl-4-methylphenol, substantially reduced the yield but did not completely suppress the reaction (Scheme 3c). These data suggest that the methoxylation reaction does not proceed via a radical mechanism. The intermolecular KIE for methoxylation reaction is 3.0 while the intramolecular KIE is 4.5. The large KIE value ruled out the possibility of a simple electrophilic aromatic substitution (SEAr) pathway and suggested that C–H cleavage is either the turnover-limiting step or the turnover-limiting step occurs prior to C–H cleavage.24
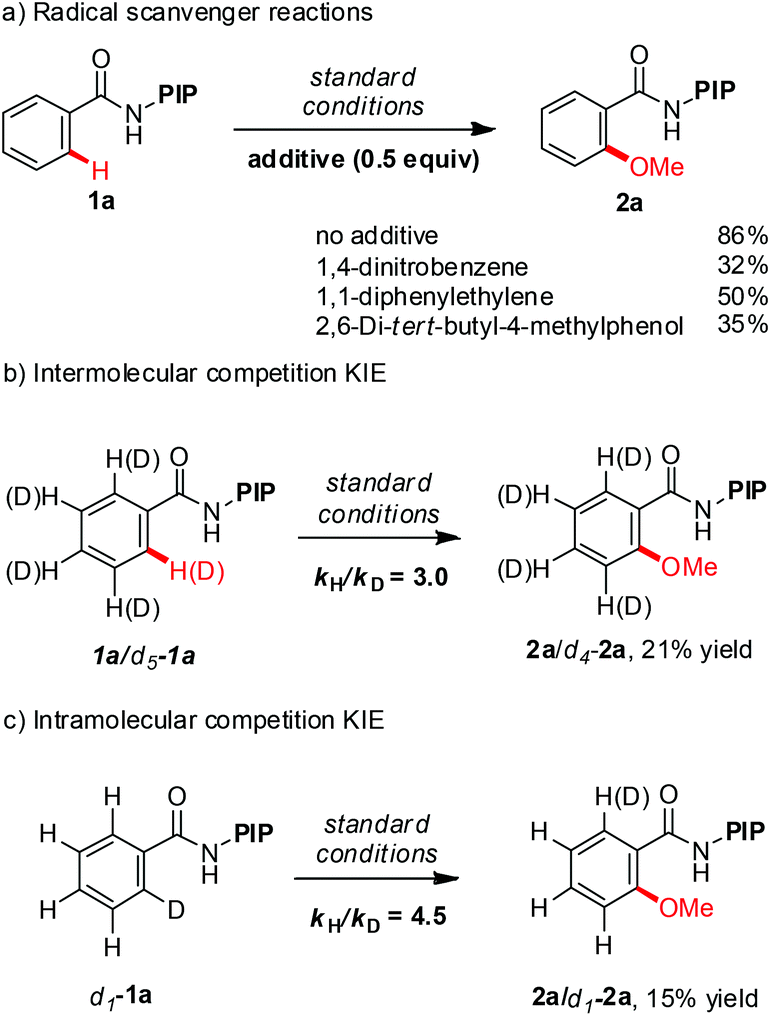 | ||
| Scheme 3 Mechanistic studies. | ||
A plausible mechanism is proposed in Fig. 2 on the basis of these preliminary mechanistic studies and earlier precedents.17,25 Complexation of benzamide 1a to the copper catalyst followed by disproportionative C–H activation affords the pincer type Cu(III) intermediate B. The Cu(III) intermediate B undergoes reductive elimination to give intermediate C, which was followed by protonation to generate the desired product 2a and Cu(I). The copper(I) species that was generated by disproportionative C–H activation and reductive elimination could be oxidized to Cu(II) by air. The use of air as the sole oxidant represents an important advancement in C–H alkoxylation reaction.
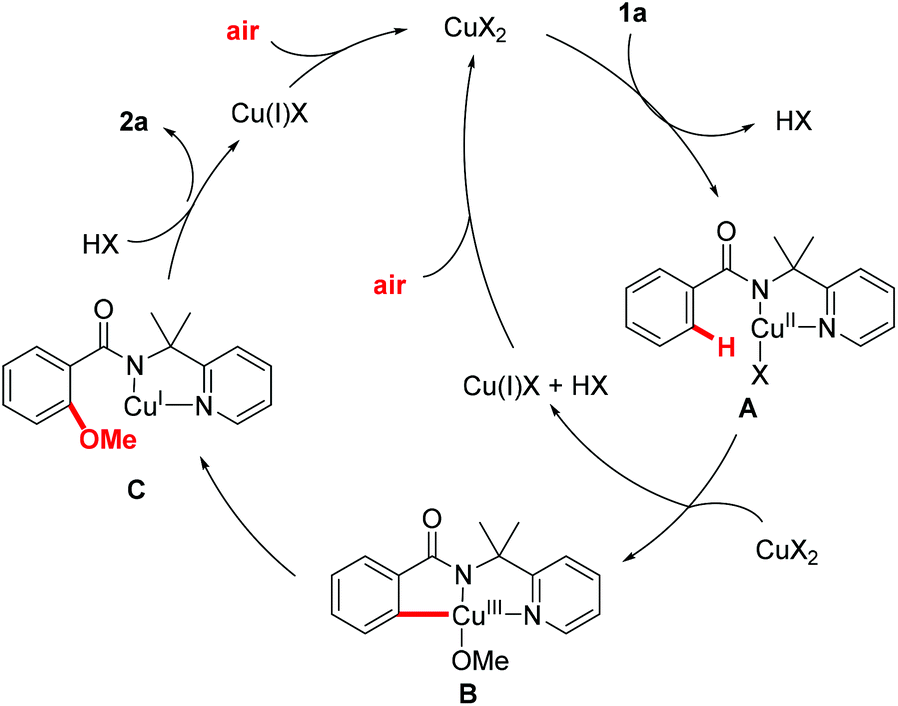 | ||
| Fig. 2 Plausible reaction mechanism. | ||
In conclusion, we have developed a copper(II)-catalyzed C–H methoxylation of arenes and heteroarenes using our newly developed PIP directing group. The reaction proceeded efficiently with low catalyst loading of copper salt under aerobic conditions. Moreover, this protocol features a broad substrate scope, high functionality tolerance and compatibility with various biologically important heteroarenes.
Acknowledgements
Financial support from the National Basic Research Program of China (2015CB856600), the NSFC (21422206, 21272206, 21002087 and J1210042), and the Fundamental Research Funds for the Central Universities (2014QNA3008) is gratefully acknowledged.Notes and references
- (a) S. Ohkawa, K. Fukatsu, S. Miki, T. Hashimoto, J. Sakamoto, T. Doi, Y. Nagai and T. Aono, J. Med. Chem., 1997, 40, 559 CrossRef CAS PubMed; (b) M. Esteki and T. Khayamian, Chem. Biol. Drug Des., 2008, 72, 409 CrossRef CAS PubMed; (c) K. Kubo, Y. Kohara, E. Imamiya, Y. Sugiura, Y. Inada, Y. Furukawa, K. Nishikawa and T. Naka, J. Med. Chem., 1993, 36, 2182 CrossRef CAS.
- For selected reviews, see: (a) J. F. Hartwig, Acc. Chem. Res., 1998, 31, 852 CrossRef CAS; (b) J. F. Hartwig, Angew. Chem., Int. Ed., 1998, 37, 2046 CrossRef CAS; (c) A. R. Muci and S. L. Buchwald, Top. Curr. Chem., 2002, 219, 131 CrossRef CAS; (d) F. Monnier and M. Taillefer, Angew. Chem., Int. Ed., 2009, 48, 6954 CrossRef CAS PubMed.
- For selected reviews, see: (a) J. X. Qiao and P. Y. S. Lam, Synthesis, 2011, 829 CrossRef CAS PubMed; (b) A. W. Thomas and S. V. Ley, in Modern Arylation Methods, ed. L. Ackermann, Wiley-VCH, Weinheim, Germany, 2009, p. 121 Search PubMed.
- For representative reviews, see: (a) S. Enthaler and A. Company, Chem. Soc. Rev., 2011, 40, 4912 RSC; (b) S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936 CrossRef CAS PubMed.
- (a) A. R. Dick, K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2004, 126, 2300 CrossRef CAS PubMed; (b) L. V. Desai, H. A. Malik and M. S. Sanford, Org. Lett., 2006, 8, 1141 CrossRef CAS PubMed.
- (a) T.-T. Yuan and G.-W. Wang, J. Org. Chem., 2010, 75, 476 CrossRef PubMed; (b) T.-S. Jiang and G.-W. wang, J. Org. Chem., 2012, 77, 9504 CrossRef CAS PubMed; (c) M. Anand and R. B. Sunoj, Org. Lett., 2011, 13, 4802 CrossRef CAS PubMed.
- (a) W. Li and P. Sun, J. Org. Chem., 2012, 77, 8362 CrossRef CAS PubMed; (b) T. Gao and P. Sun, J. Org. Chem., 2014, 79, 9888 CrossRef CAS PubMed.
- S.-Y. Zhang, G. He, Y. Zhao, K. Wright, W. A. Nack and G. Chen, J. Am. Chem. Soc., 2012, 134, 7313 CrossRef CAS PubMed.
- F. Peron, C. Fossey, J. S. O. Santos, T. Cailly and F. Fabis, Chem. – Eur. J., 2014, 20, 7507 CrossRef CAS PubMed.
- F.-J. Chen, S. Zhao, F. Hu, K. Chen, Q. Zhang, S.-Q. Zhang and B.-F. Shi, Chem. Sci., 2013, 4, 4187 RSC.
- S.-P. Shi and C.-X. Kuang, J. Org. Chem., 2014, 79, 6105 CrossRef CAS PubMed.
- For Pd-catalyzed cycloetherification, see: (a) X.-S. Wang, Y. Lu, H.-X. Dai and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 12203 CrossRef CAS PubMed; (b) B. Xiao, T.-J. Gong, Z.-J. Liu, J.-H. Liu, D.-F. Luo, J. Xu and L. Liu, J. Am. Chem. Soc., 2011, 133, 9250 CrossRef CAS PubMed; (c) H. Wang, G. Li, K. M. Engle, J.-Q. Yu and H. M. L. Davies, J. Am. Chem. Soc., 2013, 135, 6774 CrossRef CAS PubMed.
- X. Chen, X.-S. Hao, C. E. Goodhue and J.-Q. Yu, J. Am. Chem. Soc., 2006, 128, 6790 CrossRef CAS PubMed.
- (a) J. Zhao, Y. Wang, Y. He, L. Liu and Q. Zhu, Org. Lett., 2012, 14, 1078 CrossRef CAS PubMed; (b) J. Zhao, Q. Zhang, L. Liu, Y. He, J. Li, J. Li and Q. Zhu, Org. Lett., 2012, 14, 5362 CrossRef CAS PubMed.
- X.-Q. Hao, L.-J. Chen, B. Ren, L.-Y. Li, X.-Y. Yang, J.-F. Gong, J.-L. Niu and M.-P. Song, Org. Lett., 2014, 16, 1104 CrossRef CAS PubMed.
- (a) S. Bhadra, W. I. Dzik and L. J. Goossen, Angew. Chem., Int. Ed., 2013, 52, 2959 CrossRef CAS PubMed; (b) S. Bhadra, C. Matheis, D. Katayev and L. J. Goossen, Angew. Chem., Int. Ed., 2013, 52, 9279 CrossRef CAS PubMed; (c) S. Bhadra, W. I. Dzik and L. J. Goossen, J. Am. Chem. Soc., 2012, 134, 9938 CrossRef CAS PubMed.
- A. M. Suess, M. Z. Ertem, C. J. Cramer and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 9797 CrossRef CAS PubMed.
- J. Roane and O. Daugulis, Org. Lett., 2013, 15, 5842 CrossRef CAS PubMed.
- (a) Q. Zhang, K. Chen, W.-H. Rao, Y.-J. Zhang, F.-J. Chen and B.-F. Shi, Angew. Chem., Int. Ed., 2013, 52, 13588 CrossRef CAS PubMed; (b) Q. Zhang, X.-S. Yin, S. Zhao, S.-L. Fang and B.-F. Shi, Chem. Commun., 2014, 50, 8353 RSC; (c) Q. Zhang, K. Chen and B.-F. Shi, Synlett, 2014, 25, 1941 CrossRef CAS PubMed; (d) X. Li, Y.-H. Liu, W.-J. Gu, B. Li, F.-J. Chen and B.-F. Shi, Org. Lett., 2014, 16, 3904 CrossRef CAS PubMed; (e) F.-J. Chen, G. Liao, X. Li, J. Wu and B.-F. Shi, Org. Lett., 2014, 16, 5644 CrossRef CAS PubMed.
- For reviews and early reports on bidentate auxiliary, see: (a) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726 CrossRef CAS PubMed; (b) B. Zhang, H.-X. Guan, B. Liu and B. F. Shi, Chin. J. Org. Chem., 2014, 34, 1487 Search PubMed; (c) V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154 CrossRef CAS PubMed; (d) D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2010, 132, 3965 CrossRef CAS PubMed.
- For selected examples of Cu-mediated C–H functionalization directed by bidentate auxiliary, see: (a) L. D. Tran, I. Popov and O. Daugulis, J. Am. Chem. Soc., 2012, 134, 18237 CrossRef CAS PubMed; (b) T. Truong, K. Klimovica and O. Daugulis, J. Am. Chem. Soc., 2013, 135, 9342 CrossRef CAS PubMed; (c) M. Nishino, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2013, 52, 4457 CrossRef CAS PubMed; (d) B. Urones, Á. M. Martínez, N. Rodríguez, R. G. Arrayάs and J. Carretero, Chem. Commun., 2013, 49, 11044 RSC; (e) Z. Wang, J.-Z. Ni, Y. Kuninobu and M. Kanai, Angew. Chem., Int. Ed., 2014, 53, 3496 CrossRef CAS PubMed; (f) X. Wu, Y. Zhao, G. Zhang and H. Ge, Angew. Chem., Int. Ed., 2014, 53, 3706 CrossRef CAS PubMed; (g) Q. Li, S.-Y. Zhang, G. He, Z. Ai, W. A. Nack and G. Chen, Org. Lett., 2014, 16, 1764 CrossRef CAS PubMed; (h) M. Shang, S.-Z. Sun, H.-X. Dai and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 3354 CrossRef CAS PubMed; (i) J. Dong, F. Wang and J. You, Org. Lett., 2014, 16, 2884 CrossRef CAS PubMed.
- K. Chen and B.-F. Shi, Angew. Chem., Int. Ed., 2014, 53, 11950 CrossRef CAS PubMed.
- CCDC 1011905 (2p), 1011904 (2s) and 1012662 (5d) contain the supplementary crystallographic data for this paper.
- E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066 CrossRef CAS PubMed.
- (a) Z.-L. Wang, L. Zhao and M.-X. Wang, Org. Lett., 2012, 14, 1472 CrossRef CAS PubMed; (b) Z.-L. Wang, L. Zhao and M.-X. Wang, Org. Lett., 2011, 13, 6560 CrossRef CAS PubMed; (c) A. E. King, L. M. Huffman, A. Casitas, M. Costas, X. Ribas and S. S. Stahl, J. Am. Chem. Soc., 2010, 132, 12068 CrossRef CAS PubMed; (d) L. M. Hufman and S. S. Stahl, J. Am. Chem. Soc., 2008, 130, 9196 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and characterization data for new compounds. CCDC 1011904, 1011905 and 1012662. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qo00276h |
| This journal is © the Partner Organisations 2015 |