
The 2-phosphaethynolate anion: convenient synthesis and the reactivity
Zheng-Jun
Quan
*ab and
Xi-Cun
Wang
*ab
aKey Laboratory of Eco-Environment-Related Polymer Materials, Ministry of Education, China, Gansu 730070, P. R. China
bGansu Key Laboratory of Polymer Materials, College of Chemistry and Chemical Engineering, Northwest Normal University, Anning East Road 967, Lanzhou, Gansu 730070, P. R. China. E-mail: wangxicun@nwnu.edu.cn; quanzhengjun@hotmail.com
First published on 19th August 2014
Abstract
Since Becker and co-workers isolated the 2-phosphaethynolate anion (P![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–O−) in 1992, the phosphorus-containing analogue of the cyanate ion (NC–O−), the 2-phosphaethynolate anion has gained much attention because this anion is isoelectronic to small molecules such as O
C–O−) in 1992, the phosphorus-containing analogue of the cyanate ion (NC–O−), the 2-phosphaethynolate anion has gained much attention because this anion is isoelectronic to small molecules such as O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CS and F–CP as well as the thiocyanate anion SCN−. This paper highlights the recently reported methods for the synthesis of 2-phosphaethynolate anions and its utilization in the synthesis of organophosphorus compounds, especially the interesting work reported by two independent groups of J. M. Goicoechea and H. Grützmacher.
CS and F–CP as well as the thiocyanate anion SCN−. This paper highlights the recently reported methods for the synthesis of 2-phosphaethynolate anions and its utilization in the synthesis of organophosphorus compounds, especially the interesting work reported by two independent groups of J. M. Goicoechea and H. Grützmacher.
 Zheng-Jun Quan | Zheng-Jun Quan was born in 1978 in Gansu, China. He received his B.S. and Ph.D. degrees under Professor Xi-Cun Wang from Northwest Normal University in 2004 and 2007. After his B.S. he joined the chemistry faculty of Northwest Normal University, where he was promoted as Associate Professor in 2008 and Professor in 2014. His current research interests focus on transition-metal-catalyzed cross-coupling reactions, multicomponent reactions, tandem approaches and synthesis of biologically important heterocycles. |
 Xi-Cun Wang | Xi-Cun Wang was born in 1965 in Gansu, China. He obtained his Ph.D. under Professor Borisova, E. Ya from the Moscow College of Fine Chemical Engineering in 1993. After his Ph.D. he joined the chemistry faculty of the Northwest Normal University in 1994, where he was promoted as Associate Professor in 1996 and Professor in 2001. His current research interests focus on transition-metal-catalyzed cross-coupling reactions, multicomponent reactions, tandem approaches and green chemistry. |
In the early 1990s, Becker and co-workers isolated the 2-phosphaethynolate anion (P
C–O− or PCO−), the phosphorus-containing analogue of the cyanate ion (NC–O−), through the reaction of lithium bis(trimethylsilyl)phosphanide with dimethylcarbonate in 1,2-dimethoxyethane (dme) (Scheme 1).1 Thereafter, the 2-phosphaethynolate anion gained much attention because this anion is isoelectronic to small molecules such as OCS and F–CP as well as the thiocyanate anion SCN−.2 In 2002, Becker's procedure was explored to synthesize other alkali earth metal salts of the anion, such as magnesium, calcium, strontium and barium (Scheme 1).3 The salts are extremely sensitive towards moisture and air, and only [Ca(O–CP)2(dme)3] could be isolated as a crystalline solid. However, at −20 °C the dme solutions of the alkaline earth metal bis(2-phosphaethynolates) can be stored for several weeks. The natural resonance theory calculations indicate that this anion is a hybrid between the phosphaethynolate (PC–O−, 51.7%) and phosphaketenide (−PCO, 40.2%) resonance structures.4 In the last two decades the chemistry of the phosphaethynolate anion has been exploited in the metal-mediated dimerizations and oxidative tetramerizations, reaction with carbon disulfide and as a ligand in a transition-metal complex.5 Herein, we highlight recent breakthroughs in the synthesis of the 2-phosphaethynolate anions and the chemistry of the anions, especially the interesting work reported by two independent groups of J. M. Goicoechea6 and H. Grützmacher.7
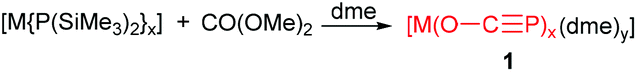 | ||
| Scheme 1 Becker's procedure for the 2-phosphaethynolate anion. M = Li, x = 1, y = 2; M = Mg–Ba, x = 2, y = 3. | ||
Pioneering studies by Grützmacher's group provided a remarkable synthesis of two new sodium bis(2-phosphaethynolates), [{Na(O–CP)(dme)2}2] (1) and [{Na(O–CP)(dioxane)2.5}∞] (2), by reaction of NaPH2 with CO conducted at 110 bar (Scheme 2).7a These sodium salts can be obtained in pure form as 1 upon concentration of the reaction solution or more conveniently by adding dioxane, whereby 2 precipitates as a colorless microcrystalline powder. Alternatively the replacement of NaPH2 for [Na(PH2)·Na(OtBu)x] as the PH2− source reacted with [Fe(CO)5] or dme led to the exclusive formation of 1 and 2 (Scheme 3). The formation of 1 and 2 was confirmed by combined crystallographic, spectroscopic, and computational methods. The computations and experimental evidence suggests that binding of CO to a transition metal complex (such as [Fe(CO)5]) to enhance its electrophilicity would significantly improve the efficiency of the reaction. In contrast to the lithium and alkaline earth salts, [Na(O–CP)] is thermally remarkably stable and does not decompose when heated to 110 °C for 2.5 days in DME or THF. In particular, they developed a most convenient procedure to make 1 and 2 through reaction of red phosphorus, ethylene carbonate and sodium at room temperature.7b Krummenacher and Cummins reported an alternative procedure via bond metathesis reaction of a terminal niobium phosphide ((L)nNbP) with CO2.8 The advantages of this protocol are that terminal niobium phosphide can readily transform carbon dioxide (1 equivalent), under mild conditions, into the phosphaethynolate anion.
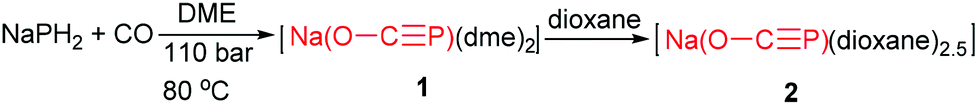 | ||
| Scheme 2 Synthesis of the 2-phosphaethynolate anion from a reaction between NaPH2 and CO. | ||
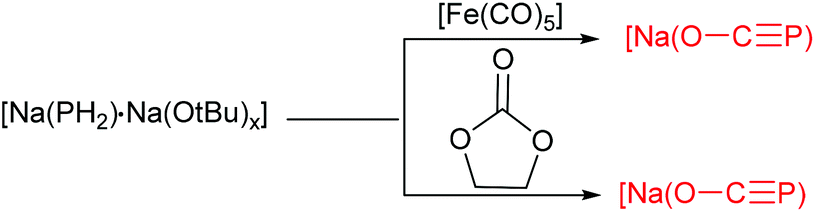 | ||
| Scheme 3 Synthesis of the phosphaethynolate anion from [Na(PH2)·Na(OtBu)x]. | ||
Another novel and straightforward strategy to generate phosphaethynolate anion from bare K3P7 and one atmosphere of CO was discovered at the end of 2013 by Jupp and Goicoechea.6a In the study, [K(O–CP)([18]crown-6)] (3) was obtained by heating a DMF solution of K3P7 and [18]crown-6 to 150 °C under one atmosphere of CO for 24 h (Scheme 4). A mechanistic rationalization for this reaction was proposed by nucleophilic attack of a phosphide vertex (P− anion) at the carbon atom of CO resulting in the formation of oxidized polyphosphide species (P162− and P213−). Unlike the reported metal salts of the PCO− anion1,7a—where forms the metal bis(2-phosphaethynolates)—the structure of 3 has a single triatomic linear anion in the asymmetric unit alongside a [K([18]crown-6)]+ cation. There is a close electrostatic contact between the K+ cation with the phosphorus and the oxygen atoms, while the C–O moiety interacts with an adjacent [K([18]crown-6)]+ moiety.
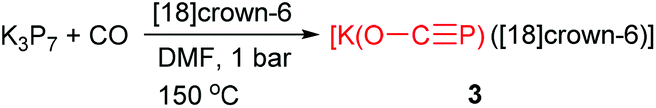 | ||
| Scheme 4 Synthesis of phosphaethynolate anion between K3P7 and CO. | ||
In the early studies on the chemistry of phosphaethynolate anions showed that the reaction between Li(OCP) and CS2 gave [Li(SCP)] (4) (Scheme 5).5c Oxidative tetramerization resulted in (P4C4O4)22− and a transition-metal-promoted dimerization led to dianionic transfer species (P2C2O2)22− (5).5d These reactions do show the high potential of 2-phosphaethynolates as reagents. The PCO− anion is a valuable building block for the synthesis of organophosphorus compounds. Much work has been devoted to explore the chemistry of 2-phosphaethynolates based on the discovery of alternative synthetic routes for the stable PCO− anions. Recently, the theme of reaction of the phosphaethynolate anion with common organic or inorganic substrates giving small molecules was independently expanded by Goicoechea's6 and Grützmacher's groups.7
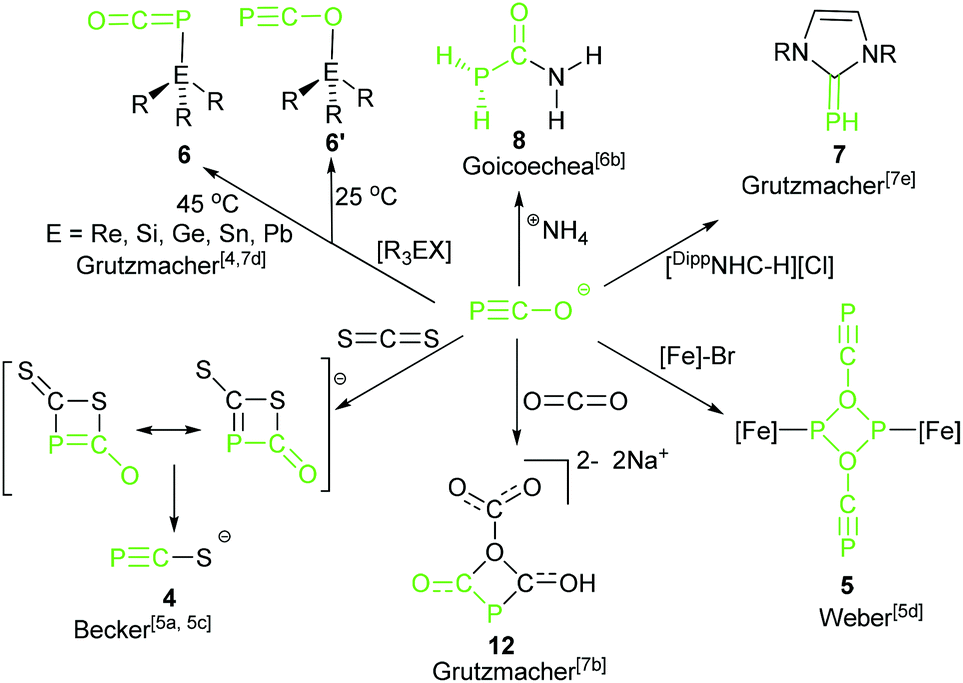 | ||
| Scheme 5 Reaction of phosphaethynolate anion with small molecules or metals. [Fe] = (C5Me5)(CO)2Fe; (1,3-t-Bu2C5H3)(CO)2Fe; (1,2,4-i-Pr3C5H2)(CO)2Fe; [Re] = [Re(OTf)(CO)2(triphos)] (triphos = (CH2PPh2)3, OTf− = F3C). | ||
The reactivity of complex 2 toward transition metals4 and triorganyl tetrel compounds R3EX was explored by Grützmacher's group7d (Scheme 5). The density functional theory (DFT) calculations show that the thermodynamic product phosphaketenes (6, attack by P) are lower in energy than the corresponding kinetic product oxyphosphaalkynes (6′, attack by O). Higher reaction temperature induced formation of the favored thermodynamic product 6. Importantly, these phosphaketenes (6, E = Si and Sn) were used as building blocks to prepare complex triphospha heterocycles by the same group (16, Scheme 6).7g Another example of application of 2 as a P transfer reagent was achieved by the same group. 2 reacted with an imidazolium salt resulting in a parent phosphinidene–carbene adduct, DippNHCPH 7, via the loss of CO.7e The mechanistic study suggested that the phosphaethynolate anion did not likely act as a nucleophile, but acted as a Brønsted base and deprotonated the imidazolium cation forming the parent phosphaketene (H–PCO).
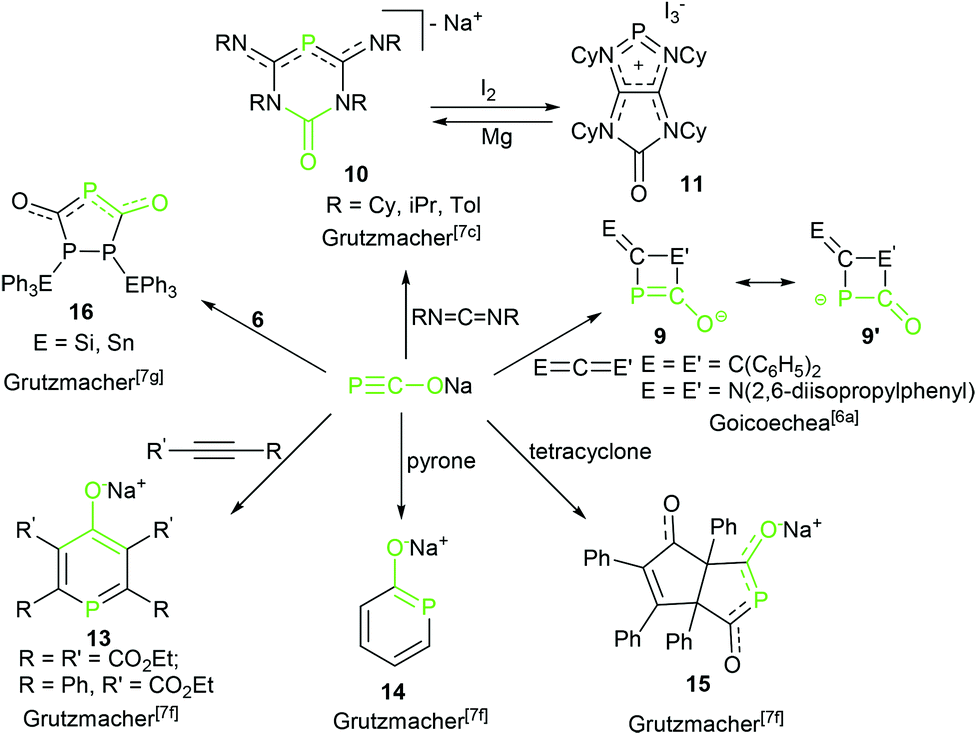 | ||
| Scheme 6 Synthesis of phosphorus-containing heterocycles by cycloadditions of phosphaethynolate anion with unsaturated substrates. | ||
In 2013, after their successful synthesis of the [K(O–CP)], Goicoechea and co-workers described an addition reaction of the 2-phosphaethynolate anion with ammonium salts to give the novel and unprecedented phosphinecarboxamide 8 (PH2C(O)NH2) under aerobic conditions in pyridine or THF (Scheme 5).7b Remarkably, Wöhler's synthesis of urea is a scientific milestone considered by many as the birth of modern organic chemistry because for the first time an organic compound was produced from inorganic reactants.9 They extended the principles of Wöhler's synthesis of urea to the chemistry of the phosphaethynolate anion with ammonium salts (NH4BPh4, NH4Cl) to afford a heavier analogue of urea, PH2C(O)NH2, where one of the primary amine groups (–NH2) is replaced by a phosphine (–PH2). In contrast to the primary phosphine, this research indicates that phosphinecarboxamide exhibits remarkable air and moisture stability. This product can be synthesized even in water, and its half-life in a pyridine-d5 solution exposed to air is approximately 9 days.
Exploratory investigations into the reactivity of ammonium salts revealed that the reaction time required for full conversion of starting materials was strongly dependent on the solubility of the ammonium salt used. When using NH4BPh4, the product can be obtained by means of trap-to-trap distillation. This result is due to the reduced hydrogen-bonding ability relative to urea. However, when employing NH4Cl, it is not viable to purify the product by distillation because the product can form a sufficiently strong hydrogen-bonding interaction with the anion.
The experimental and DFT studies conducted by the authors provided extensive spectrosopic data for the [K(O–CP)([18]crown-6)], heteroallenes and phosphinecarboxamide, and a detailed picture of the electronic structure of these species. The crystallographic characterization reveals that the phosphinecarboxamide moiety displays a pyramidalized geometry at the phosphorus atom, P1, with a planar carboxamide moiety. The structure is similar to that of urea, which is almost planar but that the amino groups are somewhat pyramidal.10,11 Compared with urea, 8 shows a more complex NMR spectrum, i.e. the 1H NMR spectrum reveals a doublet centered at 3.82 (1JP–H = 209 Hz) in addition to two broad resonances at 8.57 and 9.05 ppm. The 13C NMR spectrum also reveals the presence of a doublet at 175.8 ppm (1JP–C = 8 Hz).
Furthermore, the most interesting reactivity of the PCO anion is the cycloaddition chemistry and several phosphorus-containing heterocycles were generated by [2 + 2], [3 + 2] and [4 + 2] cycloadditions. Unlike its nitrogen analogue (NCO), the PCO− anion usually performed self-dimerization and cycloadditions that are uncommon or even unprecedented for cyanate.
In a further noteworthy reaction, Goicoechea and co-workers explored a novel [2 + 2] cycloaddition of potassium salt 3 with diphenylketene leading to the unprecedented heterocyclic species 9 (Scheme 6).6a This is the first example of an isolated monoanionic four-membered phosphorus-containing heterocycle, although the reaction affords the desired product involving the formation of other phosphorus-containing intermediates. 3 also reacted with a carbodiimide with full conversion and quantitative selectivity to give the corresponding four-membered ring. Extensive exploratory investigations into the scope and limitations of this synthetic protocol have revealed that this [2 + 2] cycloaddition strongly depends on the nature of the substituents on the carbodiimide, i.e. bis(cyclohexyl)carbodiimide cannot give the cycloaddition product in this study.
Strikingly, the reaction of sodium salt 2 with two equivalents of carbodiimides including the bis(cyclohexyl)carbodiimide was recently described by Grützmacher and co-workers that resulted in six-membered hererocyclic anions 10 (Scheme 6).7c They assumed that a four-membered heterocycle likely as product 96a is an intermediate, although it was not observed in this reaction. Theoretical calculations indicated that all activation barriers for the formation reaction of the four-membered heterocyclic intermediate are below 10 kcal mol−1 and the reaction profile is shallow. It immediately reacts to form the final product. These results explain why no intermediates 9 were observed. Treatment 10 with I2 as a two-electron oxidative reagent resulted in a bicyclic phosphenium cation 11, which was reduced by Mg to reconvert into the anion 10. Interestingly, the reduced anion 10 and the oxidized cation 11 are stable and coexist in solution due to the thermodynamically stable redox states (Epeak,ox = −0.78 V for anion 10, Epeak,red = −1.84 V for cation 11) and do not react with each other.
Complex 2 as an important building block was further utilized in the construction of mono-substituted four-membered ring 12 by combining with CO2 (Scheme 5).7b The reaction mechanism was different from a classical [2 + 2] cycloaddition. It is stepwise and initiated by a nucleophilic attack of the phosphorus center of PCO−, following attack by a second PCO− anion led to the formation of the 1,3-diphospha-2,4-dionate ring.
In addition to the [2 + 2] cycloaddition, the Grützmacher's group also studied the [3 + 2] and [4 + 2] cycloadditions of 2 with alkynes, tetracyclone and α-pyrone forming various phosphorus-containing five- and six-membered hererocycles 13–15 in excellent yields (Scheme 6).7f The reaction of 2 and two equivalents of phenylethynylcarboxylic ethylester in refluxing dme gave the sodium salt of a six-membered heterocyclic ring (13). However, the more-electron-deficient diethyl but-2-ynedioate formed a six-membered ring together with a five-membered heterocyclic anion removal of the CO in an approximate 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. Similarly, the reaction of 2 with tetracyclone and α-pyrone undergoes cycloadditions delivering functionalized hydroxy substituted phosphorus heterocycles 14 and 15, respectively. These examples impressively demonstrate not only the broad possibilities of cycloadditions for the synthesis of phosphorus-containing molecules, but also how easy these reactions are to conduct.
:1 ratio. Similarly, the reaction of 2 with tetracyclone and α-pyrone undergoes cycloadditions delivering functionalized hydroxy substituted phosphorus heterocycles 14 and 15, respectively. These examples impressively demonstrate not only the broad possibilities of cycloadditions for the synthesis of phosphorus-containing molecules, but also how easy these reactions are to conduct.
Inspired by these exciting results, Zhao and co-workers reported some interesting computational studies of the cycloadditions of PCO− anion with alkynes, ketene, carbodiimide and 2H-pyran-2-one.12 They suggested that the [2 + 2] and [3 + 2] cycloaddition of the anion favored stepwise processes, while [4 + 2] cycloaddition is a concerted fashion. More importantly, electronic effects play a key role in the regioselectivity of cycloaddition. The higher electrophilicity of PCO− compared to NCO− and a stronger tendency of the former to hamper the transformation of π-bond into a σ-bond, which provides a possible explanation for the different reactivity of the PCO− anion compared to its lighter congener.
Summary and outlook
Considering the similar structure to N-heterocycles and urea, phosphorus-containing heterocycles and phosphinecarboxamide moiety species may ultimately be used as a fundamental building block of enormous importance, such as in coordination chemistry, anion sensing, or in the synthesis of novel materials and organic compounds containing phosphorus. Sure enough, current and subsequent developments in the field of synthesis and reactivity of 2-phosphaethynolate anion hold great potential for future theoretic and academic, even industrial applications. Additional mechanistic studies and experimentation is needed to expand the scope of these transformations of the 2-phosphaethynolate anions to further exciting molecules.Acknowledgements
We thank NSFC (no. 21362032 and 21362031) for financial support.Notes and references
- G. Becker, W. Schwarz, N. Seidler and M. Westerhausen, Z. Anorg. Allg. Chem., 1992, 612, 72–82 CrossRef CAS PubMed.
- T. M. Klapötke, Angew. Chem., Int. Ed. Engl., 1994, 33, 1575–1576 CrossRef PubMed.
- M. Westerhausen, S. Schneiderbauer, H. Piotrowski, M. Suter and H. Nöth, J. Organomet. Chem., 2002, 643–644, 189–193 CrossRef CAS.
- S. Alidori, D. Heift, G. Santiso-Quinones, Z. Benkő, H. Grützmacher, M. Caporali, L. Gonsalvi, A. Rossin and M. Peruzzini, Chem. – Eur. J., 2012, 18, 14805–14811 CrossRef CAS PubMed.
- (a) G. Becker, K. Hübler and W. Schwarz, Z. Anorg. Allg. Chem., 1994, 620, 405–417 CrossRef CAS PubMed; (b) L. Weber, S. Buchwald, D. Lentz, O. Stamm, D. Preugschat and R. Marschall, Organometallics, 1994, 13, 4406–4412 CrossRef CAS; (c) G. Becker, G. Heckmann, K. Hübler and W. Schwarz, Z. Anorg. Allg. Chem., 1995, 621, 34–46 CrossRef CAS PubMed; (d) L. Weber, B. Torwiehe, G. Bassmann, H.-G. Stammler and B. Neumann, Organometallics, 1996, 15, 128–132 CrossRef CAS; (e) G. Becker, G. Ditten, K. H_bler, U. Hübler, K. Merz, M. Niemeyer, N. Seidler, M. Westerhausen and Z. Zheng, in Organosilicon Chem. II, ed. N. Auner and J. Weis, VCH, Weinheim, 1996, 161–186 Search PubMed.
- (a) A. R. Jupp and J. M. Goicoechea, Angew. Chem., Int. Ed., 2013, 52, 10064–10067 CrossRef CAS PubMed; (b) A. R. Jupp and J. M. Goicoechea, J. Am. Chem. Soc., 2013, 135, 19131–19134 CrossRef CAS PubMed.
- (a) F. F. Puschmann, D. Stein, D. Heift, C. Hendriksen, Z. A. Gal, H.-F. Grützmacher and H. Grützmacher, Angew. Chem., Int. Ed., 2011, 50, 8420–8423 CrossRef CAS PubMed; (b) D. Heift, Z. Benkő and H. Grützmacher, Dalton Trans., 2014, 43, 831–840 RSC; (c) D. Heift, Z. Benkő and H. Grützmacher, Angew. Chem., Int. Ed., 2014, 53, 6757–6761 CrossRef CAS PubMed; (d) D. Heift, Z. Benkő and H. Grützmacher, Dalton Trans., 2014, 43, 5920–5928 RSC; (e) A. M. Tondreau, Z. Benkő, J. R. Harmer and H. Grützmacher, Chem. Sci., 2014, 5, 1545–1554 RSC; (f) X. Chen, S. Alidori, F. F. Puschmann, G. Santiso-Quinones, Z. Benkő, Z. Li, G. Becker, H.-F. Grützmacher and H. Grützmacher, Angew. Chem., Int. Ed., 2014, 53, 1641–1645 CrossRef CAS PubMed; (g) D. Heift, Z. Benkő and H. Grützmacher, Chem. – Eur. J., 2014, 20 DOI:10.1002/chem.201403419.
- I. Krummenacher and C. C. Cummins, Polyhedron, 2012, 32, 10–13 CrossRef CAS PubMed.
- (a) F. Wöhler, Ann. Phys., 1828, 87, 253–256 CrossRef PubMed; (b) F. Wöhler, Ann. Chim. Phys., 1828, 37, 330–333 Search PubMed; (c) P. S. Cohen and S. M. Cohen, J. Chem. Educ., 1996, 73, 883–886 CrossRef CAS.
- P. D. Godfrey, R. D. Brown and A. N. Hunter, J. Mol. Struct., 1997, 413–414, 405–414 CrossRef CAS.
- A. Pryor and P. L. Sanger, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Cryst., 1970, 26, 543–558 CrossRef CAS.
- L. Liu, J. Zhu and Y. Zhao, Chem. Comm., 2014 10.1039/C4CC04610B.
| This journal is © the Partner Organisations 2014 |