
Palladium(II)-catalyzed direct conversion of allyl arenes into alkenyl nitriles†
Zhibin
Shu
a,
Yujing
Zhou
a,
Yan
Zhang
a and
Jianbo
Wang
*ab
aBeijing National Laboratory of Molecular Sciences (BNLMS), Key Laboratory of Bioorganic Chemistry and Molecular Engineering of Ministry of Education, College of Chemistry, Peking University, Beijing 100871, China. E-mail: wangjb@pku.edu.cn; Fax: (+86) 10-6275-7248; Tel: (+86) 10-6275-1708
bState Key Laboratory of Organometallic Chemistry, Chinese Academy of Sciences, Shanghai 200032, China
First published on 10th September 2014
Abstract
A mild palladium-catalyzed ammoxidation approach, which leads to the formation of the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N triple bond in an allyl group, has been developed to directly convert allylarenes into alkenyl nitriles.
N triple bond in an allyl group, has been developed to directly convert allylarenes into alkenyl nitriles.
Introduction
Alkenyl nitriles are both unique structural units in organic synthesis and versatile building blocks of natural products, agricultural chemicals, pharmaceuticals, and dyes.1 Due to their important applications in various fields, efforts have been devoted to the development of efficient synthetic methods for this type of nitrile compound.2 However, most of the methods so far developed are based on functional group transformations or addition reactions. To the best of our knowledge, there is only one case in which a Fe-catalyzed direct conversion of the allyl derivatives into the corresponding unsaturated nitriles was reported.3,4 Qin and Jiao have demonstrated the oxidative C–H bond transformation of allyl arenes or alkenes into the corresponding nitriles, with Me3SiN3 as the nitrogen source and 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) as the oxidant.4 The allyl radical generated through single-electron-transfer is proposed as the key step intermediate in this transformation. This allylic C–H bond transformation is related to the recent studies on the transition-metal-catalyzed direct allylic C–H functionalization of terminal alkenes, which has emerged as a powerful strategy in organic synthesis.5–9On the other hand, we have recently reported a direct synthesis of aromatic nitriles from the methyl arenes with Pd(OAc)2 and N-hydroxyphthalimide (NHPI) as the catalysts, and tert-butyl nitrite (tBuONO, TBN) as the nitrogen source.10 Benzyl radical A is proposed as the key intermediate in the reaction (Scheme 1a). As the continuation of our interest in the development of novel cyanation methods,11 we further conceived that a similar allyl radical B should also be generated in a similar catalytic system, and a direct conversion of terminal alkenes into alkenyl nitriles might be achieved. Herein, we report a Pd-catalyzed direct transformation of allyl arenes into the corresponding alkenyl nitriles, using tert-butyl nitrite as both the nitrogen source and oxidant. The reaction proceeds under mild conditions, affording moderate to good yields of alkenyl nitriles (Scheme 1b).
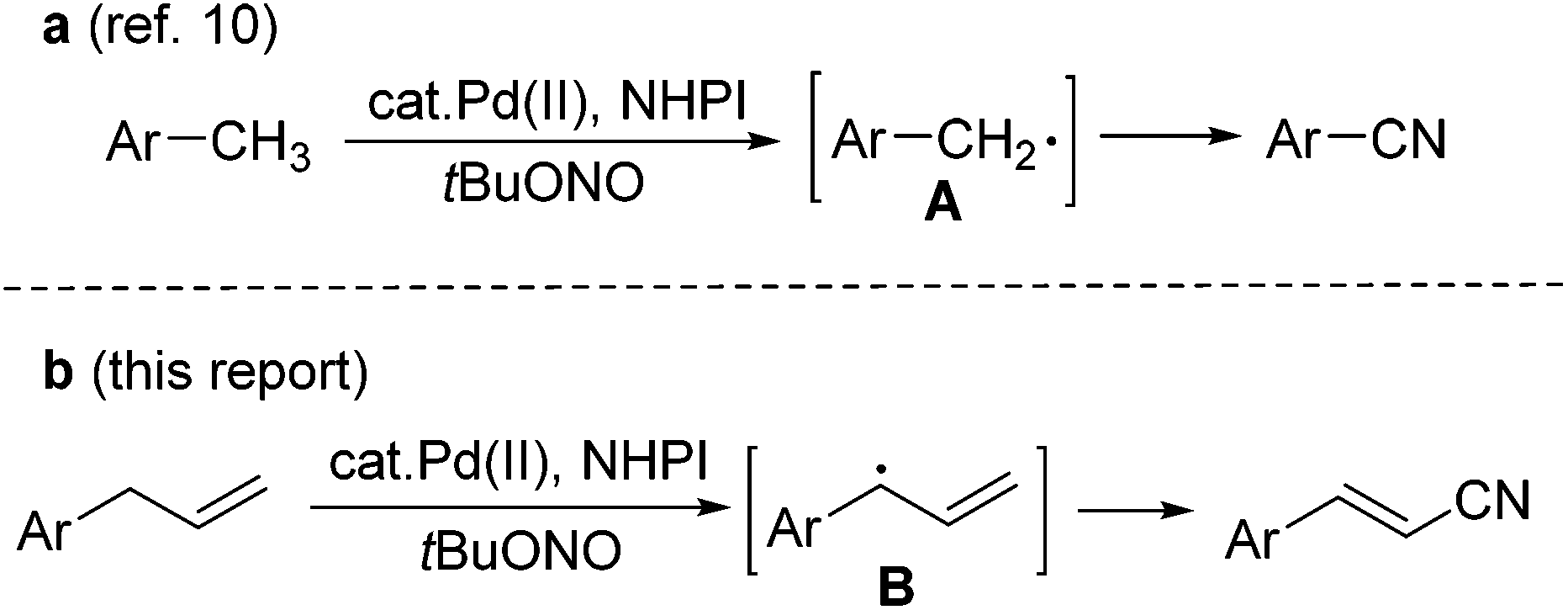 | ||
| Scheme 1 Pd(OAc)2-catalyzed cyanation of methyl arenes and allyl arenes. | ||
Results and discussion
Similar to our previous study,10 the investigation began with evaluation of the direct transformation of 1-allylbenzene 1a into the corresponding cinnamonitrile 2a under oxidative conditions (Table 1). In the absence of an additive, the reaction of 1a catalyzed by 10 mol% of Pd(OAc)2 with tert-butyl nitrite at 60 °C in DCE or THF gave only a trace amount of 2a (entries 1 and 2), whereas the reaction in 1,4-dioxane and acetonitrile produced 2a in 14% and 26% yield, respectively (entries 3 and 4). Gratifyingly, 2a was formed in 62% yield in the presence of N-hydroxyphthalimide (NHPI) as an additive in a catalytic amount (30 mol%) with 5 mol% of Pd(OAc)2 (entry 5). The reaction could be optimized using 10 mol% of Pd(OAc)2 at 50 °C (entry 6). We also examined another carbon radical producing catalyst N,N′,N′′-trihydroxyisocyanuric acid (THICA) as an additive.12 The reaction afforded 2a, albeit in diminished yield. 2,2,6,6-Tetramethyl-1-piperidinyloxy (TEMPO) has also been examined as an additive, however the reaction only gives 2a in 26% yield (entry 8). Other metal catalysts, including PdCl2(MeCN)2, Cu(OAc)2, CuCl and Fe(OAc)2, have also been examined but they only afforded a very low yield or a trace amount of the product 2a (entries 9–12).|
|
|||||
|---|---|---|---|---|---|
| Entry | Cat. (mol%) | TBN (equiv.) | Additive (mol%) | Solvent | Yieldb (%) |
a The reaction conditions: 1a (0.3 mmol), catalyst, additive, and tert-butyl nitrite (TBN) in a dry solvent with stirring under N2 for 16 h.
b Isolated yields. NHPI: N-hydroxyphthalimide; THICA: N,N′,N′′-trihydroxyisocyanuric acid. TEMPO: 2,2,6,6-tetramethyl-1-piperidinyloxy.
|
|||||
| 1 | Pd(OAc)2 (10) | 3 | None | DCE | Trace |
| 2 | Pd(OAc)2 (10) | 3 | None | THF | Trace |
| 3 | Pd(OAc)2 (10) | 3 | None | Dioxane | 14 |
| 4 | Pd(OAc)2 (10) | 3 | None | MeCN | 26 |
| 5 | Pd(OAc)2 (5) | 3 | NHPI (30) | MeCN | 62 |
| 6 | Pd(OAc)2 (10) | 2 | NHPI (30) | MeCN | 80 |
| 7 | Pd(OAc)2 (10) | 2 | THICA (10) | MeCN | 56 |
| 8 | Pd(OAc)2 (10) | 2 | TEMPO (30) | MeCN | 26 |
| 9 | PdCl2(MeCN)2 (10) | 2 | NHPI (20) | MeCN | 11 |
| 10 | Cu(OAc)2 (10) | 2 | NHPI (20) | MeCN | Trace |
| 11 | CuCl (10) | 2 | NHPI (20) | MeCN | Trace |
| 12 | Fe(OAc)2 (10) | 2 | NHPI (20) | MeCN | Trace |

With the optimized reaction conditions, various allylarenes were investigated with 10 mol% Pd(OAc)2 and 30 mol% NHPI as a co-catalyst system (Scheme 2). Electron-donating substituents, such as Me and OMe, at the para, meta, and ortho positions of the arene group did not affect the reaction, affording the corresponding alkenyl nitriles in 67–83% yields (2b–h, 2o). Remarkably, some sensitive substituents or functional groups, such as trimethylsilyl (TMS), Cl and Br, were tolerated well in this transformation (2h, 2l, 2m). Substrates substituted with electron-withdrawing groups, such as F and CF3, also worked well and afforded the desired products in moderate yields (2k, 2n).
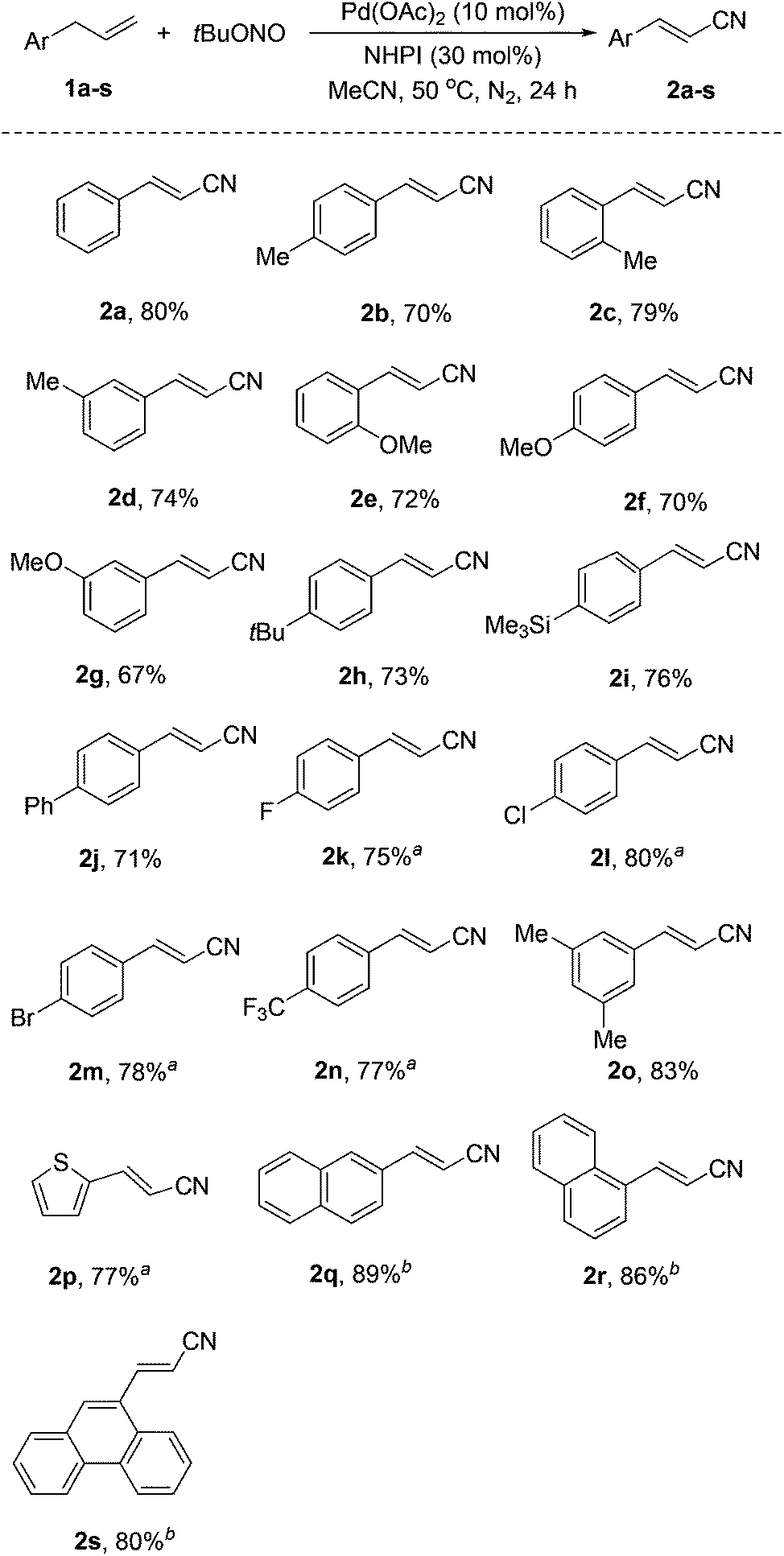 | ||
| Scheme 2 Scope of the Pd-catalyzed direct conversion of allylarenes into alkenyl nitriles. If not otherwise noted, the reaction conditions are as following: 1a–s (0.3 mmol), tert-butyl nitrite (0.6 mmol), Pd(OAc)2 (0.03 mmol), and NHPI (0.09 mmol) in MeCN (1.5 mL) at 50 °C under N2 for 24 h. Yields of isolated products are given. atert-Butyl nitrite (2.5 equiv.) was used. btert-Butyl nitrite (3.0 equiv.) was used. | ||
It is noteworthy that this reaction also worked with a heteroaryl-substituted propene, 1-allyl-2-thiophene (1p), giving 2p in 77% yield. In addition, polycyclic aromatic-substituted propenes were also successfully converted into the corresponding alkenyl nitriles in good yields (2q–s).
Similar to the transformation of methyl arenes into aromatic nitriles,10 a plausible mechanism is proposed as shown in Scheme 3. Initially, as an oxidant, tert-butyl nitrite reacts with NHPI to generate the active phthalimide N-oxyl radical (PINO). The tert-butyl nitrite itself decomposes into an NO radical and 2-methyl-2-propanol.13 Then, allyl arene 1 undergoes single-electron-transfer (SET) oxidation with PINO to produce the corresponding allyl radical A. Subsequently, radical recombination of the NO radical with A occurs to form intermediate B. Upon isomerization of B to aldoxime C, Pd(OAc)2-catalyzed dehydration of C finally leads to the desired nitrile product 2.14 To substantiate this mechanistic hypothesis, we have carried out the reaction of 1a under the standard conditions but in the absence of the Pd(OAc)2 catalyst. The reaction gave a complex mixture, from which oxime C along with the corresponding cinnamaldehyde can be identified by GC-MS.
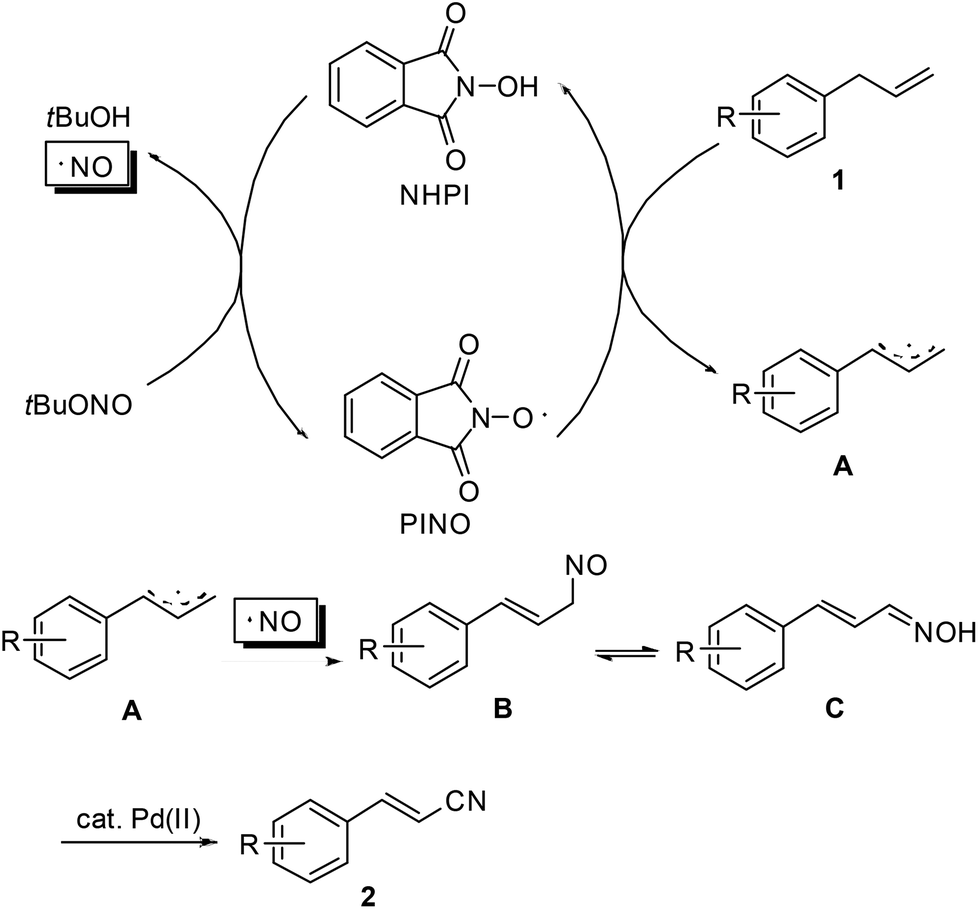 | ||
| Scheme 3 Proposed mechanism. | ||
In conclusion, we have developed a novel Pd(II)-catalyzed direct synthesis of alkenyl nitriles from the corresponding allyl arenes under mild conditions using tert-butyl nitrite as the nitrogen source and inexpensive NHPI as the co-catalyst. Notably, in this transformation, three C–H bonds are cleaved to form one CN bond. This reaction offers a novel method for the synthesis of biologically and medicinally important alkenyl nitriles.
Experimental section
General
All the palladium-catalyzed reactions were performed under a nitrogen atmosphere in a flame-dried reaction flask. All solvents were distilled under a nitrogen atmosphere prior to use. 1,4-Dioxane and THF were dried over Na with the benzophenone-ketyl intermediate as an indicator. Acetonitrile and 1,2-dichloroethane were dried over CaH2. For chromatography, 200–300 mesh silica gel (Qingdao, China) was employed. 1H and 13C NMR spectra were recorded at 400 MHz and 100 MHz with a Bruker ARX 400 spectrometer. Chemical shifts are reported in ppm using tetramethylsilane as the internal standard. IR spectra were recorded with a Nicolet 5MX-S infrared spectrometer. LRMS were obtained on an Agilent 5975C inert 350 EI mass spectrometer. HRMS were obtained on a Bruker Apex IV FTMS by ESI or a GCT CA127 Micronass UK by EI. All reactions were carried out in dry sealed tubes under an atmosphere of nitrogen. Unless otherwise noted, materials obtained from commercial suppliers were used without further purification. The starting materials 1a–o and 1q–s were prepared from the corresponding aryl bromide according to a previously reported literature.7d1p was prepared from thiophene according to a previously reported literature.15General procedure for Pd(II)-catalyzed reaction
Under a nitrogen atmosphere, allylbenzene 1a (36 mg, 0.3 mmol), tert-butylnitrite (65 mg, 0.6 mmol, 2.0 equiv.), NHPI (16 mg, 0.09 mmol, 0.3 equiv.) and Pd(OAc)2 (7 mg, 0.03 mmol, 0.1 equiv.) in MeCN (1.5 mL) were stirred at 50 °C for 16 h. After cooling, the reaction was diluted with CH2Cl2 (2 mL) and the resulting mixture was filtered, and the filtrate was concentrated. Purification by column chromatography of the mixture gave pure 2a as light yellow oil (31 mg, 80%).41H NMR (CDCl3, 400 MHz) δ 7.38–7.46 (m, 6H), 5.88 (d, J = 16.8 Hz, 1H); 13C NMR (CDCl3, 100 MHz) δ 150.5, 133.5, 131.2, 129.1, 127.3, 118.1, 96.3.Acknowledgements
The project was supported by the 973 Program (no. 2015CB856602) and the National Nature Science Foundation of China (grant 21272010 and 21332002).Notes and references
- For reviews, see: (a) F. F. Fleming and Q. Wang, Chem. Rev., 2003, 103, 2035 CrossRef CAS PubMed; (b) J. S. Miller and J. L. Manson, Acc. Chem. Res., 2001, 34, 563 CrossRef CAS PubMed; (c) A. J. Fatiadi, in Preparation and Synthetic Applications of Cyano Compounds, ed. S. Patai and Z. Rappaport, Wiley, New York, 1983 Search PubMed.
- (a) M. Alterman and A. Hallberg, J. Org. Chem., 2000, 65, 7984 CrossRef CAS PubMed; (b) K. Ishihara, Y. Furuya and H. Yamamoto, Angew. Chem., Int. Ed., 2002, 41, 2983 CrossRef CAS; (c) K. Yamaguchi, H. Fujiwara, Y. Ogasawara, M. Kotani and N. Mizuno, Angew. Chem., Int. Ed., 2007, 46, 3922 CrossRef CAS PubMed; (d) Y. Nakao, A. Yada, S. Ebata and T. Hiyama, J. Am. Chem. Soc., 2007, 129, 2428 CrossRef CAS PubMed; (e) S. Zhou, D. Addis, S. Das, K. Junge and M. Beller, Chem. Commun., 2009, 4883 RSC; (f) T. Oishi, K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2009, 48, 6286 CrossRef CAS PubMed; (g) W. Zhou, J. Xu, L. Zhang and N. Jiao, Org. Lett., 2010, 12, 2888 CrossRef CAS PubMed; (h) P. Anbarasan, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 519 CrossRef CAS PubMed.
- For selected reports on heterogeneous approaches to acrylonitrile from propene, see: (a) J. Holmberg, S. Hansen, R. Grasselli and A. Andersson, Top. Catal., 2006, 38, 17 CrossRef CAS PubMed; (b) N. Burriesci, F. Garbassi, M. Petrera and G. Petrini, J. Chem. Soc., Faraday Trans. 1, 1982, 78, 817 RSC.
- C. Qin and N. Jiao, J. Am. Chem. Soc., 2010, 132, 15893 CrossRef CAS PubMed.
- For C–O bond formation, see: (a) M. S. Chen and M. C. White, J. Am. Chem. Soc., 2004, 126, 1346 CrossRef CAS PubMed; (b) K. J. Fraunhoffer, N. Prabagaran, L. E. Sirois and M. C. White, J. Am. Chem. Soc., 2006, 128, 9032 CrossRef CAS PubMed; (c) A. N. Campbell, P. B. White, I. A. Guzei and S. S. Stahl, J. Am. Chem. Soc., 2010, 132, 15116 CrossRef CAS PubMed.
- For C–N bond formation, see: (a) S. A. Reed and M. C. White, J. Am. Chem. Soc., 2008, 130, 3316 CrossRef CAS PubMed; (b) S. A. Reed, A. R. Mazzotti and M. C. White, J. Am. Chem. Soc., 2009, 131, 11701 CrossRef CAS PubMed; (c) G. S. Liu, G. Y. Yin and L. Wu, Angew. Chem., Int. Ed., 2008, 47, 4733 CrossRef CAS PubMed.
- For C–C bond formation, see: (a) Z. Li and C. Li, J. Am. Chem. Soc., 2006, 128, 56 CrossRef CAS PubMed; (b) J. H. Delcamp, A. P. Brucks and M. C. White, J. Am. Chem. Soc., 2008, 130, 11270 CrossRef CAS PubMed; (c) A. J. Young and M. C. White, J. Am. Chem. Soc., 2008, 130, 14090 CrossRef CAS PubMed; (d) S. Lin, C. X. Song, G. X. Cai, W. H. Wang and Z. J. Shi, J. Am. Chem. Soc., 2008, 130, 12901 CrossRef CAS PubMed; (e) J. M. Howell, W. Liu, A. J. Young and M. C. White, J. Am. Chem. Soc., 2014, 136, 5750 CrossRef CAS PubMed; (f) P.-S. Wang, H.-C. Lin, X.-L. Zhou and L.-Z. Gong, Org. Lett., 2014, 16, 3332 CrossRef CAS PubMed; (g) J. T. Osberger and M. C. White, J. Am. Chem. Soc., 2014, 136, 11176 CrossRef PubMed.
- For C–CF3 bond formation, see: (a) A. T. Parsons and S. L. Buchwald, Angew. Chem., Int. Ed., 2011, 50, 9120 CrossRef CAS PubMed; (b) J. Xu, Y. Fu, D. F. Luo, Y. Y. Jiang, B. Xiao, Z. J. Liu, T. J. Gong and L. Liu, J. Am. Chem. Soc., 2011, 133, 15300 CrossRef CAS PubMed; (c) X. Wang, Y. Ye, S. Zhang, J. Feng, Y. Xu, Y. Zhang and J. Wang, J. Am. Chem. Soc., 2011, 133, 16410 CrossRef CAS PubMed.
- For C–F bond formation, see: M. Braun and A. F. Doyle, J. Am. Chem. Soc., 2013, 135, 12990 CrossRef CAS PubMed.
- Z. Shu, Y. Ye, Y. Deng, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2013, 52, 1057 CrossRef.
- (a) G. Yan, C. Kuang, Y. Zhang and J. Wang, Org. Lett., 2010, 12, 1052 CrossRef CAS PubMed; (b) Y. Yang, Y. Zhang and J. Wang, Org. Lett., 2011, 13, 5608 CrossRef CAS PubMed; (c) Z. Shu, W. Ji, X. Wang, Y. Zhou, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2014, 53, 2186 CrossRef CAS PubMed.
- N. Hirai, N. Sawatari, N. Nakamura, S. Sakaguchi and Y. Ishii, J. Org. Chem., 2003, 68, 6587 CrossRef CAS PubMed.
- For a recent review, see: (a) F. Recupero and C. Punta, Chem. Rev., 2007, 107, 3800 CrossRef CAS PubMed; for a recent example, see: (b) C.-X. Miao, B. Yu and L.-N. He, Green Chem., 2011, 13, 541 RSC.
- (a) H. S. Kim, S. H. Kim and J. N. Kim, Tetrahedron Lett., 2009, 50, 1717 CrossRef CAS PubMed; (b) X.-Y. Ma, Y. He, T.-T. Lu and M. Lu, Tetrahedron, 2013, 69, 2560 CrossRef CAS PubMed.
- Y. Zhang, C. Wang, L. Rothberg and M. Ng, J. Mater. Chem., 2006, 16, 2721 CAS.
- C. Wang, Y. Huang, S. Sheng, W. Yang and M. Cai, Synth. Commun., 2009, 39, 1282 CrossRef CAS.
- Z. Wang and S. Chang, Org. Lett., 2013, 15, 1990 CrossRef CAS PubMed.
- M. B. Andrus, C. Song and J. Zhang, Org. Lett., 2002, 4, 2079 CrossRef CAS PubMed.
- B. V. Rokade, S. K. Malekar and K. R. Prabhu, Chem. Commun., 2012, 48, 5506 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Preparation of substrates, characterization data, 1H, 13C NMR, MS and IR spectra. See DOI: 10.1039/c4qo00218k |
| This journal is © the Partner Organisations 2014 |