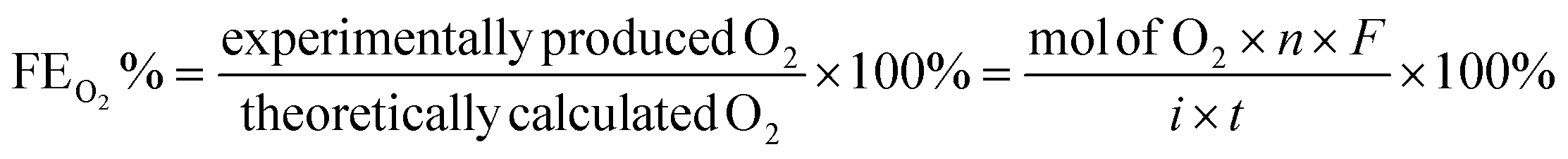Activating cobalt inverse spinel oxides via Fe substitution for enhanced water splitting reaction†
Received
6th April 2024
, Accepted 15th July 2024
First published on 16th July 2024
Abstract
Owing to their expeditious kinetics and remarkable catalytic prowess, spinel oxides have attracted significant attention as promising non-precious metal electrocatalysts for oxygen evolution reaction (OER). Modulating the oxidation state of the catalytically active B site in the spinel structure is a highly potent strategy for amplifying their OER activity. Investigations into the impact of Fe doping on the oxidation state as well as the electronic configuration of Co in NiCo2O4 revealed the generation of Co3+ ions with an electron occupancy close to one in the eg orbital, leading to an increased production of intermediate –OOH species, thereby accelerating the OER kinetics. The synergistic effect due to the optimal presence of Fe and Co in the NiCo0.5Fe1.5O4 catalyst allowed the catalyst to exhibit a reduced overpotential of 310 mV at 10 mA cm−2 with a constant current density for 22 h in the chronoamperometric study.
1. Introduction
The depletion of fossil fuels coupled with escalating concerns of global warming has attracted the attention towards the advancement of renewable and environmentally friendly energy sources. Hydrogen energy stands out as a potential alternative due to its remarkable energy density per unit weight/volume. Moreover, it can be perceived as an inherently pristine fuel, offering a pathway to carbon-free energy generation. The process of water electrolysis has emerged as a promising method for creating sustainable hydrogen feedstock, powered by renewable energy sources.1–6 Fundamentally, the process of water splitting involves a duo of half reactions within the electrolyser: the cathodic hydrogen evolution reaction (HER) and the anodic oxygen evolution reaction (OER). In contrast to HER, OER represents a multi-step process involving a four electron–proton transfer, intricately intertwined with the adsorption of different oxygen intermediates. From a thermodynamic perspective, the process of water splitting necessitates an energy input equivalent to 286 kJ mol−1 (i.e., electric potential of 1.23 V) under standard ambient conditions. However, a substantial overpotential, surpassing the theoretical potential, is generally essential to activate OER owing to its sluggish kinetics. To date, Ru- and Ir-based oxide catalysts are still considered the state-of-the-art choices.7,8 However, their inherent value and significant expense often present substantial obstacles to their widespread implementation within real-world electrolytic industries. Hence, economical substitutes exhibiting remarkable intrinsic activity are urgently desired for advancing the field of overall water electrolysis.
Presently, mixed-valence non-noble metal oxides are attracting significant attention due to their low cost, high abundance, and intriguing electronic configurations.9–15 Spinels are such mixed-valence metal oxides consisting of a closely packed array of O2− ions with the occupancy of A and B cations in tetrahedral and octahedral sites, respectively. For the ternary spinel structure AB2O4, the cation distribution formula can be represented as (A1−λBλ)Td[AλB2−λ]OhO4 (0 ≤ λ ≤ 1), where λ denotes the degree of inversion of the spinel structure. Normal spinels are represented by λ = 0 while λ = 1 are inverse spinels. Among the mixed-valence transition metal spinels, those with Co occupying the B site have attracted much research interest based on the superior OER activity of Co in octahedrally coordinated environments.16–26 For example, pristine NiCo2O4,27 supported NiCo2O4,28,29 and doped NiCo2O4 have shown remarkable OER activity.30 To elucidate the superior catalytic activity, various descriptors, such as the number of d electrons in the transition metal, electron occupancy in the eg orbital, bulk formation energy, metal–oxygen covalency, and the position of the O 2p band centre, are often considered as key factors contributing to enhanced OER catalytic efficacy. While these descriptors are well established in the perovskite family, further studies are necessary to comprehend their implications in spinel chemistry.
In general, Co cations in spinel oxides would prefer electronic configurations with a high spin in a tetrahedral coordination and low spin in an octahedral coordination, respectively.22 On the other hand, Fe cations in an octahedral coordination usually prefer a high-spin configuration.31 Interestingly, upon the incorporation of Fe3+ into octahedra, the electron configuration of octahedral Co3+ in the eg orbital is supposed to transform from a low spin to intermediate spin, thus making the electron filling in the eg orbital close to unity.32 The near unity eg occupancy would facilitate the formation of the key intermediate species –OOH, and thereby would contribute to a superior catalytic OER efficacy.33–35 To achieve this, varied amounts of Fe were doped in to NiCo2O4 with the help of a solution combustion synthesis route. The obtained materials were then morphologically and structurally characterized by FE-SEM and XRD, and the electronic configuration was thoroughly probed by XPS and EPR spectroscopy. Among the synthesized materials, NiCo0.5Fe1.5O4 exhibited a superior OER activity. The study considered the particular descriptor of electron occupancy in the eg orbital towards rationalizing the OER activity of NiCo0.5Fe1.5O4.
2. Experimental method
2.1 Synthesis
NiCo2O4, NiCo1.5Fe0.5O4, NiCoFeO4, NiCo0.5Fe1.5O4, and NiFe2O4 were synthesized using a novel solution combustion method, known for its energy efficiency and low-temperature initiation.21,36,37 The advantages of the solution combustion method are many. It is a low temperature-initiated process with a high transient temperature and produces a huge amount of gases. The combustion process is fast (instantaneous) and due to the production of the huge amount of gases, the products are homogenous nanocrystalline materials with the desired composition and structure. This process utilizes the highly exothermic redox chemical reactions between metal nitrates and fuels, as the metathetical (exchange) reactions between reactive compounds or reactions involving redox compounds/mixtures. Combustion synthesis is also known as self-propagating high-temperature synthesis. In this synthesis, water-soluble Ni(NO3)2·6H2O (S.D. Fine Chem Limited, 99%) Co(NO3)2·6H2O (S.D. Fine Chem Limited, 99%), and Fe(NO3)3·9H2O (S.D. Fine Chem Limited, 99%) were utilized as oxidizers while glycine served as the fuel. The synthesis of NiCo2O4 was carried out by dissolving 0.29 g of Ni(NO3)2·6H2O, Co(NO3)2·6H2O, and 0.16 g of glycine in 20 mL of water, respectively. The resulting homogeneous aqueous solution was then placed in a preheated furnace at 450 °C for 15 min. Initially, the solution exhibited boiling, and foaming, and the exothermic redox reaction generated a sufficient heat of combustion, compensating for the energy required for oxide formation, and resulting in a burning flame at the ignition point. Subsequently, the complete dehydration of the solution led to the formation of a solid powder product of pristine NiCo2O4, accompanied by the evolution of N2, and CO2. Similarly, NiCo2−xFexO4 (x = 0.5, 1, and 1.5) samples were synthesized with the respective varied molar ratio of Co(NO3)2·6H2O and Fe(NO3)3·9H2O. The synthesis of pristine NiFe2O4 was carried out by dissolving Ni(NO3)2·6H2O, Fe(NO3)3·9H2O, and glycine in a weight ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.8:0.6, respectively.
:2.8:0.6, respectively.
2.2 Characterization
The structure and crystalline nature of the synthesized spinels were assessed through X-ray diffraction (XRD) analysis performed on a Rigaku Ultima IV apparatus employing Cu Kα radiation. The XRD scans were conducted using a scan rate of 0.03° per minute, employing a step size of 0.01. The Debye–Scherrer formula was employed to ascertain the nanocrystalline dimensions of the synthesized spinels. The surface morphology and composition analyses were conducted using a field emission scanning electron microscopy system (FE-SEM, FEI-Apreo S) equipped with an energy dispersive spectroscopy (EDS) unit. The instrument was operated at an acceleration voltage of 20 kV. The Brunauer–Emmett–Teller (BET) method was employed to determine the surface area and pore size of the synthesized catalysts. X-Ray photoelectron spectroscopy (XPS) of the synthesized materials was performed using a Thermo Scientific K-Alpha surface analysis spectrometer with Al Kα radiation (1486.6 eV). The data profiles were subjected to a nonlinear least-squares curve fitting program with a Gaussian–Lorentzian production function and processed with Avantage software. The binding energies (BE) of all the XPS data were calibrated vs. the standard C 1s peak at 284.8 eV. The EPR spectra were measured at room temperature using a JEOL JES-FA200 EPR spectrometer. The photoluminescence (PL) spectra of the solid samples were collected using a Horiba Fluorolog FL-3C instrument at an excitation wavelength of 320 nm.
2.3 Electrocatalysis
Electrochemical tests were carried out using a conventional three-electrode system with glassy carbon (GC) as the working electrode, Hg/HgO as the reference electrode (saturated with aqueous KOH solution), and Pt wire as the counter electrode. About 3 mg of synthesized catalyst and 0.5 mg of commercial carbon nanotubes (CNTs) powder were dispersed in 1 mL methanol and 10 μL of 5% aqueous Nafion (Sigma-Aldrich) solution. The active mass ratio of the catalyst to commercial CNTs was maintained at 6:1. The solution was sonicated for 60 min to achieve a homogenized dispersion. About 5 μL of the prepared ink of the catalyst was then drop-cast on to a clean GC electrode (diameter of 0.07 cm2) with a 0.21 mg cm−2 active mass loading and was kept at room temperature for drying. Cyclic voltammetry (CV), linear sweep voltammetry (LSV), and electrochemical impedance spectroscopy (EIS) were performed in basic electrolyte medium in 1 M KOH at a scan rate of 10 mV s−1. CV cycles were performed in between 1 and 1.85 V (vs. RHE) and OER polarization curves were obtained through linear sweep voltammetry (LSV) from 1 to 1.85 V (vs. RHE) at a scan rate of 10 mV s−1. EIS was carried out in the frequency range of 100 kHz to 10 mHz by applying the corresponding overpotentials. The potentials were converted into the reversible hydrogen electrode (RHE) using the equation: ERHE = EHg/HgO + 0.059 pH + E0Hg/HgO, where ERHE is the final converted potential with respect to the RHE, EHg/HgO is the experimental potential with respect to the Hg/HgO electrode, and E0Hg/HgO is the standard reduction potential of the Hg/HgO electrode. Chronoamperometric (CA) analysis was performed to check the materials' stability in a three-electrode system for 22 h in 1 M KOH solution. The gaseous product O2 was measured using a portable gas chromatography system (Mayura Analytical, India) with different potentials.
3. Results and discussion
3.1 Structures and surfaces of the catalysts
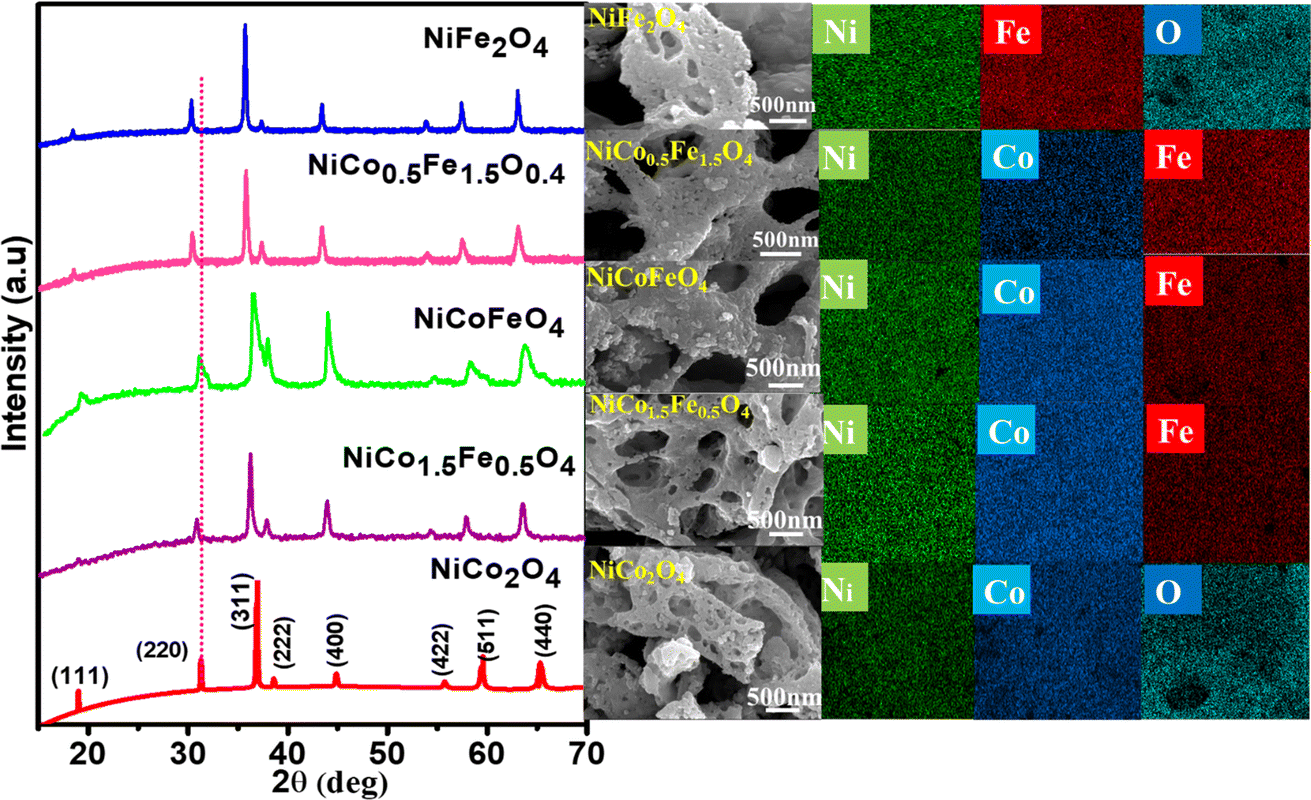
The powder XRD patterns are presented in Fig. 1 and illustrate the phase purity and structure of the synthesized oxides. The indexed peaks correspond to the face-centered cubic inverse spinel structure with the space group Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m. Notably, no additional peaks, beyond the expected reflections, could be observed in the XRD patterns, confirming the formation of phase-pure solid solutions. While the reflections appeared intense for pristine NiCo2O4 and NiFe2O4, the solid solutions displayed peak broadening attributed to their nanocrystalline nature. The crystallite size from the plane (311), calculated using Scherrer's formula and presented in Table 1, supports the observed changes in crystallinity in both the solid solutions and endmembers. A dotted line was introduced in the powder XRD pattern to emphasize the peak shifts towards lower 2θ angles with the increase in Fe doping. This shift was likely a result of lattice substitution, where the lower atomic radius Co was replaced by the higher atomic radius Fe. The corresponding lattice expansion due to the incorporation of Fe is detailed in Table 1. It should be noted that the pristine NiFe2O4 exhibited the highest cell parameters. The Co2+ and Co3+ in tetrahedral environment displayed ionic radii of 0.58 and 0.4 Å, whereas Fe2+ and Fe3+ in tetrahedral environment displayed ionic radii of 0.63 and 0.49 Å. A similar trend was also observed in the octahedral environment. The higher ionic radii of Fe compared to Co rationalizes the increased cell parameters in NiFe2O4. Further, Rietveld refinements of the pristine and doped oxides were also carried out, resulting in a good profile fit. The reliability factors along with refined lattice parameters and constraints are provided in Table S1.† The refined lattice parameters corroborated the similar trend in variation with respect to doping shown in Table 1. The FE-SEM micrographs displaying the surface morphology of the synthesized spinels are presented in Fig. 1 The micrographs clearly reveal the porosity in all the materials with an average pore diameter greater than 100 nm. Notably, NiCo0.5Fe1.5O4 exhibited the highest porosity compared to the other spinels. The EDS elemental mapping revealed a significant and widespread distribution of the elements across the matrix. The surface elemental atomic percentages calculated from the EDS data (Fig. S2†) matched well with the theoretically calculated amount of the bulk solid solutions and XRF analysis results (Table 2).
m. Notably, no additional peaks, beyond the expected reflections, could be observed in the XRD patterns, confirming the formation of phase-pure solid solutions. While the reflections appeared intense for pristine NiCo2O4 and NiFe2O4, the solid solutions displayed peak broadening attributed to their nanocrystalline nature. The crystallite size from the plane (311), calculated using Scherrer's formula and presented in Table 1, supports the observed changes in crystallinity in both the solid solutions and endmembers. A dotted line was introduced in the powder XRD pattern to emphasize the peak shifts towards lower 2θ angles with the increase in Fe doping. This shift was likely a result of lattice substitution, where the lower atomic radius Co was replaced by the higher atomic radius Fe. The corresponding lattice expansion due to the incorporation of Fe is detailed in Table 1. It should be noted that the pristine NiFe2O4 exhibited the highest cell parameters. The Co2+ and Co3+ in tetrahedral environment displayed ionic radii of 0.58 and 0.4 Å, whereas Fe2+ and Fe3+ in tetrahedral environment displayed ionic radii of 0.63 and 0.49 Å. A similar trend was also observed in the octahedral environment. The higher ionic radii of Fe compared to Co rationalizes the increased cell parameters in NiFe2O4. Further, Rietveld refinements of the pristine and doped oxides were also carried out, resulting in a good profile fit. The reliability factors along with refined lattice parameters and constraints are provided in Table S1.† The refined lattice parameters corroborated the similar trend in variation with respect to doping shown in Table 1. The FE-SEM micrographs displaying the surface morphology of the synthesized spinels are presented in Fig. 1 The micrographs clearly reveal the porosity in all the materials with an average pore diameter greater than 100 nm. Notably, NiCo0.5Fe1.5O4 exhibited the highest porosity compared to the other spinels. The EDS elemental mapping revealed a significant and widespread distribution of the elements across the matrix. The surface elemental atomic percentages calculated from the EDS data (Fig. S2†) matched well with the theoretically calculated amount of the bulk solid solutions and XRF analysis results (Table 2).
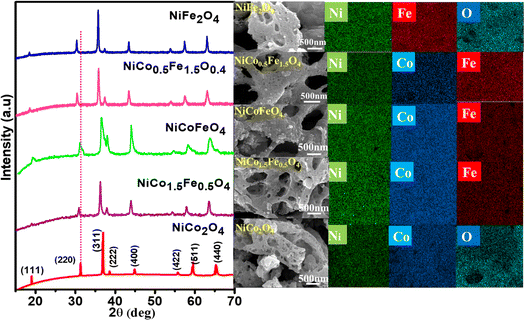 |
| | Fig. 1 Powder XRD patterns, FE-SEM, and image mapping of the synthesized spinels. | |
Table 1 Crystalline sizes, cell parameters, and pore sizes of the synthesized catalysts
| Catalysts |
Crystalline size (nm) |
Cell parameter (Å) |
Pore size from BJH analysis (nm) |
| NiCo2O4 |
33.52 |
8.07 |
49.01 |
| NiCo1.5Fe0.5O4 |
26.13 |
8.21 |
66.29 |
| NiCoFeO4 |
12.16 |
8.20 |
68.84 |
| NiCo0.5Fe1.5O4 |
24.57 |
8.34 |
69.24 |
| NiFe2O4 |
29.38 |
8.40 |
48.0 |
Table 2 Observed bulk atomic percentages of the elements from XRF in comparison to the calculated percentages, and surface atomic percentages of the elements derived from EDS and the XPS survey spectra in the synthesized catalysts
| Catalysts |
Element |
Theoretically calculated atomic % |
Bulk atomic % from XRF |
Surface atomic % from EDS |
Surface atomic % from XPS |
| NiCo2O4 |
Ni |
33.29 |
30.96 |
34.1 |
22.44 |
| Co |
66.71 |
69.04 |
65.9 |
77.56 |
| NiCo1.5Fe0.5O4 |
Ni |
33.54 |
39.93 |
43.94 |
18.54 |
| Co |
50.49 |
41.98 |
36.95 |
72.56 |
| Fe |
15.97 |
18.09 |
19.11 |
8.9 |
| NiCoFeO4 |
Ni |
33.84 |
31.57 |
30.86 |
16.38 |
| Co |
33.96 |
39.42 |
33.76 |
36.88 |
| Fe |
32.2 |
29.01 |
35.38 |
46.74 |
| NiCo0.5Fe1.5O4 |
Ni |
34.14 |
33.05 |
37.1 |
19.8 |
| Co |
17.14 |
21.41 |
18.8 |
29.91 |
| Fe |
48.72 |
45.33 |
44.1 |
50.29 |
| NiFe2O4 |
Ni |
34.4 |
32.01 |
33.3 |
20.24 |
| Fe |
65.6 |
67.99 |
66.7 |
79.76 |
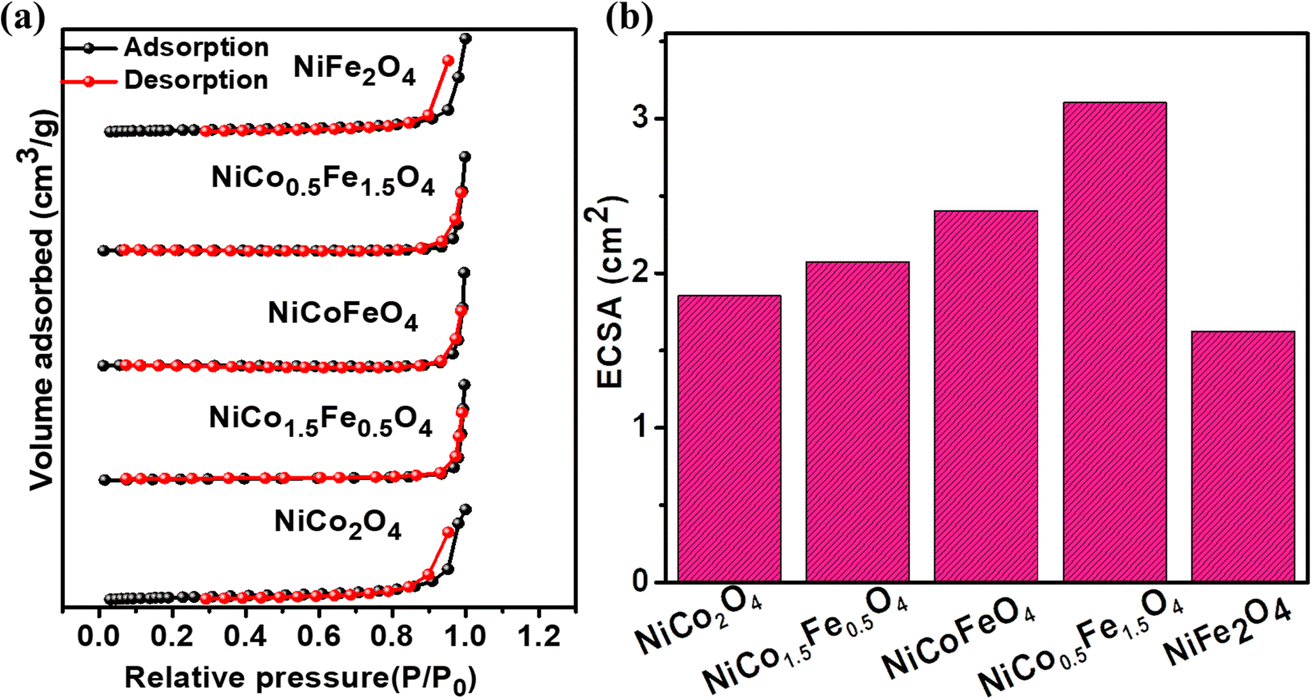
Electrocatalysis is inherently connected with the surface of a material, and so investigations were extended to the surface area analysis, and we found that the materials possessed a type II adsorption–desorption isotherm with minimum hysteresis (Fig. 2a). The average surface areas estimated from the BET equation were found to be 3–7 m2 g−1, and the pore diameters according to BJH analysis ranged from 48–70 nm (Table 1). Further, the electrochemical active surface area (ECSA) of the synthesized spinels was determined using the following equation ECSA = Cdl/Cs, where Cdl represents the electrochemical double-layer capacitance, and Cs stands for the specific capacitance.38–41Cdl was extracted from the slope of half of the capacitive current derived from the CV traces within the non-faradaic range (Fig. S3†) against the scan rate, as shown in Fig. S4.†42 Also, CS was determined using the following equation: 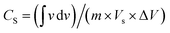 , where
, where 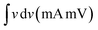 represents the area under the CV curve in the faradaic region obtained with a scan rate of 10 mV s−1 (Fig. S6†), m (0.21 mg) is the active mass of the catalyst loaded onto the electrode (with an area of 0.07 cm2), VS is the scan rate (mV s−1), and ΔV is the potential window difference (mV). The calculated ECSA plotted in Fig. 2b shows NiCo0.5Fe1.5O4 possessed a superior electrochemical surface area compared to the other synthesized spinels.
represents the area under the CV curve in the faradaic region obtained with a scan rate of 10 mV s−1 (Fig. S6†), m (0.21 mg) is the active mass of the catalyst loaded onto the electrode (with an area of 0.07 cm2), VS is the scan rate (mV s−1), and ΔV is the potential window difference (mV). The calculated ECSA plotted in Fig. 2b shows NiCo0.5Fe1.5O4 possessed a superior electrochemical surface area compared to the other synthesized spinels.
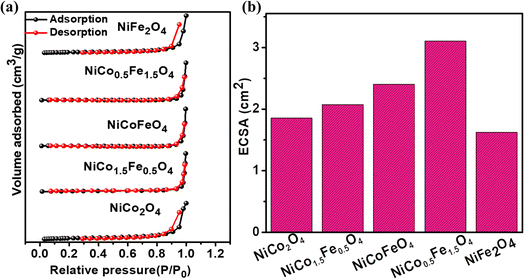 |
| | Fig. 2 (a) Adsorption–desorption isotherms, and (b) ECSAs of the synthesized catalysts. | |
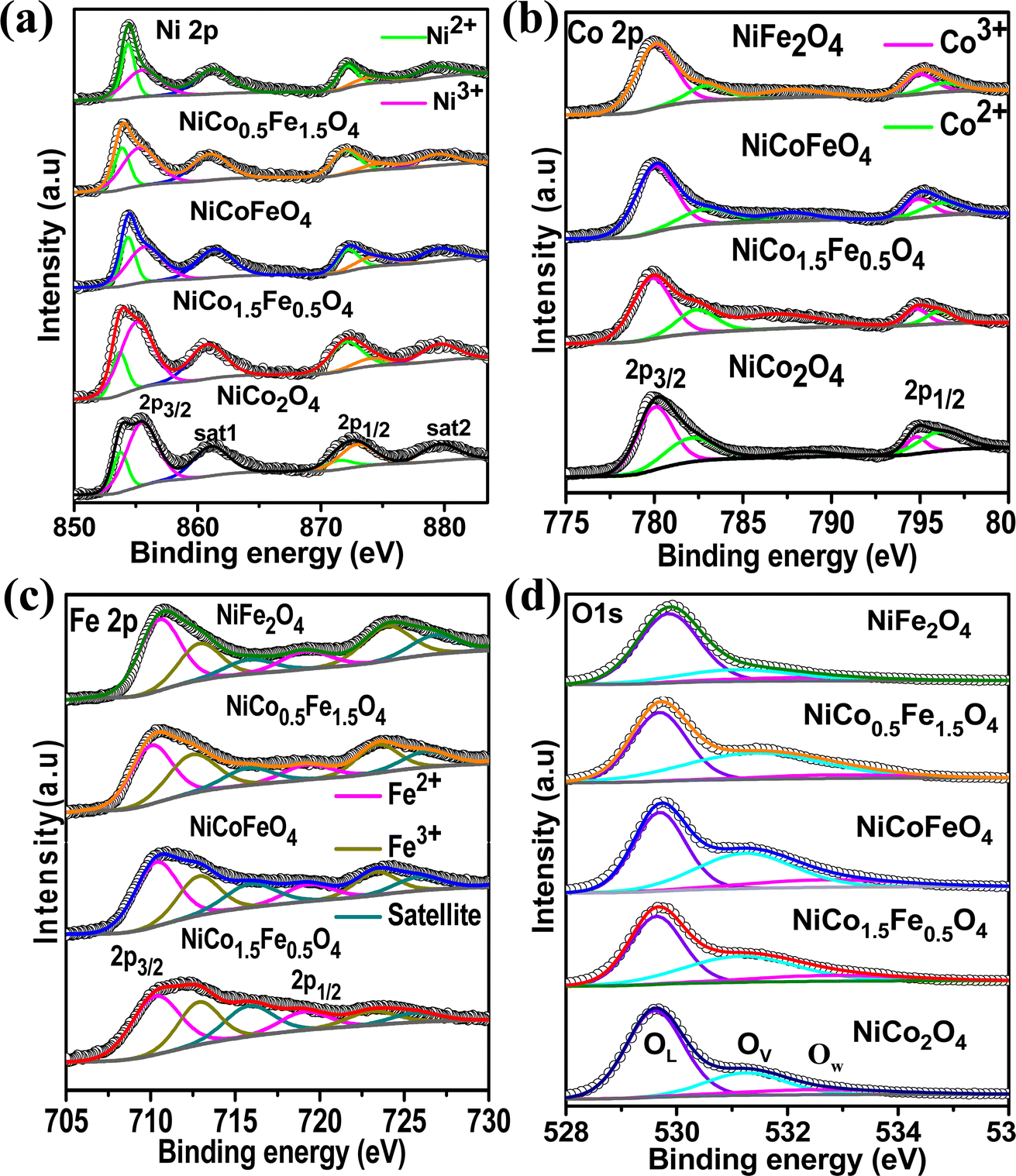
Next, XPS was employed to investigate the surface elemental composition and oxidation states of the synthesized catalysts. The survey spectra in Fig. S6† of the synthesized catalysts exhibited all the elements present, but with the surface enrichment of the B site elements (Table 2). Fig. 3a displays the spin–orbit doublet peaks of the high-resolution Ni 2p core-level spectra for all five catalysts. The deconvoluted Ni 2p demonstrated 2p3/2 and 2p1/2 peaks associated with Ni2+ as well as Ni3+ ions. The Ni3+ peak appeared at a higher binding energy (2p3/2–2p1/2 at 855.5–873.2 eV) compared to the Ni2+ (2p3/2–2p1/2 at 853.4–871.1 eV).43–47 Two satellite peaks were observed at 861 and 879 eV. The calculated Ni2+/Ni3+ ratio presented in Table 3 suggests a predominance of Ni2+ ions, which further rose with increasing the Fe doping. The core-level spectra of Co 2p for all the cobalt-containing catalysts are presented in Fig. 3b and show the spin–orbit doublet peaks of 2p3/2 and 2p1/2 associated with Co2+ (2p3/2–2p1/2 at 781.9–795.9 eV) and Co3+ (2p3/2–2p1/2 at 780.1–794.5 eV).48–50 A notable shift to higher binding energies was observed with the Fe doping, and NiCo0.5Fe1.5O4 exhibited the maximum shifts. The calculated Co3+/Co2+ ratios presented in Table 3 showed a gradual increase in the ratio with increasing the Fe concentration. Fig. 3c illustrates the Fe 2p core-level spectra with 2p3/2 and 2p1/2 peaks arising from Fe2+ (2p3/2–2p1/2 at 710.4–719.3 eV) and Fe3+ (2p3/2–2p1/2 at 712.9–723.8 eV).33,51–53 The calculated Fe3+/Fe2+ ratio derived from the area under the deconvoluted peaks in Table 3 suggests a monotonic increase in the Fe3+ concentration with a higher doping of Fe. It is important to note that with higher Fe doping, the lower oxidation states of Ni were stabilized, while the higher oxidation states of Co and Fe were stabilized in the spinels. From the bivalent and trivalent ratios of the cations for A and B sites, we derived the stoichiometric molecular formula of the endmembers of the synthesized inverse spinels as [Co0.462+Co0.273+]Td[Ni0.62+Ni0.43+Co0.273+]OhO4 and [Fe0.422+Fe0.293+]Td[Ni0.772+Ni0.233+Fe0.293+]OhO4. The details stoichiometric molecular formula provided in Table 3 show that with Fe doping in NiCo2O4, the concentration of Co3+ in the octahedral geometry (B site) gradually increased. It has been reported in the literature that the electron configuration of Co3+ in the octahedral environment undergoes a transition from a low spin state to an intermediate spin state of t52ge1g in the presence of Fe3+.29–34 While the electron occupancy of the d orbital in the tetrahedral geometry does not matter, as neither the high-lying t2 nor the low-lying e align effectively with the O 2p, the near unity electron occupancy in the eg orbital in the octahedral geometry plays a crucially pivotal role in the OER mechanism.54,55 It should be noted that the Co3+ t52ge1g configuration was found to be the maximum in the case of NiCo0.5Fe1.5O4 (Table 3). The core-level deconvoluted spectra of the O 1s region of the oxides are presented in Fig. 3d. The peak at 529.56 eV was ascribed to the lattice oxygen (OL), whereas the peak at 531.27 eV was assigned to the adsorbed oxygen species in the defect sites or oxygen vacancies (Ov), while the feeble peak at 532.5 eV was assigned to adsorbed water molecule (Ow).56–58 It should be noted that the O 1s binding energy gradually shifted to a higher value with Fe doping. The lowering in the higher valency (3+) cations with Fe doping in the synthesized spinels (videTable 3) rationalizes the shift in the binding energy of O 1s. Also, the peak associated with OV exhibited a gradual increase in intensity with higher levels of Fe doping. The OV/OT, where OT = OL + OV + OW, as presented in Table 3, displayed the maximum oxygen vacancies in the case of NiCo0.5Fe1.5O4. Oxygen vacancies are also known to play an important role in the OER efficacy.19,49
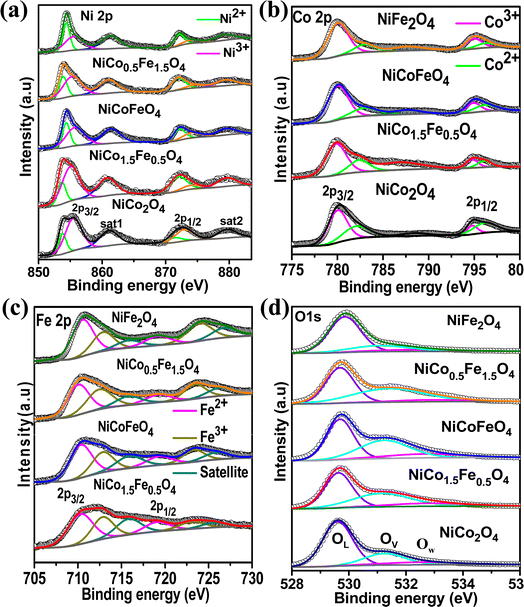 |
| | Fig. 3 (a) Ni 2p core-level spectra; (b) Co 2p core-level spectra; (c) Fe 2p core-level spectra; and (d) O 1s core-level spectra of the synthesized catalysts. | |
Table 3 Metal oxidation state ratio, oxygen vacancy, and stoichiometric molecular formula
| Catalyst |
Ni2+/Ni3+ |
Co3+/Co2+ |
Fe3+/Fe2+ |
OV/OT |
[B2+/3+ B′2+/3+]Td[A2+/3+B3+B′3+]OhO4−δ |
| NiCo2O4 |
1.5 |
1.22 |
— |
0.25 |
[Co0.462+Co0.273+]Td[Ni0.62+Ni0.43+Co0.273+]OhO4 |
| NiCo1.5Fe0.5O4 |
2.23 |
2.12 |
0.61 |
0.35 |
[Co0.322+Fe0.622+Co0.343+Fe0.193+]Td [Ni0.692+Ni0.313+Co0.343+Fe0.193+]OhO4 |
| NiCoFeO4 |
2.57 |
2.22 |
0.72 |
0.39 |
[Co0.32+Fe0.582+Co0.353+Fe0.213+]Td [Ni0.722+Ni0.283+Co0.353+Fe0.213+]OhO4 |
| NiCo0.5Fe1.5O4 |
3.1 |
3.34 |
1.5 |
0.57 |
[Co0.232+Fe0.42+Co0.383+Fe0.33+]Td [Ni0.762+Ni0.243+Co0.383+Fe0.33+]OhO4 |
| NiFe2O4 |
3.34 |
— |
1.38 |
0.2 |
[Fe0.422+Fe0.293+]Td[Ni0.772+Ni0.233+Fe0.293+]OhO4 |
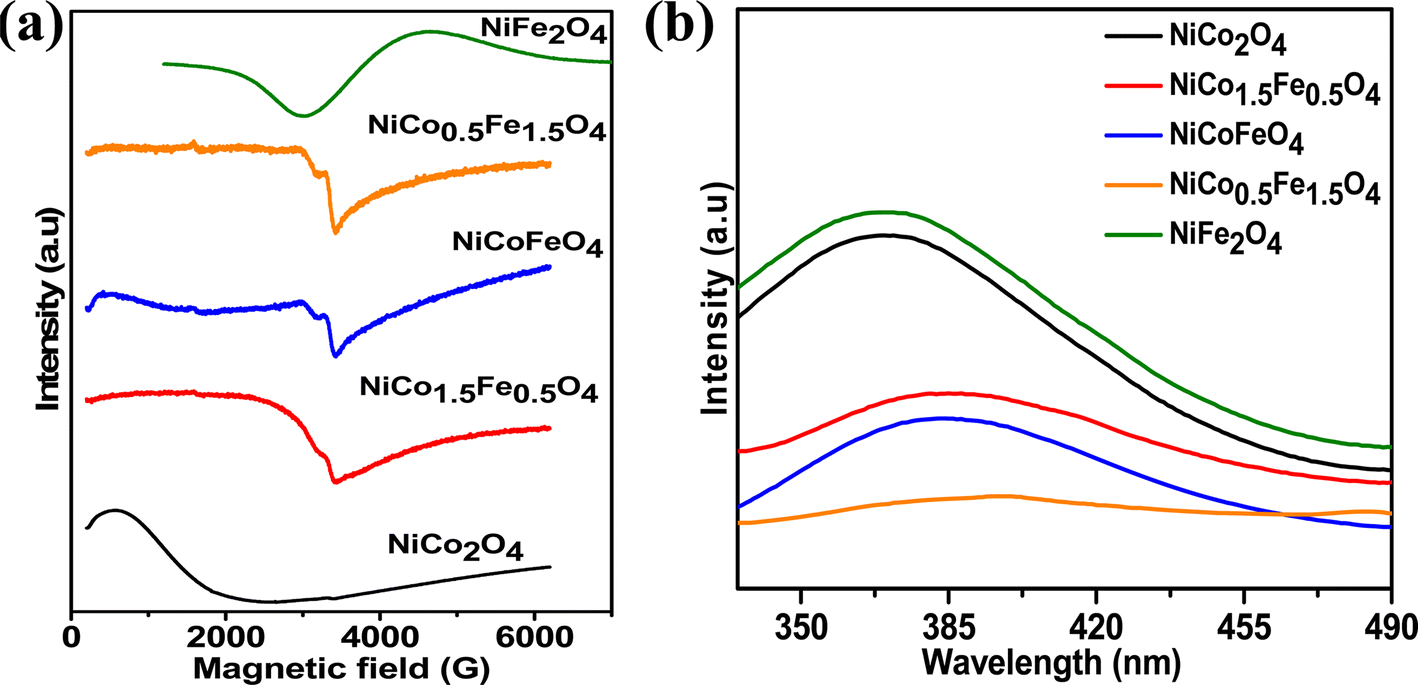
Fig. 4a presents the EPR data, which were collected to explore the number of unpaired electrons in the synthesized materials. The Fe-doped NiCo2O4 samples showed a sharp EPR signal, whereas broad spectra were found for the endmembers NiFe2O4 and NiCo2O4. The sharp signal from the doped samples could be due to the unpaired electrons in the intermediate electronic configuration of Co3+(t52ge1g), corroborating the XPS results. Next, the photoluminescence of the synthesized materials was recorded to obtain additional information on the oxygen vacancies. Fig. 4b shows the highest PL intensity for the pristine endmembers of NiCo2O4 and NiFe2O4. However, the doped solid solutions showed reduced PL intensities, and the lowest PL intensity was observed with NiCo0.5Fe1.5O4. The intermediate states generated between the valence and conduction bands due to the oxygen vacancies reduce the electron–hole recombination59 and it was apparent from the PL spectra that the doped samples contained higher degrees of oxygen vacancies than the pristine samples. The PL spectra suggested the maximum presence of oxygen vacancies over NiCo0.5Fe1.5O4, corroborating the XPS findings.
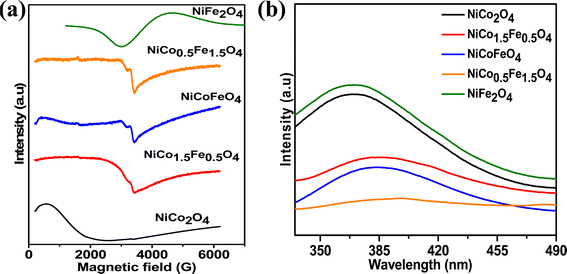 |
| | Fig. 4 (a) EPR and (b) fluorescence spectra of the synthesized spinels. | |
3.2 OER
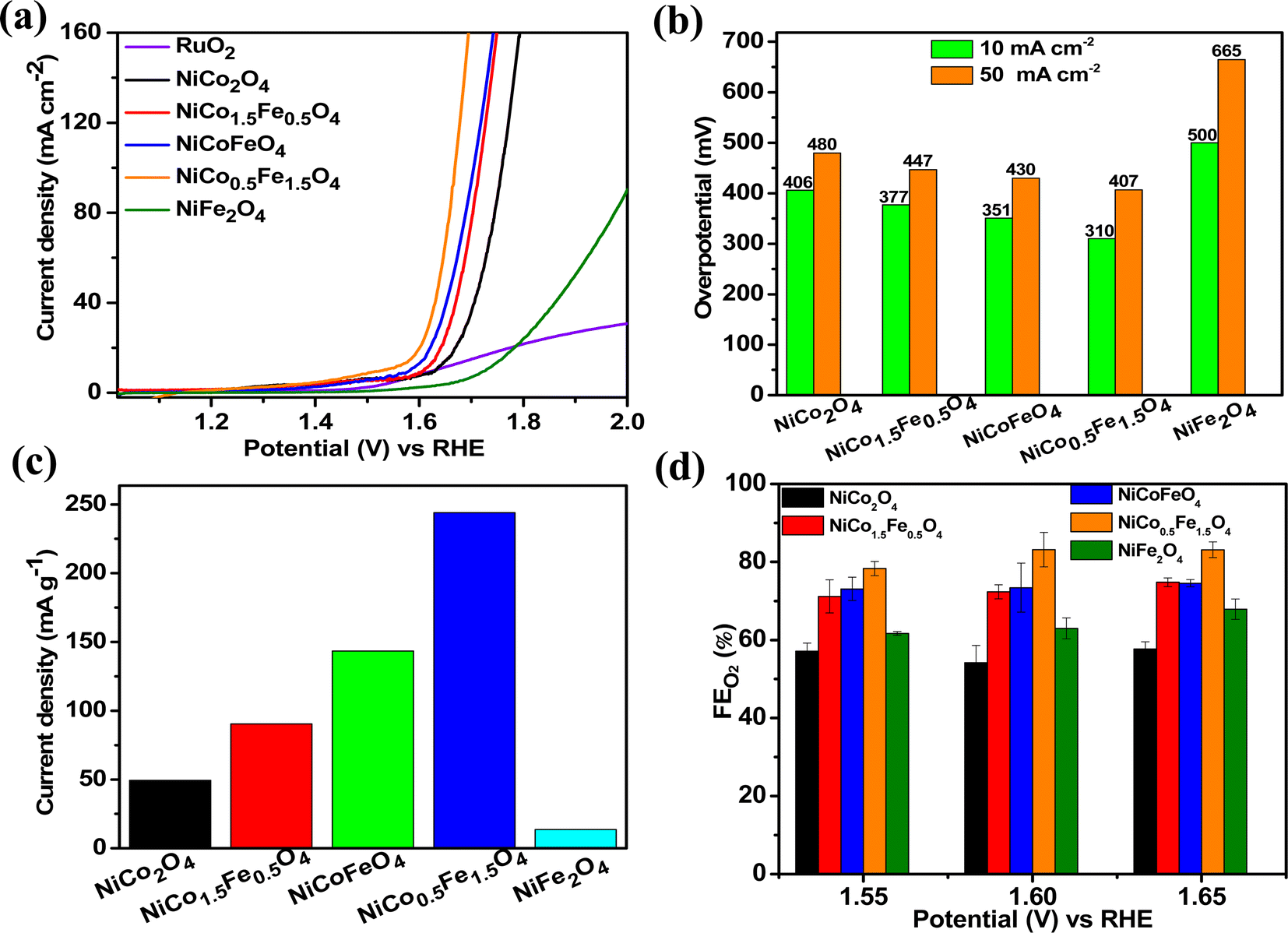
The catalytic OER activity of the synthesized materials on a GC electrode was measured in 1 M KOH using the LSV polarization curves, as shown in Fig. 5a. The pristine NiCo2O4 displayed a feeble peak at approximately 1.5 V vs. RHE prior to reaching the OER onset potential corresponding to the oxidation of Co2+ to Co3+.21 According to Fig. 5a, the doped spinel NiCo0.5Fe1.5O4 exhibited the best catalytic activity towards the OER. The overpotential at a current density of 10 mA cm−2 is a metric relevant to solar fuel production. Therefore, the overpotentials of all five catalysts were mapped at a current density of 10 as well as 50 mA cm−2 and are plotted in Fig. 5b. Apparently, a decreasing trend of overpotentials could be observed with the higher doping of Fe in NiCo2O4, and interestingly the pristine NiFe2O4 exhibited the poorest activity. Among the materials, NiCo0.5Fe1.5O4 exhibited the best catalytic efficacy with the lowest overpotential of 310 mV at 10 mA cm−2 among the doped spinels. For benchmarking, a commercial RuO2 sample was also measured under similar conditions, and apparently NiCo0.5Fe1.5O4 showed a lower overpotential than RuO2. For a global comparison and practical applicability, it is highly desirable to find out the mass activity of a catalyst. The mass activity of electrocatalysts is determined by normalizing the current density by the loaded amount of catalytic material. Fig. 5c shows the mass activity of the spinel catalysts at the OER onset potential, where it could be evidently seen that NiCo0.5Fe1.5O4 exhibited a far superior OER catalytic activity than the other spinels. The intrinsic activity, which is defined by normalizing the current density by the ECSA, is another global comparison metric. The intrinsic activity of all the catalysts is shown in Fig. S7,† also suggesting a similar trend of catalytic efficacy, with NiFe1.5Co0.5O4 showing superior performance among the synthesized spinels. We further compared the OER activity data with other reported NiCo2O4-based catalysts, and Table 4 demonstrates that exhausted NiCo0.5Fe1.5O4 exhibited comparable or even superior performance to the other NiCo2O4-based catalysts. Further, the oxygen evolved during the OER was measured with the help of gas chromatography, and the faradaic efficiency of oxygen at three different potentials above the onset potential was plotted, as shown in Fig. 5d. The faradaic efficiency of oxygen evolution was calculated using the following equation:
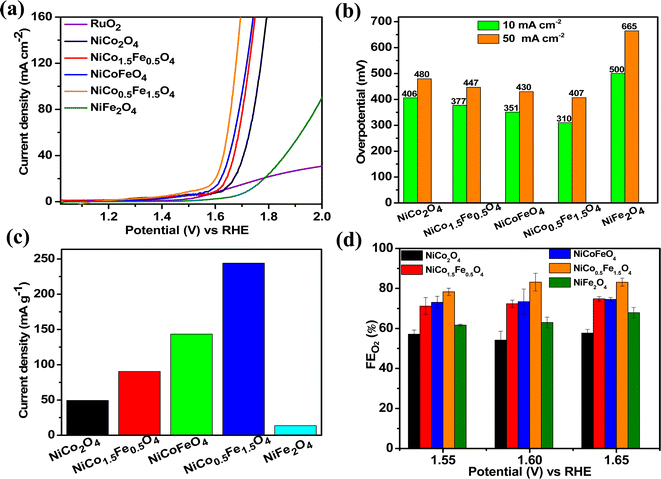 |
| | Fig. 5 (a) LSV polarisation plots; (b) overpotential at 10 and 50 mA cm−2 current densities; (c) mass activities, and (d) FE(%) of oxygen evolution over the synthesized catalysts. | |
Table 4 Comparison of the OER activity data of NiCo2O4-based catalysts with previous works
| Catalyst |
Overpotential (mV) vs. RHEa |
Working electrode |
References |
|
10 mA cm−2 current.
|
| Hierarchical NiCo2O4 |
290 |
Ni foam |
60
|
| NiCo2O4/carbon black |
420 |
Glassy carbon |
61
|
| NiCo2O4/vanadium nitrides |
395 |
| Cu/NiCo2O4 |
570 |
Graphite felt |
62
|
| Template free NiCo2O4 |
380 |
Glassy carbon |
28
|
| Pluronic-123 NiCo2O4 |
350 |
| NiCo2O4 nanowire @NiCo2O4 nanoflake |
320 |
Carbon cloth |
63
|
| Reduced-NiCo2O4 |
370 |
Ni foam |
64
|
| Hierarchical rosettes-like NiCo2O4 |
480 |
Rotating disc glassy carbon tip |
65
|
| Ir/NiCo2O4 |
300 |
Glassy carbon |
66
|
| Residual NiCo2O4 (NiCo2O4-R) |
361 |
Glassy carbon |
67
|
| NiCo2O4 fabricated on Ni foam (NiCo2O4-N) |
363 |
Ni foam |
| NiCoO2/NiCo2O4 |
351 |
Indium tin oxide |
68
|
| g-C3N4/NiCo2O4 |
294 |
Glassy carbon |
69
|
| Porous nitrogen graphene/NiCo2O4 |
470 |
PNG |
70
|
| Fe-doped NiCo2O4 |
310 |
Glassy carbon |
This work |
The faradaic efficiency (FE) describes the overall selectivity of an electrochemical process and is defined as the amount (moles) of collected product relative to the amount that could be produced from the total charge passed, expressed as a fraction or a percentage. The highest OER catalytic activity apparently facilitated NiCo0.5Fe1.5O4 exhibiting the highest FEO2 (%) over the other materials.
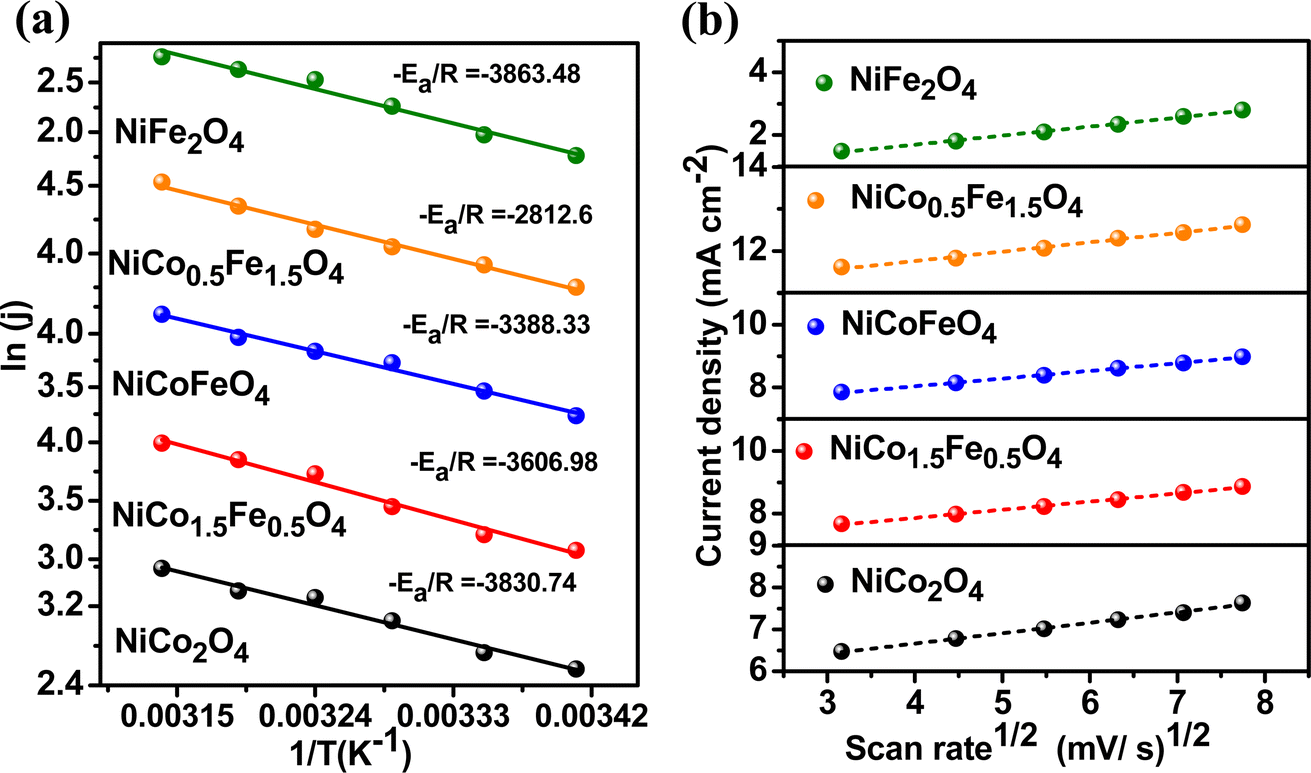
To explore the kinetics of the OER over the spinels, the activation energy was determined from an Arrhenius plot of the current density vs. temperature at the OER potential. The temperature ranged from 25 °C to 50 °C with a gap of 5 °C (Fig. S8†). The activation energy plot in Fig. 6a shows a similar trend for the mass activity and intrinsic activity of the catalysts, with the lowest activation energy of 23.38 kJ mol−1 observed over NiFe1.5Co0.5O4, indicating the fastest kinetics over the optimally doped material. We further evaluated the scan rate dependence of the OER activity over the materials by changing the scan rate from 10 to 50 mV s−1 in the presence of a 1 M KOH electrolyte. Fig. 6b reveals that the OER current density linearly increased with the square root of the scan rate, indicating a diffusion-controlled electrocatalytic OER over the spinel catalysts.
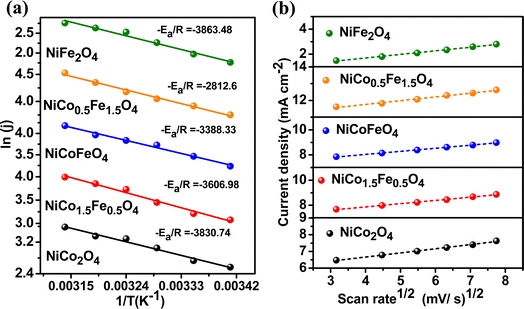 |
| | Fig. 6 (a) Activation energy plots of OER and (b) scan-rate-dependent study of the synthesized materials. | |
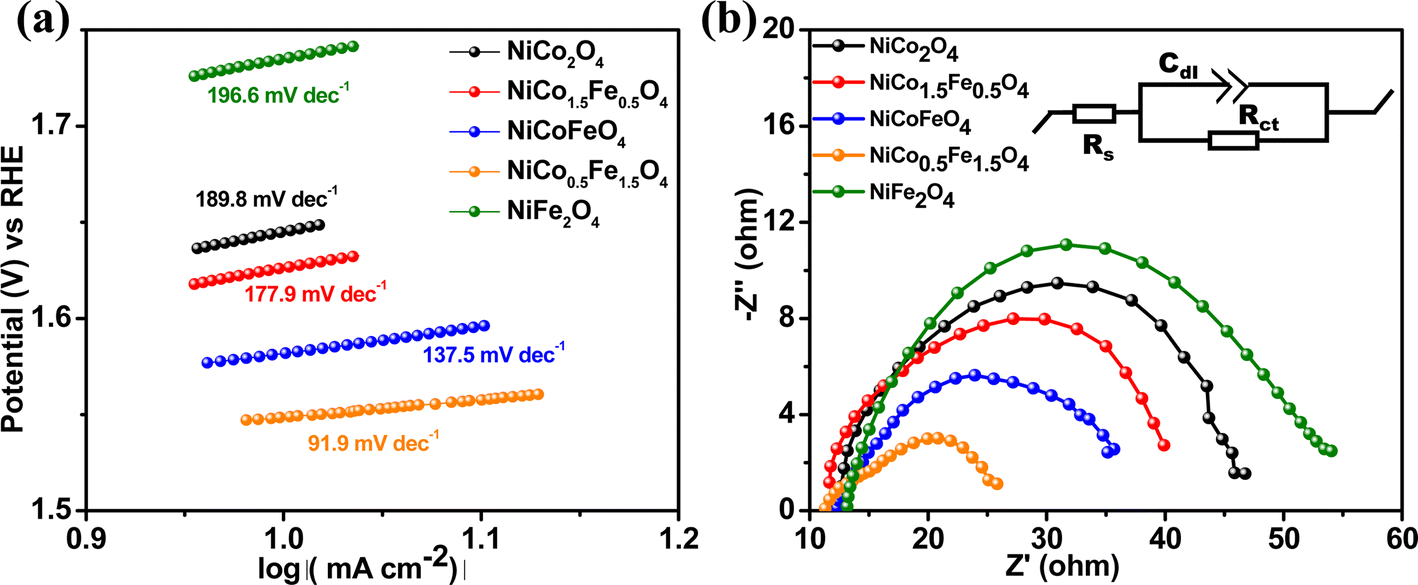
The Tafel slope is another parameter to assess the activity based on the overpotential vs. kinetic current relationship. The lowest Tafel slope of 91.9 mV dec−1 was observed over NiCo0.5Fe1.5O4, as shown in Fig. 7a. A low Tafel slope indicates improved charge transfer between the electrode and electrolyte over a doped material, rationalizing its efficient OER activity. Nyquist impedance plots further help assessing the OER efficacy. EIS measurements were thus done under OER conditions with an applied frequency of 1 MHz to 100 mHz, and the results were fitted to an equivalent circuit, as shown in the inset of Fig. 7b. NiCo0.5Fe1.5O4 showed the lowest semi-circle, suggesting the fastest shuttling of charges, i.e. higher conductivity, over the optimally doped spinel. The improved charge transfer and the consequent higher conductivity over NiCo0.5Fe1.5O4 could be due to the occurrence of a high intermediate electronic configuration of Co3+(t52ge1g), as was observed from the XPS and EPR studies. The near unity electron occupancy in the eg of Co3+ presents a strong hybridization between Co(eg)–O(p) states at Fermi energy to enhance the covalency as well as the conductivity of the material, i.e. reduces the charge transfer resistance, as was corroborated by the lowest Tafel slope or impedance results.71–73
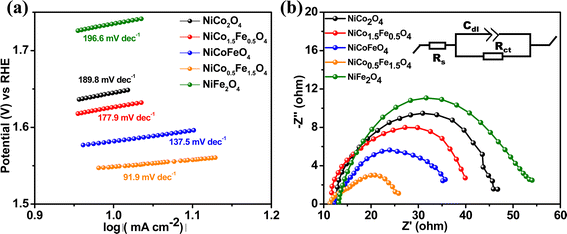 |
| | Fig. 7 (a) Tafel plots and (b) Nyquist plots with the equivalent circuit (shown in the inset) of the synthesized materials. | |
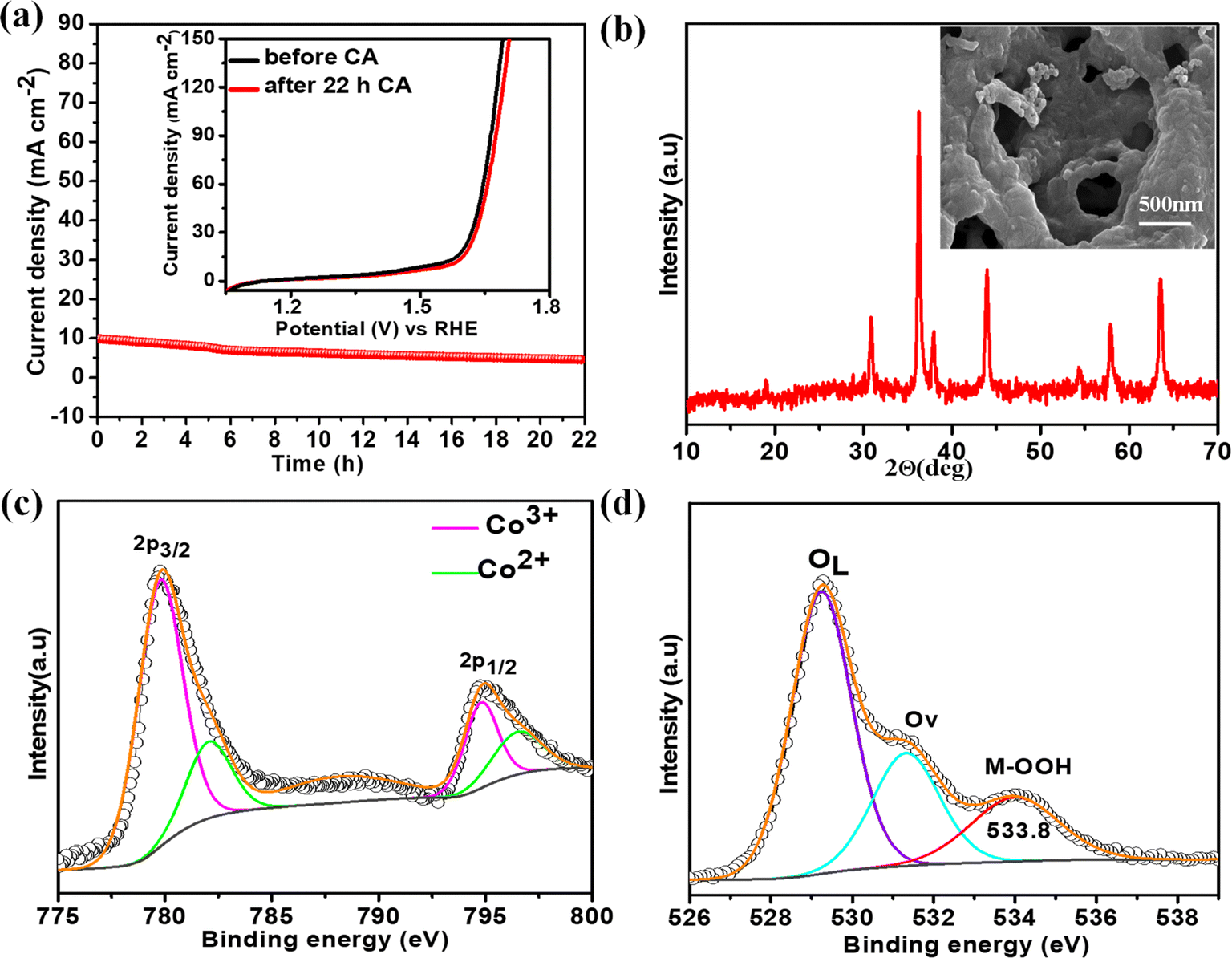
The electrochemical stability of the best catalyst was evaluated by chronoamperometry (CA) studies in 1 M KOH solution for 22 h at the corresponding OER overpotential. A remarkably stable OER current density was observed over NiCo0.5Fe1.5O4 (Fig. 8a). To corroborate this further, we also collected the LSV traces of NiCo0.5Fe1.5O4 before and after the 22 h CA study. The inset in Fig. 8a demonstrated an almost identical current density for the fresh and exhausted catalyst, signifying the outstanding stability of the catalyst even in considerably high alkaline conditions. The XRD pattern of the exhausted NiCo0.5Fe1.5O4 with the FE-SEM image in the inset in Fig. 8b exhibited the unchanged bulk crystalline structure and the surface porosity. However, the core-level spectra of Co 2p from the exhausted NiCo0.5Fe1.5O4 indicated there was a significant increase in Co3+ compared to the fresh catalysts (Fig. 8c). The core-level deconvoluted spectra of the O 1s region of the exhausted NiCo0.5Fe1.5O4 presented in Fig. 8d evidently showed an extra peak at 533.8 eV, which was absent in the fresh catalyst. This could be attributed to the formed intermediate Cooh3+–OOH during the OER.21 It could be concluded that the near unity electron filling in the eg orbital and the significant oxygen vacancies as observed from the XPS and PL studies of the fresh catalyst would lead to stabilisation of the Cooh3+–OOH bond, and thereby would contribute to the superior catalytic OER efficacy of NiCo0.5Fe1.5O4.
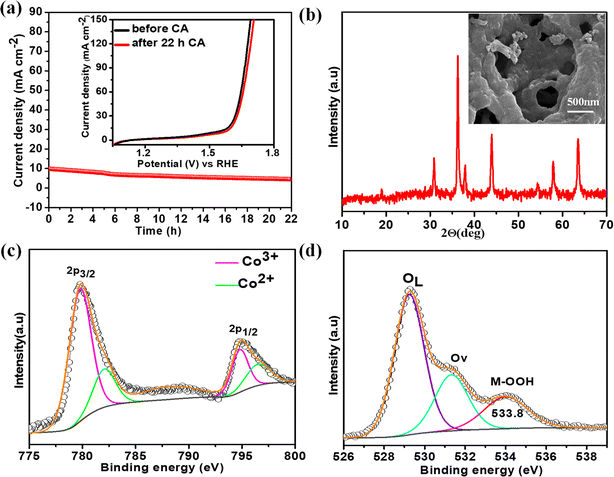 |
| | Fig. 8 (a) CA plots with polarisation curves of the fresh and exhausted NiCo0.5Fe1.5O4 in the inset; (b) XRD pattern with FE-SEM image of the exhausted NiCo0.5Fe1.5O4; (c) CO 2p and (d) O 1s spectra of the exhausted NiCo0.5Fe1.5O4. | |
4. Conclusion
Combustion-synthesized solid solutions of NiCo2−xFexO4 (x = 0, 0.5, 1, 1.5, 2) were crystallized in a phase-pure cubic inverse spinel structure. While the pristine NiCo2O4 and NiFe2O4 showed a highly crystalline nature, the doped solid solutions displayed nanocrystallinity. The FE-SEM and BET surface analyses exhibited a high degree of porosity in all the materials. However, the electrochemical surface area was found to be highest for NiCo0.5Fe1.5O4. The XPS as well as the EPR data concluded that with optimal Fe doping, the intermediate spin state of Co3+ with a t52ge1g configuration was stabilized in NiCo0.5Fe1.5O4. The surface oxygen vacancies were also found to be the maximum over NiCo0.5Fe1.5O4, as established from the XPS and PL studies. The near unity electron occupancy in the eg of Co3+ in NiCo0.5Fe1.5O4 enhanced the covalency, as corroborated by its lowest Tafel slope and impedance results. The combined effect of the higher covalency, typical electronic configuration of Co3+, and high degree of oxygen vacancies facilitated NiCo0.5Fe1.5O4 to exhibit a superior OER efficacy, with an overpotential of 310 mV at 10 mA cm−2, through the formation of the key intermediate species of Cooh3+–OOH. The near unity electron occupancy in the eg orbital of the B site of AB2O4, high degree of oxygen vacancies, and the ability of oxygen vacancies to facilitate the formation of the intermediate species of –OOH were the key to the rational design of a superior OER electrocatalyst. This particular study highlights the importance of the electronic configuration of the active sites as well as the role of oxygen vacancies in advancing the kinetics of the oxygen evolution reaction.
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
The authors declare that they have no conflicts of interest.
Acknowledgements
Authors thank BITS Pilani Hyderabad Campus for the financial support.
References
- Z. A. Lan, Y. Fang, Y. Zhang and X. Wang, Angew. Chem., Int. Ed., 2018, 57, 470–474 CrossRef CAS PubMed.
- Y. Fang, Y. Xu, X. Li, Y. Ma and X. Wang, Angew. Chem., Int. Ed., 2018, 57, 9749–9753 CrossRef CAS PubMed.
- M. Zhao, T. Li, L. Jia, H. Li, W. Yuan and C. Ming, ChemSusChem, 2019, 12, 5041–5055 CrossRef CAS PubMed.
- M. Zhao, H. Li, W. Yuan and C. Ming, ACS Appl. Energy Mater., 2020, 3, 3966–3977 CrossRef CAS.
- P. C. Meenu, S. P. Datta, S. A. Singh, S. Dinda, C. Chakraborty and S. Roy, J. Power Sources, 2020, 461, 228150 CrossRef.
- S. Roy, S. Payra, S. Challagulla, R. Arora, S. Roy and C. Chakraborty, ACS Omega, 2018, 3, 17778–17788 CrossRef CAS PubMed.
- L. C. Seitz, C. F. Dickens, K. Nishio, Y. Hikita, J. Montoya, A. Doyle, C. Kirk, A. Vojvodic, H. Y. Hwang, J. K. Norskov and T. F. Jaramillo, Science, 2016, 353, 1011–1014 CrossRef CAS PubMed.
- T. Reier, H. N. Nong, D. Teschner, R. Schlögl and P. Strasser, Adv. Energy Mater., 2017, 7, 1601275 CrossRef.
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem, 2017, 1, 0003 CrossRef CAS.
- Y. Yan, B. Y. Xia, B. Zhao and X. Wang, J. Mater. Chem. A, 2016, 4, 17587–17603 RSC.
- A. Ashok, A. Kumar, R. R. Bhosale, F. Almomani, M. A. H. Saleh Saad, S. Suslov and F. Tarlochan, Int. J. Hydrogen Energy, 2019, 44, 436–445 CrossRef CAS.
- F. Švegl, B. Orel, I. Grabec-Švegl and V. Kaučič, Electrochim. Acta, 2000, 45, 4359–4371 CrossRef.
- A. La Rosa-Toro, R. Berenguer, C. Quijada, F. Montilla, E. Morallón and J. L. Vázquez, J. Phys. Chem. B, 2006, 110, 24021–24029 CrossRef CAS PubMed.
- M. De Koninck, S. C. Poirier and B. Marsan, J. Electrochem. Soc., 2006, 153, A2103 CrossRef CAS.
- B. Chi, H. Lin and J. Li, Int. J. Hydrogen Energy, 2008, 33, 4763–4768 CrossRef CAS.
- A. Badruzzaman, A. Yuda, A. Ashok and A. Kumar, Inorg. Chim. Acta, 2020, 511, 119854 CrossRef CAS.
- Y. Li, F. M. Li, X. Y. Meng, S. N. Li, J. H. Zeng and Y. Chen, ACS Catal., 2018, 8, 1913–1920 CrossRef CAS.
- T. Y. Ma, S. Dai, M. Jaroniec and S. Z. Qiao, J. Am. Chem. Soc., 2014, 136, 13925–13931 CrossRef CAS PubMed.
- J. Qi, W. Zhang and R. Cao, Chem. Commun., 2017, 53, 9277–9280 RSC.
- A. Moysiadou, S. Lee, C. S. Hsu, H. M. Chen and X. Hu, J. Am. Chem. Soc., 2020, 142, 11901–11914 CrossRef CAS PubMed.
- S. Roy, N. Devaraj, K. Tarafder, C. Chakraborty and S. Roy, New J. Chem., 2022, 46, 6539–6548 RSC.
- L. Xu, Q. Jiang, Z. Xiao, X. Li, J. Huo, S. Wang and L. Dai, Angew. Chem., Int. Ed., 2016, 55, 5277–5281 CrossRef CAS PubMed.
- C. Alex, S. C. Sarma, S. C. Peter and N. S. John, ACS Appl. Energy Mater., 2020, 3, 5439–5447 CrossRef CAS.
- S. K. Jung, H. Gwon, J. Hong, K. Y. Park, D. H. Seo, H. Kim, J. Hyun, W. Yang and K. Kang, Adv. Energy Mater., 2014, 4, 1300787 CrossRef.
- Z. Chen, C. X. Kronawitter and B. E. Koel, Phys. Chem. Chem. Phys., 2015, 17, 29387–29393 RSC.
- H. Yang, H. Sun, X. Fan, X. Wang, Q. Yang and X. Lai, Mater. Chem. Front., 2021, 5, 259–267 RSC.
- J. Bao, X. Zhang, B. Fan, J. Zhang, M. Zhou, W. Yang, X. Hu, H. Wang, B. Pan and Y. Xie, Angew. Chem., Int. Ed., 2015, 54, 7399–7404 CrossRef CAS PubMed.
- S. V. Devaguptapu, S. Hwang, S. Karakalos, S. Zhao, S. Gupta, D. Su, H. Xu and G. Wu, ACS Appl. Mater. Interfaces, 2017, 9, 44567–44578 CrossRef CAS PubMed.
- A. Wang, Y. Hu, H. Wang, Y. Cheng, T. Thomas, R. Ma and J. Wang, Mater. Today Phys., 2021, 17, 100353 CrossRef CAS.
- R. Nasser, X. L. Wang, A. B. G. Trabelsi, F. H. Alkallas, H. Elhouichet and J.-M. Song, J. Energy Storage, 2022, 52, 104619 CrossRef.
- H. Li, S. Sun, S. Xi, Y. Chen, T. Wang, Y. Du, M. Sherburne, J. W. Ager, A. C. Fisher and Z. J. Xu, Chem. Mater., 2018, 30, 6839–6848 CrossRef CAS.
- Z. Yu, S. Wang, Y. Huang, Y. Zou, F. Xu, C. Xiang, J. Zhang, J. Xie and L. Sun, J. Energy Storage, 2022, 55, 105486 CrossRef.
- A. Grimaud, O. Diaz-Morales, B. Han, W. T. Hong, Y. L. Lee, L. Giordano, K. A. Stoerzinger, M. T. M. Koper and Y. Shao-Horn, Nat. Chem., 2017, 9, 828 CrossRef CAS PubMed.
- Y. Duan, S. Sun, S. Xi, X. Ren, Y. Zhou, G. Zhang, H. Yang, Y. Du and Z. J. Xu, Chem. Mater., 2017, 29, 10534–10541 CrossRef CAS.
- Y. Sun, S. Gao, F. Lei, J. Liu, L. Liang and Y. Xie, Chem. Sci., 2014, 5, 3976–3982 RSC.
- A. Gayen, T. Baidya, K. Biawas, S. Roy and M. S. Hegde, Appl. Catal., A, 2006, 315, 135–146 CrossRef CAS.
- R. Rameshan, P. Pentyala, S. A. Singh, P. A. Deshpande and S. Roy, J. Environ. Chem. Eng., 2022, 10, 108966 CrossRef CAS.
- Y. Zheng, S. Chen, K. A. I. Zhang, J. Zhu, J. Xu, C. Zhang and T. Liu, ACS Appl. Mater. Interfaces, 2021, 13, 13328–13337 CrossRef CAS PubMed.
- X. Jiang, Y. Dong, Z. Zhang, J. Li, J. Qian and D. Gao, J. Alloys Compd., 2021, 878, 160433 CrossRef CAS.
- A. Valipour, N. Hamnabard, S. M. H. Meshkati, M. Pakan and Y. H. Ahn, Dalton Trans., 2019, 48, 5429–5443 RSC.
- S. Roy, P. Dahiya, T. Mandal and S. Roy, Dalton Trans., 2024, 53, 5484–5494 RSC.
- M. Cheng, H. Fan, Y. Song, Y. Cui and R. Wang, Dalton Trans., 2017, 46, 9201–9209 RSC.
- R. Gao, L. Pan, H. Wang, Y. Yao, X. Zhang, L. Wang and J. J. Zou, Adv. Sci., 2019, 6, 1900054 CrossRef PubMed.
- P. C. Meenu and S. Roy, ACS Appl. Mater. Interfaces, 2023, 15, 36154–36166 CrossRef PubMed.
- P. C. Meenu and S. Roy, ACS Appl. Energy Mater., 2023, 6, 11212–11225 CrossRef CAS.
- P. C. Meenu, P. K. Samanta, T. Yoshida, N. J. English, S. P. Datta, S. A. Singh, S. Dinda, C. Chakraborty and S. Roy, ACS Appl. Energy Mater., 2022, 5, 503–515 CrossRef.
- L. Zhuang, L. Ge, Y. Yang, M. Li, Y. Jia, X. Yao and Z. Zhu, Adv. Mater., 2017, 29, 1606793–1606800 CrossRef PubMed.
- S. Y. Zhang, T. T. Li, H. L. Zhu and Y. Q. Zheng, J. Mater. Sci., 2018, 53, 4323–4333 CrossRef CAS.
- S. Payra, S. Challagulla, C. Chakraborty and S. Roy, J. Electroanal. Chem., 2019, 853, 113545–113554 CrossRef CAS.
- J. Pu, J. Wang, X. Jin, F. Cui, E. Sheng and Z. Wang, Electrochim. Acta, 2013, 106, 226–234 CrossRef CAS.
- Z. Xue, H. Su, Q. Yu, B. Zhang, H. Wang, X. Li and J. Chen, Adv. Energy Mater., 2017, 7, 1602355 CrossRef.
- J. Zhang, J. Shi and M. Gong, J. Solid State Chem., 2009, 182, 2135–2140 CrossRef CAS.
- P. C. Meenu, P. K. Samanta, S. P. Datta, S. A. Singh, S. Dinda, C. Chakraborty and S. Roy, Inorg. Chem., 2024, 63, 526–536 CrossRef CAS PubMed.
- C. Wei, Z. Feng, G. G. Scherer, J. Barber, Y. Shao-Horn and Z. J. Xu, Adv. Mater., 2017, 29, 1606800 CrossRef PubMed.
- S. Sun, Y. Sun, Y. Zhou, S. Xi, X. Ren, B. Huang, H. Liao, L. P. Wang, Y. Du and Z. J. Xu, Angew. Chem., 2019, 131, 6103–6108 CrossRef.
- D. Y. Yoon, E. Lim, Y. J. Kim, J. H. Kim, T. Ryu, S. Lee, B. K. Cho, I.-S. Nam, J. W. Choung and S. Yoo, J. Catal., 2014, 319, 182–193 CrossRef CAS.
- A. Hezam, K. Namratha, Q.
A. Drmosh, D. Ponnamma, J. Wang, S. Prasad, M. Ahamed, C. Cheng and K. Byrappa, ACS Appl. Nano Mater., 2020, 3, 138–148 CrossRef CAS.
- T. Ye, W. Huang, L. Zeng, M. Li and J. Shi, Appl. Catal., B, 2017, 210, 141–148 CrossRef CAS.
- S. Payra, S. K. Ganeshan, S. Challagulla and S. Roy, Adv. Powder Technol., 2020, 31, 510–520 CrossRef CAS.
- X. Gao, H. Zhang, Q. Li, X. Yu, Z. Hong, X. Zhang, C. Liang and Z. Lin, Angew. Chem., Int. Ed., 2016, 55, 6290–6294 CrossRef CAS PubMed.
- Z. Zheng, X. Du, Y. Wang, C. M. Li and T. Qi, ACS Sustainable Chem. Eng., 2018, 6, 11473–11479 CrossRef CAS.
- H. Park, B. H. Park, J. Choi, S. Kim, T. Kim, Y. S. Youn, N. Son, J. H. Kim and M. Kang, Nanomaterials, 2020, 10, 1727 CrossRef CAS PubMed.
- R. Chen, H. Y. Wang, J. Miao, H. Yang and B. Liu, Nano Eng., 2015, 11, 333–340 CAS.
- C. Zhu, S. Fu, D. Du and Y. Lin, Chem.–Eur. J., 2016, 22, 4000–4007 CrossRef CAS PubMed.
- J. Bejar, L. A. Contreras, J. L. Garcia, N. Arjona and L. G. Arriaga, J. Electroanal. Chem., 2019, 847, 113190 CrossRef CAS.
- H. J. Lee, D. H. Park, W. J. Lee, S. B. Han, M. H. Kim, J. H. Byeon and K. W. Park, Appl. Catal., A, 2021, 626, 118377 CrossRef CAS.
- L. Yang, B. Zhang, B. Fang and L. Feng, Chem. Commun., 2018, 54, 13151–13154 RSC.
- P. S. Hung, W. A. Chung, S. C. Chou, K. C. Tso, C. K. Chang, G. R. Wang, W. Q. Guo, S. C. Weng and P. W. Wu, Catal. Sci. Technol., 2020, 10, 7566–7580 RSC.
- X. Zeng, Q. Zhang, Z. Y. Shen, H. Zhang, T. Wang and Z. Liu, ChemNanoMat, 2022, 8, e202200191 CrossRef CAS.
- S. Chen and S. Z. Qiao, ACS Nano, 2013, 7, 10190–10196 CrossRef CAS PubMed.
- M. Ahmed, B. Xi, Y. Gu and S. Xiong, Inorg. Chem. Front., 2021, 8, 3740–3747 RSC.
- Q. Xu, H. Jiang, H. Zhang, H. Jiang and C. Li, Electrochim. Acta, 2018, 259, 962–967 CrossRef CAS.
- Y. Xiao, Y. Wang, M. Xiao, C. Liu, S. Hou, J. Ge and W. Xing, NPG Asia Mater., 2020, 12, 73 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: EDS, CV curves in non-faradaic and faradaic region at different scan rates, XPS, LSV polarisation curve at different temperatures 20° to 45°, intrinsic activity of catalysts. See DOI: https://doi.org/10.1039/d4se00468j |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab
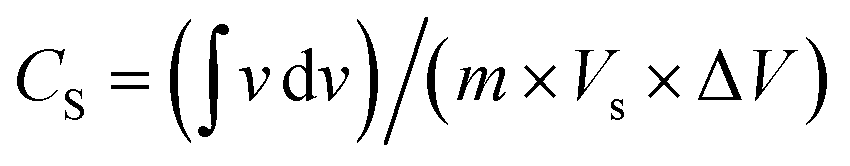 , where
, where 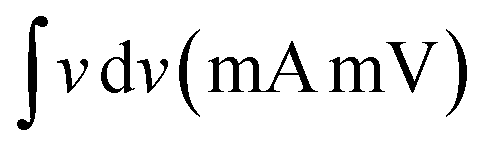 represents the area under the CV curve in the faradaic region obtained with a scan rate of 10 mV s−1 (Fig. S6†), m (0.21 mg) is the active mass of the catalyst loaded onto the electrode (with an area of 0.07 cm2), VS is the scan rate (mV s−1), and ΔV is the potential window difference (mV). The calculated ECSA plotted in Fig. 2b shows NiCo0.5Fe1.5O4 possessed a superior electrochemical surface area compared to the other synthesized spinels.
represents the area under the CV curve in the faradaic region obtained with a scan rate of 10 mV s−1 (Fig. S6†), m (0.21 mg) is the active mass of the catalyst loaded onto the electrode (with an area of 0.07 cm2), VS is the scan rate (mV s−1), and ΔV is the potential window difference (mV). The calculated ECSA plotted in Fig. 2b shows NiCo0.5Fe1.5O4 possessed a superior electrochemical surface area compared to the other synthesized spinels.