
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polymer-templated films of ordered mesoporous carbon: preparation, characterization and applications
Martina
Huber
 ,
Patricia
Sonnenberg
and
Stefan
Naumann
*
,
Patricia
Sonnenberg
and
Stefan
Naumann
*
University of Freiburg, Institute of Macromolecular Chemistry, 79104 Freiburg, Germany. E-mail: stefan.naumann@makro.uni-freiburg.de
First published on 11th March 2025
Abstract
Ordered mesoporous carbons (OMCs), typically prepared as fine powders, provide a striking combination of beneficial properties, which can be considered as crucial for many current and future technological applications. These properties, which include light weight, high surface areas, tunable pore sizes and pore arrangements, variable surface chemistry and electric conductivity, can be further boosted if more complex OMC morphologies are realized. Perhaps most rewarding in this regard are film-like structures, either as defined layers on specific surfaces or even as self-supporting, free-standing films/membranes. Such materials are of high relevance, yet their synthesis and characterization is also significantly more demanding than powder formation. As a consequence, this research field is only just emerging and the number of publications describing self-supporting and well-defined OMC film structures is still rather limited. The presented mini-review thus aims to highlight this exciting type of material in a compact manner, focusing on aspects of synthesis, characterization and applications, with the overall aim of encouraging further research efforts. Such efforts are particularly dependent on the polymer community, as the realization of well-defined OMC properties very much depends on polymers (e.g., as templates) and polymerization processes (e.g., cross-linking of carbon precursors). The translation of well-defined template block copolymers into well-defined OMC properties (especially pore size and pore geometry/connectivity) is an ongoing research focus, which is highly important for film morphologies.
 Martina Huber | Martina Huber received her Diploma in Chemistry and Chemical Technology in 2023 from the Johannes Kepler University in Linz, Austria. She then commenced her PhD in 2023 within the Nauman Research Group at the University of Stuttgart, Germany, and relocated with the group in 2024 to the University of Freiburg, Germany. Her research focuses on the development of polymerization catalysts for the synthesis of polymeric SDAs, as well as the optimization of OMC materials. |
 Patricia Sonnenberg | Patricia Sonnenberg completed her M.Sc. in Chemistry at the University of Stuttgart in 2022. Currently, she is pursuing her doctoral degree at the Macromolecular Institute at the University of Freiburg under the supervision of Prof. Stefan Naumann. Within the framework of collaborative research council 1333 (DFG), her research focuses on the synthesis and characterization of ordered mesoporous carbon materials, with particular emphasis on OMC film preparation and the development of highly defined support materials with selectively immobilized metal catalysts. |
 Stefan Naumann | Stefan Naumann received his PhD in 2014 as a member of the Buchmeiser Group (University of Stuttgart, Germany). He was granted a DFG research stipend to join Prof. Andrew P. Dove at the University of Warwick (UK) as a postdoc. After returning to Stuttgart in 2015, he worked as an independent Research Group Leader and successfully concluded his habilitation in polymer chemistry in 2021. Since 2018 he has been PI in collaborative research council (CRC) 1333 (“Molecular Heterogeneous Catalysis in Confined Geometries”). Since 2024, he has been a full professor of macromolecular chemistry at the University of Freiburg, Germany. His current research interests encompass the development of polymerization catalysts as well as the preparation of ordered mesoporous carbon materials and their application in catalysis. |
Introduction
The preparation and application of nanostructured, porous carbon materials, including in particular ordered mesoporous carbons (OMCs), has attracted considerable interest in recent years.1–3 This is down to a straightforward synthesis, an array of beneficial properties and numerous applications, which together make these materials attractive. Applications include, among others, the employment of mesoporous carbons as sensors,4,5 drug delivering matrices,6,7 electrodes,8 catalyst supports9,10 or adsorbents for purification processes.11,12Such broad usage is only possible since many of the OMC key properties are also well tuneable. This is true for surface areas,13,14 pore sizes (2–50 nm, by definition mesopores),15–17 pore arrangements (e.g., hexagonal, gyroidal, etc.),16–19 pore volumes,20 surface chemistry and surface polarity,21,22 electric conductivity (heavily dependent on heteroatom content and carbonization temperature)23–25 and density.26,27 While adaption of these properties is not always trivial – precise control can be defeated by distributions (such as pore size distributions) or gradients (of, e.g., surface functional groups) – these tuning sites nonetheless allow for highly specialized and optimized OMC products.
On top of these advantageous aspects, the preparation of OMCs can also be relatively simple, at least from the chemical point of view. For example, the popular soft-templating28,29 approach requires only a carbon precursor (most frequently based on very well-established phenolic resin chemistry or readily available carbohydrates), a template (typically an amphiphilic block copolymer, which may be commercial) and a suitable solvent (e.g., ethanol). Further preparation is then largely covered by thermal treatment, which can also be done on a somewhat larger scale. Interest in OMC materials is therefore well founded and will certainly continue to grow.
One particularly promising area of OMC research is that the corresponding materials can be realized as powders,30,31 discrete particles,32,33 monoliths19,34 or films.25,35 Especially the latter type seems rewarding for a number of reasons.
Firstly, successful OMC film preparation can render the use of so-called binders obsolete.25,35,36 Binders are typically polymers, which are used to glue OMC powders together in film-like layers or coatings (e.g., for making electrodes37–39). Examples of binders include Nafion and other fluorinated polymers such as polyvinylidene fluoride (PVDF). These polymers exhibit elevated mechanical strength, chemical resistance and low thermal conductivity.40 However, in such a setup the mesopores or parts thereof may be blocked.41 Furthermore, chemical interactions between the binding polymer and reaction media, educts or catalysts cannot always be excluded, leading to further uncertainty regarding the observed results. From a practical point of view, binders can also be considered dead weight, reducing the loading of active carbon materials for a given application. Additionally, PVDF and other fluorinated binder polymers can suffer from degradation, for example within Li–O2 batteries under certain conditions.42 The current regulatory situation for fluorinated chemicals casts further doubt on their future suitability in many products.
A pure film morphology without any binders, on the other hand, would enable a much more detailed correlation of carbon material properties and results. This is particularly valuable for the study of so-called confinement effects (understanding the impact of mesopore sizes and geometries on catalyzed reactions) or the investigation of model structures.43,44
Most desired in this regard is the preparation of fully self-supporting, free-standing OMC films. If practical handling can be ensured, the corresponding films can potentially be functionalized in many ways and then be applied as active surface modifications or as true membranes for separation and analytical purposes.25,35
While examples of the above exist, these are still relatively rare. One reason for this is that the transition from OMC powder synthesis to crack-free and mechanically stable OMC film preparation requires some specific procedural adaptions. Furthermore, suitable characterization of OMC films is more challenging than for powders, yet at the same time even more decisive. For example, accessibility to the inner volume of the film is an issue, which may be solved by 3D-connected pore networks. At the same time, isolated pore channels with hexagonal symmetry (very often oriented parallel to the film plane24) would lead to a very significant loss of addressable pore volume. Detailed characterization is thus a key requirement for OMC film development and applications.
The present mini-review thus intends to summarize current knowledge on this intriguing type of material in a very condensed manner, focusing on film synthesis, specifics of their characterization and selected examples of their application. Rather than provide an all-encompassing compilation, it is hoped to highlight the potential of this research area and encourage research groups to further extend the current state of the art.
Expectedly, the level of detail with which different publications have examined film properties and/or applications varies considerably, and a full set of properties is rarely given. Nonetheless, wherever possible, film properties (pore sizes, surface areas etc.) are provided. Both substrate-supported and self-supported films are included in this work. In some cases, OMC powders are considered for comparison purposes.
Preparation of OMC films
In principle, OMC film morphologies can be accessed via soft-templating,15,45,46 hard-templating47,48 or even via template-free49 approaches.Hard templating strategies involve the use of solid, porous materials (e.g. silica) coated and filled with a carbon precursor. The template is then etched to yield the desired porous carbon material as an inverse image of the template structure.47,48 However, adjusting the pore size and structure is more complicated because the hard template first has to be adapted, which is often time-consuming. Etching the template also leads to defects and instabilities in the porous structure, which impacts film formation.47,48 The etching process is chemically rather harsh and difficult to conduct on a larger scale.
In contrast, template-free methods employ the direct carbonization of citrate salt to yield mesoporous carbon50,51 or use methods like molten coassembly52 (see also below). This is intriguing, yet more detailed investigations are needed for confident OMC film production this way.
Hence, for well-defined and especially for self-supporting carbonaceous membranes, soft-templating is the superior and preferred methodology. Accordingly, the following focuses on this synthesis route, discussing the major aspects of (a) chemical setup and self-assembly, (b) (film) casting and (c) carbonization. Soft-templating for generating OMCs, developed and pioneered by Dai45 and Zhao53 about 20 years ago, is usually employed to prepare powders, so particular attention is paid to modifications of the traditional approach for the formation of film-like layers or self-supporting, crack-free OMC membranes.
Chemical setup and self-assembly
The typical composition of a soft-templating mixture for the preparation of OMCs includes (a) the template, also referred to as a structure-directing agent (SDA), (b) the carbon precursor and (c) a suitable solvent. Additional additives,35,54,55 catalysts56,57 or dopants58,59 may also be added.As templates, amphiphilic block copolymers are usually employed. Thus, these polymers display hydrophilic and lipophilic blocks of varying lengths; apart from diblocks, triblock architectures are also popular choices for this application. Fig. 1 displays some of the more frequently employed SDAs. It should be noted that the examples given are a selection of a much wider field of templates. The polar blocks are commonly based on poly(ethylene oxide) (PEO)/poly(ethylene glycol) (PEG),15,23,60–62 poly(acrylonitrile) (PAN)63–70 or polymers with ionic functionalities (e.g., sulfonic groups).71,72 PAN, unlike the others, can fulfill the dual function of being a carbon precursor and the polar block of the SDA.66
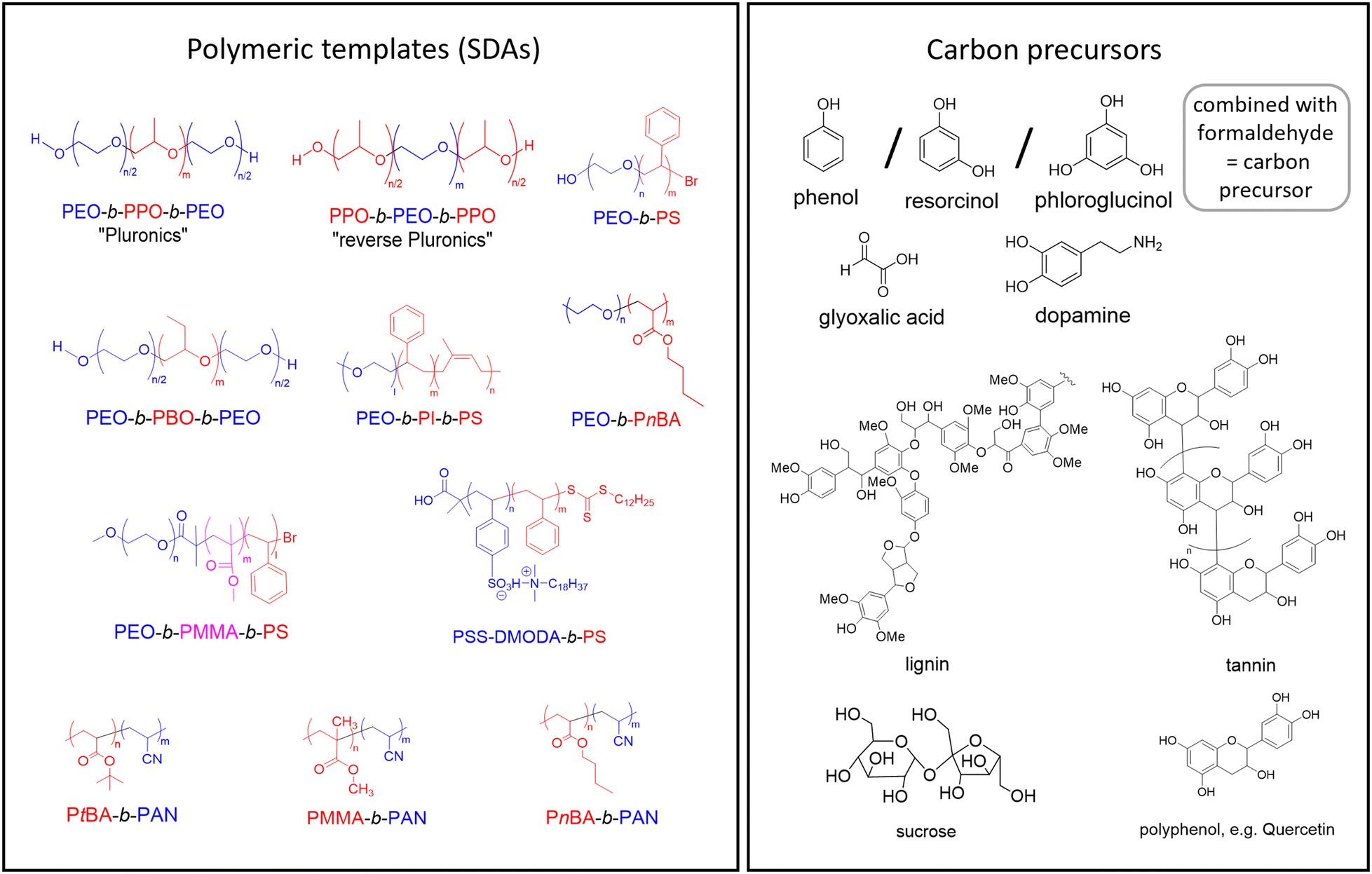 | ||
| Fig. 1 Left: SDAs that have been employed for OMC preparation via soft-templating. Red = lipophilic, blue = hydrophilic moieties. PEO: poly(ethylene oxide), PPO: poly(propylene oxide), PS: polystyrene, PEA: poly(ethylene adipate), PSS-DMODA: poly(N,N-dimethyl-n-octadecylammonium p-styrenesulfonate), PBA: poly(n-butyl acrylate), PtBA: poly(tert-butyl acrylate), PAN: polyacrylonitrile, PMMA: poly(methyl methacrylate), PI: poly-isoprene. Right: Frequently applied carbon precursors. | ||
For the lipophilic blocks, the constitutional variety is significantly larger, ranging from substituted epoxides (propylene oxide (PO),15,23,60–62 1-butylene oxide (BO))73 to various acrylic monomers and poly(styrene) (PS)71,72 or even poly(isoprene).19
Some of the above examples are commercially available, in particular the popular Pluronic® triblock copolyethers (Fig. 1). These are available in a number of grades (e.g., F127 or P123, among many others) with different HLB (hydrophilic-to-lipophilic balance) values and varying molar masses.74,75 Also reversed Pluronic® architectures (Fig. 1), which display more complex self-assembly behavior, can be used to template OMCs.76–79 Another approach for a simple and robust option to create hierarchically mesoporous carbon materials can be achieved by using thermoplastic elastomer blends. By incorporating a triblock copolymer into the matrix of another polymer, followed by crosslinking, mesopores are generated through the controlled removal of the polystyrene component.80
The choice of the SDA is a crucial step for successful soft-templating. One of the more obvious requirements is that the template and carbon precursor fit together, regarding polarity and interactions (see below). Moreover, it is essential that the template polymer is well-defined, meaning it should display a narrow molar mass distribution (ĐM), as well as highly controlled architectures and end groups. Indeed, the impact of dispersity effects, which have been discussed by O'Reilly and co-workers in detail, can be rather severe for polymeric self-assembly.81 In terms of OMC formation, such dispersity effects could negatively impact the clean formation of pore arrangements or broaden pore size distributions. Consequently, well-behaved polymerization techniques, such as anionic polymerization or controlled radical polymerization, have proved to be best suited for the synthesis of tailored polymeric SDAs.79,82–85 Apart from molar mass distribution, the architectural purity is also crucial: homopolymer impurities (such as PPO in commercial Pluronics® or diblock impurities in triblock copolymers86) can act as swelling agents for the micellar structures formed during self-assembly or modify the ordered gel phases.
When designing an SDA, the HLB and the packing parameter can provide the first orientation of the desired polymer constitution. The HLB value is defined as the ratio of the hydrophilic mass proportion of the polymer to the total polymer mass.87,88 It can thus serve as a quick tool for comparing different SDAs or to undergo systematic variations of the template, for example in order to adjust pore sizes.15 The well-known packing parameter,89 which connects the molecular structure of a surfactant (= SDA in this context) and the geometry of the self-assembled structures, can be useful in a general sense to categorize template polymers. However, it must be considered that the actual structure formation (see discussion below and Fig. 2) occurs in conjunction with the carbon precursor, so looking at the SDA alone is not enough to predict the phase separations and resulting pore structure in the OMC. Also, standard application of the packing parameter may underestimate the impact of some structural features of the template.90
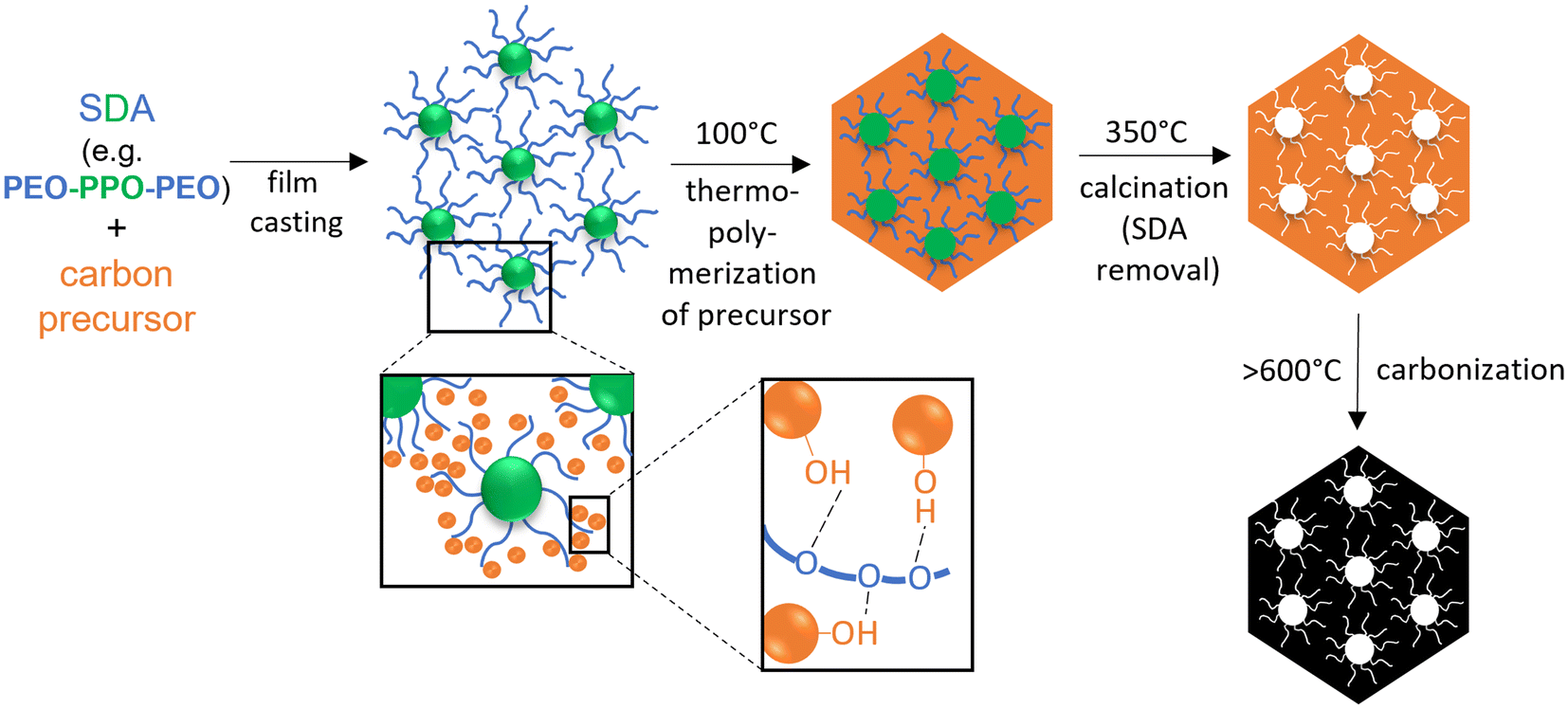 | ||
| Fig. 2 Scheme of SDA/carbon precursor self-assembly through the soft-templating process, according to Dai.45 | ||
The carbon precursor is the second crucial addition to the soft-templating process. While carbon precursors are usually chemically simple and well-available components (Fig. 1), their role is actually quite complex, because they have several requirements to fulfill. Firstly, the carbon precursor must interact with the SDA in a selective manner, in order to enable well-defined self-assembly. Selective in this regard typically refers to an exclusive interaction with the polar part of the template polymer (e.g. PEO), for example via H-bonds45 (Fig. 2). Thus, the carbon precursor will not migrate into the lipophilic, micellar cores but rather assemble with the hydrophilic “arms” in the micellar corona and between micellar structures. This in turn means that it must be a polar component itself, which is frequently achieved by the introduction of hydroxyl moieties on the precursor. In order to enable efficient assembly, the molar mass of the precursor must be relatively low. Additionally, it must be able to transition to an sp2-carbon rich matrix upon heating – which leads to oligomerized phenol/formaldehyde mixtures or derivatives thereof (“resols”) being the most widely employed carbon precursors15,25,57 (Fig. 1). Alternatives include for example glyoxalic acid,91 dopamine92,93 or plant-based compounds such as lignins,94 tannins95 or polyphenols,96 alongside many others.
From a synthetic point of view, resol generation is not trivial. One major issue is reproducibility, because during oligomerization small changes in pH, temperature or educt quality can entail different, e.g., molar masses of the carbon precursor. Also, it is advisable to quickly use the resol after preparation and avoid longer storage as further creeping oligomerization may set in.97–99 The carbon precursor, as an external component, can be replaced by PAN with its dual functionality as part of a diblock copolymer SDA. During thermo-induced stabilization (240 °C, 10 h), PAN forms a six-membered ring structure network via cyclization and dehydrogenation of the nitrile groups.100,101
In a landmark publication, Zhao and co-workers97 demonstrated that, using commercial Pluronic® templates and resol-type carbon precursor, the resulting pore symmetry (hexagonal, gyroidal and others) could be neatly addressed by adapting the template/precursor ratio. The overall phase separation can be rationalized by considering that the carbon precursor belongs to the polar phase, as do the PEG blocks of the SDA, while the lipophilic phase is constituted by the PPO blocks. This strategy has since been employed numerous times to tailor pore arrangements,18,102 including for OMC films (see below), but also to modulate pore sizes.103
It should be noted that in some cases the addition of an external carbon precursor (such as resol) can be avoided. This also circumvents complications in the prediction of the pore morphologies formed, as discussed above. One strategy that can be employed utilizes block copolymers for templating in the bulk state, whereby one block of the compound can undergo decomposition under certain conditions.104 To illustrate this point, consider the previously mentioned PAN-containing copolymers. Following the thermal stabilization of the PAN segment, its non-stabilized counterpart (e.g. PMMA) undergoes decomposition under heat.100,101 Another example is poly(D,L-lactide) (PLA), which is easily removed under basic conditions. In block architectures with poly(4-fluorostyrene), a gyroidal morphology in the bulk phase was demonstrated by Steiner, Snaith and colleagues.105
The third critical component of soft-templating OMCs is the applied solvent. The most straightforward way to create a film-like structure that can later be carbonized is soft-templating via evaporation-induced self-assembly (EISA).106 This requires the mixing of the SDA and carbon precursor in a solvent that readily evaporates and competently dissolves the other components. Very frequently, ethanol35,102 is used for this purpose. After casting the mixture via a suitable method on a fitting substrate (next section), the solvent evaporates, entailing a growing concentration of SDA and carbon precursor, upon which self-assembly (Fig. 2) takes place. The residue is typically a soft film that later needs to be thermally stabilized and subsequently carbonized. While polymer self-assembly and soft-templating have been discussed in excellent reviews28,31,107,108 and more detailed aspects shall not be reiterated here, it must be noted that the specifics of this process very much depend on the polymer employed. For example, with regard to well-established F127 Pluronic®, it has been demonstrated that actual self-assembly is thermally induced109 (not occurring at the evaporation stage). Rather subtle changes, such as a higher molar mass of the SDA or blocks with a more pronounced bias in hydrophilicity and lipophilicity, can be expected to favor self-assembly at room temperature. In situ investigations, for example via (GI)SAXS, of the self-assembly processes and their kinetics will certainly prove highly valuable for further development of the field.
Film casting
After a suitable soft-templating mixture has been created according to the considerations briefly touched above, film casting is the next step. Thereby, a number of techniques can be employed (Fig. 3), depending on the type of desired substrate, viscosity of the casting solution and intended application of the OMC afterwards. | ||
| Fig. 3 Different film casting techniques. | ||
Perhaps the most versatile of these casting techniques is the dip coating process (Fig. 3 (1)). This technique involves the immersion of a device in the soft-templating suspension or solution, thereby enabling the coating of a diverse range of devices. Such devices may include wires,110 plates,30,111 or foils of varying length.24 The formation of free-standing films represents a challenging issue using this technique, as the entire device is coated during the process, which can complicate delamination of the OMC film formed later in the process. Thus, dip-coating is often used for forming OMC film layers rather than self-supporting structures and is especially interesting for soft/flexible substrates. An instructive example in this regard was published by Vogt and co-workers,24 who coated a sheet of Kapton (polyimide) in this way. Subsequent thermal treatment and carbonization delivered a defined OMC layer on the carbonized substrate, as revealed by SAXS and nitrogen sorption. The whole structure was mechanically robust, allowing for gentle bending or cutting. Nevertheless, the most frequently referenced technique in the scientific literature for the fabrication of OMC films is spin coating23,25,71,112–114 (Fig. 3 (2)). The process uses centrifugal forces to disperse the soft-templating mixture across the surface of the device through the application of a specific rotational velocity, very often moderate ones such as 500 rpm.115 In general, spin coating can deliver films with higher uniformity than dip-coating, and by adjusting the spin speed it is possible to accommodate very different coating liquid viscosities and to realize different coating thicknesses. Obviously, the substrate scope is limited to flat, rigid structures such as silicon wafers.25
An alternative method for the production of OMC films is the tape casting process35 (Fig. 3 (3)). This technique has also been employed in the context of ceramics,116 within the field of battery research117,118 and innumerous other applications. The templating mixture is placed on a flat substrate and distributed through a doctor blading process. The uniform spreading of the suspension across the device is achieved through the application of the blade under constant speed and pressure. This can be used for relatively large surfaces, and defined coating thicknesses can be addressed by regulating the blade–substrate distance.119 For the specific case of OMC films, Mylar®-foil has been described as a beneficial substrate choice,35 as it is easily available, flexible (important for OMC film delamination, see below) and, as a polyester (based on poly(ethylene-terephthalate), PET), provides balanced polarity. Moreover, it is thermally stable enough to endure during the cross-linking process, which facilitates easy application, allowing for the film to be effortlessly transferred afterwards. The foil can also be reused.120
Techniques that are analogous to the aforementioned process but exhibit a less uniform distribution of material include drop casting94,96 (Fig. 3 (4)) and zone casting24 (Fig. 3 (5)). Both of these techniques are based on the pouring of a suspension/solution into or onto a device, and both have been employed to obtain OMC films, for example by Tang et al.65 The differentiating factor between zone casting and drop casting is the movement of the device on which the suspension/solution is poured. In zone casting, the device is in motion in a single direction, whereas in drop casting, the device remains stationary.
The above methods require casting mixtures containing considerable amounts of solvent. It must be acknowledged that some drawbacks are associated with that, including sustainability and safety aspects of the organic solvents employed, and higher costs in comparison with solvent-free approaches.121 Established ways to circumvent the use of solvents via chemical ball-milling strategies121,122 cannot be used if film structures are targeted. Thus, novel approaches are needed. In this regard, obtaining porous polymer films via melt shear organization of core–shell particles is promising. An example of this approach for OMC film preparation was published by Gallei and coworkers.127
Finally, it must be considered that the thickness of the film that is created by one of the above methods can also impact the self-assembly process (e.g., “thin film confinement”). In a very thin film (ca. 100 nm and below), these templating polymer chains are constrained in their ability to arrange freely due to surface and substrate interactions.128 The properties and structures of thin films vary with the type of substrate support and interfaces/surfaces. As a consequence, the process is rather complex.129,130
Thermal treatment and carbonization
After casting, the solvent is evaporated, typically for 2–72 h,24,25,35 depending on the specific setup and whether information about the self-assembly kinetics is known. If the carbon precursor is of the resol type, which is true for the majority of cases known in the literature, the resulting soft film and the nanophase separated structure is then stabilized by mild thermal treatment. This is usually conducted at 80–120 °C for 24 h24,25,35 and results in the cross-linking and more complete polymerization of the oligomeric carbon precursor. Chemically, this corresponds to the hardening of phenolic resins.This cross-linking step is a delicate matter, in particular if self-assembly and phase separation processes are occurring in parallel. As cross-linking proceeds, the molecular mobility of all components involved is increasingly restricted. The conflicting kinetics of phase separation and cross-linking can therefore negatively impact clear structure formation.109
Such cross-linking delivers a fixed and stable nanostructure, very often in the shape of off-white or orange-colored layers. These can be removed from the substrate and further processed. This is also where the OMC powder synthesis and OMC film formation fully diverge. For OMC powders, the polymeric, cross-linked film is subsequently shredded or ball-milled and then carbonized.30
For films, on the other hand, it is important that the polymeric layer is uniform and crack-free. This stage is also a good opportunity to delaminate the film from its substrate prior to carbonization.25 Delamination at this point is not a strict requirement for OMC film preparation (including those examples where free-standing films are targeted35). However, carbonizing a structure that is not fixed to a surface has the fundamental advantage of facilitating a crack-free shrinking process (see below).
The polymeric and, in the case of resol carbon precursors, cross-linked film structure is then subjected to further thermal treatment at higher temperatures under protective conditions (e.g., nitrogen or argon atmosphere). Samples treated to 300–400 °C are often referred to as calcined.61,71 This process ensures that the template polymers are thermally degraded (forming the mesopores) and a significant mass-loss alongside shrinkage is observed.112 Importantly, the bulk of the material is not carbonized, but still rather polymeric in nature. Treatment to at least 600 °C,61,71 but frequently considerably higher, then results in carbonization, forming the black, often shiny OMC films.
During carbonization, the sp2-configured carbon bulk is formed,131,132 whereby the mass loss from the cross-linked film to the carbonized structure can easily amount to 75%. The subsequent (anisotropic) volume shrinkage puts the film structure under stress, which can result in cracks or even complete fragmentation. This issue is exacerbated when the film strongly adheres to the substrate surface or very fast carbonization is conducted. For carbonization directly on a substrate, the latter must be either able to withstand high temperatures (for example, Si wafers) or degrade in a desired way (e.g., carbonize). Carbonization of a film that has previously been delaminated, on the other hand, often requires protection and “flattening” during the process, which can be ensured by placing the film between carbon papers and ceramic plates.25 This way, rolling-up of the film during carbonization can be avoided.
The specific carbonization temperature has a major impact on the resulting OMC. A higher carbonization temperature generally entails fewer heteroatoms and more sp2-configured carbon,103 smaller pores,103,133 fewer functionalities on the surface (such as hydroxyls) and thus a less hydrophilic surface,22 higher electrical conductivity103 and higher danger of structural collapse.109 The surface can also be strongly influenced by altering the ratio of reactive to protective gases. For example, introducing varying amounts of oxygen can change the surface functionalities or increase the surface area during pyrolysis.103 The majority of papers cited in this review have applied carbonization temperatures in the range of 600–1200 °C.24,25,31,35 Importantly, this also means that amorphous carbon is formed (and 5–15% of heteroatoms, usually oxygen, may still be present in the material), in contrast to true graphitization.
Calcination and carbonization are often conducted together in one programmed heating cycle. Very often, these contain plateaus for several hours at 300–400 °C.25,35 In particular for film preparation, the dimensions of the gas-tight oven apparatus are important. Especially tube furnaces with a small tubular diameter may be unsuitable to prepare the larger OMC films (typical areas for the self-supporting structures as discussed in this review are 4 cm2).25
After carbonization, the resulting films can be very brittle, which is a major stumbling block for their preparation and application. Zhao and colleagues demonstrated that film thickness is a critical factor determining flexibility and, consequently, mechanical strength. Their experiments involved synthesizing films with thicknesses ranging from 90 to 3000 nm. Their observations indicated that films with high thickness were often rather brittle, while very thin films were obviously fragile.25 In this context, Hou's group demonstrated an alternative method for producing flexible, elastic carbon material through the direct carbonization of polymer foam.134
Selected practical examples for OMC film preparation
To illustrate the more general aspects discussed above, Table 1 lists successful published examples of OMC film preparation and some of their respective chemical and procedural specifics. It should be noted that very different film types result from these efforts, including free-standing films, substrate-attached OMC layers, very thin/thick film structures, various pore geometries and different mesopore sizes. These differences are often down to skillful variation of common themes in the soft-templating and carbonization process, details of which can be found in the references given. It is instructive to consider some of these examples more closely. In a noteworthy study (Table 1, entry 1), Zhao and colleagues25 employed a spin coating technique to produce films with dimensions of 2 × 2 cm2 and a thickness of 500 nm. To achieve this, a pretreated silicon wafer (using Piranha solution at 90 °C, 30 min) was used as the substrate. The suspension of F127/phenolic resin/EtOH (1/2/8 ratio) was spin coated at 4000 rpm for 60 s. The resulting film was then subjected to a drying process for a period of 5–8 h at room temperature, followed by curing at 100 °C for 24 h. Subsequently, the film was pyrolyzed in order to obtain the OMC film (still attached to the silicon wafer) at 600 °C (1 °C min−1 heating rate) for a period of 3 h under a flow of nitrogen (60 cm3 min−1) (Fig. 4). | ||
| Fig. 4 Top: Schematic preparation process for free-standing mesoporous carbon thin films with a highly ordered Fmmm mesostructure by using a coating–etching approach. Bottom: (a) Free-standing OMC film (inset: film transferred onto substrate). (b) SEM images of the free-standing mesoporous carbon films (inset: cross-section). (c)–(e) SEM images from different viewpoints, and (f) cross-section of the free-standing OMC film after carbonization. Reproduced from ref. 25 with permission from ACS, copyright 2025. | ||
| SDA | Carbon precursor | Solventa | Casting method | Carbonizaton T | Pore structure and size | Ref. | |
|---|---|---|---|---|---|---|---|
| a EtOH = ethanol, DMF = dimethylformamide, THF = tetrahydrofuran, MEK = methyl ethyl ketone. b PAN also acted as a carbon precursor. c TMB = trimethylbenzene | |||||||
| 1 | F127 | Phenol + formaldehyde | EtOH | Spin | 600 °C | Fmmm; 4.3 nm | 25 |
| 2 | F127 | Phloroglucinol + formaldehyde | EtOH | Tape | 850 °C | Worm-like; 5.7–6.4 nm | 35 |
| 3 | F127 | Phenol + formaldehyde | EtOH | Dip, tape | 800 °C, 1000 °C, 1200 °C | Hexagonal packed cylinders; 5.8 nm | 24 |
| 4 | F127 | Phenol + formaldehyde | EtOH | Drop | 800 °C | — | 123 |
| 5 | PBA-b-PAN | DMF | Zone | 800 °C | Lamellar; — | 65 | |
| 6 | F127 | Resorcinol + phloroglucinol + formaldehyde | H2O + EtOH | Dip | 200–800 °C | Fmmm; — | 30 |
| 7 | PEO-b-PEA-b-PS | Phenol + formaldehyde | MEK | Spin | 800 °C |
Ia![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) d; 12 ± 2 nm d; 12 ± 2 nm |
113 |
| 8 | PS-b-PSS-DMODA | Phenol + formaldehyde | Not given | Spin | 800 °C | Hexagonal packed cylinders; 6–9.2 nm | 71 |
| 9 | F127 | TMBc | H2O + EtOH | Dip | 600 °C | Hexagonal honeycomb; 10 nm | 124 |
| 10 | F127 | Resorcinol | EtOH | Dip | 800 °C | Different structures (cubic, tetragonal, etc.); 8 nm | 125 |
| 11 | PAN-b-PMMA | DMF | Spin | 800 °C | 40–50 nm | 63 | |
| 12 | F127 | Resorcinol + phloroglucinol | H2O + EtOH | Spin | 800 °C | Fmmm; 3.7–4.7 nm | 126 |
As a crucial step, a free-standing OMC membrane could then be obtained by immersing the silicon wafer in a potassium hydroxide solution at room temperature for a period of 5–8 h. This slowly detached the OMC film (BET surface area (powdered film) 700 m2 g−1), which ended up floating on the aqueous solution and could then be harvested crack-free and was used in several applications (see below). However, the fragility of these films was noted, and in some cases required spin-coating with PMMA for stabilization. This enhanced flexibility of the film is at the cost, however, of losing the free-standing and “binder-free” criterion.
In a study conducted in 2010 (Table 1, entry 2), Dai and colleagues35 employed a tape casting technique to create free-standing films. In order to synthesize the OMC, the researchers employed the self-assembly of in situ formed phloroglucinol-formaldehyde resins and F127 under acidic conditions, dissolved in a hexamethylenetetramine (HMTA)–ethanol (EtOH) solution (1.2 wt%). The use of the additive HMTA (as a source of additional formaldehyde) in this approach was described as being essential for the production of a flexible and crack-free film.
In contrast to the previous example, the mixture was tape-casted on Mylar® foil and, after drying at room temperature overnight, cured for 24 h at 80 °C. Crucially, the film was detached from the foil substrate at this point.
The carbonization process of the polymeric, cross-linked film was conducted in a sandwich-like configuration (Fig. 5), with the sample placed between two layers of carbon paper and quartz stripes. This resulted in large, crack-free and flat OMC films (surface area (powdered film) of 429 m2 g−1).
 | ||
| Fig. 5 Left: Sandwich-type-carbonization. Right: Photograph (A) and cross-sectional SEM image (B) of the OMC film. STEM images of as-made (C and D) and surface-etched (E and F) OMC. Reproduced from ref. 35 with permission from Elsevier, copyright 2025. | ||
Another illustrative example (Table 1, entry 3) for OMC film preparation was published by Vogt and colleagues24 in 2013. Here, rather than aiming for a self-supporting film, enhanced flexibility of the OMC structure was targeted. In contrast to the approach taken by Zhao and co-workers25 mentioned previously, who used a PMMA layer to enhance flexibility and avoid fragmentation of the film, in this case a layer of Kapton (polyimide, 7.5 μm thickness) was employed as a substrate for casting. A previously reported synthesis135 of the OMC was modified to include a mixture of F127 as an SDA, tetraethylorthosilicate (TEOS) as an additive (to template micropores, silica was later etched away), phenolic resol as a carbon precursor, and ethanol as a solvent. The aforementioned mixture was then applied to Kapton via a dip coating technique (Fig. 6), at a rate of 6 mm s−1.
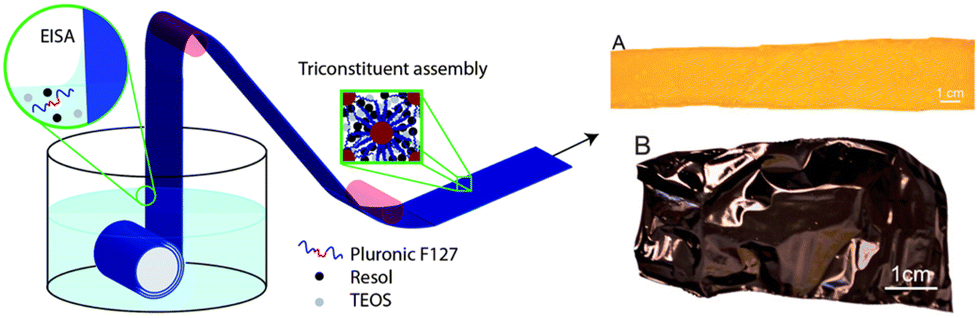 | ||
| Fig. 6 Left: Schematic synthesis of OMC films on plastic substrate. The Kapton substrate is submersed in a solution containing TEOS (silica source), resol (carbon precursor) and Pluronic F127 (template). Drawing the Kapton through the surface of the solution deposits a nanostructured coating through EISA. Right: Kapton after dip coating (A) and after carbonization (B). Reproduced from ref. 24 with permission from ACS, copyright 2025. | ||
Following the casting of the films, they were dried for a period of 2 h at room temperature and subsequently annealed at 100 °C for 24 h to facilitate cross-linking of the resol. In order to obtain thicker films, multiple coatings were also applied to Kapton using a tape coating technique. These samples were then annealed at 200 °C for 1 h.24 The carbonization process of the coated Kapton was conducted at a heating rate of 1 °C min−1 to 600 °C and 4 °C min−1 above 600 °C, utilizing three distinct maximal temperatures: 800, 1000, and 1200 °C, under a nitrogen atmosphere. The resulting sheets, designated as OMC film-coated Kapton (surface area up to 2000 m2 g−1), demonstrated a remarkable degree of flexibility, allowing for gentle bending and cutting to achieve the desired shape.
OMC film characterization
Suitable characterization of the thus synthesized OMC material is a key requirement for optimizing preparative methods and investigating structure–property correlations for the numerous applications that OMC (films) can be used for. Typically, this is achieved by combining different methods, for example infrared/Raman analysis (for identifying surface functionalities or the degree of graphitization) or elemental analysis (for, e.g., investigating doping or catalyst immobilization). Most important, however, are small angle X-ray scattering (SAXS), transmission electron microscopy (TEM) and gas sorption measurements (e.g., surface area, pore sizes, micro/mesopore volume). Together, these techniques can give a realistic impression of the carbon material where one method alone (e.g., electron microscopy) could be misleading.For OMC films, the situation is more demanding than for OMC powders. Of course, such film structures can be broken down or milled and then be subjected to various analytical investigations. This simplifies handling and procedures (for example during SAXS or nitrogen sorption), but can also entail the loss of very significant information. The latter can include defining aspects, such as pore orientation (parallel to the film plane or perpendicular) and uniformity thereof, accessible pore volume/pore blockages, permeability of gases and liquids or the existence of “skin layers”.102,109
The following tries to highlight some of these OMC film specifics with regard to material characterization. For traditional OMC powder characterization, excellent reviews can be recommended.15,53
Shrinkage, skin and gradients
The carbonization temperature significantly impacts various material properties, including significant mass loss. The ensuing shrinkage can be employed as a method for controlling pore size.103 Hence, by varying the carbonization temperature and adjusting the combination of protective gases (such as nitrogen, argon) with possible trace amounts of reactive gases (e.g. oxygen), the resulting properties can be fine-tuned.53 In this context, it is important to note that for OMC films, shrinkage is largely anisotropic. Apart from potential film rupture, this can also entail deformation of the symmetry of the pore arrangement and of the pore shape (elliptic distortion, see Fig. 7).35,112 | ||
| Fig. 7 Effects induced by anisotropic shrinkage of the OMC film during carbonization, (a) distortion of the pore shape from spherical to ellipsoidal, (b) some examples of symmetry-allowed transitions of pore arrangements. (b) Reproduced from ref. 137 with permission from ACS, copyright 2025. | ||
An illustrative example is given by Zhao, Zheng and co-workers (Fig. 8).25 In this work, the distortion of body-centered cubic Im3m symmetry via treatment at 600 °C and ensuing anisotropic contraction was highlighted (finally resulting in Fmmm symmetry). For complete characterization, such shrinkage-induced distortion phenomena usually mean that analysis via SAXS or TEM can become more complicated and assignment of the correct pore symmetry – assuming it is valid for the whole bulk of the film – can be challenging. In fact, the occurrence of defects is common and closely connected to the preparative method. EISA entails different interfaces (liquid–substrate, liquid–air) and corresponding gradients of surfactants in the templating solution/suspension, which can lead to the generation of different mesophases (pore arrangements) in the same film layer.136
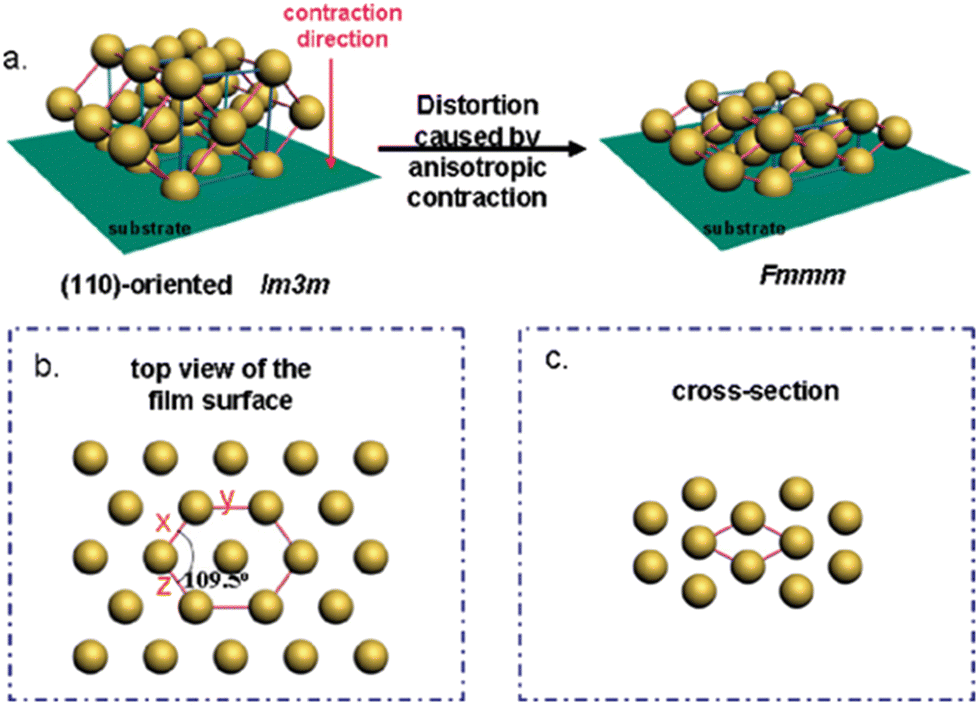 | ||
| Fig. 8 Illustration of (a) structural transformation of the OMC from Im3m to Fmmm symmetry during carbonization; distorted lattice from (b) the top and (c) cross-section. Reproduced from ref. 25 with permission from ACS, copyright 2025. | ||
Additionally, it must be considered that mesoporous films are not “monocrystalline” in a way that pore orientation stays truly aligned over large length scales. Rather, different domains and corresponding grain boundaries can be observed (Fig. 9); this is well-described for mesoporous silica materials137 and can be assumed to be true for most OMC films, too.
 | ||
| Fig. 9 2D hexagonal ordered porous material with (a) alignment defects and (b) uniform parallel orientation to the substrate. Reproduced from ref. 137 with permission from ACS, copyright 2025. | ||
In addition to shrinkage, the formation of a skin layer during the heating process alters the exposed surface both visually and in terms of surface functionality and porosity.35,138 This skin layer, which is frequently microporous or nonporous, can have a strong impact on characterization results and film applicability (e.g. permeability, sieving effects, catalyst loading), so it is advisable to check for its existence via suitable techniques, for example TEM. Importantly, it has been shown that oxygen plasma etching, a straightforward and relatively simple method, can be employed to remove the skin layer and reveal the underlying pore system.97,139
In summary, the above means that, when reporting OMC film properties, care must be taken to properly portray such deviations from ideal behavior and structure. If possible, average grain/domain sizes should be determined and the actual occurrence of the dominant pore symmetry (as determined by SAXS) should be established in different parts of the film (e.g., middle part versus surface) by a complementary technique (usually TEM).
(GI)SAXS
Various types of 2D- and 3D-ordered mesostructures have been successfully realized for carbon thin films, including hexagonal (p6mm),61,113,140 gyroidal (Ia3d)82,113 or cubic (Im3m)102,113 pore arrangements, among others. The type of pore connectivity and symmetry has a major impact on the applicability of the corresponding OMC film and is thus a key interest for material characterization and optimization.In contrast to powder samples, which can be directly analyzed in capillaries, films require special holders and specific techniques for SAXS analysis.141 In this regard, grazing incidence small-angle X-ray scattering (GI-SAXS) is especially beneficial, as it allows for direct measurement of the film, either applied to a support material (e.g., silicon wafer) or without (Fig. 10).
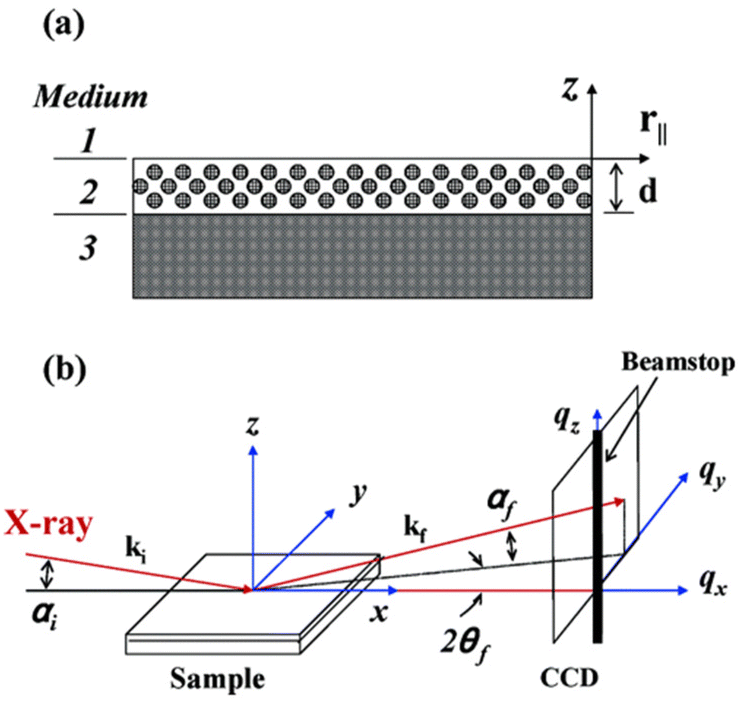 | ||
| Fig. 10 General GI-SAXS setup: (a) medium 1 = vacuum; medium 2 = mesostructured thin film with thickness d; medium 3 = substrate with infinite thickness. (b) αi = incident angle; αf and 2θf = exit angles of the X-ray beam. ki and kf = wave vector of the incident and scattered X-ray beams. qx, qy, and qz = components of the scattering vector q. Reproduced from ref. 142 with permission from ACS, copyright 2025. | ||
The measured film is thus neither cut nor milled and film-specific information (such as pore alignment or grain sizes) is best preserved. GI-SAXS allows for a detailed structural understanding of the mesoporous structure.109 For optimal results, it is recommended to use a film thickness between 60 nm and 250 nm, with a sample size up to 1 cm2. The incident angle should be set relatively high (around 0.3°),109 to ensure penetration of the full film thickness. Typically, the sample is placed on a flat, smooth silicon wafer or a grooved copper plate109 for measurement. Using rough substrates will lead to significant diffuse scattering.
Due to the small angle of incidence, the analysis typically focuses on a specific region of the sample, which is defined by the elongated profile of the X-ray beams on the sample surface.142,143 The arrangement of a sample can be quickly recognized by the GI-SAXS signature. Parallel channels to the substrate will be measured as diffuse sheets, perpendicular channels will be received as scattering rods and a random arrangement (with ordered channels) or thick films will lead to a powder ring. There may also be a superposition of two arrangements where e.g. a powder ring and a scattering rod are visible.143Fig. 11 displays typical patterns obtained via GI-SAXS.
 | ||
| Fig. 11 GI-SAXS pattern of the mesoporous film before (a) and after (b) template removal. Reproduced from ref. 112 with permission from Elsevier, copyright 2025. | ||
Very importantly, the sample holder can also be equipped with heating coils for in situ GI-SAXS measurements, allowing for observation of the self-assembly/structure formation (and the corresponding kinetics) at different temperatures. Additionally, rotational GI-SAXS can be performed by rotating the sample stage through a range of 180°, allowing a complete scan of the in-plane mesostructured orientation.24
Surface area, pore volume and pore sizes
Physisorption analysis of OMC materials is crucial for obvious reasons; applicability and structure–property correlations can only be meaningful if the key parameters of specific surface area, pore size (diameters) and pore volumes are known, ideally also differentiating between micro- and mesoporous regimes. Doing so correctly can be a challenging task, in particular for film morphologies. Apart from choosing the right measurement conditions (and correct algorithm for data interpretation) for determining the parameters mentioned above, which can also be a difficulty, using actual film samples rather than powdered samples is only possible with specific adaptions.144 Certain techniques require specially manufactured tubes or measurements under cryogenic conditions to keep the uniform film intact and reduce the limit of detection, respectively.145Also, interpreting isotherms can be complex, due to the quadrupole moment of N2 and its specific interactions with surface groups or metal ions. Kr at 77 K provides exceptional sensitivity for detecting low surface areas as small as <0.05 m2. Since the low vapor pressure reduces the number of molecules in the measurement cell by a factor of 300 compared to N2 (or Ar), sensitivity for samples with small surface areas is thereby greatly enhanced.146 However, adsorption is limited to the filling of pores smaller than 10 nm in thin films.147
In spite of these challenges, it is very informative to keep the film structure intact in order to gain highly relevant insights, especially regarding deviations from the perceived, ideal structural order. This can encompass pore blockages or smaller than expected pore volumes, multimodal pore size distributions (indicative of, among other things, different pore arrangements), specific hysteresis curves (e.g. regarding bottlenecks or cage-window structures) or excessive microporosity resulting from high carbonization temperatures.
SEM/TEM
Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) are valuable tools for the characterization of mesoporous materials; again, for film morphologies their contribution is especially valuable.Thus, SEM is a straightforward method to determine film thicknesses (which can go down to few nanometers)124,148 and their respective uniformity. Also, smoothness and integrity of the film (occurrence of cracks, larger defects) is quickly established through this approach.
TEM is a useful tool for structural analysis, as it reveals a more detailed pore structure and pore orientation (Fig. 12).111
 | ||
| Fig. 12 TEM images of carbon films with well-ordered hexagonal structure (a–c). Reproduced from ref. 114 with permission from Elsevier, copyright 2025. | ||
Such analysis can substantiate results from (GI)-SAXS and physisorption investigations (pore symmetry, pore sizes) and together provide an informative picture of the respective OMC material.149 For films, it is highly recommendable to have a look via TEM at different sections of the film (middle, upper/lower surfaces) to identify structural defects such as the transition from one type of pore arrangement to another, which might be difficult to spot via other techniques. As pointed out above, it is also vital to check for the existence of skin layers, otherwise a lack of the desired applicability might be wrongly interpreted. Energy-dispersive X-ray spectroscopy (EDX)-type measurements can be helpful for investigating the uniformity of doping or catalyst loadings of the OMC film.150
For OMC film measurement, a TEM specimen can be prepared using the focused ion beam (FIB) method.125 A small section of the film is laser-cut and inserted into a TEM sample holder. Alternative preparation methods include crushing the sample and using a drop-coating technique for TEM grids.64 Additionally, a microtome can be employed to prepare the film by embedding it in resin and performing ultrathin sectioning with a diamond knife. The cut piece is then glued parallel to the slot in the grid.112 However, using a microtome may not be the optimal preparation method, as it can cause the carbon films to break into pieces, preventing accurate determination of the mesostructure orientation. Furthermore, the sample orientation may not align with the primary axis of the ordered lattice, which can lead to misinterpretation.24 In the case of carbon-based materials, standard carbon-coated TEM grids often lead to a noticeable loss of contrast. This issue can be resolved by employing holey or lacey grids.151
While highly ordered structures in films have been achieved, attaining full coherence and unidirectional orientation over macroscopic dimensions continues to be a significant challenge for self-assembled films. This is especially evident in TEM images, where changes in the alignment are visually observed, depending on the selected area in the mesostructured film.102 In certain TEM images, a series of interconnected small pores can be observed between the main stripe-like channels of the mesoporous film, which can be attributed to varying shrinkage rates.152
Applications of OMC films
Interest in OMC films, both as coatings and self-supporting membranes, stems largely from the broad range of applications for which they offer intriguing perspectives. The combination of electrical conductivity, high surface area, tuneable pore sizes and absence of necessary polymeric binder is particularly relevant for electrochemistry and energy applications.Beyond that, the relative chemical inertness of the carbon material is beneficial for biocompatibility; in combination with pore-size-dependent aspects (release of payloads, sieving effects), OMC films are also suitable for sensing and drug delivery. True membrane applications are still limited, partly because the number of free-standing OMC films with suitable mechanic stability and suitable permeability is still low. Successful examples of the above, however, display remarkable results, for example regarding the nanofiltration of proteins.25
In the following, selected publications are summarized according to the respective area of research (Fig. 13). It should be noted that this discussion mainly focuses on materials derived from soft-templating, but setups based on hard-templating are also discussed. Wherever possible, the structural parameters of the OMC films are given and correlated with the resulting properties.
 | ||
| Fig. 13 Application fields of OMC films as discussed in this review. | ||
While the following strictly concentrates on film morphology, for a comprehensive overview of functional porous polymeric materials and polymer-based porous carbons, as well as their applications in environmental treatment and energy storage, a 2020 review article by the Matyjaszewski group can be recommended.153
Catalytic applications
The possibility of generating hydrogen gas via the hydrogen evolution reaction (HER) represents a significant area of interest within contemporary academic discourse.154,155 The catalyst that displays the greatest activity and stability within the HER is platinum, a rare and expensive metal. This presents a significant challenge within the field.156 One strategy for enhancing performance is the dispersion of platinum in the form of nanoparticles on a support material. In this context, porous carbon is a valuable resource, offering a high surface area and reasonable accessibility. Bernsmeier and co-workers36 explored a synthesis pathway for platinum nanoparticles (Pt NPs) dispersed in a soft-templated OMC film. This approach circumvents the challenges associated with blocked pores and active sites provided by binders such as Nafion, which are encountered in other methods where the powders must be processed as coatings.157 Instead, this work employed the simplicity of the soft-templating process and incorporated the Pt NPs into the carbon material precursor before film casting (Fig. 14). The morphology of a 440 nm thick film was determined, exhibiting 75% contracted body-centered cubic (bcc) interconnected ellipsoidal mesopores with a high surface area and pore connectivity. The ellipsoidal pore geometry is a consequence of the shrinkage that occurs during the removal of the template and carbonization, which is observed as a common phenomenon in soft templated films.28,126,158 The pore diameter was determined to be 6 nm parallel and 4 nm normal to the surface of the film. The resulting current densities were observed to reach levels that were twice as high as those achieved by commercially available Pt/C catalysts.36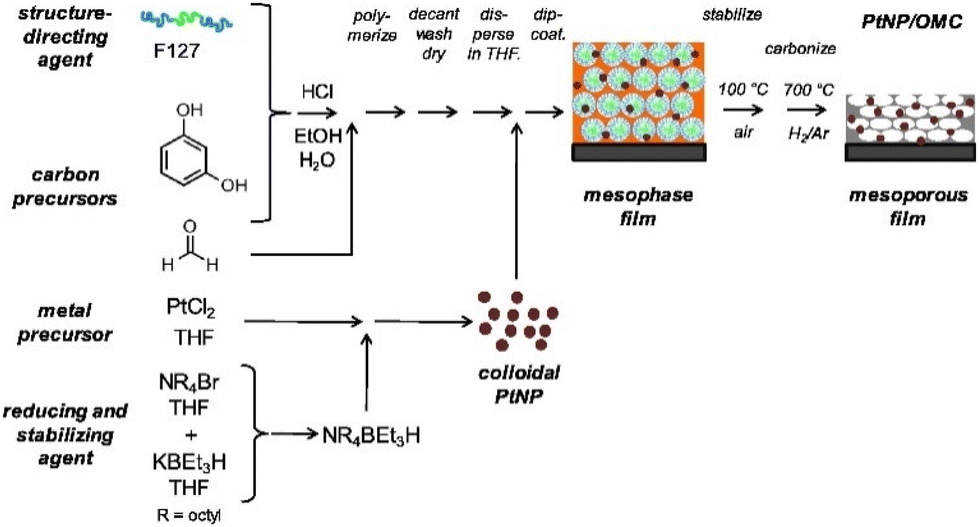 | ||
| Fig. 14 Preparative strategy as employed for mesoporous PtNP/OMC catalyst films. Reproduced from ref. 36 with permission from Elsevier, copyright 2025. | ||
A similar process was also employed by the same group for the incorporation of RuPt56,159 and Pd57 nanoparticles into OMC films. The RuPt-NP film comprises 0.85 wt% of Pt and 4.3 wt% of Ru, embedded in a 200 nm thick film. The mesopores of this film were of an ellipsoidal shape with a diameter of 9 nm. The Pd-NP film had a thickness of approximately 430 nm and exhibited elliptical pores with sizes of 8 nm parallel and 6 nm perpendicular to the surface, structured in a 50% contracted bcc organization. The Pd loading was 1.15 μg cm−2. This film exhibited activity not only in the HER but also in the hydrogenation of butadiene. It demonstrated a threefold increase in yield for the HER and a markedly higher space–time yield for the hydrogenation of butadiene compared to a Pd/C system.57 Kraehnert and co-workers employed the aforementioned process to incorporate rhenium and ruthenium nanoparticles into OMC films for utilization in the HER, also demonstrating high platinum mass-based activities.56,159 The 174 nm-thick film exhibited contracted bcc-ordered mesopores as a consequence of the 75% perpendicular shrinkage that resulted from template removal and carbonization, as previously indicated.
An alternative approach for the HER was adopted by the group of Li.160,161 Both studies employed the deposition of soft templated OMC films on carbon cloth as a 3D self-supported electrode as a support for the catalytic species (Fig. 15). Huang et al.161 employed cobalt-doped iron disulfide as the catalytic species, resulting in exceptional HER activity and rapid kinetics. This was attributed to the utilization of the OMC film mesopores, which served as nanoreactors to restrict the growth of sulfide particles and prevent agglomeration during the electrochemical reaction, thereby maximizing the exposure of active sites. The deposited OMC film exhibited an ordered hexagonal arrangement of one-dimensional mesopores with a diameter of 7 nm. These mesopores were oriented parallel to the surface of the carbon cloth. Liu et al.,160 on the other hand, synthesized FeP nanocrystals as catalytic species directly on the substrate. Their device exhibited a notable HER activity and favorable kinetics, in addition to excellent stability under acidic conditions. The OMC film deposited in this study has an approximate thickness of 200 nm and exhibits pores with a uniform diameter of approximately 6 nm in an interconnected three-dimensional arrangement.
 | ||
| Fig. 15 Metal catalysts intended for HER incorporated into OMC-coated carbon cloth. Reproduced from ref. 161 with permission from ACS, copyright 2025. | ||
A hard-templated film as a catalytic material, created as a replica of a mesoporous silica film, was developed by Kwon and colleagues.162 They conducted research on nitrogen-doped films for use in electrocatalytic iodine/iodide redox reactions. To achieve nitrogen doping, they employed polypyrrole as a carbon precursor through electrodeposition in the silica pores, resulting in the formation of predominantly active pyridine groups on the OMC film. The porosity of the OMC film could not be determined due to strong adhesion to the substrate, which is a general issue for hard-templated OMC film structures. Another illustrative example of the catalytic application of an OMC film is the electrocatalytic oxidation of estrogens.163 In a study conducted by Luo, a hard-templated OMC was created using SBA-15 as the template and sucrose as the carbon source. The utilization of the aforementioned material as a catalytic film on a glassy carbon electrode (GCE) was achieved through a two-step process.
Initially, L-proline and the OMC were suspended in phosphate buffered saline (PBS). Thereafter, the GCE was immersed in this suspension, and OMC/L-proline was deposited on the GCE through an electrochemical technique (10 segments at a scan rate of 100 mV s−1, with a potential range of −0.8–2.4 V). The device demonstrated effective electrocatalytic activity towards natural estrogens and high sensitivity for the analysis of E2 (estradiol, estra-1,3,5-(10)-triene-3β,17β-diol) in real samples.
Energy storage/conversion
One of the most common applications of carbon materials is their use as electrodes in electrochemical devices, including batteries and capacitors.16,164,165 To provide a representative overview, in Table 2 three studies23–25 that report on the use of soft-templated OMC films and three studies162,166,167 that report on the use of hard-templated OMC films are summarized. Direct comparison of different films is challenging, not only because of the various preparation routes, but also because pore sizes, pore arrangements and electrolytes vary considerably. Also, some of the entries represent fully self-supporting OMC films (entry 6) while others refer to coatings that are not removed from their substrate (entry 3). However, all these examples serve as successful proof-of-concept studies. Indeed, the capacities achieved seem very comparable to OMC powder-type examples25 – with the added benefit of being able to avoid binders and thus provide much better access to structure–activity correlations for future systematic investigations.| Templating | Doping | Morphology | Geometric C (mF cm−1) | Gravimetric C (F g−1) | Electrolyte | Ref. | |
|---|---|---|---|---|---|---|---|
| a Depending on the carbonization temperature (400–800 °C). b Assumed. The weight of the OMC coating could not be determined. | |||||||
| 1 | Hard | N | 12 nm pores | 252 | H2SO4 | 166 | |
| 2 | Hard | Loss of structural order compared to silica, 2–3 nm pores | 7 | H2SO4 | 167 | ||
| 3 | Hard | N | TEM: 7 nm pores | 3.92 | 140–250b | HClO4, HCl | 162 |
| 4 | Soft | Bcc, 6.3–8.2 nma | 3.5 | H2SO4 | 23 | ||
| 5 | Soft | Hexagonal, 5.8 nm pores | 85 | Na2SO4 | 24 | ||
| 6 | Soft | Hexagonal, 20 nm and 8 nm pores | 136 | KOH | 25 | ||
It is instructive to notice the different strategies the authors employ to overcome the challenges connected with the preparation of crack-free and well-defined OMC films for supercapacitors and similar applications. Thus, Cheng and colleagues26 employed a methodology that integrated two distinct SDAs to generate a double layer film with varying mesopores. Another approach for overcoming the difficulties presented by the brittle nature of OMC films was demonstrated by Vogt and co-workers24via dip coating of Kapton (also mentioned above) to obtain flexible electrodes. While the capacity of this device was moderate, the main focus of this study was on simplifying the process and handling of the device, which was convincingly achieved.
Compared to non-doped, non-film OMC materials, the latter exhibited gravimetric capacitance broadly within the same range.168
A number of studies have been conducted with the objective of investigating the utilization of OMCs in fuel cell applications. In their 2007 review,169 Chang, Joo and Pak conducted a comparative analysis of some of the aforementioned studies. Expectedly, the majority of these studies utilize OMC powder.170 An instructive, hard-templated example is the utilization of an OMC thin film in a direct methanol fuel cell, as demonstrated by Mou and colleagues.171 In this study, the synthesis of a mesoporous thin film with 4 nm pore sizes oriented perpendicular to the film plane was achieved. A PtRu catalyst was then deposited inside the OMC, which served as the anodic material. Interestingly, the relative shortness of the mesoporous channels was found to be beneficial, as the setup was less vulnerable to pore blockage than long-channel alternatives.
The utilization of OMC films as a corrosion-resistant coating to prevent the formation of a passivation layer on the stainless-steel bipolar plate in proton exchange fuel cells (PEMFCs) represents another promising avenue for further investigation. The doping of carbon with nitrogen, phosphorous and boron has been demonstrated to alter its electronic properties and generate additional functional groups on its surface.172–174 In this instance, the utilization of PAN-containing SDAs is advantageous due to the direct incorporation of N-doping via the SDA.68 In view of the preceding findings, He and colleagues58 synthesized a boron-modified OMC film by incorporating a specified quantity of boric acid into the soft templating components, which were subsequently spin-coated onto a clean type 304 stainless steel surface. The morphology of the boron-modified OMC films was analyzed as an ordered two-dimensional hexagonal structure by TEM and small-angle XRD. The pore size was analyzed by BET analysis, with the results indicating a diameter of 3–4 nm. This approach results in enhanced protective performance within a 0.5 M H2SO4 environment with a positive corrosion potential and a reduced corrosion current. In a related study, the group of He59 investigated the effects of doping the OMC film coatings (soft-templated via a F127/resol setup) with graphene or carbon nanotubes. This resulted in the carbon film exhibiting excellent protective performance in a 0.5 M H2SO4 corrosion system, while also demonstrating enhanced electrochemical performance.
Analytics
OMC films are emerging materials for a variety of analytical techniques that utilize the porous nature of the material for the purpose of separation. One potential application is the use of the material as a membrane for the separation of gases. An example of the optimization of OMC films in this application was demonstrated by Dai and colleagues.175 They synthesized a soft-templated OMC membrane and demonstrated that treatment with ammonia at high temperatures for 10 min following the carbonization process increased the gas permeance values.Furthermore, by adjusting the temperature (900 °C), the selectivity for CO2/N2 and C3H6/N2 mixtures increased by a factor of two. OMC film sieving effects can also be used in the liquid state, as elegantly shown by the group of Dai.176 This working group investigated two phenomena in diffusion: simple diffusion and electrochemical-aided diffusion (making use of the inherent electrical conductivity of the material). A self-supporting OMC film membrane was produced in a straightforward process, resulting in a three-dimensional pore network, which engendered permeability to the material. Rather well-defined pore sizes centered at 7.2 nm in diameter were obtained this way. The setup depicted in Fig. 16 was utilized for measuring diffusion, with and without applied external electrical fields.
 | ||
| Fig. 16 Schematic setup used by Hou et al.176 Reproduced from ref. 176 with permission from ACS, copyright 2025. | ||
The findings illustrate the molecular sieving effect on molecules transported through the pores of OMC film membranes, with a particular focus on the relationship between pore size and molecular dimensions. Thus, a high selectivity of α = 56.9 for anilinium over the larger rhodamine B molecule was found. The utilization of an external applied electric field enables the adjustment of ion selectivity, thereby facilitating the selective separation of ions of varying sizes.176
In the context of the separation of aqueous solutions of proteins, another example was reported in 2011 by Zhao,25 whereby free-standing OMC films (distorted body-centered cubic pore arrangement, uniform mesopores with 4.3 nm diameter) were also employed. The film served as a membrane for the filtration of a solution comprising cytochrome c and bovine serum albumin (BSA), whereby the OMC film was sandwiched between commercial, macroporous membranes for mechanical support. This resulted in an almost quantitative permeation of cytochrome c, while only 53% of BSA managed to diffuse though the film. This difference can be explained by the sizes of the respective proteins (2.5 × 2.5 × 3.7 nm for the former and 4.0 × 4.0 × 14.0 nm for the larger BSA). The fact that BSA could still partially overcome the OMC film barrier, in spite of it having at least one dimension that was larger than the mesopore window, was explained by defects. The direct usage of OMC films is also shown in solid phase microextraction (SPME), which is employed as a sample preparation method for the analysis of organic compounds at trace levels,177 whereby a variety of polymer coatings178–181 are utilized on silica fibers.180,182
Inherently, this leads to certain limitations: the polymer–silica composite can only be applied in a low-temperature environment (240–280 °C)183 and is unstable in organic solvents,184 which can result in removal of the coating.185 Consequently, carbon materials have been investigated as potential alternatives.186–189 Among these, OMC coatings warrant particular attention. In their 2015 study, the groups of Li and Wang110 investigated a simplified method for producing OMC-coated graphite fibers through soft-templating and dip coating. This approach was in contrast with the previous method, which involved hard templating and coating with OMC powder and chemical glue or polymeric adhesive.183 The OMC film was characterized by various techniques, whereby a crack-free layer of 8.5 μm thickness was found (hexagonal pore arrangement, 4.5 nm pore size, 630 m2 g−1 BET surface). The utilization of this coated fiber was demonstrated through the examination of five benzene series in aqueous samples, revealing wide linear ranges of detection and low detection limits. Interestingly, the extraction efficiency of the OMC coated fiber was found to be improved by a factor of two compared to commercial setups.
Biomedical applications
OMC materials have been employed in a variety of biomedical applications37,190–192 due to their relative inertness, high surface area and electrical conductivity. Crucially, the biocompatibility of OMCs is comparable to or higher than that observed for other biomaterials.193 Vogt, Zhao and co-workers investigated the effects of the pore morphology of OMC films on cell adhesion for tissue engineering.140 Interestingly, the calcined samples (treated at 350 °C, which ensures pore formation but not carbonization) displayed a significantly higher cell adhesion than that observed for a dense film analogue without mesopores. Conversely, the same samples, but now carbonized at 800 °C, showed a lower adhesion of osteoblasts – in particular, if a cubic mesostructure was applied. This effect was attributed to the ability of the carbonized samples to adsorb adhesion promoters or nutrients vital for cell growth. Overall, these findings suggest carbonization temperature and pore morphology as being potentially valuable for tuning cell adhesion.In 2012, Vogt and colleagues194 highlighted an application of OMC films as a possible drug delivery system as an alternative to mesoporous silicates.195–199 They showed that loading with mitoxantrone as a model drug was possible, although they concluded from their results that smaller drug molecules would be better suited for achieving high loading. They inferred this from the fact that loading did not increase with thicker OMC films, which they attributed to the hydrophobic nature of the carbons, resulting in only the pores near the surface being filled with the mitoxantrone molecules. The drug delivery system is comprised of two distinct steps: an initial burst release, followed by a slow release over a period of 5–8 days. However, only a fraction of the loaded drug was released. This phenomenon was attributed to the strong interactions between the carbon surface within the pores and the mitoxantrone molecule. These interactions can be tuned slightly through the carbonization temperature, with lower temperatures leading to a more hydrophilic surface and, consequently, a higher release rate.194
Optimization of the carbon properties for similar drug delivery applications thus seems readily possible, since adjustment of the pore size, pore connectivity and surface chemistry are well established concepts for OMCs.15,103
Another application field that has emerged in recent years is electrochemical sensing. One key employment is in the sensing of amino acids, which is of obvious interest in the context of medical diagnostics. The electrochemical sensing of dopamine with an OMC film electrode was published by Zhao and co-workers124 in 2021, detailing the use of swelling agents to achieve precisely controlled vertical alignment in the OMC film. The latter was utilized as a sensor to attain enhanced efficacy and, consequently, an ultralow limit (50 nmol L−1) and high sensitivity for dopamine detection.124 Wang employed a combination of soft and hard templates to generate stable, ultrasonic-regenerable sensors.200 Similarly, an L-tryptophan sensor was developed using a hard-templated, nitrogen-doped OMC film.
Conclusion and outlook
OMC films provide access to a unique combination of beneficial properties. As outlined above, this has led to a number of exciting applications, ranging from electrochemistry to the biomedical field, and encompassing both highly specialized and (prospective) high-volume end uses.Since this is coupled to a straightforward chemical setup and typically cheap starting compounds, it is very realistic that at some point OMC films will break out from lab use and enter commercial production; interest to do so is certainly there.
In order to further this desired development, interdisciplinary efforts are required, in particular involving the polymer field (for precisely tailored, yet well-accessible templates), process technology (for the development of semi- or even fully continuous OMC film production) and materials science (for optimizing OMC properties and, maybe most importantly, well-understood structure–property correlations).
More specifically, research areas that seem very relevant for further advances in the field include a better understanding of the self-assembly kinetics, a more successful suppression of ubiquitous structural defects, better mechanical properties in particular for the self-supporting structures and a more wholesome insight into the confinement-related (catalytic) effects, which are inherently present in OMC films, yet to date only addressable to a very limited degree.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. N. gratefully acknowledges financial support from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Project-ID 358283783 – SFB 1333/2 2022. M. H. and P. S. contributed equally to this work.References
- W. Xin and Y. Song, RSC Adv., 2015, 5, 83239–83285 CAS.
- C. Liang, Z. Li and S. Dai, Angew. Chem., Int. Ed., 2008, 47, 3696–3717 CAS.
- M. R. Benzigar, S. N. Talapaneni, S. Joseph, K. Ramadass, G. Singh, J. Scaranto, U. Ravon, K. Al-Bahily and A. Vinu, Chem. Soc. Rev., 2018, 47, 2680–2721 CAS.
- J. Ju and L. P. Guo, Chin. J. Anal. Chem., 2013, 41, 681–686 CAS.
- M. Zhou, L. Shang, B. Li, L. Huang and S. Dong, Electrochem. Commun., 2008, 10, 859–863 CAS.
- Q. Zhao, Y. Lin, N. Han, X. Li, H. Geng, X. Wang, Y. Cui and S. Wang, Drug Delivery, 2017, 24, 94–107 CAS.
- C. Karavasili, E. P. Amanatiadou, L. Sygellou, D. K. Giasafaki, T. A. Steriotis, G. C. Charalambopoulou, I. S. Vizirianakis and D. G. Fatouros, J. Mater. Chem. B, 2013, 1, 3167–3174 CAS.
- J. B. Park, J. Lee, C. S. Yoon and Y. K. Sun, ACS Appl. Mater. Interfaces, 2013, 5, 13426–13431 CrossRef CAS PubMed.
- S. H. Joo, H. I. Lee, D. J. You, K. Kwon, J. H. Kim, Y. S. Choi, M. Kang, J. M. Kim, C. Pak, H. Chang and D. Seung, Carbon, 2008, 46, 2034–2045 CrossRef CAS.
- M. Gao, L. Wang, Y. Yang, Y. Sun, X. Zhao and Y. Wan, ACS Catal., 2023, 13, 4060–4090 CrossRef CAS.
- S. Moon, J. Ryu, J. Hwang and C. G. Lee, Chemosphere, 2023, 313, 137448 CAS.
- M. Barczak, K. Michalak-Zwierz, K. Gdula, K. Tyszczuk-Rotko, R. Dobrowolski and A. Dąbrowski, Microporous Mesoporous Mater., 2015, 211, 162–173 CrossRef CAS.
- W. Gao, Y. Wan, Y. Dou and D. Zhao, Adv. Energy Mater., 2011, 1, 115–123 CrossRef CAS.
- A. Vinu, Adv. Funct. Mater., 2008, 18, 816–827 CrossRef CAS.
- F. Markus, C. Vogler, J. R. Bruckner and S. Naumann, ACS Appl. Nano Mater., 2021, 4, 3486–3492 CrossRef CAS.
- M. J. Bleda-Martínez, D. Lozano-Castelló, D. Cazorla-Amorós and E. Morallón, Energy Fuels, 2010, 24, 3378–3384 CrossRef.
- J. Jin, N. Nishiyama, Y. Egashira and K. Ueyama, Microporous Mesoporous Mater., 2009, 118, 218–223 CAS.
- F. Sakina and R. T. Baker, Carbon Trends, 2021, 4, 100051 CAS.
- J. G. Werner, T. N. Hoheisel and U. Wiesner, ACS Nano, 2014, 8, 731–743 CAS.
- A. H. Lu, W. Schmidt, B. Spliethoff and F. Schüth, Adv. Mater., 2003, 15, 1602–1606 CAS.
- P. A. Bazuła, A. H. Lu, J. J. Nitz and F. Schüth, Microporous Mesoporous Mater., 2008, 108, 266–275 Search PubMed.
- Z. Li, W. Yan and S. Dai, Langmuir, 2005, 21, 11999–12006 Search PubMed.
- T. Mitome, Y. Uchida, Y. Egashira and N. Nishiyama, Colloids Surf., A, 2014, 449, 51–56 CAS.
- J. Xue, C. Henry, J. Lee and B. D. Vogt, RSC Adv., 2013, 4, 3675–3683 Search PubMed.
- D. Feng, Y. Lv, Z. Wu, Y. Dou, L. Han, Z. Sun, Y. Xia, G. Zheng and D. Zhao, J. Am. Chem. Soc., 2011, 133, 15148–15156 CAS.
- W. Xing, S. Z. Qiao, R. G. Ding, F. Li, G. Q. Lu, Z. F. Yan and H. M. Cheng, Carbon, 2006, 44, 216–224 CAS.
- Z. Peng, D. Zhang, L. Shi and T. Yan, J. Mater. Chem., 2012, 22, 6603–6612 CAS.
- L. Chuenchom, R. Kraehnert and B. M. Smarsly, Soft Matter, 2012, 8, 10801–10812 CAS.
- K. W. Kim, B. Park, J. Kim, C. Jo and J. K. Kim, J. Mater. Chem. A, 2023, 11, 7358–7386 CAS.
- S. Tanaka, A. Doi, N. Nakatani, Y. Katayama and Y. Miyake, Carbon, 2009, 47, 2688–2698 CAS.
- T. Y. Ma, L. Liu and Z. Y. Yuan, Chem. Soc. Rev., 2013, 42, 3977–4003 CAS.
- M. Li and J. Xue, J. Colloid Interface Sci., 2012, 377, 169–175 CrossRef CAS PubMed.
- J. E. Hampsey, Q. Hu, Z. Wu, L. Rice, J. Pang and Y. Lu, Carbon, 2005, 43, 2977–2982 CrossRef CAS.
- W. Libbrecht, A. Verberckmoes, J. W. Thybaut, P. Van Der Voort and J. De Clercq, Carbon, 2017, 116, 528–546 CrossRef CAS.
- X. Wang, Q. Zhu, S. M. Mahurin, C. Liang and S. Dai, Carbon, 2010, 48, 557–560 CrossRef CAS.
- D. Bernsmeier, R. Sachse, M. Bernicke, R. Schmack, F. Kettemann, J. Polte and R. Kraehnert, J. Catal., 2019, 369, 181–189 CAS.
- L. Zhu, C. Tian, R. Yang and J. Zhai, Electroanalysis, 2008, 20, 527–533 CAS.
- M. Zhou, L. P. Guo, Y. Hou and X. J. Peng, Electrochim. Acta, 2008, 53, 4176–4184 CAS.
- D. Zheng, J. Ye, L. Zhou, Y. Zhang and C. Yu, J. Electroanal. Chem., 2009, 625, 82–87 CrossRef CAS.
- R. Dallaev, T. Pisarenko, D. Sobola, F. Orudzhev, S. Ramazanov and T. Trčka, Polymers, 2022, 14, 4793 CAS.
- C. C. Huang and J. C. He, Chem. Eng. J., 2013, 221, 469–475 CAS.
- J. K. Papp, J. D. Forster, C. M. Burke, H. W. Kim, A. C. Luntz, R. M. Shelby, J. J. Urban and B. D. McCloskey, J. Phys. Chem. Lett., 2017, 8, 1169–1174 CAS.
- S. T. Emmerling, F. Ziegler, F. R. Fischer, R. Schoch, M. Bauer, B. Plietker, M. R. Buchmeiser and B. V. Lotsch, Chem. – Eur. J., 2022, 28, e202104108 CAS.
- H. H. Nguyen, M. Högler, N. Schnabel, N. Hansen, T. Sottmann and D. P. Estes, ACS Catal., 2024, 14, 11252–11261 CAS.
- C. Liang and S. Dai, J. Am. Chem. Soc., 2006, 128, 5316–5317 CAS.
- Y. Yan, F. Zhang, Y. Meng, B. Tu and D. Zhao, Chem. Commun., 2007, 2867–2869 CAS.
- M. Chen, H. Xuan, X. Zheng, J. Liu, X. Dong and F. Xi, Electrochim. Acta, 2017, 238, 269–277 CrossRef CAS.
- A. Ramesh, R. Manigandan, B. M. Ali, K. Dhandapani, C. T. Da and M. T. Nguyen-Le, J. Alloys Compd., 2022, 918, 165729 CrossRef CAS.
- D. Morales-Acosta, F. J. Rodríguez-Varela and R. Benavides, Int. J. Hydrogen Energy, 2016, 41, 3387–3398 CrossRef CAS.
- G. A. Ferrero, A. B. Fuertes, M. Sevilla and M. M. Titirici, Carbon, 2016, 106, 179–187 CrossRef CAS.
- G. A. Ferrero, M. Sevilla and A. B. Fuertes, Carbon, 2015, 88, 239–251 CrossRef CAS.
- X. Liu, Y. Zhou, C. L. Wang, Y. Liu and D. J. Tao, Chem. Eng. J., 2022, 427, 130878 CAS.
- F. Zhang, Y. Meng, D. Gu, Y. Yan, C. Yu, B. Tu and D. Zhao, J. Am. Chem. Soc., 2005, 127, 13508–13509 CAS.
- L. Liu, F. Y. Wang, G. S. Shao, T. Y. Ma and Z. Y. Yuan, Carbon, 2010, 48, 2660–2664 CAS.
- Y. Deng, J. Liu, C. Liu, D. Gu, Z. Sun, J. Wei, J. Zhang, L. Zhang, B. Tu and D. Zhao, Chem. Mater., 2008, 20, 7281–7286 CAS.
- D. Bernsmeier, M. Bernicke, E. Ortel, R. Schmack, J. Polte and R. Kraehnert, J. Catal., 2017, 355, 110–119 CAS.
- D. Bernsmeier, L. Chuenchom, B. Paul, S. Rümmler, B. Smarsly and R. Kraehnert, ACS Catal., 2016, 6, 8255–8263 CAS.
- T. Wang, C. Zhang, X. Sun, Y. Guo, H. Guo, J. Tang, H. Xue, M. Liu, X. Zhang, L. Zhu, Q. Xie and J. He, J. Power Sources, 2012, 212, 1–12 CAS.
- H. Xue, T. Wang, H. Guo, X. Fan, Z. Zhu, X. Pan and J. He, RSC Adv., 2014, 4, 57724–57732 CAS.
- B. Zhang, Z. Yu, Y. An, Y. Wu, Y. Shi, Z. Liu and T. Wang, Mater. Res. Innovations, 2014, 18, 294–299 CAS.
- B. D. Vogt, V. L. Chavez, M. Dai, M. R. C. Arreola, L. Song, D. Feng, D. Zhao, G. M. Perera and G. E. Stein, Langmuir, 2011, 27, 5607–5615 CAS.
- P. Li, Y. Song, Q. Lin, J. Shi, L. Liu, L. He, H. Ye and Q. Guo, Microporous Mesoporous Mater., 2012, 159, 81–86 CAS.
- Z. Zhou and G. Liu, Small, 2017, 13, 1603107 Search PubMed.
- Y. Lin, X. Wang, G. Qian and J. J. Watkins, Chem. Mater., 2014, 26, 2128–2137 CAS.
- C. Tang, A. Tracz, M. Kruk, R. Zhang, D. M. Smilgies, K. Matyjaszewski and T. Kowalewski, J. Am. Chem. Soc., 2005, 127, 6918–6919 CAS.
- C. Tang, B. Dufour, T. Kowalewski and K. Matyjaszewski, Macromolecules, 2007, 40, 6199–6205 CAS.
- T. Kowalewski, N. V. Tsarevsky and K. Matyjaszewski, J. Am. Chem. Soc., 2002, 124, 10632–10633 CrossRef CAS PubMed.
- M. Kopeć, R. Yuan, E. Gottlieb, C. M. R. Abreu, Y. Song, Z. Wang, J. F. J. Coelho, K. Matyjaszewski and T. Kowalewski, Macromolecules, 2017, 50, 2759–2767 CrossRef.
- T. Kowalewski, R. D. McCullough and K. Matyjaszewski, Eur. Phys. J. E:Soft Matter Biol. Phys., 2003, 10, 5–16 CrossRef CAS PubMed.
- Y. Song, G. Wei, M. Kopeć, L. Rao, Z. Zhang, E. Gottlieb, Z. Wang, R. Yuan, G. Ye, J. Wang, T. Kowalewski and K. Matyjaszewski, ACS Appl. Nano Mater., 2018, 1, 2536–2543 CrossRef CAS.
- Z. Qiang, J. Xue, K. A. Cavicchi and B. D. Vogt, Langmuir, 2013, 29, 3428–3438 CrossRef CAS PubMed.
- Z. Qiang, Y. Zhang, Y. Wang, S. M. Bhaway, K. A. Cavicchi and B. D. Vogt, Carbon, 2015, 82, 51–59 CrossRef CAS.
- N. P. Wickramaratne and M. Jaroniec, Carbon, 2013, 51, 45–51 CrossRef CAS.
- A. Naharros-Molinero, M. Á. Caballo-González, F. J. de la Mata and S. García-Gallego, Pharmaceutics, 2022, 14, 2628 CrossRef CAS PubMed.
- N. A. Di Spirito, N. Grizzuti and R. Pasquino, Phys. Fluids, 2024, 36, 111302 CrossRef CAS.
- E. Larrañeta and J. R. Isasi, Langmuir, 2013, 29, 1045–1053 Search PubMed.
- Y. Huang, H. Cai, T. Yu, F. Zhang, F. Zhang, Y. Meng, D. Gu, Y. Wan, X. Sun, B. Tu and D. Zhao, Angew. Chem., Int. Ed., 2007, 46, 1089–1093 CrossRef CAS PubMed.
- K. Mortensen, W. Brown and E. Jørgensen, Macromolecules, 1994, 27, 5654–5666 CrossRef CAS.
- A. Balint, M. Papendick, M. Clauss, C. Müller, F. Giesselmann and S. Naumann, Chem. Commun., 2018, 54, 2220–2223 RSC.
- A. Griffin, M. Robertson, P. Frame, G. Ma, K. A. Green, Z. Han, S. E. Morgan, X. Gu, M. Wang and Z. Qiang, J. Mater. Chem. A, 2024, 12, 13139–13152 RSC.
- K. E. B. Doncom, L. D. Blackman, D. B. Wright, M. I. Gibson and R. K. O'Reilly, Chem. Soc. Rev., 2017, 46, 4119–4134 RSC.
- C. J. Zhang, H. Y. Duan, L. F. Hu, C. H. Zhang and X. H. Zhang, ChemSusChem, 2018, 11, 4209–4213 CrossRef CAS PubMed.
- Y. Chen, J. Shen, S. Liu, J. Zhao, Y. Wang and G. Zhang, Macromolecules, 2018, 51, 8286–8297 CrossRef CAS.
- S. Naumann, A. W. Thomas and A. P. Dove, Angew. Chem., Int. Ed., 2015, 54, 9550–9554 CrossRef CAS PubMed.
- C. Vogler and S. Naumann, RSC Adv., 2020, 10, 43389–43393 RSC.
- K. Mortensen, W. Batsberg and S. Hvid, Macromolecules, 2008, 41, 1720–1727 CrossRef CAS.
- C. Rodríguez-Abreu, J. Surfactants Deterg., 2019, 22, 1001–1010 CrossRef.
- H. Schott, J. Pharm. Sci., 1995, 84, 1215–1222 CrossRef CAS PubMed.
- J. N. Israelachvili, D. J. Mitchell and B. W. Ninham, J. Chem. Soc., Faraday Trans. 2, 1976, 72, 1525–1568 RSC.
- R. Nagarajan, Langmuir, 2002, 18, 31–38 CrossRef CAS.
- C. Matei Ghimbeu, L. Vidal, L. Delmotte, J. M. Le Meins and C. Vix-Guterl, Green Chem., 2014, 16, 3079–3088 RSC.
- H. Tian, Z. Lin, F. Xu, J. Zheng, X. Zhuang, Y. Mai and X. Feng, Small, 2016, 3155–3163 CrossRef CAS PubMed.
- L. Peng, C. Te Hung, S. Wang, X. Zhang, X. Zhu, Z. Zhao, C. Wang, Y. Tang, W. Li and D. Zhao, J. Am. Chem. Soc., 2019, 141, 7073–7080 CrossRef CAS PubMed.
- D. Saha, K. E. Warren and A. K. Naskar, Carbon, 2014, 71, 47–57 CrossRef CAS.
- A. Sanchez-Sanchez, M. T. Izquierdo, G. Medjahdi, J. Ghanbaja, A. Celzard and V. Fierro, Microporous Mesoporous Mater., 2018, 270, 127–139 CrossRef CAS.
- X. Liu, Y. Wang, Y. Tang, R. L. Smith, Y. Xu, Y. Liang and X. Qi, Carbon, 2023, 202, 90–100 CrossRef CAS.
- Y. Meng, D. Gu, F. Zhang, Y. Shi, L. Cheng, D. Feng, Z. Wu, Z. Chen, Y. Wan, A. Stein and D. Zhao, Chem. Mater., 2006, 18, 4447–4464 CrossRef CAS.
- J. K. Joseph, V. Naiker, P. Sreeram, F. Mampulliyalil, P. J. George Varghese, P. V. Dhawale, S. P. Sasidharan, V. K. Thakur and P. Raghavan, Handbook of Thermosetting Foams, Aerogels, and Hydrogels: From Fundamentals to Advanced Applications, 2024, pp. 383–420 Search PubMed.
- A. Knop and L. A. Pilato, Phenolic Resins, Springer, 1985 Search PubMed.
- S. Heinz, L. Gemmer, O. Janka and M. Gallei, Polymers, 2024, 16, 2142 CAS.
- R. C. Houtz, Text. Res. J., 1950, 20, 786–801 CrossRef CAS.
- L. Song, D. Feng, N. J. Fredin, K. G. Yager, R. L. Jones, Q. Wu, D. Zhao and B. D. Vogt, ACS Nano, 2010, 4, 189–198 CrossRef CAS PubMed.
- S. Naumann, Org. Mater., 2021, 3, 283–294 CrossRef CAS.
- F. H. Schacher, P. A. Rupar and I. Manners, Angew. Chem., Int. Ed., 2012, 51, 7898–7921 CrossRef CAS PubMed.
- E. J. W. Crossland, M. Kamperman, M. Nedelcu, C. Ducati, U. Wiesner, D. M. Smilgies, G. E. S. Toombes, M. A. Hillmyer, S. Ludwigs, U. Steiner and H. J. Snaith, Nano Lett., 2009, 9, 2807–2812 CrossRef CAS PubMed.
- C. J. Brinker, Y. Lu, A. Sellinger and H. Fan, Adv. Mater., 1999, 11, 579–585 CrossRef CAS.
- N. D. Petkovich and A. Stein, Chem. Soc. Rev., 2013, 42, 3721–3739 RSC.
- Y. Xia, Z. Yang and R. Mokaya, Nanoscale, 2010, 2, 639–659 RSC.
- J. Schuster, R. Köhn, M. Döblinger, A. Keilbach, H. Amenitsch and T. Bein, J. Am. Chem. Soc., 2012, 134, 11136–11145 CrossRef CAS PubMed.
- H. Jiang, J. Li, M. Jiang, R. Lu, J. Shen, X. Sun, W. Han and L. Wang, Anal. Chim. Acta, 2015, 888, 85–93 CAS.
- C. Zhang, Z. Geng and J. Ma, Microporous Mesoporous Mater., 2013, 170, 287–292 CAS.
- N. Zhao, W. Hua, Y. Wang, C. Yang, X. Ouyang, B. Yuan and F. Bian, Appl. Surf. Sci., 2019, 479, 776–785 CAS.
- G. Deng, Y. Zhang, C. Ye, Z. Qiang, G. E. Stein, K. A. Cavicchi and B. D. Vogt, Chem. Commun., 2014, 50, 12684–12687 RSC.
- F. S. H. Simanjuntak, J. Jin, N. Nishiyama, Y. Egashira and K. Ueyama, Carbon, 2009, 47, 2531–2533 CAS.
- M. D. Tyona, Adv. Mater. Res., 2013, 2, 195–208 CrossRef.
- M. Jabbari, R. Bulatova, A. I. Y. Tok, C. R. H. Bahl, E. Mitsoulis and J. H. Hattel, Mater. Sci. Eng., B, 2016, 212, 39–61 CrossRef CAS.
- B. Emley, Y. Liang, R. Chen, C. Wu, M. Pan, Z. Fan and Y. Yao, Mater. Today Phys., 2021, 18, 100397 CrossRef CAS.
- N. G. Orsetti, E. Rosado, A. Alonso, G. Lorenzo, R. Moreno and G. Suárez, J. Eur. Ceram. Soc., 2023, 43, 6956–6966 CrossRef CAS.
- A. Berni, M. Mennig and H. Schmidt, 2.2.8 Doktor Blade, 2004 Search PubMed.
- I. Goodman and J. A. Rhys, Polyesters,Vol. I: Saturated Polymers, American Elsevier Publishing Company, Inc., New York, NY, 1965, vol. 1, p. 158 Search PubMed.
- Q. Wang, Y. Mu, W. Zhang, L. Zhong, Y. Meng and Y. Sun, RSC Adv., 2014, 4, 32113–32116 RSC.
- J. Zhu, J. Yang, R. Miao, Z. Yao, X. Zhuang and X. Feng, J. Mater. Chem. A, 2016, 4, 2286–2292 RSC.
- H. L. Ye, Y. Ping Zheng, R. Xue and Y. L. Xie, Int. J. Electrochem. Sci., 2021, 16, 1–15 Search PubMed.
- R. Wang, K. Lan, R. Lin, X. Jing, C. Te Hung, X. Zhang, L. Liu, Y. Yang, G. Chen, X. Liu, C. Fan, A. M. El-Toni, A. Khan, Y. Tang and D. Zhao, ACS Nano, 2021, 15, 7713–7721 CrossRef CAS PubMed.
- S. Kataoka, T. Yamamoto, Y. Inagi, A. Endo, M. Nakaiwa and T. Ohmori, Carbon, 2008, 46, 1358–1360 CrossRef CAS.
- S. Tanaka, Y. Katayama, M. P. Tate, H. W. Hillhouse and Y. Miyake, J. Mater. Chem., 2007, 17, 3639–3645 RSC.
- A. M. B. Schlander and M. Gallei, ACS Appl. Mater. Interfaces, 2019, 11, 44764–44773 CrossRef CAS PubMed.
- D. Mu, J. Q. Li and S. Y. Feng, Phys. Chem. Chem. Phys., 2017, 19, 21938–21945 CAS.
- C. Tang, J. Bang, G. E. Stein, G. H. Fredrickson, C. J. Hawker, E. J. Kramer, M. Sprung and J. Wang, Macromolecules, 2008, 41, 4328–4339 CAS.
- J. N. L. Albert and T. H. Epps, Mater. Today, 2010, 13, 24–33 CAS.
- T. Kimura, A. M. Emre, K. Kato and Y. Hayashi, J. Mater. Chem. A, 2013, 1, 15135–15141 CAS.
- Y. Yang, Z. Wang, Z. Liang, L. Shen, C. Guo, Y. Shi, H. Tan, Z. Lu and C. Yan, ACS Appl. Mater. Interfaces, 2022, 14, 43690–43700 CrossRef CAS PubMed.
- J. Górka, C. Fenning and M. Jaroniec, Colloids Surf., A, 2009, 352, 113–117 Search PubMed.
- S. Chen, G. He, H. Hu, S. Jin, Y. Zhou, Y. He, S. He, F. Zhao and H. Hou, Energy Environ. Sci., 2013, 6, 2435–2439 CAS.
- R. Liu, Y. Shi, Y. Wan, Y. Meng, F. Zhang, D. Gu, Z. Chen, B. Tu and D. Zhao, J. Am. Chem. Soc., 2006, 128, 11652–11662 CAS.
- N. Yao, A. Y. Ku, N. Nakagawa, T. Lee, D. A. Saville and I. A. Aksay, Chem. Mater., 2000, 12, 1536–1548 CAS.
- P. Innocenzi, L. Malfatti, T. Kidchob and P. Falcaro, Chem. Mater., 2009, 21, 2555–2564 CAS.
- P. Vandezande, L. E. M. Gevers and I. F. J. Vankelecom, Chem. Soc. Rev., 2008, 37, 365–405 CAS.
- K. Xiao, Y. Liu, P. Hu, G. Yu, X. Wang and D. Zhu, Appl. Phys. Lett., 2003, 83, 150–152 CAS.
- V. L. Chavez, L. Song, S. Barua, X. Li, Q. Wu, D. Zhao, K. Rege and B. D. Vogt, Acta Biomater., 2010, 6, 3035–3043 CAS.
- Anton Paar GmbH, 2020, preprint.
- B. Lee, I. Park, J. Yoon, S. Park, J. Kim, K. W. Kim, T. Chang and M. Ree, Macromolecules, 2005, 38, 4311–4323 CAS.
- D.-M. Smilgies, The SAXS Guide, Anton Paar Company, 4th edn, 2017.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CAS.
- K. A. Cychosz, X. Guo, W. Fan, R. Cimino, G. Y. Gor, M. Tsapatsis, A. V. Neimark and M. Thommes, Langmuir, 2012, 28, 12647–12654 CAS.
- T. Stassin, R. Verbeke, A. John Cruz, S. Rodríguez-Hermida, I. Stassen, J. Marreiros, M. Krishtab, M. Dickmann, W. Egger, I. F. J. Vankelecom, S. Furukawa, D. De Vos, D. Grosso, M. Thommes and R. Ameloot, Adv. Mater., 2021, 33, 2006993 CAS.
- M. Thommes, N. Nishiyama and S. Tanaka, Stud. Surf. Sci. Catal., 2007, 165, 551–554 CAS.
- Z. Tian and M. A. Snyder, Langmuir, 2014, 30, 12411–12420 CAS.
- W. Teng, Z. Wu, J. Fan, H. Chen, D. Feng, Y. Lv, J. Wang, A. M. Asiri and D. Zhao, Energy Environ. Sci., 2013, 6, 2765–2776 CAS.
- C. Puttichaem, G. P. Souza, K. C. Ruthe and K. Chainok, Coatings, 2021, 11, 729 CAS.
- F. Salver-Disma, J. M. Tarascon, C. Clinard and J. N. Rouzaud, Carbon, 1999, 37, 1941–1959 CrossRef CAS.
- M. Si, D. Feng, L. Qiu, D. Jia, A. A. Elzatahry, G. Zheng and D. Zhao, J. Mater. Chem. A, 2013, 1, 13490–13495 RSC.
- B. Zheng, X. Lin, X. Zhang, D. Wu and K. Matyjaszewski, Adv. Funct. Mater., 2020, 30, 1907006 CrossRef CAS.
- D. Strmcnik, P. P. Lopes, B. Genorio, V. R. Stamenkovic and N. M. Markovic, Nano Energy, 2016, 29, 29–36 CrossRef CAS.
- A. Lasia, Int. J. Hydrogen Energy, 2019, 44, 19484–19518 CAS.
- M. A. Qadeer, X. Zhang, M. A. Farid, M. Tanveer, Y. Yan, S. Du, Z. F. Huang, M. Tahir and J. J. Zou, J. Power Sources, 2024, 613, 234856 CAS.
- C. Y. Ahn, J. Y. Cheon, S. H. Joo and J. Kim, J. Power Sources, 2013, 222, 477–482 CAS.
- M. Dai, L. Song, J. T. Labelle and B. D. Vogt, Chem. Mater., 2011, 23, 2869–2878 CAS.
- R. Sachse, D. Bernsmeier, R. Schmack, I. Häusler, A. Hertwig, K. Kraffert, J. Nissen and R. Kraehnert, Catal. Sci. Technol., 2020, 10, 2057–2068 CAS.
- Z. Liu, Z. Gao, F. Luo, S. Yuan, K. Wang, N. Li and X. Li, ChemCatChem, 2018, 10, 3441–3446 CAS.
- G. Huang, S. Xu, Z. Liu, S. Yuan, C. Zhang, J. Ai, N. Li and X. Li, ChemCatChem, 2020, 12, 788–794 CAS.
- K. Y. Park, J. H. Jang, J. E. Hong and Y. U. Kwon, J. Phys. Chem. C, 2012, 116, 16848–16853 CAS.
- L. Luo, F. Li, L. Zhu, Y. Ding and D. Deng, Sens. Actuators, B, 2013, 187, 78–83 CAS.
- T. Chen and L. Dai, J. Mater. Chem. A, 2014, 2, 10756–10775 CAS.
- D.-W. Wang, F. Li, M. Liu, G. Q. Lu and H.-M. Cheng, Angew. Chem., Int. Ed., 2008, 373–376 CAS.
- L. Fan, P. Sun, L. Yang, Z. Xu and J. Han, Korean J. Chem. Eng., 2020, 37, 166–175 CAS.
- S. Leyva-García, D. Lozano-Castelló, E. Morallón and D. Cazorla-Amorós, J. Mater. Chem. A, 2016, 4, 4570–4579 Search PubMed.
- J. A. Fernández, S. Tennison, O. Kozynchenko, F. Rubiera, F. Stoeckli and T. A. Centeno, Carbon, 2009, 47, 1598–1604 Search PubMed.
- H. Chang, S. H. Joo and C. Pak, J. Mater. Chem., 2007, 17, 3078–3088 CAS.
- S. H. Joo, C. Pak, D. J. You, S. A. Lee, H. I. Lee, J. M. Kim, H. Chang and D. Seung, Electrochim. Acta, 2006, 52, 1618–1626 CAS.
- M. L. Lin, C. C. Huang, M. Y. Lo and C. Y. Mou, J. Phys. Chem. C, 2008, 112, 867–873 CrossRef CAS.
- R. Liu, D. Wu, X. Feng and K. Müllen, Angew. Chem., Int. Ed., 2010, 49, 2565–2569 CrossRef CAS PubMed.
- D. Hulicova-Jurcakova, M. Kodama, S. Shiraishi, H. Hatori, Z. H. Zhu and G. Q. Lu, Adv. Funct. Mater., 2009, 19, 1800–1809 CrossRef CAS.
- J. Zhang, X. Liu, R. Blume, A. Zhang, R. Schlögl and S. S. Dang, Science, 2008, 322, 73–77 CrossRef CAS PubMed.
- S. M. Mahurin, J. S. Lee, X. Wang and S. Dai, J. Membr. Sci., 2011, 368, 41–47 CrossRef CAS.
- C. H. Hou, X. Wang, C. Liang, S. Yiacoumi, C. Tsouris and S. Dai, J. Phys. Chem. B, 2008, 112, 8563–8570 CAS.
- M. Azenha, P. Nogueira and A. Fernando-Silva, Anal. Chim. Acta, 2008, 610, 205–210 CAS.
- Z. Zhang and J. Pawliszyn, Anal. Chem., 1995, 67, 34–43 CAS.
- M. R. Lee, Y. C. Yeh, W. S. Hsiang and C. C. Chen, J. Chromatogr. B: Biomed. Sci. Appl., 1998, 707, 91–97 CAS.
- C. L. Arthur and J. Pawliszyn, Anal. Chem., 1990, 62, 2145–2148 CAS.
- I. Arambarri, M. Lasa, R. Garcia and E. Millán, J. Chromatogr. A, 2004, 1033, 193–203 CAS.
- Z. Li, R. Ma, S. Bai, C. Wang and Z. Wang, Talanta, 2014, 119, 498–504 CAS.
- A. Rahimi, P. Hashemi, A. Badiei, P. Arab and A. R. Ghiasvand, Anal. Chim. Acta, 2011, 695, 58–62 CAS.
- F. Zhu, J. Guo, F. Zeng, R. Fu, D. Wu, T. Luan, Y. Tong, T. Lu and G. Ouyang, J. Chromatogr. A, 2010, 1217, 7848–7854 CAS.
- H. Liu, J. Li, X. Liu and S. Jiang, Talanta, 2009, 78, 929–935 CAS.
- J. M. Jiménez-Soto, S. Cárdenas and M. Valcárcel, J. Chromatogr. A, 2010, 1217, 3341–3347 Search PubMed.
- J. Fan, Z. Dong, M. Qi, R. Fu and L. Qu, J. Chromatogr. A, 2013, 1320, 27–32 Search PubMed.
- W. Zhang, Y. Sun, C. Wu, J. Xing and J. Li, Anal. Chem., 2009, 81, 2912–2920 CAS.
- N. Rastkari, R. Ahmadkhaniha and M. Yunesian, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2009, 877, 1568–1574 CAS.
- J. Zhu, L. Liao, X. Bian, J. Kong, P. Yang and B. Liu, Small, 2012, 8, 2715–2720 CAS.
- A. Vinu, K. Z. Hossian, P. Srinivasu, M. Miyahara, S. Anandan, N. Gokulakrishnan, T. Mori, K. Ariga and V. V. Balasubramanian, J. Mater. Chem., 2007, 17, 1819–1825 Search PubMed.
- J. Gu, S. Su, Y. Li, Q. He and J. Shi, Chem. Commun., 2011, 47, 2101–2103 Search PubMed.
- M. F. Gencoglu, A. Spurri, M. Franko, J. Chen, D. K. Hensley, C. L. Heldt and D. Saha, ACS Appl. Mater. Interfaces, 2014, 6, 15068–15077 Search PubMed.
- A. Labiano, M. Dai, D. Taylor, W. S. Young, T. H. Epps, K. Rege and B. D. Vogt, Microporous Mesoporous Mater., 2012, 160, 143–150 CAS.
- Y. Salinas, C. Hoerhager, A. García-Fernández, M. Resmini, F. Sancenón, R. Matínez-Máñez and O. Brueggemann, ACS Appl. Mater. Interfaces, 2018, 10, 34029–34038 CAS.
- Y. Salinas, M. Kneidinger, C. Fornaguera, S. Borrós, O. Brüggemann and I. Teasdale, RSC Adv., 2020, 10, 27305–27314 RSC.
- V. Poscher and Y. Salinas, Materials, 2020, 13, 3668 CrossRef CAS PubMed.
- Y. Salinas, O. Brüggemann, U. Monkowius and I. Teasdale, Nanomaterials, 2020, 10, 1030 CrossRef CAS PubMed.
- J. Salonen, A. M. Kaukonen, J. Hirvonen and V. P. Lehto, J. Pharm. Sci., 2008, 97, 632–653 CrossRef CAS PubMed.
- S. Wang, P. Guo, G. Ma, J. Wei, Z. Wang, L. Cui, L. Sun and A. Wang, Electrochim. Acta, 2020, 360, 137016 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |