
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReaction acceleration at the surface of a levitated droplet by vapor dosing from a partner droplet†
Lingqi Qiu ,
Xilai Li,
Dylan T. Holden and
R. Graham Cooks*
,
Xilai Li,
Dylan T. Holden and
R. Graham Cooks*
Department of Chemistry, Purdue University, 560 Oval Drive, West Lafayette, Indiana 47907, USA. E-mail: cooks@purdue.edu
First published on 2nd July 2024
Abstract
Chemical reactions in micrometer-sized droplets can be accelerated by up to six orders of magnitude. However, this acceleration factor (ratio of rate constants relative to bulk) drops to less than 10 for millimeter-sized droplets due to the reduction in surface/volume ratio. To enhance the acceleration in millimeter-sized droplets, we use a new synthesis platform that directly doses reagent vapor onto the reaction droplet surface from a second levitated droplet. Using Katritzky transamination as a model reaction, we made quantitative measurements on size-controlled vapor-dosed droplets, revealing a 31-fold increase in reaction rate constants when examining the entire droplet contents. This enhancement is attributed to a greater reaction rate constant in the droplet surface region (estimated as 105 times greater than that for the bulk). The capability for substantial reaction acceleration in large droplets highlights the potential for rapid synthesis of important chemicals at useful scales. For example, we successfully prepared 23 pyridinium salts within minutes. This efficiency positions droplets as an exceptional platform for rapid, in situ catalyst synthesis. This is illustrated by the preparation of pyridinium salts as photocatalysts and their subsequent use in mediation of amine oxidation both within the same droplet.
Introduction
Microdroplets under ambient conditions constitute a versatile and robust platform for accelerating chemical reactions,1–4 with applications in the rapid synthesis of small molecules,5–12 nanoparticles,13,14 as well as in protein degradation15 and enzymatic reactions.16 Despite achieving up to a 106-fold acceleration in reaction rates,17 there is ongoing debate about the degree to which this acceleration results from droplet shrinking, which increases concentration and enhances the reaction rate,18,19 or from the unique air/liquid interfacial properties of microdroplets, which modify the rate constants and thus accelerate reactions.2,20–24 Efforts to quantitatively describe microdroplet reactions have been made to support the latter hypothesis,25–29 but few studies have successfully addressed the challenges associated with controlling droplet size.25,30 These challenges include (i) polydispersed microdroplets generated from nozzles through spray have a wide size distribution, and (ii) droplet sizes change when suspended in air due to solvent evaporation and/or droplet fission. Generating monodispersed droplets using microfluidic systems31 or directly measuring each droplet's size and related chemistry32 are possible solutions. However, the reported studies of this type were performed in oil; the differences between oil/water (liquid/liquid interface) and the air/liquid interface discussed here, cannot be neglected.An alternative way to address this problem is to study size-controlled single droplets.33 Individual microdroplets can be trapped easily using electric fields;30,34 however, controlling solvent evaporation is crucial. This necessitates the use of water or other non-volatile solvents, like glycols,35 and even high concentrations of salts,30 to minimize evaporation. Unfortunately, these conditions are not applicable to many reported accelerated chemical reactions in microdroplets, where common organic solvents such as methanol, ethanol and acetonitrile are used. Additionally, trapping droplets in electric fields requires a net charge, which can complicate the mechanism due to possible field effects.36
Larger droplets (millimeter-scale) can be levitated using acoustic fields37,38 or by the Leidenfrost effect,39,40 and it is easier to control droplet size. However, they typically exhibit much lower acceleration factors (less than one order of magnitude). In addition to having an intrinsically smaller surface-to-volume ratio, diffusion limits reaction acceleration in large droplets. According to Fick's second law of diffusion, a reagent needs just 0.3 ms to reach the surface in a 1 μm-radius droplet, but 5 minutes in a 1 mm-radius droplet. This problem can be mitigated by directly dosing the reagents onto the droplet surface (Fig. 1).37
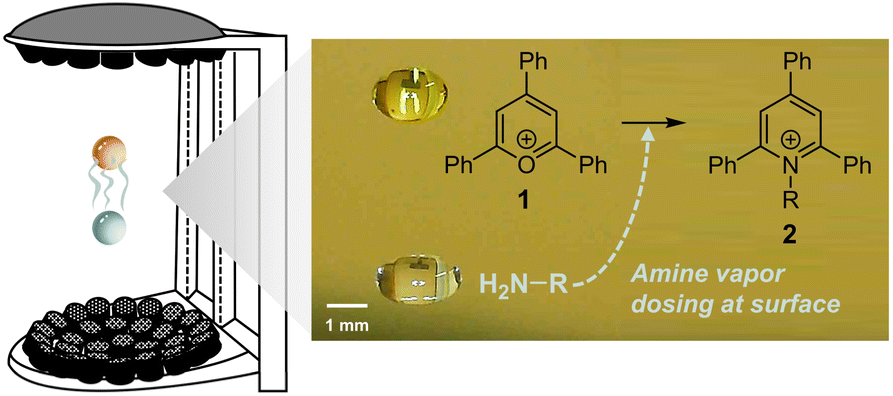 | ||
| Fig. 1 Schematic diagram of vapor-dosed droplet in acoustic levitator showing the reacting droplet (gold) and the reagent reservoir droplet (blue; in this case, pure amine liquid). The distance between the two droplets is roughly 4 mm. | ||
In this study, we developed a new method for accelerating reactions within a size-controlled millimeter-scale droplet that is acoustically levitated and captures vaporized reagents on its surface from a contactless partner droplet (serving as vapor reservoir). In contrast to the behavior of homogeneous droplets containing both reagents, this vapor-dosed droplet exhibited significantly higher reaction rates in the Katritzky transamination, with a 31-fold increase in overall reaction rate constant compared to the conventional bulk reaction. Through quantitative kinetics analysis and surface sampling studies, we determined that the rate constant at or near the surface layer was amplified by more than five orders of magnitude, demonstrating the role of the interfacial effect in accelerating reactions. The water adsorbed by the droplet from the air affected the kinetics, too, being responsible for at maximum an 8-fold enhancement. Using this acceleration system, 23 distinct pyridinium salts were successfully prepared within just two minutes each. Furthermore, a proof-of-concept experiment was conducted in this droplet system, showcasing a streamlined process involving accelerated in situ catalyst preparation and subsequent one-droplet photocatalyzed amine oxidation.
Results and discussion
Apparatus and performance of the vapor-dosed droplet
A single-axis acoustic levitator41 was used to trap two millimeter-sized droplets (3 μL each, approximately 1.8 mm in diameter) at different positions. To control droplet size and simplify the system, several measures were implemented. First, the Katritzky transamination of 2,4,6-triphenylpyrylium (TPP, 1) and tert-octylamine (3) was chosen as a model reaction (Fig. 2a). The pre-charged moiety makes 1 (m/z 309) surface-active and easily quantified using mass spectrometry, with a pre-charged non-reactive internal standard (1-methyl-2,4,6-triphenylpyridinium, m/z 322). Second, the steric hindrance of the α-methyl groups on amine 3 halted the reaction at the addition stage, simplifying the kinetics to cover a single step by preventing further dehydration to the final pyridinium product. Note that the amine used in the two-droplet system must be volatile to provide amine vapor. Third, acetonitrile was selected as the solvent, with the entire reaction time being limited to 30 seconds (including five seconds for reagent dosing) to avoid significant droplet size changes. Lastly, the reaction and detection processes were strictly separated, ensuring that comparisons among different acceleration systems used identical analytical procedures.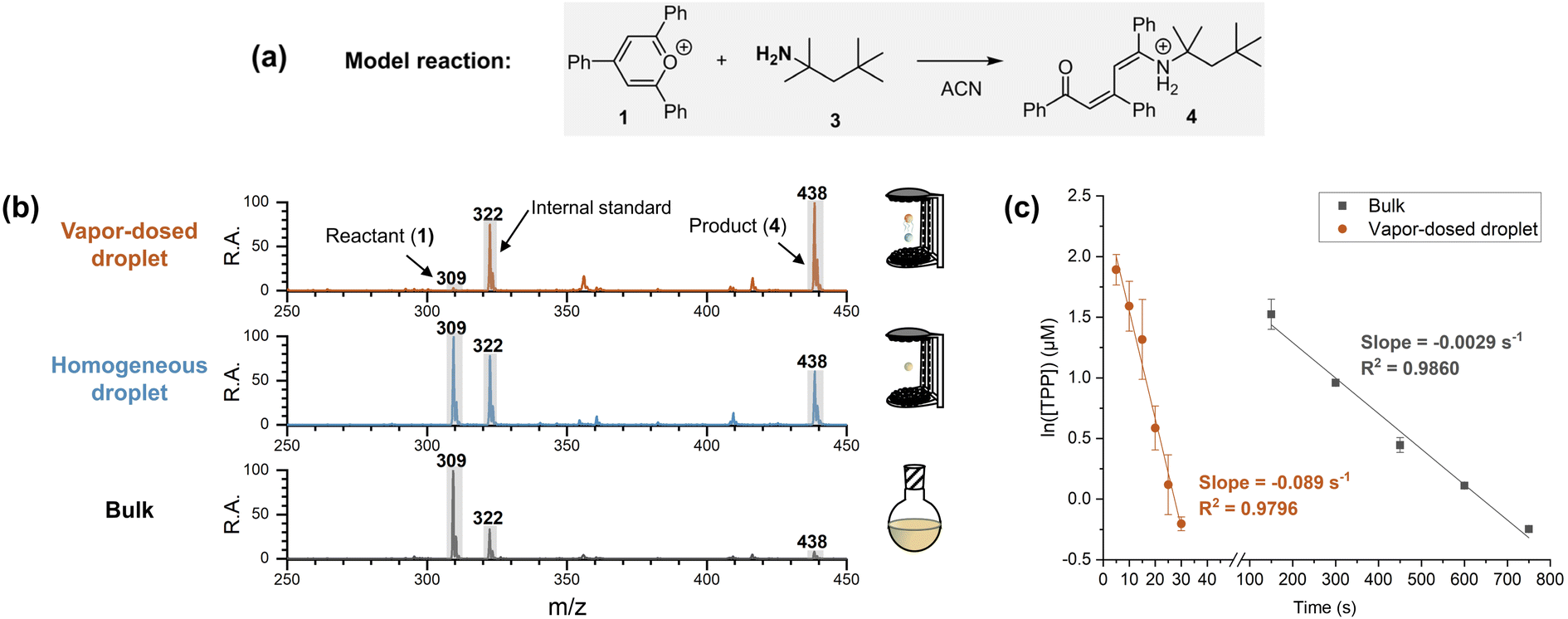 | ||
| Fig. 2 Reaction acceleration in vapor-dosed droplet. (a) Model reaction. (b) Comparison of vapor-dosed droplet (top panel), individual homogeneous droplet (middle panel), and bulk (bottom panel). The reaction time was 30 s. (c) Kinetics plots for vapor-dosed droplet and bulk at room temperature (note dramatically different time scales). | ||
To examine the performance of the vapor-dosed droplet, the same reactions were carried out in bulk and in an individual homogeneous droplet (containing both the amine and TPP) for 30 seconds. Note that the amount of captured amine in the reacting droplet (top droplet, with initial concentration of 10 μM of TPP) in the five-second dosing experiment was measured to be 5 mM (see experimental details in Section 2.3.2 of ESI†). As shown in Fig. 2b, the TPP signal at m/z 309 was significantly reduced in the amine-dosed droplet, with a 57-fold decrease in the ratio of TPP to the internal standard (I309/I322) compared to the corresponding bulk reaction, with initial concentrations of 10 μM of TPP and 5 mM of amine 3. In contrast, the corresponding individual homogeneous droplet only showed a two-fold change. This outcome demonstrates a clear increase in reaction extent when the amine is dosed onto the droplet surface. The detailed kinetics of the reaction in the amine-dosed droplet are illustrated in Fig. 2c. The pseudo-first-order kinetic model for the linear regression is justified by use of excess amine (5 mM) compared to TPP. The rate constants for the vapor-dosed droplet and the bulk reaction were determined to be 18 M−1 s−1 and 0.58 M−1 s−1, respectively, resulting in a rate acceleration factor (RAF) of 31.
Mechanistic investigation of reaction acceleration in the vapor-dosed droplet
The core–shell model, which posits that surface (shell) reactions occur faster than core-region reactions (which are equal in rate to bulk reactions), is thought to govern chemical reactions in droplets (Fig. 3d).3,39 At lower reagent concentrations, greater acceleration is achieved because a higher fraction of reagents can be located at the surface with its limited capacity, viz. the surface is saturated. This effect was more pronounced when [TPP] was less than 5 μM in the amine-dosed droplet (Fig. 3a). Notably, the TPP consumption rates at 1, 2, and 5 μM were identical, suggesting surface-dominated reactions when reactants are concentrated at the surface, making core-region conversion negligible. This hypothesis was further supported by the pseudo-zero-order kinetics observed with 5 μM of TPP in both the amine-dosed and homogeneous droplets (Fig. 3b and c, left panel). Furthermore, reducing the dosing time from 5 seconds to 3 seconds, which altered the dosed amine amount (total amine concentration was 2 mM; see Table S1 in ESI†), resulted in similar reaction rates in the amine-dosed droplet (Fig. 3b). By contrast, homogeneous droplets with varying amine concentrations showed distinct rates (Fig. 3c). This differing behavior in these two systems can be explained by different models of amine distribution.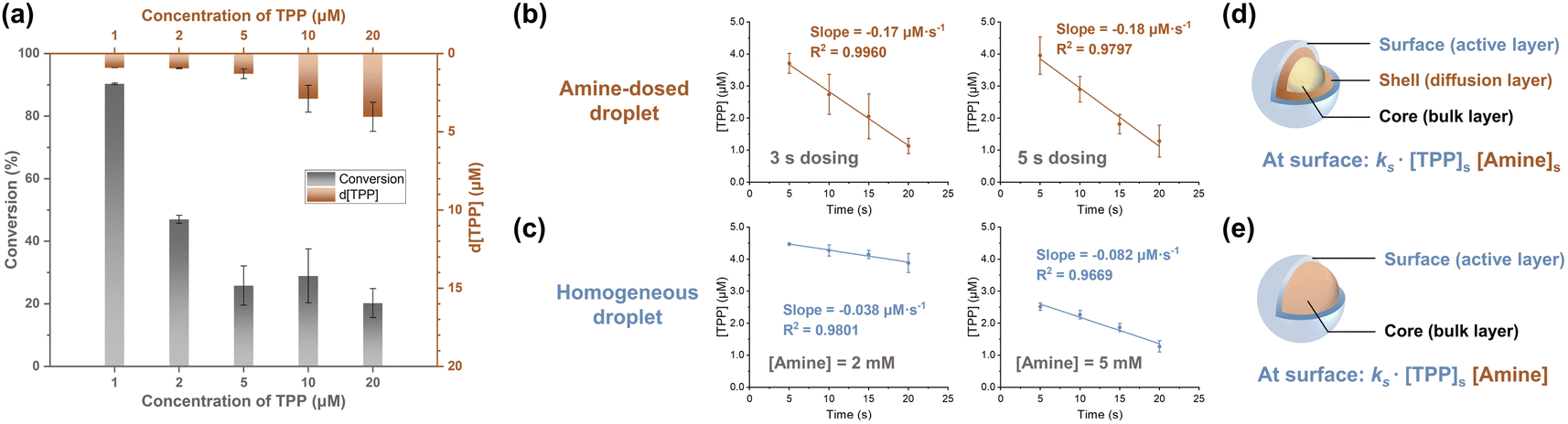 | ||
| Fig. 3 Kinetics of accelerated Katritzky transamination in amine-dosed droplet. (a) Effect of initial TPP concentration (1 through 20 μM) on the reaction conversion (left ordinate) and decreased TPP concentration (right ordinate) in the amine-dosed droplet after 5 s dosing. Comparison of (b) amine-dosed droplet and (c) homogeneous droplet when surface reactions are the major contributors to conversion. (d) Three-layer model that depicts the chemistry in an amine-dosed droplet, with a radial amine concentration gradient. Both amine and TPP concentrations remain constant when the surface is amine-saturated and TPP-saturated, and the surface reaction then follows pseudo zero-order kinetics. (e) Classic core–shell model for the homogeneous droplet, with evenly distributed amine throughout the droplet. The much greater concentration of amine than TPP causes the surface reaction also to exhibit pseudo zero-order kinetics. | ||
A core–shell-surface model42 was developed to describe the reaction scenario in the vapor-dosed droplet (Fig. 3d). In this model, a reagent (A; amine in this case) is dosed onto the surface of a homogeneous solution of another reagent (R; TPP in this case) at time zero, ideally as an infinitely narrow pulse, though in practice the dosing time spans several seconds. The droplet is divided into three regions: (i) the surface, considered the primary reaction site where reaction occurs fastest, (ii) a “shell” region, with significant concentrations of the dosed reagent due to diffusion during the experiment, and (iii) the core (bulk) region, into which the dosed reagent A has not diffused significantly. Three key processes are recognized: (i) radial diffusion of A from the interface, treated as linear diffusion, (ii) reaction of A and R in the surface region to form the product, and (iii) diffusion of the product into the core region. The reaction is considered irreversible. The distribution of amine is therefore the primary difference between the vapor-dosed droplet and homogeneous droplet (Fig. 3e), for which a classic core–shell model was applied. The former system exhibits an amine gradient across the droplet, whereas the latter case assumes uniform amine dispersion. Note that although rapid global mixing in acoustically levitated droplets has been reported, usually triggered by extrinsic events,43 it is not a concern in our system. Moreover, internal vortex flow is generated in each layer (Reynolds number estimated as 117, indicating laminar flow),44 which does not disturb the radial distribution.
Given that surface reaction dominates droplet reactions at 5 μM of TPP, the actual surface reaction rate constant can be determined once the surface concentrations of TPP and amine are known. It is crucial to understand that the reaction rates shown in Fig. 3b and c do not reflect the true surface rates, as the analysis encompasses the entire droplet, thereby diluting the surface rate with the large volumes of the core (and shell) regions. The surface layer has an estimated volume of 10−5 μL, assuming a monolayer of molecules resides at the surface (the TPP diameter45 and air–water interface thickness46 are both estimated as 10 Å), compared to the droplet's total volume of 3 μL. Consequently, the actual surface reaction rates are significantly higher than the measured values.
The saturated amine concentration, [amine]s in the vapor-dosed droplet can be derived by multiplying the rate ratio between the two droplet systems by the amine concentration in the corresponding individual droplet, [amine]. In the 3 second and 5 second dosing experiments, the calculated [amine]s values were 9 mM and 11 mM, respectively (see Section 2.3.3 in ESI† for the calculation). These consistent results further validate application of the core–shell-surface model. To measure the saturated concentration of TPP at the droplet surface, a near-surface sampling method was developed using a glass tip with short taper to touch a levitated TPP droplet, yielding a near-surface concentration measurement of TPP below 38 μM (see Section 2.4 in ESI†). Therefore, in the surface region (10−5 μL), the rate constant of the vapor-dosed droplet is calculated to be greater than 1.4 × 105 M−1 s−1 (see Section 2.5 in ESI† for the calculation), with the RAF surpassing 2.4 × 105. This outcome is comparable to the reported acceleration factor in sprayed microdroplets, where diffusion is efficient enough to provide great acceleration even when the entire droplets are analyzed (see Section 2.6 in ESI† for more discussion).
Water adsorption is an inevitable process when experiments are conducted under ambient conditions, and water has been found to accelerate bulk reactions through “on-water” catalysis.47,48 Additionally, water-driven microdroplet reactions have been reported, via generation of reactive species (e.g. superacid/superbase,49–51 redox species52–56), even when using organic solvents.57,58 Interestingly, we discovered that water can significantly accelerate the studied reaction even in bulk (Fig. S9 in ESI†). Thus, to assess the adsorbed water's impact in our system, we employed a recently established method using a pre-charged molecular probe (4-formyl-1-methylpyridinium)59 to react with and quantify water adsorption within the 30 seconds of reaction time. An increase of 2% in water uptake was observed at a relative humidity of about 45% (Table S5†). The corresponding bulk reaction with this additional 2% water exhibited a rate constant of 4.8 M−1 s−1 (Fig. S10†), representing an 8-fold increase in reaction rate constant. Considering that water adsorption is an cumulative process, its effect on acceleration in vapor-dosed droplets should be less than 8-fold. (A summary of all measured rate constants is provided in Table 1.) Hence, the higher rate constants in vapor-dosed droplets are attributed to the amplified interfacial rate constant and to a small effect of water absorbed from ambient air.
| Reaction system | Rate constant (M−1 s−1) | RAF |
|---|---|---|
| a RAFs were calculated as the ratio of droplet rate constant vs. that in bulk. | ||
| Bulk | 0.58 | 1 |
| Bulk with additional 2% water | 4.8 | 8 |
| Vapor-dosed droplet (entire) | 18 | 31 |
| Vapor-dosed droplet (surface) | 1.4 × 105 | 2.4 × 105 |
Rapid synthesis of pyridinium salts using vapor-dosed droplets
To investigate the efficacy of vapor-dosed droplets in rapidly synthesizing pyridinium salts, we evaluated 25 versatile primary amines (no conversion of TPP was observed in the cases of secondary amines and aniline; 6% conversion when reacting with phenyl hydrazine; see Fig. S11†), as depicted in Scheme 1. The studied process involves two steps: the addition-induced dearomatization of TPP to form the intermediate 6, followed by its dehydration to generate pyridinium 2. Initially, a one-minute reaction was conducted in vapor-dosed droplets with continuous amine dosing (condition a). Monoamines exhibited good conversion for both steps, ranging from 40% to 95% (5a to 5t), while some diamines gave lower yields, possibly due to the relatively high basicity of the additional tertiary amino group (5v to 5y). By contrast, 70% overall conversion is observed in 5u which contains two primary amino groups. Interestingly, the addition step was minimally affected by steric hindrance, whereas the elimination step was significantly affected. Linear fatty amines (5a to 5e) or a less bulky branched amine (5f) still yielded the product 2, albeit with a low ratio for compounds 6 and 2 (less than 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :89, seen in agemine 5d). Bulky amines barely produced any pyridinium (5h to 5o). By contrast, diamines showed obviously higher ratios for 2 to 6, but the overall conversion was unsatisfactory, as noted earlier. To enhance synthetic efficiency, an additional 1 minute droplet reaction time was introduced to facilitate the conversion from 6 to 2, with the assistance of acid.60 Under this condition, significantly higher overall conversion was achieved, ranging from 49% to 100%, with up to 100:0 ratio of 2:6. The exception is for very bulky amines, like 5j and 5k, where decomposition was observed, and no further product was generated. Overall, 23 distinct pyridinium salts could be easily prepared within 2 minutes each using the droplet system on a microgram scale, an advantageous quantity for early phase drug discovery.61,62
:89, seen in agemine 5d). Bulky amines barely produced any pyridinium (5h to 5o). By contrast, diamines showed obviously higher ratios for 2 to 6, but the overall conversion was unsatisfactory, as noted earlier. To enhance synthetic efficiency, an additional 1 minute droplet reaction time was introduced to facilitate the conversion from 6 to 2, with the assistance of acid.60 Under this condition, significantly higher overall conversion was achieved, ranging from 49% to 100%, with up to 100:0 ratio of 2:6. The exception is for very bulky amines, like 5j and 5k, where decomposition was observed, and no further product was generated. Overall, 23 distinct pyridinium salts could be easily prepared within 2 minutes each using the droplet system on a microgram scale, an advantageous quantity for early phase drug discovery.61,62
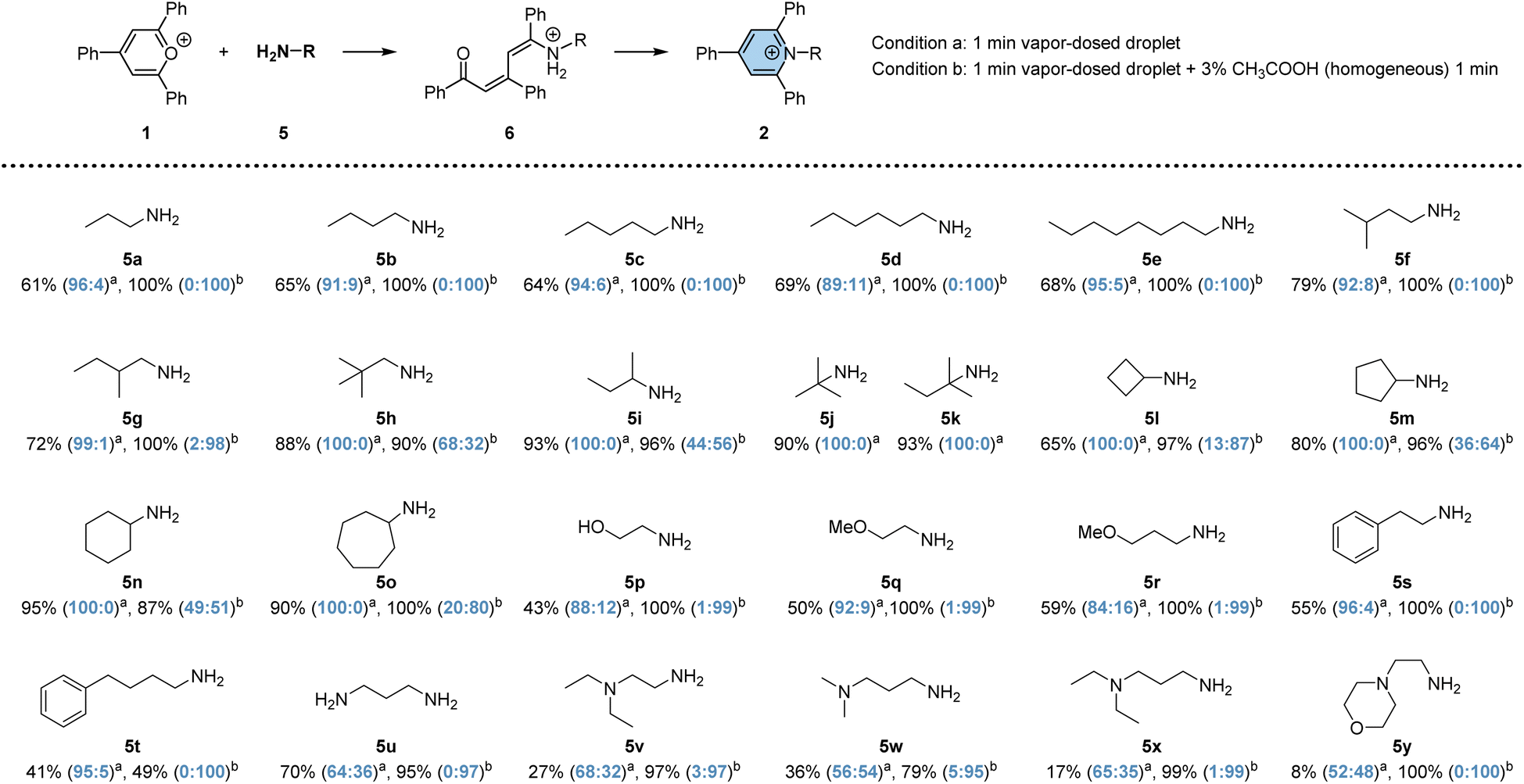 | ||
| Scheme 1 Substrate scope of Katritzky reactions in vapor-dosed droplets. The overall yield of intermediate 6 and final product 2 was obtained by performing the reactions under conditions a or b. The ratio between product 6 and 2 is shown in brackets, annotated in blue. A three-microliter (3 μL) droplet was used in each case, yielding the product on a microgram scale. | ||
Photocatalytic amine oxidation mediated by in situ generated pyridinium
Pyridinium salts act as versatile scaffolds in organic synthesis, functioning as substrate, reagent, catalyst and even as reaction medium (e.g. ionic liquid).63 Specifically, pyridinium salts have been reported to work as photocatalysts, promoting alcohol oxidation in the presence of air by converting molecular oxygen into singlet oxygen and superoxide.64 Since singlet oxygen is also capable of oxidizing amines,65 there is potential for conducting amine oxidation in vapor-dosed droplets under ambient conditions, benefiting from the rapid, in situ synthesis of the photocatalyst, pyridinium, as illustrated in Fig. 4a.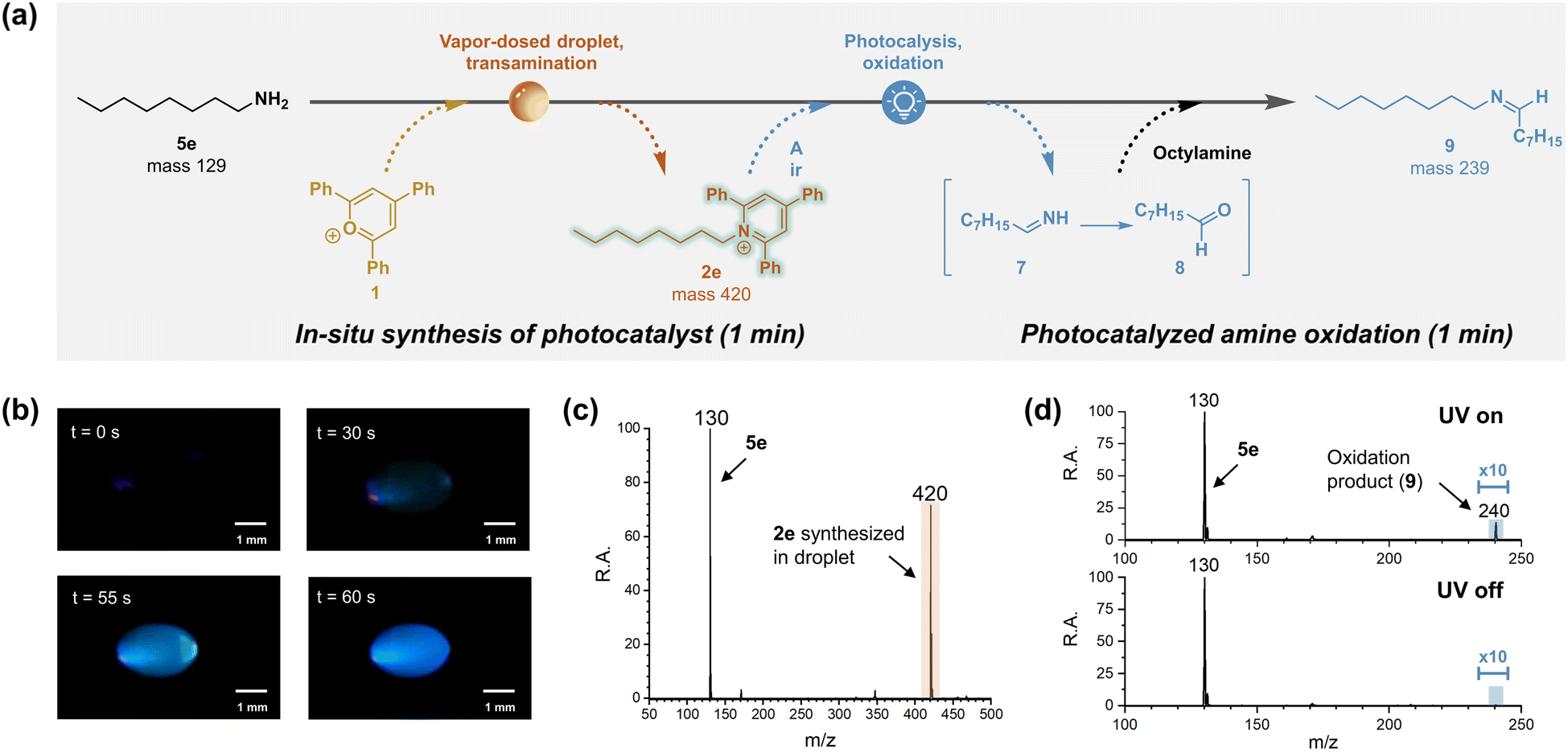 | ||
| Fig. 4 Photocatalyzed amine oxidation mediated by rapid, in situ synthesis of pyridinium photocatalyst in vapor-dosed droplet. (a) Schematic illustration showing transformation of octylamine to resulting oxidation product 9. Pyridinium salt 2e was generated in a vapor-dosed droplet, and then it served as photocatalyst to oxidize the excess amine in the same droplet. (b) Microphotograph of pyridinium synthesis under UV illumination. (c) Mass spectrum of collected droplet containing excess amine 5e and pyridinium salt 2e. (d) Detection of photocatalyzed amine oxidation in droplet, with and without exposure to 365 nm UV. To avoid hydrolysis of generated imine 9, no acid was added. | ||
A proof-of-concept experiment was carried out using n-octylamine (5e) as starting material. The initial step involved vapor-dosing of amine to transaminate pyrylium 1 and form pyridinium 2e. Optical monitoring of this process clearly demonstrated the synthesis of 2e under UV lamp illumination (Fig. 4b). As the reaction progressed, a blue emission was observed and intensified over time, consistent with the reported emission spectrum of similar pyridinium structures.66 Interestingly, intermittently a green light was observed, for example, at t = 55 s. This red-shifted emission might be attributed to water adsorption altering the solvent environment,67 but more probably it is due to aggregation-induced enhanced emission (AIEE) resulting from the rotor structure of 2e, which features three phenyl rings.68 The formation of the pyridinium photocatalyst was further confirmed by mass spectrometry, revealing a peak at m/z 420 (Fig. 4c). Meanwhile, signal corresponding to excess amine was observed at m/z 130, this compound serving as the substrate of the subsequent photocatalyzed reaction. This reaction was easily initiated by removing the bottom amine droplet and exposing the top droplet to 365 nm UV light for 1 min. The resulting oxidation product, imine 9, was confirmed by tandem mass spectrometry, when compared to the standard reference (Fig. S36†). By contrast, no product was detected when the UV lamp was turned off (Fig. 4d). It is noteworthy that although the direct oxidation product 7 was observed in the UV-on droplet, this signal was weak (Fig. S37†), likely because of its rapid hydrolysis to octanal (8), which would further react with excess octylamine in the same droplet to form product 9.
Conclusions
We have developed a novel reaction acceleration system utilizing a size-controlled, vapor-dosed droplet pair that is acoustically levitated. The millimeter-sized reacting droplet exhibits a significant increase in reaction acceleration, unprecedented at this scale. Quantitative analysis of reaction kinetics and surface sampling experiments reveal five orders of magnitude increase in the reaction rate constant at the surface, comparable to the acceleration observed in micron-sized droplets. This result, along with the lower overall acceleration in large (millimeter-scale) droplets compared to small (micrometer-scale) droplets, highlights the influence of reagent diffusion, which significantly impacts the distribution of reagents at or near the droplet surface. It also raises a question as to the relative importance of the electric field vs. partial solvation in reaction acceleration. In the mm sized droplets studied here, the field effect should be much smaller than in the previously much-studied sprayed microdroplets. Additionally, water adsorption by droplets from the air plays a role; introducing an additional 2% water in the bulk solution results in an eight-fold increase in rate constants. While the water effect on droplet acceleration has been noted in previous studies, it requires further attention, particularly in the quantification of organic microdroplets.This study demonstrates that millimeter-sized droplets can achieve significant reaction acceleration, facilitating the rapid synthesis of chemically important compounds on a significant scale. As an example, we synthesized 23 different pyridinium salts each within two minutes each using vapor-dosed droplets. This high efficiency allows for a streamlined process, utilizing the in situ synthesized pyridinium salts as photocatalysts to mediate subsequent one-droplet (analogous to one-pot in conventional synthetic chemistry) photocatalyzed reactions. A proof-of-concept experiment was successfully conducted in a photocatalyzed amine oxidation. Therefore, this system (in elaborated form) has the potential to be used in a high-throughput manner for rapid screening of catalyst synthesis and for use in concurrent catalytic reactions.
Data availability
All data on which this publication is based appears either in the main text or in the ESI.† The underlying raw data is available on request to the authors.Author contributions
Dr R. G. Cooks and L. Qiu designed the experiments. Dr L. Qiu, X. Li and D. T. Holden performed the experiments. Dr R. G. Cooks and Dr L. Qiu wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the Multi-University Research Initiative (MURI) of the Air Force Office of Scientific Research (FA9550-21-1-0170) via Stanford University (sub-award 62741613-204669). We particularly thank Dr Brett Marsh and Dr Eric T. Dziekonski for assistance in fabricating the acoustic levitator and custom electronics.References
- J. K. Lee, S. Banerjee, H. G. Nam and R. N. Zare, Q. Rev. Biophys., 2015, 48, 437–444 CrossRef CAS PubMed.
- X. Yan, R. M. Bain and R. G. Cooks, Angew. Chem., Int. Ed., 2016, 55, 12960–12972 CrossRef CAS PubMed.
- Z. Wei, Y. Li, R. G. Cooks and X. Yan, Annu. Rev. Phys. Chem., 2020, 71, 31–51 CrossRef CAS PubMed.
- X. Yan, Int. J. Mass Spectrom., 2021, 468, 116639 CrossRef CAS.
- T. Müller, A. Badu-Tawiah and R. G. Cooks, Angew. Chem., Int. Ed., 2012, 51, 11832–11835 CrossRef PubMed.
- C. Liu, J. Li, H. Chen and R. N. Zare, Chem. Sci., 2019, 10, 9367–9373 RSC.
- H. Nie, Z. Wei, L. Qiu, X. Chen, D. T. T. Holden and R. G. G. Cooks, Chem. Sci., 2020, 11, 2356–2361 RSC.
- K. Huang, N. M. Morato, Y. Feng and R. G. Cooks, Angew. Chem., Int. Ed., 2023, 62, e202300956 CrossRef CAS PubMed.
- W. Zhang, S. Yang, Q. Lin, H. Cheng and J. Liu, J. Org. Chem., 2019, 84, 851–859 CrossRef CAS PubMed.
- X. Yan, Y. H. Lai and R. N. Zare, Chem. Sci., 2018, 9, 5207–5211 RSC.
- C. Salvitti, A. Troiani, F. Mazzei, C. D'Agostino, R. Zumpano, C. Baldacchini, A. R. Bizzarri, A. Tata and F. Pepi, Int. J. Mass Spectrom., 2021, 468, 116658 CrossRef CAS.
- M. Girod, E. Moyano, D. I. Campbell and R. G. Cooks, Chem. Sci., 2011, 2, 501–510 RSC.
- D. Sarkar, M. K. Mahitha, A. Som, A. Li, M. Wleklinski, R. G. Cooks and T. Pradeep, Adv. Mater., 2016, 28, 2223–2228 CrossRef CAS PubMed.
- P. Basuri, J. Shantha Kumar, K. Unni, S. Manna and T. Pradeep, Chem. Commun., 2022, 58, 12657–12660 RSC.
- H. P. Gunawardena, R. N. Zare, H. Chen, P. Zhao and X. Zhong, Anal. Chem., 2021, 93, 3997–4005 CrossRef PubMed.
- X. Zhong, H. Chen and R. N. Zare, Nat. Commun., 2020, 11, 1–9 CrossRef PubMed.
- N. Sahota, D. I. Abusalim, M. L. Wang, C. J. Brown, Z. Zhang, T. J. El-Baba, S. P. Cook and D. E. Clemmer, Chem. Sci., 2019, 10, 4822–4827 RSC.
- K. R. R. Wilson, A. M. M. Prophet, G. Rovelli, M. D. D. Willis, R. J. J. Rapf and M. I. I. Jacobs, Chem. Sci., 2020, 11, 8533–8545 RSC.
- C. J. Chen and E. R. Williams, Chem. Sci., 2023, 4704–4713 RSC.
- K. D. Judd, S. W. Parsons, D. B. Eremin, V. V. Fokin and J. M. Dawlaty, Chem. Sci., 2024, 15, 8346–8354 RSC.
- H. Hao, I. Leven and T. Head-Gordon, Nat. Commun., 2022, 13, 280 CrossRef CAS PubMed.
- D. Ben-Amotz, J. Chem. Phys., 2024, 160, 084704 CrossRef CAS PubMed.
- C. F. Chamberlayne and R. N. Zare, J. Chem. Phys., 2022, 156, 054705 CrossRef CAS PubMed.
- M. F. Ruiz-López and M. T. C. Martins-Costa, Phys. Chem. Chem. Phys., 2022, 24, 29700–29704 RSC.
- M. Li, C. Boothby, R. E. Continetti and V. H. Grassian, J. Am. Chem. Soc., 2023, 145, 22317–22321 CrossRef CAS PubMed.
- B. Zheng, L. Xue, C. Dai, J. Liu and H. Cheng, J. Org. Chem., 2022, 87, 5287–5295 CrossRef CAS PubMed.
- Y. H. Lai, S. Sathyamoorthi, R. M. Bain and R. N. Zare, J. Am. Soc. Mass Spectrom., 2018, 29, 1036–1043 CrossRef CAS PubMed.
- B. M. Marsh, K. Iyer and R. G. Cooks, J. Am. Soc. Mass Spectrom., 2019, 30, 2022–2030 CrossRef CAS PubMed.
- L. Qiu, Z. Wei, H. Nie and R. G. Cooks, ChemPlusChem, 2021, 86, 1362–1365 CrossRef CAS PubMed.
- M. I. Jacobs, J. F. Davies, L. Lee, R. D. Davis, F. Houle and K. R. Wilson, Anal. Chem., 2017, 89, 12511–12519 CrossRef CAS PubMed.
- A. Fallah-Araghi, K. Meguellati, J. C. Baret, A. El Harrak, T. Mangeat, M. Karplus, S. Ladame, C. M. Marques and A. D. Griffiths, Phys. Rev. Lett., 2014, 112, 028301 CrossRef PubMed.
- K. J. Vannoy, I. Lee, K. Sode and J. E. Dick, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2025726118 CrossRef CAS PubMed.
- U. K. Krieger, C. Marcolli and J. P. Reid, Chem. Soc. Rev., 2012, 41, 6631 RSC.
- K. R. Wilson, A. M. Prophet and M. D. Willis, J. Phys. Chem. A, 2022, 126, 7291–7308 CrossRef CAS PubMed.
- R. K. Kohli and J. F. Davies, Anal. Chem., 2021, 93, 12472–12479 CrossRef CAS PubMed.
- J. P. Heindel, R. A. LaCour and T. Head-Gordon, Nat. Commun., 2024, 15, 3670 CrossRef CAS PubMed.
- R. L. Grimm, R. Hodyss and J. L. Beauchamp, Anal. Chem., 2006, 78, 3800–3806 CrossRef CAS PubMed.
- E. A. Crawford, C. Esen and D. A. Volmer, Anal. Chem., 2016, 88, 8396–8403 CrossRef CAS PubMed.
- Y. Li, T. F. Mehari, Z. Wei, Y. Liu and R. G. Cooks, J. Mass Spectrom., 2021, 56, 2e4585 Search PubMed.
- R. M. Bain, C. J. Pulliam, F. Thery and R. G. Cooks, Angew. Chem., Int. Ed., 2016, 55, 10478–10482 CrossRef CAS PubMed.
- A. Marzo, A. Barnes and B. W. Drinkwater, Rev. Sci. Instrum., 2017, 88, 085105 CrossRef PubMed.
- J. F. Davies and K. R. Wilson, Chem. Sci., 2015, 6, 7020–7027 RSC.
- E. T. Chainani, W.-H. Choi, K. T. Ngo and A. Scheeline, Anal. Chem., 2014, 86, 2229–2237 CrossRef CAS PubMed.
- Z. L. Yan, W. J. Xie and B. Wei, Phys. Lett. A, 2011, 375, 3306–3309 CrossRef CAS.
- P. Bonhôte, E. Gogniat, S. Tingry, C. Barbé, N. Vlachopoulos, F. Lenzmann, P. Comte and M. Grätzel, J. Phys. Chem. B, 1998, 102, 1498–1507 CrossRef PubMed.
- R. Kusaka, S. Nihonyanagi and T. Tahara, Nat. Chem., 2021, 13, 1–7 CrossRef PubMed.
- D. C. Rideout and R. Breslow, J. Am. Chem. Soc., 1980, 102, 7816–7817 CrossRef CAS.
- Y. Jung and R. A. Marcus, J. Am. Chem. Soc., 2007, 129, 5492–5502 CrossRef CAS PubMed.
- P. Basuri, L. E. Gonzalez, N. M. Morato, T. Pradeep and R. G. Cooks, Chem. Sci., 2020, 11, 12686–12694 RSC.
- K.-H. Huang, Z. Wei and R. G. Cooks, Chem. Sci., 2021, 12, 2242–2250 RSC.
- K. Gong, J. Ao, K. Li, L. Liu, Y. Liu, G. Xu, T. Wang, H. Cheng, Z. Wang, X. Zhang, H. Wei, C. George, A. Mellouki, H. Herrmann, L. Wang, J. Chen, M. Ji, L. Zhang and J. S. Francisco, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2219588120 CrossRef CAS PubMed.
- J. K. Lee, K. L. Walker, H. S. Han, J. Kang, F. B. Prinz, R. M. Waymouth, H. G. Nam and R. N. Zare, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 19294–19298 CrossRef CAS PubMed.
- L. Qiu and R. G. Cooks, Angew. Chem., Int. Ed., 2022, 61, e202210765 CrossRef CAS PubMed.
- S. Jin, H. Chen, X. Yuan, D. Xing, R. Wang, L. Zhao, D. Zhang, C. Gong, C. Zhu, X. Gao, Y. Chen and X. Zhang, JACS Au, 2023, 3, 1563–1571 CrossRef CAS PubMed.
- L. Qiu and R. G. Cooks, Angew. Chem., Int. Ed., 2024, e202400118 CAS.
- Z. Rao, X. Li, Y.-G. Fang, J. S. Francisco, C. Zhu and C. Chu, J. Am. Chem. Soc., 2023, 145, 10839–10846 CrossRef CAS PubMed.
- D. B. Eremin and V. V. Fokin, J. Am. Chem. Soc., 2021, 143, 18374–18379 CrossRef CAS PubMed.
- L. Xue, B. Zheng, J. Sun, J. Liu and H. Cheng, ACS Sustain. Chem. Eng., 2023, 11, 12780–12789 CrossRef CAS.
- L. Qiu, M. Saha, S. Kraft, E. T. Dziekonski, C. J. Welch, Y. Dai, A. Kaerner and R. G. Cooks, Angew. Chem., Int. Ed., 2023, 62, e202310884 CrossRef CAS PubMed.
- A. R. Katritzky and R. H. Manzo, J. Chem. Soc., Perkin Trans. 2, 1981, 571 RSC.
- T. Cernak, N. J. Gesmundo, K. Dykstra, Y. Yu, Z. Wu, Z.-C. Shi, P. Vachal, D. Sperbeck, S. He, B. A. Murphy, L. Sonatore, S. Williams, M. Madeira, A. Verras, M. Reiter, C. H. Lee, J. Cuff, E. C. Sherer, J. Kuethe, S. Goble, N. Perrotto, S. Pinto, D.-M. Shen, R. Nargund, J. Balkovec, R. J. DeVita and S. D. Dreher, J. Med. Chem., 2017, 60, 3594–3605 CrossRef CAS PubMed.
- D. Perera, J. W. Tucker, S. Brahmbhatt, C. J. Helal, A. Chong, W. Farrell, P. Richardson and N. W. Sach, Science, 2018, 359, 429–434 CrossRef CAS PubMed.
- S. Sowmiah, J. M. S. S. Esperança, L. P. N. Rebelo and C. A. M. Afonso, Org. Chem. Front., 2018, 5, 453–493 RSC.
- S. Ma, J.-W. Cui, C.-H. Rao, M.-Z. Jia, Y.-R. Chen and J. Zhang, Green Chem., 2021, 23, 1337–1343 RSC.
- G. Jiang, J. Chen, J.-S. Huang and C.-M. Che, Org. Lett., 2009, 11, 4568–4571 CrossRef CAS PubMed.
- J.-W. Cui, S. Ma, C.-H. Rao, M.-Z. Jia and J. Zhang, ACS Sustain. Chem. Eng., 2022, 10, 2148–2157 CrossRef CAS.
- U. Narang, C. F. Zhao, J. D. Bhawalkar, F. V. Bright and P. N. Prasad, J. Phys. Chem., 1996, 100, 4521–4525 CrossRef CAS.
- D. Devadiga and T. N. Ahipa, Soft Matter, 2022, 18, 8008–8016 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc03528c |
| This journal is © The Royal Society of Chemistry 2024 |