
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent progress in biochar-supported photocatalysts: synthesis, role of biochar, and applications
Md Manik Mian ab and
Guijian Liu*ab
ab and
Guijian Liu*ab
aCAS Key Laboratory of Crust-Mantle Materials and Environments, School of Earth and Space Sciences, University of Science and Technology of China, Hefei 230026, China. E-mail: lgj@ustc.edu.cn; Fax: +86 551 63621485; Tel: +86 551 63603714
bState Key Laboratory of Loess and Quaternary Geology, Institute of Earth Environment, The Chinese Academy of Sciences, Xi’an, Shaanxi 710075, PR China
First published on 17th April 2018
Abstract
Incorporating photocatalytic nanoparticles with biochar templates can produce biochar-supported photocatalysts (BSPs) and combine the advantages of biochar with catalytic nanoparticles. The obtained composite exhibits excellent surface properties, crystallinity, chemical stability, recoverability, and higher photocatalytic competency than the bare semiconductor photocatalyst. The literature and advances in BSPs based on the combination of low-cost biochar and catalytic nanoparticles are presented in this review. Various synthetic techniques and physicochemical properties of BSPs are summarized. The article then discusses in detail the important role of biochar in influencing the photocatalytic performance of BSPs such as supporting nanoparticles, increasing the surface area and the number of active sites, shuttling electrons, acting as an electron reservoir, increasing charge separation, and reducing band gap energy. Furthermore, the synergistic effects of adsorption and photodegradation of organic pollutants by BSPs are discussed with in-depth mechanistic evidence. Finally, the application of BSPs in various fields and constructive suggestions for their future development are reported.
1. Introduction
Nowadays, environmental pollution and insufficient sources of natural clean energy are serious global problems faced by humankind. The high growth rate of the world’s population, industrialization, and urbanization accelerates the consumption of non-renewable energy and causes the release of pollution into the air and waterways. According to a UN water report, on average 70%, 28–38%, and 8% municipal wastewater is treated before discharge in high-income, middle-income, and low-income countries, respectively, and about 80% of total global wastewater is discharged into the environment without any treatment.1 Untreated or improperly treated wastewater discharges certainly impose a detrimental effect on the natural ecosystem and public health. Therefore, a diverse group of researchers devoted themselves to find an environmentally friendly, cost-effective, and sustainable way to resolve this adverse situation. From that perspective, photocatalysis, in which naturally available, safe, and clean solar energy has been utilized by semiconductor photocatalysts to degrade harmful organic pollutants, is a major advancement.Up until now, various nanoparticles such as RuO2, TiO2, Fe2O3, SiO2, Al2O3, ZrO2, ZnO, CdS, and ZnS have been utilized as semiconductor photocatalysts.2–6 Among them, different forms of TiO2 nanoparticles have received significant attention due to their chemical stability, low toxicity, inexpensiveness, availability, and high sensitivity to UV light illumination.7 However, the large band gap energy and the separation of TiO2 nanoparticles are the major drawbacks for their applications. Furthermore, TiO2 cannot utilize most of the solar energy spectrum due to its large band gap energy.4 Therefore, an external UV light source is required for their activation, which is not a cost-effective method. However, various hybrid composites synthesized by combining doping of transition metals (Ti, Fe, Ru, Au, and Mo)8–11 and/or non-metals (C, N, and S)12–14 can improve the photocatalytic performance, but, high preparation costs, non-renewability, and risks of secondary pollution prevent their large-scale application. Therefore, more and more attention has been applied to giving a stable platform for photocatalytic nanoparticles by incorporating secondary materials, which can improve the recoverability and visible light sensitivity of the photocatalysts. Biochar, as a supporting material for catalytic nanoparticles, receives significant attention in this direction. The unique physicochemical properties of biochar are beneficial for incorporating various photocatalytic nanoparticles.2,15
Biochar is a low-cost, stable, environmentally friendly, and sustainable material produced from available waste biomass via pyrolysis, hydrolysis, gasification, and carbonization methods.16,17 The prominent fractions of plant- and animal-originated biomass used for biochar production are illustrated in Fig. 1. The major streams of biomass are lignocellulose, starch, animal protein, and polysaccharides. Starch and lignocellulose are the forms of plant-driven biomass used for the synthesis of biochar and different chemicals. Starch is composed of a large number of glucose units connected by glycosidic bonds and consists of two molecules such as amylose and amylopectin. Starch is commonly used as a pore-forming agent in ceramic technology.18 The composition of lignocellulose, especially lignin and hemicellulose structures, is highly dependent on its origins such as hardwood, softwood, or grasses. Lignin is a constituent of the cell wall, and is the second most abundant polymer after cellulose. Lignin, composed of three different phenyl propane monomers, is electronegative and has a strong affinity for various electropositive metal ions.2 Among animal-originated polysaccharides, chitin is the most abundant natural polysaccharide, and can be found in the exoskeleton of insects and crustacean cell walls.19 The amino (chitin, chitosan, gelatin, and collagen) and hydroxyl (chitin, chitosan, and glycogen) functional groups of animal-origin biochar possess strong adsorption capacities for various heavy metals,20 dyestuffs,20 and pathogens.21 Moreover, the –OH and –NH2 functional groups of biomass can easily be tuned with other functional materials.
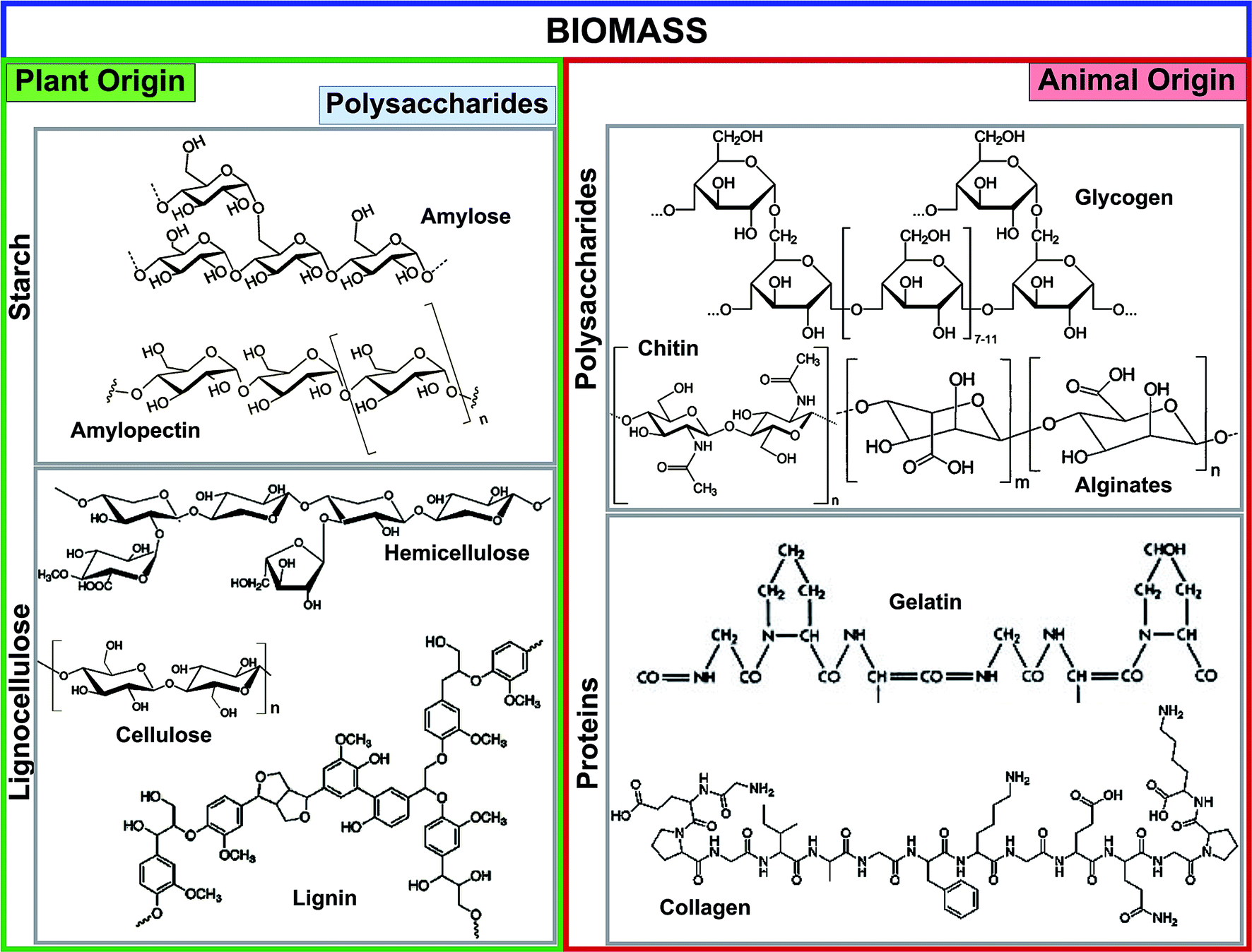 | ||
| Fig. 1 Prominent fractions of biomass feedstocks derived from plants and animals. | ||
Biochar can be used as an excellent platform for supporting various catalytic nanoparticles due to its unique surface properties, easily tunable functional groups, chemical stability, and electrical conductivity.22–24 The electron-conductive nature of biochar can reduce the quick recombination of the e−/h+ pair during photocatalysis25 and suitable surface functional groups enable immobilization of different pollutants, which is favorable for photocatalysis.26,27 An excellent review article published recently describes the TiO2-immobilized carbon from different renewable sources and introduces the application of biochar in the synthesis of photocatalysts as well.2 Consequently, various biochar-supported photocatalysts have been synthesized and reported in recent years. The reported BSPs show promising improvements in visible light sensitivity, chemical stability, recollection ability, and photocatalytic degradation of various pollutants. Some of those recently reported BSPs are TiO2–SPW, TiO2–MSP,28 TiO2–waste plum,29 TiO2–corn cob,25,30 TiO2–chitosan,31 TiO2–wheat husks,32 TiO2–bamboo,33 TiO2–reed straw,34 g-C3N4/FeVO4–pine needles,35 ZrO2–wheat husks,36 g-C3N4–chestnut,37 TiO2–wood charcoal,38 and BiOX–biochar.39
We believe that it is high time to give a comprehensive review on the recent advancements of biochar-supported photocatalysts, in particular, on their syntheses, applications, functionality, and future prospects based on environmental sustainability. Therefore this review provides major scientific insight into BSPs synthesized from low-cost biochar combined with catalytic nanoparticles, summarizes their syntheses and physicochemical properties, discusses in depth the role of biochar in BSPs driving photocatalysis, highlights their applications in various fields, and provides useful suggestions for the future development of efficient and novel BSPs.
2. BSP synthesis methods
2.1 Sol–gel
Sol–gel is the most common method for synthesizing BSPs. In TiO2-BSP, granular-shaped anatase and/or rutile TiO2 is agglomerated on the biochar surface via a sol–gel synthetic method.25 The crystal phase structure, average size, and dispersion of TiO2 upon the biochar depends on the sol–gel decomposition temperature. Usually, calcination temperatures below 700 °C can create anatase TiO2 crystals. The dispersion of TiO2 and changes in biochar structure increase with the calcination temperature.34 At high temperatures above 700 °C, TiO2 starts forming the rutile crystalline structure and the percentages of rutile increase with the rising temperature.40 BSP synthesis through sol–gel methods shows significant alteration of oxygen-containing functional groups due to their involvement in TiO2 doping with biochar. A typical sol–gel method follows three sequential formation steps: (1) prepare biochar templates via thermal decomposition of biomass, (2) increase surface oxides and reduce pH of biochar via acid treatment, then deposit catalytic nanoparticles on the biochar surface, and finally (3) calcine the obtained biochar-laden catalytic nanoparticles to give a stable structure.25,34,41 The schematic of the BSP synthetic process using the sol–gel method is illustrated in Fig. 2A. Recently, Zhang et al.34 synthesized an efficient TiO2–reed straw BSP using the sol–gel method for the photodegradation of sulfamethoxazole. They produced the biochar template by heating at a temperature of 500 °C for 6 h with a heating rate of 20 °C min−1. The obtained biochar was crushed and sieved through a 0.15 mm mesh, and rinsed with HCl acid (1 mol L−1) for 2.5 h to increase the number of surface oxides and reduce the pH at the point of zero charge. The acid-treated biochar was used as the precursor for TiO2 doping. Later, the precursor was immersed in 50 mL ethanol and 20 mL Ti(OBu)4 solution, and 2.5 mL acetic acid was added dropwise into that solution. The prepared mixture was stirred for 2 h at room temperature. Then, 2.5 mL of ultra-pure water (pH = 2) was added and stirring was continued for a further 4 h. After stirring the obtained mixture, it was dried in an oven at 105 °C. The dried sample of Ti(OBu)4-laden biomass was further calcined in a muffle furnace at different temperatures (300–500 °C) to obtain the TiO2–biochar.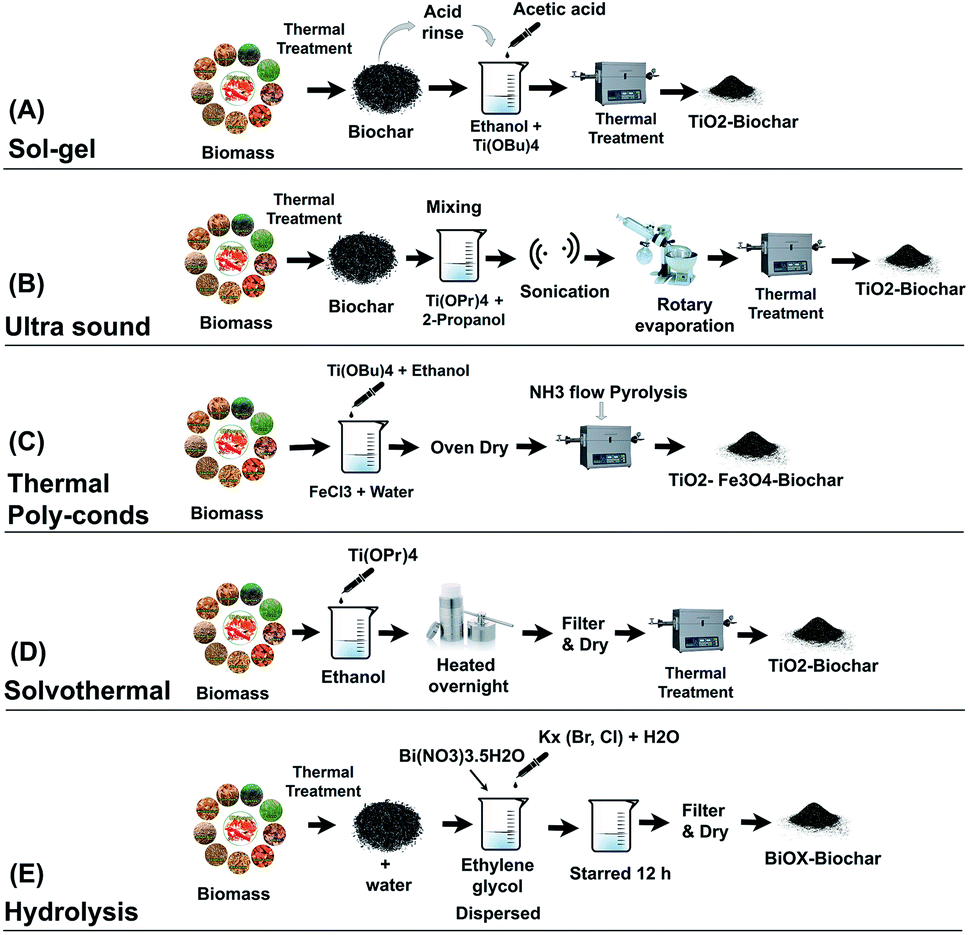 | ||
| Fig. 2 Schematic of BSP synthetic methods followed by interpretation of ref. 28, 31, 34 and 39. | ||
2.2 Ultrasound
Ultrasound (US) is a very new technique for the synthesis of BSPs. Recently, Lisowski et al.28 followed a multistep synthetic process with ultrasound mechanisms for the preparation of TiO2–softwood pellets and TiO2–miscanthus straw pellet biochar. The BSPs obtained contain enormous cracked structures on the surface that confirm the formation of a mesoporous structure. Using US treatment, anatase TiO2 crystal structures formed on the BSPs, and the growth of the TiO2 crystal size and structure is significantly higher than that of BSPs formed by a non-US method. A local trapping state due to Ti3+ below the TiO2 conduction band reduces the band gap of TiO2, and therefore the obtained material shows visible light sensitivity. US methods can act as interfacial mediators to improve the visible light response of the photocatalyst. The schematic of the multistep process with US mechanisms for the synthesis of BSPs is illustrated in Fig. 2B. Firstly, biochar was prepared through a thermal decomposition process at 500–700 °C. Then, the obtained biochar was processed for further treatment by crushing, sieving, washing, and drying. The clean and dry biochar (500 g) was mixed with a titanium(IV) isopropoxide and propanol solution, and that mixture was ultrasonicated for 1 h. After sonication, the materials were extracted using a rotary vacuum evaporator assisted by sonication. The sonicated material was dried and further calcined at 400 °C to prepare the TiO2–biochar.2.3 Thermal polycondensation
Thermal polycondensation is a simple and single-step heating synthesis of BSPs. Recently Liu et al.37 and our research group synthesized BSPs using a thermal polycondensation process. Liu et al. followed a multistep calcination process where biochar was obtained firstly from a thermal decomposition process. Then, the obtained biochar was blended with melamine in different ratios and ground. Later, the mixture of melamine–biochar was placed in a furnace and calcined at 300 °C for 2 h with a heating rate of 5 °C min−1 for the synthesis of BSPs. However, our research group produced an agar biochar-based catalyst (N–TiO2–Fe3O4–biochar) via a single-step thermal polycondensation process that is shown in Fig. 2C. The natural polysaccharide agar was used due to its thermally responsive sol–gel properties. At low temperatures (30–40 °C), agar formed a gel network, which is beneficial for doping with other elements.42,43 The nitrogen functional groups were activated on the catalyst via NH3 ambiance calcination. The obtained BSPs (N–TiO2–Fe3O4–biochar) had a significantly reduced band gap energy, and therefore, the material is able to perform excellent visible light photocatalysis for methylene blue degradation. It is well-known that the appropriate proportion of transition metal Fe- and N-doping with TiO2 nanoparticles can significantly reduce the band gap energy44 and improve visible light photocatalysis.7,45,46 Furthermore, the Fe3O4 of the composite can activate the magnetic properties of the BSPs, which is suitable for easy collection from the aqueous media.2.4 Solvothermal
TiO2-aggregated hollow spheres supported by carbon structures of furfural, saccharose, or chitosan can be prepared via a solvothermal method. Mainly anatase and some rutile TiO2 crystal structures formed on the composites through solvothermal synthesis. The crystal size of the sphere increased after calcination. The typical solvothermal synthesis follows several sequential steps (Fig. 2D). Firstly, catalytic nanoparticles such as TiO2 are deposited on the biomass templates via mixing them in an ethanol solution. For example, 1 g of carbon supporting materials such as furfural,47 chitosan, or saccharose31,48,49 and 1 g of titanium(IV) isopropoxide are dissolved in 18 mL of ethanol. Secondly, the prepared solution is sealed into a Teflon-lined stainless steel autoclave followed by solvothermal treatment at ∼175 °C overnight. The resultant solid after treatment is collected by filtration, washed several times, and dried. Finally, that dry sample is calcined at 600–800 °C for 6 h to remove the inner materials and form TiO2 hollow spheres. Composites synthesised by solvothermal methods show a reduction in the band gap energy and an improvement in visible light photocatalysis.2.5 Hydrolysis
Some researchers have reported the synthesis of BSPs using a simple hydrolysis process, and improvement in visible light sensitivity during photocatalysis of organic pollutants. Min et al.39 incorporated BiOX (X = Br or Cl) with biochar through a hydrolysis method for the degradation of methyl orange. The schematic for the hydrolysis process for the synthesis of BSPs is given in Fig. 2E. Firstly, 2 mmol Bi(NO3)3·5H2O was dispersed in 20 mL ethylene glycol at ambient temperature. Meanwhile, pre-prepared biochar was evenly dispersed into 20 mL pure water by sonication. Later, the biochar solution was added dropwise into the Bi(NO3)3·5H2O solution. Afterwards, a pre-prepared KX (Br, Cl) solution (2 mmol KX with 20 mL pure water) was poured slowly into the mixture. The prepared mixture was stirred for a further 12 h at ambient temperature. Finally, the solid residue of BiOX (Br, Cl)–biochar in the mixture was collected, washed, dried, and stored for further use. Except for the heating required for biochar production, further heating is not required for BSP synthesis by the hydrolysis method, which is energy-saving.3. Role of biochar in BSPs and photocatalysis
3.1 Supporting nanoparticles
Biochar can give an excellent platform for supporting a large number of nanoparticles. Simple thermochemically converted biochar contains available surface functional groups (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, C–O, COOH, COO–, OH etc.) and an enormous pore structure. The surface functional groups of biochar can easily be reacted with other functional materials. Moreover, some metallic and non-metallic mineral-containing biomass produced by thermal conversion shows the existence of N, S, P, K, Ca, and Mg content in biochar,50 which can directly act as an adsorbent, catalyst, or catalyst support.51 A recent review article by Tan et al.52 stated the general overview of different nanoparticle incorporation with biochar templates, their synthesis, and applications for environmental pollution decontamination. The tuning of biochar with different nanoparticles improves biochar functionality and pollutant removal competency via adsorption and/or catalytic degradation. Nanoparticles can combine with biochar in two ways: (1) thermal decomposition of nanoparticle-loaded biomass, and (2) tuning nanoparticles with prepared biochar functional groups through chemical modification. For example, Yang et al.53 synthesized Fe3O4-deposited biochar via a single step pyrolysis of FeCl3-laden sawdust biochar. Initially, the FeCl3-laden biochar hydrolyzed to form Fe(OH)3 and FeO(OH) during the drying process. After subjecting the FeCl3 biomass to pyrolysis, the biomass components such as lignocellulose decomposed quickly into volatile components and biochar. Meanwhile, FeO(OH) was reduced to form Fe3O4 by the reducing agents H2, CO, and amorphous carbon which were produced by the pyrolysis reaction. The advantages of metal-laden biomass pyrolysis are: a single step heating process, simultaneously producing nanoparticle-loaded biochar, and metal compounds which can have some catalytic effect on biochar pyrolysis which improves bio-oil and biochar textural properties.22
O, C–O, COOH, COO–, OH etc.) and an enormous pore structure. The surface functional groups of biochar can easily be reacted with other functional materials. Moreover, some metallic and non-metallic mineral-containing biomass produced by thermal conversion shows the existence of N, S, P, K, Ca, and Mg content in biochar,50 which can directly act as an adsorbent, catalyst, or catalyst support.51 A recent review article by Tan et al.52 stated the general overview of different nanoparticle incorporation with biochar templates, their synthesis, and applications for environmental pollution decontamination. The tuning of biochar with different nanoparticles improves biochar functionality and pollutant removal competency via adsorption and/or catalytic degradation. Nanoparticles can combine with biochar in two ways: (1) thermal decomposition of nanoparticle-loaded biomass, and (2) tuning nanoparticles with prepared biochar functional groups through chemical modification. For example, Yang et al.53 synthesized Fe3O4-deposited biochar via a single step pyrolysis of FeCl3-laden sawdust biochar. Initially, the FeCl3-laden biochar hydrolyzed to form Fe(OH)3 and FeO(OH) during the drying process. After subjecting the FeCl3 biomass to pyrolysis, the biomass components such as lignocellulose decomposed quickly into volatile components and biochar. Meanwhile, FeO(OH) was reduced to form Fe3O4 by the reducing agents H2, CO, and amorphous carbon which were produced by the pyrolysis reaction. The advantages of metal-laden biomass pyrolysis are: a single step heating process, simultaneously producing nanoparticle-loaded biochar, and metal compounds which can have some catalytic effect on biochar pyrolysis which improves bio-oil and biochar textural properties.22
In recent years, natural polysaccharides and their derivatives have received significant interest in the synthesis of biochar-based materials due to their availability, low toxicity, biocompatibility, biodegradability, and easy modifiability with various nanoparticles.54 In drug delivery systems, natural polysaccharides are commonly used for carrying nanoparticles.55 Among different natural polysaccharides, chitosan is the most widely used polysaccharide in different fields of research as well as for biochar production.56 The nanoparticles can incorporate with chitosan easily by beading, crosslinking, grafting, and surface impregnation.57 Chitosan is soluble in acidic water due to the protonation of NH2. Acid-soluble chitosan can turn back into a flock structure by adjusting the pH to 7.5–9 with NaOH due to deprotonation and insolubility of the polymer at natural pH.56 Using this mechanism, an excellent iron-based γ-Fe2O3 biochar has been synthesized from water hyacinths and applied for the removal of Cr(VI).58 Recently, our research group used agar for the synthesis of a N–TiO2–Fe3O4 doped photocatalyst. The gel matrix of agar at low temperature gives an excellent platform for supporting TiO2 and Fe3O4 nanoparticles. The enormous number of OH or NH functional groups in natural polysaccharides is easily tunable with the catalytic nanoparticles.
3.2 Increasing surface area and number of active sites
Usually, catalytic nanoparticles have a small surface area. When nanoparticles are dispersed on the biochar surface, the synthesized material (BSP) surface area can have an increased surface area compared to the bare nanoparticles. However, the overall surface area of biochar may decrease due to aggregation of metallic nanoparticles.37 Nanoparticles can homogeneously disperse on the biochar surface via suitable modification processes. Well-dispersed surface nanoparticles can improve light scattering and increase the number of active sites, which enhances photodegradation of organic pollutants. Lisowski et al.,28 Balu et al.,59 and Wang et al.60 stated that the larger surface area and adequate number of active sites of TiO2–biochar composites can lead to the materials exhibiting greater photocatalytic effects due to the presence of more photocatalytic surface-active site centers. Moreover, biochar templates can provide an aromatic carbon structure and different hydrophilic and hydrophobic surface functional groups that can favor adsorption of organic pollutants via π–π stacking, hydrogen bonding, and/or electrostatic interactions. The higher adsorption competencies of the catalyst can facilitate the photodegradation. Nawi et al.61 reported that the synergies of adsorption–photodegradation of aqueous phenol by TiO2–chitosan can increase the degradation competency and reduce charge carrier recombination. Similar mechanisms were also reported by Zhang et al.34 for photocatalytic degradation of sulfamethoxazole by TiO2–reed straw biochar. Biochar-supported TiO2 prevented e−/h+ recombination and increased sulfamethoxazole photodegradation from 58.47% (commercial TiO2) to 91.27%.3.3 Shuttling electrons
Some amorphous biochars have semiconductor characteristics, which helps to improve the reactivity when they are incorporated with nanoparticles via transferring electrons. A large number of studies have provided significant evidence about biochar electrical conductivity in different systems.62 Biochars facilitated the electron transformation from a bulk chemical electron donor to the receptor.63,64 Biochar surface redox-active moieties mediate the mechanisms of electron shuttling between different reactants. The condensed aromatic ring structure of biochar can promote the electron transfer process.62,63 Wu et al.65 stated that Fe3O4 nanoparticle-incorporated carbonaceous gel networks derived from watermelon biomass allow continuous and stable shuttling of electrons and electrolyte ions to the electrode surface. The excellent electrochemical behavior of Ag nanoparticle-covered waste biochar was also reported by Yao and Wu.66 In the TiO2–biochar or any BSP, the dense aromatic structure of biochar was able to shuttle electrons from the activation sites to the acceptors such as organic pollutants. This electron shuttling can reduce the quick recombination of e−/h+ pairs and enhance the photodegradation of pollutants.3.4 Electron reservoir
Biochar also can act as an electron reservoir. It is important to note that the electron storage and shuttling mechanisms of biochar are different processes. The electron shuttling process requires the conductive domain of black carbon (graphene) or the dense aromatic structure of biochar, while electron storage is the function of oxidation and reduction of biochar quinone moieties. A small number of studies have been conducted to investigate the electron reservoir potential of biochar. Klüpfel et al.67 reported that plant-based biochar has the capacity to store electrons up to 2 mmol e− per g. However, the electron storage capacity (ESC) of biochar depends on the types of biomass and the pyrolysis conditions. Biochar synthesized by intermediate to high heat temperatures shows a higher electron storage capacity via electron donation–acceptance. The electron donation and acceptance mechanisms by quinone moieties are illustrated in Fig. 3. Similar to humic acid, biochar has an enormous quinone content and can act as both electron donor and acceptor. Further, Saquing et al.68 found that wood-driven black carbon biochar has 0.85 and 0.87 mmol e− per g ESC in the acetate oxidation and nitrate reduction system. The electron sink capacity of biochar can be investigated by electron balance calculations. Saquing et al. showed that a portion of the electrons were missing during ethanol oxidation by Geobacter metallireducens, indicating the electron sink behaviour by the biochar. Xu et al.69 synthesized TiO2–carbon nanotubes (CNTs) and stated that CNTs can act as electron reservoirs during the photodegradation of benzene and methyl orange. In TiO2–biochar composites, carbon can act as an electron reservoir that conducts away e− from the e−/h+ pair and activated TiO2 during photocatalysis. Acting as an electron reservoir increases the efficiency of charge separation from TiO2 and hinders the e−/h+ pair recombination.2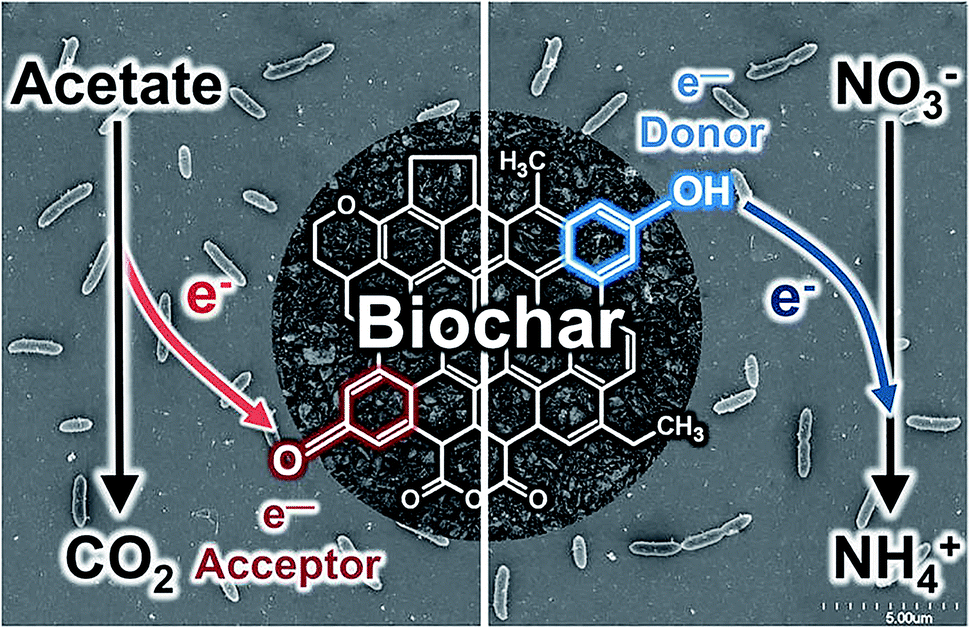 | ||
| Fig. 3 Electron storage mechanisms of biochar via quinone moiety electron acceptance and donation. Reproduced with permission from ref. 68. | ||
3.5 Improving charge separation
The charge separation due to the e−/h+ pair is one of the important aspects of photocatalysis. Usually, bare semiconductor photocatalysts are activated by charge separation after light irradiation. However, they could recombine sooner with each other and achieve deactivation. In that case, a secondary component may maintain or even strengthen the activated state of the semiconductor photocatalyst, shown in reactions (R1)–(R4) following the interpretation of Shi.70| P → P* | (R1) |
| P* → P | (R2) |
| P–C → P*–C | (R3) |
The single catalyst is activated after light irradiation (R1), while without any secondary element, the activated catalyst achieves deactivation again (R2). However, after a secondary element is incorporated into the catalyst, the activated state of the catalyst is well-maintained. Combination leads to activation of the catalyst (P) by other catalysts or non-catalysts (C) (R3). This accelerates the catalytic kinetic reaction and in some special cases both components can be mutually activated (R4).
| P + C → P*/C* | (R4) |
In the BSP, electron shuttling from the catalytic nanoparticle to carbon significantly improves charge separation. The charge separation competency of the photocatalyst can be detected by photoluminescence (PL) spectroscopy and the photocurrent response. The lower PL spectrum intensity and the higher photocurrent density indicates the higher efficiency of the electron–hole pair separation.28,39 Lisowski et al.28 synthesized TiO2–biochar through a simple ultrasound sonication process and indicated that the charge separation significantly increases when biochar is incorporated with TiO2. The formation of heterojunctions between the TiO2 and biochar templates is responsible for the photoinduced charge separation. The similar increase in charge separation due to Fe and FeVO4 heterojunctions in the g-C3N4–FeVO4–Fe@NH2–biochar were reported by Kumar et al.35 Further, Zhang et al. synthesized TiO2@SiO2 wrapped in a GO layer and state that the thin GO layer improves the charge separation. However, the thick layer of GO negatively affects the charge separation due to adsorbance or scattering of photon energy, and therefore the photocatalytic activity decreases. The similar masking effects of a thick carbon layer on photocatalysis also reported by Wu et al.33
3.6 Reducing band gap energy
The band gap energy of semiconductor photocatalysts can be reduced by combining them with biochar. There are three major ways that have been reported in the literature for reducing the TiO2–BSP band gap: (1) sensitize the photocatalytic nanoparticle with another compound, (2) create a mid-gap energy state via doping non-metals such as N, S, C, O etc., and (3) formation of a local trapping state below the TiO2 conduction band. Usually, composites with small band gaps are used to sensitize the larger band gap semiconductor photocatalyst (Fig. 4a). Carbon can act as a sensitizer when doping with TiO2. The larger band gap of TiO2 can be sensitized by carbon doping,71 and therefore, the synthesized composite light sensitivity shifts from the UV region to the visible light region. For efficient sensitization, the conduction band of the sensitizer material should be placed higher than the conduction band of TiO2.72 Another important aspect of the TiO2–BSP is the energy level of any impurities which form Ti–O–C bonds.73,74 This can create a delocalized state in the band gap of TiO2 without promoting charge carrier recombination.7 Furthermore, the incorporation of non-metals N or S with the TiO2–biochar significantly reduces the band gap of the composite by forming a mid-gap energy state. The excellent N- and S-doped biochar-based TiO2 semiconductor photocatalyst has been synthesized by Matos et al.29 and Bandosz et al.75 Due to N- and S-doping, the photocatalytic activity of the TiO2-composite increases up to 2 and 5 times more than the bare TiO2 photocatalyst. However, the photocatalytic activity of the composites is largely influenced by the texture and the content of S and N in the BSP.29,75 N- or S-doped BSPs not only reduce the energy gap of TiO2 but also act as electron shuttles from the π-orbital of the N/S-doped biochar to the conduction band of TiO2. The general procedures for reducing the band gap energy level by C-, substitutional N-, and interstitial N-doping in TiO2–biochar are illustrated in Fig. 4b. An excellent review article by Ansari et al.76 describes in detail the reduction of the band gap by N-doping semiconductor photocatalysts. The local trapping state below TiO2 can also be produced in TiO2–biochar as shown in Fig. 4c. Lisowski et al.28 reported that a TiO2–biochar synthesized by an ultrasound method contained a local trapping state corresponding to Ti3+, which reduced the band gap of the BSP as well as enhancing the photocatalytic redox reaction.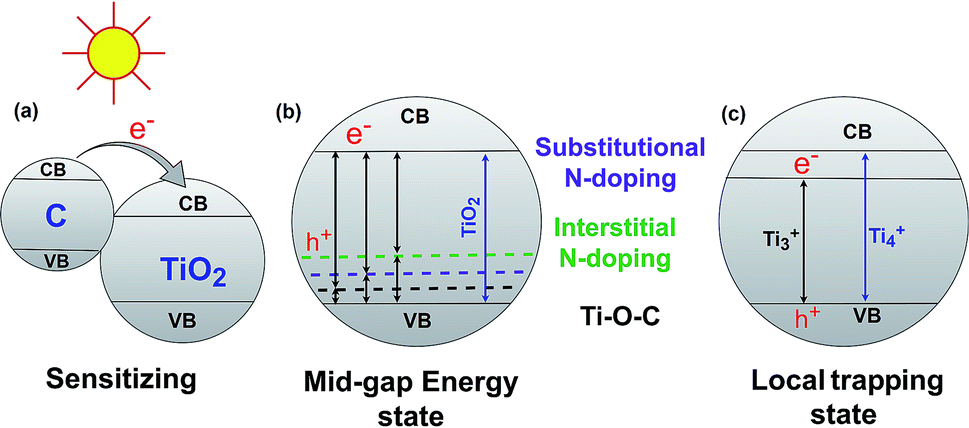 | ||
| Fig. 4 Reduction of the TiO2–BSP band gap energy via sensitizing (a), forming a mid-gap energy state (b) and forming a local trapping state (c). | ||
4. Applications of BSPs
Many excellent new technologies can be understood via applications of BSPs, extending the existing wide range of photocatalytic nanoparticle applications. The most prominent application of BSPs is in the degradation of various hazardous organic pollutants in air and waterways. Furthermore, these BSPs can also be applied as film electrodes in solar cells, and as photocatalytic H2 generators in water splitting. The list of BSPs which have been synthesized and their applications in various fields are summarized in Table 1. Organic pollutants whose photodegradation by BSPs have been published at least once include phenol,28,77 bisphenol A,38 methylene blue,29,33,37,75 safranin T,41 anthraquinone,32 malic acid,59 sulfamethoxazole,25,30,34 methyl paraben,35 2-chlorophenol,35 Reactive Yellow 39,36 methyl orange39 in the liquid phase, and methanol in the gas phase.28 Among the listed pollutants, methylene blue is the most commonly studied pollutant followed by sulfamethoxazole and phenol. The photodegradation of methylene blue by various BSPs ranges from 38% of 4.8 ppm to 100% of 400 ppm, and the degradation time varies from 1.5 h to 4 h. Phenol degradation capacity ranges from 64% to 75% of 50 ppm within 4 h, and sulfamethoxazole degradation capacity is ∼90% of 10 ppm within 3 h. The intermediate product analysis during photodegradation can give a clear understanding of the degradation pathway and evidence of mineralization. For example, the quantification of malic acid and methanol mineralization in water and air media by TiO2–guanidine–(Ni,Co)–Fe2O4 (ref. 59) and TiO2–SWP700,28 respectively give a clear understanding of the pollutant photodegradation process. It shows that the overall conversion rate of malic acid and methanol was 60% and 88%, respectively. Similarly, methyl paraben, 2-chlorophenol, and sulfamethoxazole photodegradation pathways were investigated by Kumar et al.35 and Zhang et al.,34 respectively. Biochar-supported photocatalysts are also applicable for the degradation of pollutants in the gaseous phase. Lisowski et al.28 passed a mixture of methanol and air (0.9% + 99.1% vol) at a flow rate of 25 cm3 min−1 through an author-designed BSP-containing photo-reactor. The reaction output products were measured online by gas chromatography and offline by GC-MS under conditions both with and without UV light illumination. The UV illumination and presence of TiO2–SWP700 significantly mineralized methanol into CO2 and methyl formate. Besides, organic pollutant degradation, BSPs also show good performance as film electrodes in solar cells and as H2 producers in water splitting. Matos et al.31 synthesized TiO2–C hollow spheres via a solvothermal and post-calcination process of TiO2–chitosan/furfural/saccharose precursors. The obtained BSPs were successfully applied as film electrodes in a solar cell under visible light catalysis. Furthermore, Matos et al. synthesized78 Au–TiO2 biochar through multi-step heating and slurry methods and applied them for H2 production from water. The author states that the biochar-supported Au–TiO2 performs 3 times better photocatalysis than the commercial semiconductor photocatalyst and the O functional groups of biochar enhance the photocatalytic H2 production. It also worth mentioning that BSPs possess high recyclability during organic pollutant photodegradation. In most cases, their organic removal capacity decreased only 2–10% after recycling the BSPs 3–6 times (Table 1), which indicates the higher stability, recyclability and cost effectiveness of the composites.| Name of catalyst | Biomass | Metal | Application | Initial pollutant concentration | Removal capacity (Re) and recycle capacity (Rc) | Time | Process of removal | BSP synthetic process | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| TiO2–biomass | Starbon | Ti | Phenol photodegradation | 150 mL – 50 ppm | Re: 75% | 4 h | Adsorption and photodegradation | Ultrasound sonication | 77 |
| TiO2–SWP700 | Soft wood pellets | Ti | Phenol photodegradation | 150 mL – 50 ppm | Re: 64% | 4 h | Adsorption and UV + Vis light irradiation | Ultrasound sonication | 28 |
| Rc: (∼59–67%)/5 times | |||||||||
| TiO2–wood charcoal | Pine wood | Ti | Bisphenol A photodegradation | 50 mL – 20 ppm | Re: 80% | 18 h | Adsorption and photodegradation | Dip-sol-gel method | 38 |
| N–TiO2–C | Waste plum stones | Ti | Methylene blue photodegradation | 125 mL – 25 ppm | Re: 100% | 3 h | Adsorption and photodegradation | Multi-step pyrolysis and slurry method | 29 |
| TiO2–bamboo | Bamboo | Ti | Methylene blue photodegradation | 200 mL – 30 ppm | Re: 99% | 1.5 h | Adsorption and UV + Vis light irradiation | Hydrothermal carbonization and sol–gel methods | 33 |
| Rc: ∼100%/4 times | |||||||||
| g-C3N4–biochar | Chestnut leaf biomass | Methylene blue photodegradation | 20 mL – 4.8 ppm | Re: 38% | 4 h | Photodegradation | Multi-step thermal polycondensation | 37 | |
| S-activated carbons | Activated carbon | Methylene blue photodegradation | 125 mL – 25 ppm | Re: 100% | 4 h | Adsorption and photodegradation | Multi-step heating process | 75 | |
| N–TiO2–Fe3O4–biochar | Agar | Fe, Ti | Methylene blue photodegradation | 10 mL – 400 ppm | Re: 100% | 3 h | Adsorption, photodegradation, and Fenton-like degradation | Single-step thermal polycondensation | Our study |
| Rc: (89–100%)/5 times | |||||||||
| TiO2-coated biochar | Ramie bar | Ti | Safranin T photodegradation | Re: 231.9 mg g−1 | 2 h | Adsorption and photodegradation | Sol–gel method | 41 | |
| Rc: (167–222 mg g−1)/6 times | |||||||||
| TiO2–sludge and wheat husks | Sludge and wheat husks | Ti | Reactive Blue 69 photodegradation | 20 ppm/1.5 g L−1 dose | 98% | 1.3 h | Ultrasound irradiation | Sol–gel method | 32 |
| TiO2–guanidine–(Ni,Co)–Fe2O4 | Biomass | Ti, Ni, Co, Fe | Malic acid photodegradation | 0.5 mmol/50 mL water | Re: conversion = 60%, acetic acid = 10%, formic acid = 77%, oxalic acid = 7%, CO2 = 8% | 1.5 h | Photodegradation | Hydrothermal process | 59 |
| TiO2–corn cob | Corn cob | Ti | Sulfamethoxazole photodegradation | 100 mL – 10 ppm | Re: 90% | 3 h | Adsorption and UV light irradiation | Sol–gel method | 25 |
| Rc: (90–92%)/3 times | |||||||||
| TiO2–reed straw | Reed straw | Ti | Sulfamethoxazole photodegradation | 160 mL – 10 ppm | Re: 91% | 3 h | Adsorption and UV light irradiation | Sol–gel method | 34 |
| Rc: (86–91%)/5 times | |||||||||
| g-C3N4–FeVO4–Fe@NH2–biochar | Pine needles | Fe, V | Methyl paraben (MeP) and 2-chlorophenol (2-CP) photodegradation | 100 mL – 20 ppm | Re: 98.4% of MeP | 1.5 h | Adsorption, photocatalysis, and photo-ozonation | Multi-step thermal treatment, acid treatment, and ammonia treatment | 35 |
| Rc: (97–98%)/6 times | |||||||||
| Re: 90.7% of 2-CP | |||||||||
| Rc: (89–91%)/6 times | |||||||||
| ZrO2–sludge and wheat husks | Sludge and wheat husks | Zr | Reactive Yellow 39 photodegradation | 20 ppm/1.5 g L−1 | Re: 98% | 1.2 h | Ultrasound irradiation | Modified sonochemical and sol–gel method | 36 |
| BiOX (X = Cl or Br)–biochar | Bi | Methyl orange photodegradation | 50 mL (0.03 mM) | Re: 10% BiOBr = 81% | 2.5 | Photodegradation | Hydrolysis method | 39 | |
| Re: 5% BiOCl = 38% | |||||||||
| TiO2–SWP700 | Soft wood pellets | Ti | Methanol oxidation | (0.9% + 99.1%) phenol + air-flow rate of 25 cm3 min−1 | Re: conversion = 88%, CO2 = 20%, methyl formate yield = 88% | UV + Vis light irradiation | Ultrasound sonication | 28 | |
| TiO2–chitosan | Chitosan | Ti | Film electrodes | Solvothermal | 31 | ||||
| Au–TiO2/AC | Waste plum stones | Au, Ti | H2 production | −22.5 mM at visible light, and −33 at UV light | UV light irradiation | Multi-step heating and slurry method | 78 |
5. BSP-driven photodegradation mechanisms
5.1 Adsorption
Incorporating catalytic nanoparticles with biochar can increase the BSP affinity for adsorbing a variety of organic pollutants in an aqueous medium. A higher adsorption potentiality of BSPs enhances photodegradation of organic pollutants to a greater extent than bare nanoparticles.33 Colmenares et al.77 reported that similar Ti-doping with different biochar components significantly varies their pollutant removal competency. As shown in Fig. 5, 15Ti/GO, 15 Ti/Norit, and 15Ti/Starbon exhibit different phenol reduction competencies, even though a similar synthetic process and amount of Ti has been applied in their syntheses. The higher phenol reduction competency shown by 15Ti/Starbon is due to its higher adsorption competency, its hybrid interphase proximity, and the presence of a highly-crystalline anatase phase.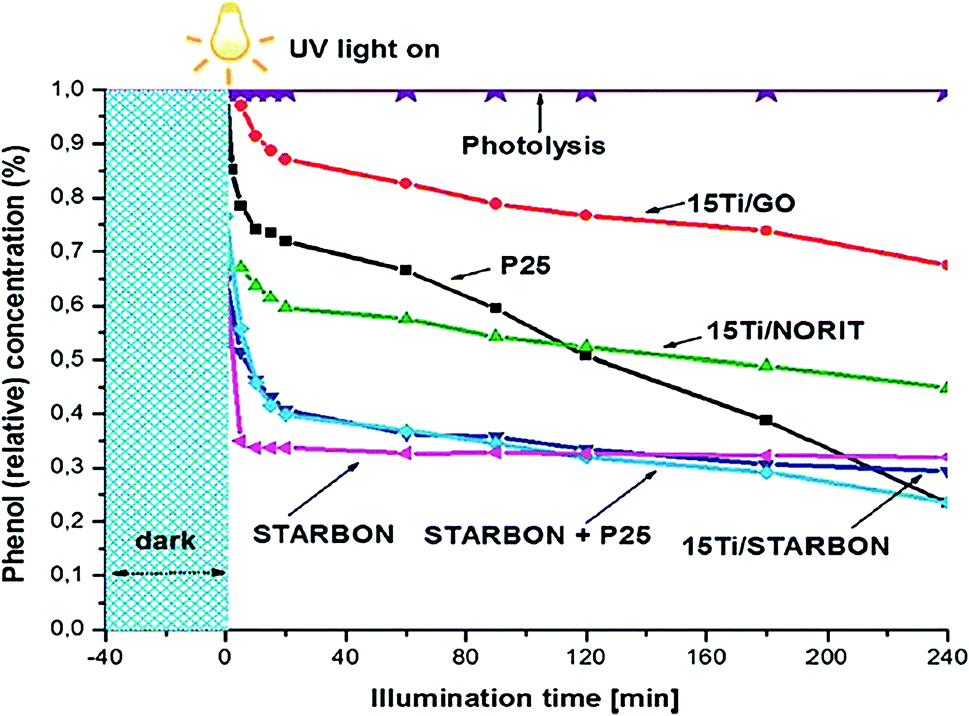 | ||
| Fig. 5 Photocatalytic degradation of phenol by different photocatalysts. Reproduced with permission from ref. 77. | ||
A similar example of higher photodegradation due to higher adsorption capacity of the composite was reported by Luo et al.38 The commercial P25 and author-synthesized TiO2–WC-450 exhibits a similar ratio of anatase and rutile TiO2 (80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20). However, TiO2–WC-450 exhibits 1.85 times higher removal of bisphenol A than P25, which was due to higher adsorption and mass transfer competency of biochar-supported TiO2. The biochar contents in the photocatalyst play a key role in the adsorption as well as in photodegradation. Due to surface adsorption capabilities, organic pollutants can deposit on the catalyst surface before transfer to the decomposition center. Therefore, the reactive oxygen species generated due to light illumination do not need to migrate so far from the activation site. However, some fractions of organic pollutants are adsorbed on the catalyst without coming into contact with TiO2. Those particles also undergo attack by ROS produced on the surface of TiO2. It is assumed that the ROS could diffuse over a sub-millimeter distance from the activation site to the reactant site located on the surface of the composites.28 The synergistic effect of adsorption and photodegradation give rise to an efficient photodegradation process.34,60 Kumar et al.35 reported that the simultaneous adsorption–photodegradation allow high and rapid degradation of methyl paraben (MeP) and 2-chlorophenol (2-CP) due to the immediate attack of ROS after being adsorbed onto the photocatalyst. However, adsorption followed by photodegradation reduces the degradation competency due to covering most of the surface active sites with the pollutants.35
:20). However, TiO2–WC-450 exhibits 1.85 times higher removal of bisphenol A than P25, which was due to higher adsorption and mass transfer competency of biochar-supported TiO2. The biochar contents in the photocatalyst play a key role in the adsorption as well as in photodegradation. Due to surface adsorption capabilities, organic pollutants can deposit on the catalyst surface before transfer to the decomposition center. Therefore, the reactive oxygen species generated due to light illumination do not need to migrate so far from the activation site. However, some fractions of organic pollutants are adsorbed on the catalyst without coming into contact with TiO2. Those particles also undergo attack by ROS produced on the surface of TiO2. It is assumed that the ROS could diffuse over a sub-millimeter distance from the activation site to the reactant site located on the surface of the composites.28 The synergistic effect of adsorption and photodegradation give rise to an efficient photodegradation process.34,60 Kumar et al.35 reported that the simultaneous adsorption–photodegradation allow high and rapid degradation of methyl paraben (MeP) and 2-chlorophenol (2-CP) due to the immediate attack of ROS after being adsorbed onto the photocatalyst. However, adsorption followed by photodegradation reduces the degradation competency due to covering most of the surface active sites with the pollutants.35
5.2 Photodegradation
The photodegradation of organic pollutants using BSPs is the function of reactive oxygen species such as ˙OH and O2˙−, and h+. Identification of specific ROS functions during photocatalysis is essential to understand the photocatalytic redox reaction.79 Generally, the photocatalyst adsorbed a photon with an energy which is equal to or less than the band gap, and excited the electron in the valence band to the conduction band. Therefore, an electron–hole is created in the upper edge of the valence band and an electron occupies a phase on the lower edge of the conduction band. Later on, the h+ and e− can either recombine or diffuse to the acceptor. The e(CB)− and h(VB)+ can scavenge the –OH, O2, and H2O molecules in the liquid phase and successively produce ˙OH and O2˙− species, which further react with organic pollutants leading to complete mineralization. The fundamentals of organic pollutant photodegradation by ROS are illustrated in Fig. 6. The role of specific ROS in various pollutant photodegradation using various BSPs is different. Recently, many researchers have been investigating the mechanisms of organic pollutant photodegradation using specific ROS identification and degradation product analysis. In most cases, ROS scavenging by different inhibitors and mass spectroscopic methods have been used.33,80,81 A comprehensive list of different ROS-function identification processes for organic pollutant photodegradation by BSPs is summarized in Table 2. Scavenger benzoquinone and TEMPOL for O2˙−, ammonium oxalate, triethanolamine, and EDTA 2Na for h+, and tert-butyl alcohol and isopropanol for ˙OH identification are very popular in ROS scavenging experiments. As shown in Table 2, the organic pollutants were degraded by the action of multiple ROS. The ranking was made based on specific ROS pollutant removal competencies determined by scavenging experiments. An excellent review article has recently been published with extensive discussion about the generation and detection process of ROS in photocatalysts.79 According to Nosaka and Nosaka, MCLA chemiluminescence, lucigenin chemiluminescence, 1270 nm emission and coumarin fluorescence probing are the most recommended methods for the detection of O2˙−, H2O2, O2, and ˙OH.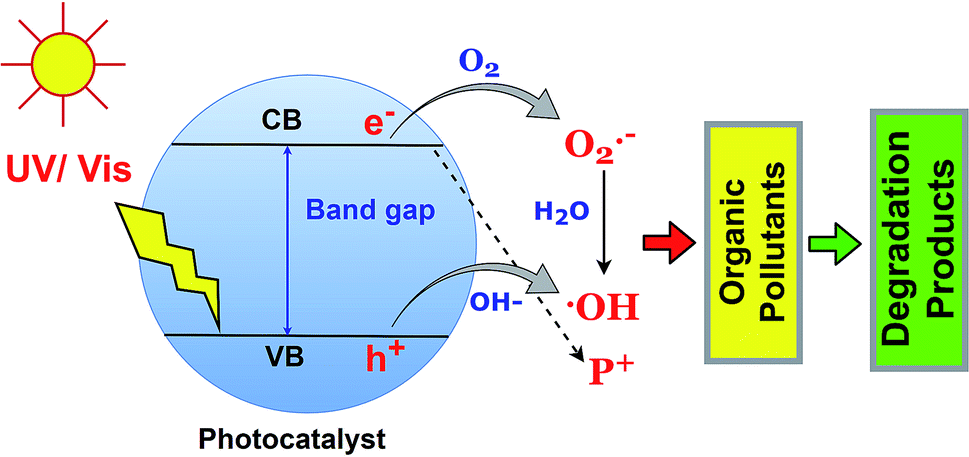 | ||
| Fig. 6 Fundamentals of organic pollutant photodegradation by ROS. | ||
| Name of catalyst | Pollutants | ROS responsible for degradation | ROS function identification process | Ref. |
|---|---|---|---|---|
| TiO2–SWP700 | Phenol | All | Scavenged by benzoquinone for O2˙−, ammonium oxalate for h+, and tert-butylalcohol for ˙OH | 28 |
| Ranking: h+ > O2˙− > ˙OH | ||||
| TiO2–bamboo | Methylene blue | All | Scavenged by benzoquinone for O2˙−, ammonium oxalate for h+, and tert-butylalcohol for ˙OH | 33 |
| Ranking: O2˙− > ˙OH > h+ | ||||
| g-C3N4–biochar | Methylene blue | O2˙− and h+ | Scavenged by benzoquinone for O2˙−, triethanolamine for h+, tert-butylalcohol for ˙OH, and sodium azide for O2 | 37 |
| Ranking: O2˙− > h+ | ||||
| BiOX (X = Cl or Br)–biochar | Methyl orange | All | Scavenged by benzoquinone for O2˙−, ethylene diaminetetraacetic acid disodium salt (EDTA 2Na) for h+, and isopropanol for ˙OH | 39 |
| Ranking: O2˙− > h+ > ˙OH | ||||
| g-C3N4–FeVO4–Fe@NH2–biochar | Methyl paraben (MeP) and 2-chlorophenol (2-CP) | All | Scavenged by 4-hydroxy-2,2,6,6-tetramethylpiperidinyloxy (TEMPOL) for O2˙−, ethylene diaminetetraacetic acid disodium salt (EDTA 2Na) for h+, and tert-butylalcohol for ˙OH | 35 |
| Ranking: ˙OH > O2˙− > h+ | ||||
| TiO2–reed straw | Sulfamethoxazole | ˙OH and O2˙− | HPLC-PDA chromatograms | 34 |
5.3 Ozonization
Ozonization during photocatalytic degradation of organic pollutants by BSPs influences reaction rate and speed. The combined mechanisms of adsorption, photodegradation, and ozonization enhance organic pollutant removal competencies to a greater extent.35 During the combined treatment, ozone leads to the production of ˙OH and H2O2 due to exposure to photon energy. Meanwhile, the electron which is unable to recombine due to the heterojunction in the BSP, substantially increases the production of O2˙− and ˙OH by reaction with O2 and H2O2. Therefore, the reaction rate between ROS and pollutants increases, which results in a rapid and high reduction of pollutants. Furthermore, Fe-doped BSPs can give Fenton-like reaction competency. Our synthesized biochar-supported photocatalyst (N–TiO2–Fe3O4–biochar) exhibited very high methylene blue reduction under visible light irradiation due to injecting H2O2 in the reaction system. Similarly, graphene oxide-encapsulated Fe3O4–TiO2 spheres synthesized by Yang et al.,8 shows good Fenton-like degradation competency of methylene blue under visible light irradiation as a result of rapid redox reactions between Fe2+ and Fe3+.Finally, it is also required to mention that the dose of organic pollutants and catalyst has a significant influence on the photodegradation process. Unlike adsorption of pollutants, photodegradation and simultaneous adsorption–photodegradation do not proportionally increase with the concentration of catalyst dose. Therefore, a specific dose of catalyst or pollutants may require determining to achieve optimum pollutant degradation. High concentrations of pollutants can prevent the light being able to reach the reactant photocatalyst. Therefore, the production of ROS can be reduced. Similarly, the dose of a catalyst has an optimum limit to reach maximum photocatalysis. An excessive photocatalyst dose can cause light scattering, which hinders the specific activity of the photocatalyst.82,83 Moreover, the number of active sites on the reactants can be reduced due to agglomeration of homogeneous particles at a high dosage.84 Therefore, an optimum dose of BSPs and pollutants should be measured to achieve optimum performance of the photocatalyst.
6. Conclusion and outlook
This review gives a detailed overview of the recent progress in the field of BSPs, summarized their synthetic processes and physicochemical characteristics, explicitly discussed the biochar role in the BSP, and pollutant photodegradation. It has revealed that the incorporation of transition metals/catalytic nanoparticles and/or non-metals with biochar templates are effective techniques to produce high-performance semiconductor photocatalysts. The catalytic nanoparticles can easily be incorporated with biochar through sol–gel, solvothermal, ultrasound, thermal polycondensation, and hydrolysis methods. The photocatalytic performance of BSPs significantly increases due to the unique physicochemical properties of biochar. Biochar can support a large number of nanoparticles, increase the number of active sites on the photocatalyst, act as an electron reservoir which conducts away the electron from the e−/h+, shuttle electrons through a graphene-like skeleton, improve charge separation, and reduce band gap energy by carbon or other non-metal doping with catalytic nanoparticles. Unlike bare catalytic nanoparticles, BSP adsorption capacity during photocatalysis enhances photodegradation performance. Simultaneous adsorption and photodegradation perform rapid and higher organic pollutant degradation due to the immediate attack of nearby ROS. Moreover, BSPs are more convenient to recover from aqueous solution than bare CNPs. Up until now, BSPs have mainly been applied for the photodegradation of organic pollutants in the aqueous phase. However, only a small portion of research focuses on BSP applications for clean energy production, air pollutant treatment, and anti-bacterial sterilization. It has already been proven that BSPs are an effective material for photocatalytic applications due to their cost-effectiveness, sustainability, and higher visible light photoactivity. However, there are still a number of aspects that need to be considered for the future development of BSPs.• Prior to the synthesis of BSPs, it is worth considering the cost-effectiveness and the availability of natural biomass, a facile synthetic process to incorporate catalytic nanoparticles, and an efficient modification technique to enhance optical properties in the visible light region.
• Applying a wide variety of biomass and nanoparticles in the synthesis of BSPs and examining the synergistic effects of biochar and catalytic nanoparticle incorporation.
• The key controlling factors that stimulate BSP performance characteristics should be investigated such as heating operating conditions, selection and ratio of biomass and nanoparticles, and incorporation processes.
• Various hazardous waste materials such as sewage sludge, manure, and various organic and inorganic municipal waste materials can be used in the synthesis of BSPs. The waste material contains various catalytic nanoparticles that could improve the BSP photoresponse.85 Furthermore, natural polysaccharides have unique physicochemical properties such as formation of gels, flocking, and complete dissolution in water, which are beneficial to the incorporation of various compounds. This is a novel, sustainable and environmentally friendly way to manage municipal waste.
• Introduction of BSPs in many other photocatalytic applications and investigation of the photocatalysis mechanisms and performance.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Basic Research Program of China (973 Program, 2014CB238903) and the National Natural Science Foundation of China (No. 41672144, 41173032, and 41373110). We thank the editors and anonymous reviewers for giving us many constructive comments that significantly improved the paper.References
- WWAP (United Nations World Water Assessment Programme), The United Nations World Water Development Report 2017: Wastewater, The Untapped Resource, UNESCO, Paris, 2017 Search PubMed.
- J. C. Colmenares, R. S. Varma and P. Lisowski, Green Chem., 2016, 18, 5736–5750 RSC.
- G. Jayanthi Kalaivani and S. K. Suja, Carbohydr. Polym., 2016, 143, 51–60 CrossRef CAS PubMed.
- J. Schneider, M. Matsuoka, M. Takeuchi, J. Zhang, Y. Horiuchi, M. Anpo and D. W. Bahnemann, Chem. Rev., 2014, 114, 9919–9986 CrossRef CAS PubMed.
- Y. Yamada, S. Shikano and S. Fukuzumi, J. Phys. Chem. C, 2013, 117, 13143–13152 CAS.
- M. T. Uddin, O. Babot, L. Thomas, C. Olivier, M. Redaelli, M. D’Arienzo, F. Morazzoni, W. Jaegermann, N. Rockstroh, H. Junge and T. Toupance, J. Phys. Chem. C, 2015, 119, 7006–7015 CAS.
- J. Chung, J. W. Chung and S.-Y. Kwak, Phys. Chem. Chem. Phys., 2015, 17, 17279–17287 RSC.
- X. Yang, W. Chen, J. Huang, Y. Zhou, Y. Zhu and C. Li, Sci. Rep., 2015, 5, 10632 CrossRef PubMed.
- P. Kumar, C. Joshi, N. Labhsetwar, R. Boukherroub and S. L. Jain, Nanoscale, 2015, 7, 15258–15267 RSC.
- A. Gołąbiewska, W. Lisowski, M. Jarek, G. Nowaczyk, A. Zielińska-Jurek and A. Zaleska, Appl. Surf. Sci., 2014, 317, 1131–1142 CrossRef.
- M. Cui, S. Pan, Z. Tang, X. Chen, X. Qiao and Q. Xu, Chem. Speciation Bioavailability, 2017, 29, 60–69 CrossRef CAS.
- P. Wang, P.-S. Yap and T.-T. Lim, Appl. Catal., A, 2011, 399, 252–261 CrossRef CAS.
- K. Kalantari, M. Kalbasi, M. Sohrabi and S. J. Royaee, Ceram. Int., 2016, 42, 14834–14842 CrossRef CAS.
- H. Tian, J. Ma, K. Li and J. Li, Ceram. Int., 2009, 35, 1289–1292 CrossRef CAS.
- I. Delidovich, K. Leonhard and R. Palkovits, Energy Environ. Sci., 2014, 7, 2803–2830 CAS.
- S.-H. Kong, S.-K. Loh, R. T. Bachmann, S. A. Rahim and J. Salimon, Renewable Sustainable Energy Rev., 2014, 39, 729–739 CrossRef CAS.
- J. Han, X. Wang, J. Yue, S. Gao and G. Xu, Fuel Process. Technol., 2014, 122, 98–106 CrossRef CAS.
- E. Gregorová, W. Pabst and I. Bohačenko, J. Eur. Ceram. Soc., 2006, 26, 1301–1309 CrossRef.
- C. D. Tran, S. Duri, A. Delneri and M. Franko, J. Hazard. Mater., 2013, 252–253, 355–366 CrossRef CAS PubMed.
- D. H. K. Reddy and S.-M. Lee, Adv. Colloid Interface Sci., 2013, 201–202, 68–93 CrossRef CAS PubMed.
- S. Mallick, S. Sharma, M. Banerjee, S. S. Ghosh, A. Chattopadhyay and A. Paul, ACS Appl. Mater. Interfaces, 2012, 4, 1313–1323 CAS.
- W.-J. Liu, H. Jiang and H.-Q. Yu, Chem. Rev., 2015, 115, 12251–12285 CrossRef CAS PubMed.
- Y. Xia and R. C. Larock, Green Chem., 2010, 12, 1893–1909 RSC.
- S. Dutta, A. Bhaumik and K. C.-W. Wu, Energy Environ. Sci., 2014, 7, 3574–3592 CAS.
- J. R. Kim and E. Kan, J. Environ. Manage., 2016, 180, 94–101 CrossRef CAS PubMed.
- M. Dahl, Y. Liu and Y. Yin, Chem. Rev., 2014, 114, 9853–9889 CrossRef CAS PubMed.
- H.-P. Jing, C.-C. Wang, Y.-W. Zhang, P. Wang and R. Li, RSC Adv., 2014, 4, 54454–54462 RSC.
- P. Lisowski, J. C. Colmenares, O. Mašek, W. Lisowski, D. Lisovytskiy, A. Kamińska and D. Łomot, ACS Sustainable Chem. Eng., 2017, 5, 6274–6287 CrossRef CAS.
- J. Matos, M. Hofman and R. Pietrzak, Carbon, 2013, 54, 460–471 CrossRef CAS.
- Q. Liu, F. Yang, X. Sun, Z. Liu and G. Li, J. Mater. Cycles Waste Manage., 2017, 19, 134–143 CrossRef CAS.
- J. Matos, P. Atienzar, H. García and J. C. Hernández-Garrido, Carbon, 2013, 53, 169–181 CrossRef CAS.
- A. Khataee, B. Kayan, P. Gholami, D. Kalderis and S. Akay, Ultrason. Sonochem., 2017, 39, 120–128 CrossRef CAS PubMed.
- F. Wu, W. Liu, J. Qiu, J. Li, W. Zhou, Y. Fang, S. Zhang and X. Li, Appl. Surf. Sci., 2015, 358, 425–435 CrossRef CAS.
- H. Zhang, Z. Wang, R. Li, J. Guo, Y. Li, J. Zhu and X. Xie, Chemosphere, 2017, 185, 351–360 CrossRef CAS PubMed.
- A. Kumar, A. Kumar, G. Sharma, M. Naushad, F. J. Stadler, A. A. Ghfar, P. Dhiman and R. V. Saini, J. Cleaner Prod., 2017, 165, 431–451 CrossRef CAS.
- A. Khataee, B. Kayan, P. Gholami, D. Kalderis, S. Akay and L. Dinpazhoh, Ultrason. Sonochem., 2017, 39, 540–549 CrossRef CAS PubMed.
- L. Pi, R. Jiang, W. Zhou, H. Zhu, W. Xiao, D. Wang and X. Mao, Appl. Surf. Sci., 2015, 358, 231–239 CrossRef CAS.
- L. Luo, Y. Yang, M. Xiao, L. Bian, B. Yuan, Y. Liu, F. Jiang and X. Pan, Chem. Eng. J., 2015, 262, 1275–1283 CrossRef CAS.
- M. Li, H. Huang, S. Yu, N. Tian, F. Dong, X. Du and Y. Zhang, Appl. Surf. Sci., 2016, 386, 285–295 CrossRef CAS.
- Y. Li, S. Zhang, Q. Yu and W. Yin, Appl. Surf. Sci., 2007, 253, 9254–9258 CrossRef CAS.
- X. Cai, J. Li, Y. Liu, Z. Yan, X. Tan, S. Liu, G. Zeng, Y. Gu, X. Hu and L. Jiang, J. Chem. Technol. Biotechnol., 2018, 93, 783–791 CrossRef CAS.
- Y. Wang, M. Dong, M. Guo, X. Wang, J. Zhou, J. Lei, C. Guo and C. Qin, Mater. Sci. Eng., C, 2017, 77, 293–299 CrossRef CAS PubMed.
- V. Normand, D. L. Lootens, E. Amici, K. P. Plucknett and P. Aymard, Biomacromolecules, 2000, 1, 730–738 CrossRef CAS PubMed.
- Z. Zhang, Z. Luo, Z. Yang, S. Zhang, Y. Zhang, Y. Zhou, X. Wang and X. Fu, RSC Adv., 2013, 3, 7215–7218 RSC.
- M. Pelaez, B. Baruwati, R. S. Varma, R. Luque and D. D. Dionysiou, Chem. Commun., 2013, 49, 10118–10120 RSC.
- J. Song, X. Wang, J. Huang, J. Ma, X. Wang, H. Wang, R. Ma, P. Xia and J. Zhao, Electrochim. Acta, 2016, 222, 1–11 CrossRef CAS.
- J. Matos, A. García, L. Zhao and M. M. Titirici, Appl. Catal., A, 2010, 390, 175–182 CrossRef CAS.
- J. Matos and A. Corma, Appl. Catal., A, 2011, 404, 103–112 CrossRef CAS.
- J. Matos, A. Borodzinski, A. M. Zychora, P. Kedzierzawski, B. Mierzwa, K. Juchniewicz, M. Mazurkiewicz and J. C. Hernández-Garrido, Appl. Catal., B, 2015, 163, 167–178 CrossRef CAS.
- A. E. Creamer, B. Gao and M. Zhang, Chem. Eng. J., 2014, 249, 174–179 CrossRef CAS.
- D. Woolf, J. E. Amonette, F. A. Street-Perrott, J. Lehmann and S. Joseph, Nat. Commun., 2010, 1, 56 Search PubMed.
- X. Tan, Y. Liu, Y. Gu, Y. Xu, G. Zeng, X. Hu, S. Liu, X. Wang, S. Liu and J. Li, Bioresour. Technol., 2016, 212, 318–333 CrossRef CAS PubMed.
- J. Yang, Y. Zhao, S. Ma, B. Zhu, J. Zhang and C. Zheng, Environ. Sci. Technol., 2016, 50, 12040–12047 CrossRef CAS PubMed.
- J. Yang, S. Han, H. Zheng, H. Dong and J. Liu, Carbohydr. Polym., 2015, 123, 53–66 CrossRef CAS PubMed.
- H. Jonassen, A.-L. Kjøniksen and M. Hiorth, Biomacromolecules, 2012, 13, 3747–3756 CrossRef CAS PubMed.
- M. Rinaudo, Prog. Polym. Sci., 2006, 31, 603–632 CrossRef CAS.
- M. Vakili, M. Rafatullah, B. Salamatinia, A. Z. Abdullah, M. H. Ibrahim, K. B. Tan, Z. Gholami and P. Amouzgar, Carbohydr. Polym., 2014, 113, 115–130 CrossRef CAS PubMed.
- M. Zhang, Y. Liu, T. Li, W. Xu, B. Zheng, X. Tan, H. Wang, Y. Guo, F. Guo and S. Wang, RSC Adv., 2015, 5, 46955–46964 RSC.
- A. M. Balu, B. Baruwati, E. Serrano, J. Cot, J. Garcia-Martinez, R. S. Varma and R. Luque, Green Chem., 2011, 13, 2750–2758 RSC.
- X. Wang, Y. Liu, Z. Hu, Y. Chen, W. Liu and G. Zhao, J. Hazard. Mater., 2009, 169, 1061–1067 CrossRef CAS PubMed.
- M. A. Nawi, A. H. Jawad, S. Sabar and W. S. W. Ngah, Desalination, 2011, 280, 288–296 CrossRef CAS.
- L. Yu, Y. Yuan, J. Tang, Y. Wang and S. Zhou, Sci. Rep., 2015, 5, 16221 CrossRef CAS PubMed.
- W. Xu, J. J. Pignatello and W. A. Mitch, Environ. Sci. Technol., 2013, 47, 7129–7136 CrossRef CAS PubMed.
- X. Yu, W. Gong, X. Liu, L. Shi, X. Han and H. Bao, J. Hazard. Mater., 2011, 198, 340–346 CrossRef CAS PubMed.
- X.-L. Wu, T. Wen, H.-L. Guo, S. Yang, X. Wang and A.-W. Xu, ACS Nano, 2013, 7, 3589–3597 CrossRef CAS PubMed.
- Y. Yao and F. Wu, ACS Appl. Mater. Interfaces, 2017, 9, 31907–31912 CAS.
- L. Klüpfel, M. Keiluweit, M. Kleber and M. Sander, Environ. Sci. Technol., 2014, 48, 5601–5611 CrossRef PubMed.
- J. M. Saquing, Y.-H. Yu and P. C. Chiu, Environ. Sci. Technol. Lett., 2016, 3, 62–66 CrossRef CAS.
- Y.-J. Xu, Y. Zhuang and X. Fu, J. Phys. Chem. C, 2010, 114, 2669–2676 CAS.
- J. Shi, Chem. Rev., 2013, 113, 2139–2181 CrossRef CAS PubMed.
- W. Wang, P. Serp, P. Kalck and J. L. Faria, J. Mol. Catal. A: Chem., 2005, 235, 194–199 CrossRef CAS.
- Y. Qu and X. Duan, Chem. Soc. Rev., 2013, 42, 2568–2580 RSC.
- Y. H. Ng, S. Ikeda, M. Matsumura and R. Amal, Energy Environ. Sci., 2012, 5, 9307–9318 CAS.
- Y. Li, D.-S. Hwang, N. H. Lee and S.-J. Kim, Chem. Phys. Lett., 2005, 404, 25–29 CrossRef CAS.
- T. J. Bandosz, J. Matos, M. Seredych, M. S. Z. Islam and R. Alfano, Appl. Catal., A, 2012, 445–446, 159–165 CrossRef CAS.
- S. A. Ansari, M. M. Khan, M. O. Ansari and M. H. Cho, New J. Chem., 2016, 40, 3000–3009 RSC.
- J. C. Colmenares, P. Lisowski and D. Lomot, RSC Adv., 2013, 3, 20186–20192 RSC.
- J. Matos, Top. Catal., 2016, 59, 394–402 CrossRef CAS.
- Y. Nosaka and A. Y. Nosaka, Chem. Rev., 2017, 117, 11302–11336 CrossRef CAS PubMed.
- A. Molla, M. Sahu and S. Hussain, J. Mater. Chem. A, 2015, 3, 15616–15625 CAS.
- Q. Lu, Y. Zhang and S. Liu, J. Mater. Chem. A, 2015, 3, 8552–8558 CAS.
- W. Liu, Q. Hu, F. Mo, J. Hu, Y. Feng, H. Tang, H. Ye and S. Miao, J. Mol. Catal. A: Chem., 2014, 395, 322–328 CrossRef CAS.
- X. Wang, P. Wu, Y. Lu, Z. Huang, N. Zhu, C. Lin and Z. Dang, Sep. Purif. Technol., 2014, 132, 195–205 CrossRef CAS.
- M. Ahmadi, H. Ramezani Motlagh, N. Jaafarzadeh, A. Mostoufi, R. Saeedi, G. Barzegar and S. Jorfi, J. Environ. Manage., 2017, 186, 55–63 CrossRef CAS PubMed.
- H. Huang, T. Yang, F. Lai and G. Wu, J. Anal. Appl. Pyrolysis, 2017, 125, 61–68 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2018 |