
A rapid genomic DNA extraction method and its combination with helicase dependent amplification for the detection of genetically modified maize†
Eric Gonzalez
Garcia
a,
Andreas H.
Farnleitner
b,
Robert L.
Mach
b,
Rudolf
Krska
c and
Kurt
Brunner
*a
aVienna University of Technology – IFA Tulln, Center for Analytical Chemistry, Konrad Lorenz Str. 20, 3430, Tulln, Austria. E-mail: kurt.brunner@romerlabs.com
bVienna University of Technology, Institute of Chemical Engineering, Gumperdorfer Str. 1a, 1060, Vienna, Austria
cUniversity of Natural Resources and Life Sciences, Vienna (BOKU), Department IFA-Tulln, Konrad Lorenz Str. 20, 3430, Tulln, Austria
First published on 19th November 2015
Abstract
It is compulsory for many crop plants to be tested for genetic modifications when entering the European Union. In this regard, a DNA extraction is performed and later a qualitative or quantitative PCR to detect and quantify any artificial inserts into the genome. Unfortunately, PCR needs specialized equipment and skilled personnel. This disadvantage has been solved by using isothermal DNA amplification methods, which are as sensitive as the PCR itself and can be performed at constant temperatures and therefore PCR thermocyclers are not needed. Nonetheless, those methods require high quality DNA and the isolation of it is still considered to be an elaborate process. Conventional DNA extraction methods are highly time consuming and tedious. In addition, they cannot be performed on-site as a centrifuge is always required. With this work, the development of a rapid method for DNA extraction from maize was carried out to overcome these problems. The method is based on the employ of an aqueous buffer system in combination with a proteinase K digestion and followed by a later filtration over a polypropylene membrane. Detection was carried out by helicase dependent amplification, as an alternative to PCR for the detection of transgenic maize. Data obtained are similar to those achieved with the more complex standard CTAB extraction method or the Promega Wizard DNA purification kit. The proposed DNA extraction method can be performed on-site, is inexpensive, simple and time saving.
Introduction
It is mandatory for many crop plants to be tested for genetic modifications when entering the European Union. In this sense, this international organization adopted the regulation (EC) no. 1829/2003 to set a limit of 0.9% for food containing authorized genetically modified organisms (GMOs); products exceeding this limit have to be declared.1 Generally, methods based on the polymerase chain reaction for nucleic acid detection are applied to quantify the GMO content of a sample. When compared to protein based techniques such as enzyme-linked immunosorbent assay (ELISA) and lateral flow devices, these techniques offer remarkable benefits, since DNA molecules show higher stability and better extraction yields than proteins (even from highly processed foods).2,3 Moreover, the amplification by polymerase chain reaction (PCR) guarantees the required sensitivity, which makes it the gold standard for the analysis of nucleic acids.4With the increasing commercialization of GMO events, their screening and identification has become a challenge for their monitoring and inspection, as they can be time consuming and their procedures tend to be complex. For this reason, on-site tests facilitating the screening process that can be carried out in a simple, fast and inexpensive ways are in great demand. Overall, there are three major steps in the GMO detection using a nucleic acid based approach. First, the DNA in the sample has to be extracted, so as to later amplify the sequence of interest and identify the amplified target. In this regard, an improvement to any of these steps would contribute to improve the entire testing procedure. The least common denominator still used in many GM plants is the cauliflower mosaic virus 35S promoter. PCR assays targeting this promoter sequence are able to detect a wide range of GMOs, including many events of maize, rice, canola and cotton.5
When extracting DNA, commonly applied methods include the use of the surfactant cetyltrimethylammonium bromide (CTAB)6–8 or commercial kits.9,10 However, they have several disadvantages as they can be time consuming and laborious, including numerous pipetting and centrifugation steps. These steps make the methods prone to errors, while carrying a certain risk of cross-contamination. Furthermore, the use of laboratory equipment, such as centrifuges, can considerably limit their usage for on-site applications.
When amplifying the target sequence, PCR techniques such as real time PCR (qPCR) for quantitative analysis have been extensively employed.11–14 Nonetheless, this technique has its limitations, as it requires expensive thermal cyclers and trained personnel to be performed. To overcome this problem, the use of other enzymes to mimic DNA replication in vivo has emerged as a solution to conventional PCR polymerases,15,16 thus remarkably lowering the cost and time for target DNA amplification. Different isothermal amplification techniques such as loop mediated isothermal amplification (LAMP),16,17 helicase dependent amplification (HDA),18,19 rolling circle amplification (RCA)20 and recombinant polymerase amplification (RPA)21 have been developed.
HDA uses the unwinding activity of the helicase to separate two complementary DNA strands into each single stranded DNA during replication.22 During this process, heat denaturation and subsequent thermocycling are not necessary, as the helicase is also able to displace DNA strands alongside the exponential amplification reaction. As a consequence, the entire HDA process can be performed at one temperature, which provides a very useful tool to amplify DNA under isothermal conditions. In addition, it offers all the advantages of PCR, such as high sensitivity, simple primer design and the possibility of multiplexing.23
In this work, a simple, inexpensive and efficient DNA method of extraction for GMO analysis has been developed. The method is then compared to a common CTAB protocol and a commercial genomic DNA purification kit for PCR efficiency and yield. In addition, its successful combination with a previously published HDA proved the applicability of the procedure with isothermal amplification methods for on-site applications.
Experimental
Materials
The non-GM maize variety RWA38 was kindly provided by the Institute of Biotechnology in Plant Production at IFA-Tulln, Austria and ground to a fine powder with a Retsch ball mill MM301 (Haan, Germany). GM maize varieties NK-603, Bt-11 and MON-810 were produced by the Institute for Reference Materials and Measurements of the European Union. Samples were stored at 4 °C when received until further usage. All chemicals were bought from Carl Roth (Karlsruhe, Germany), unless otherwise specified.DNA extraction methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 rpm for 10 min. The centrifugation step was later substituted by filtration over a polypropylene syringe filter (pore size = 0.45 μm), as it was simpler and suitable for on-site testing. Afterwards, 100 μL of filtrated sample were retrieved and analyzed via PCR. All experiments were carried out in triplicates.
000 rpm. 100 μL of supernatant were then transferred into a 1.5 mL Eppendorf vial and analyzed later via qPCR.
000 rpm. 100 μL of supernatant were then transferred into a 1.5 mL Eppendorf vial and analyzed later via qPCR.
500 rpm for 10 min. The centrifugation step was later substituted by filtration over a polypropylene syringe filter (pore size = 0.45 μm), as it was simpler and suitable for on-site testing. Afterwards, 100 μL of filtrated sample were retrieved and analyzed via PCR. All experiments were carried out in triplicates.
000 rpm. 100 μL of supernatant were then transferred into a 1.5 mL Eppendorf vial and analyzed later via qPCR.
000 rpm. 100 μL of supernatant were then transferred into a 1.5 mL Eppendorf vial and analyzed later via qPCR.
Real-time PCR assays
The real-time PCR was performed on a RotorGene-Q cycler (Qiagen, Hilden, Germany). For each reaction, 2 μL template DNA were mixed with 13 μL reaction mix containing 7.5 μL Kapa™ Probe® Fast (PeqLab, Erlangen, Germany), 4.78 μL sterile and nuclease free water and 0.24 μL for JOE-labeled probe (Eurofins MWG Operon, Ebersberg, Germany) and forward and reverse primers (each at 100 pM μL−1) (Sigma-Aldrich, St. Louis; USA).Two genes were used as a target for amplification: the ADH1 gene encoding for an alcohol dehydrogenase, which is present in GM and non-GM maize and the cauliflower mosaic virus (CaMV) promoter P35S for the detection of the genetically modified sequence in maize.
The real time PCR program for detecting the ADH1 gene contained a denaturation and polymerase activation step at 95 °C for 2 min, followed by 45 cycles of 58.6 °C for 15 s and 66.3 °C for 25 s. For the detection of the P35S promoter, the PCR program was performed according to the following cycling conditions: an initial step at 95 °C for 2 min and 45 cycles of 95 °C for 15 s and 60 °C for 60 s. All PCR programs were optimized and published by the European Union Reference Laboratory for GM Food and Feed, 2011.25
In addition, calibration curves with 4 points following five fold dilutions were performed and linearity and PCR efficiencies were calculated to assess the presence of inhibitors that might hinder the amplification process during the qPCR. qPCR efficiency was determined as:
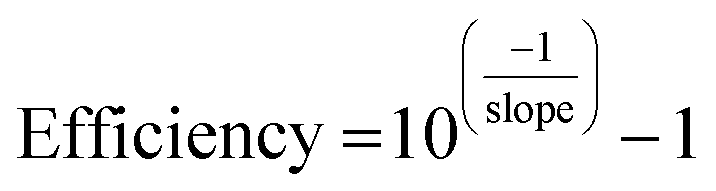
The concentration of the DNA extracts were photometrically measured in a NanoVue Plus spectrophotometer (GE Healthcare, Little Chalfont, USA). The yield was calculated as follows:
| Yield = 10c(DNA)V |
HDA
HDA assays were performed according to ref. 18 with some modifications. The method followed a two steps protocol were 12.5 μL of mix A (sterile and nuclease free water, 10× annealing buffer, 0.12 mM forward and reverse primers and 1 μL DNA (40 ng μL−1)) was overlaid with mineral oil and heated at 95 °C for 5 min for an initial DNA denaturation process. Reactions were afterwards temperature equilibrated at 63 °C for 5 min, followed by an incubation at 63 °C for 90 min after adding mix B (sterile and nuclease free water, 1× annealing buffer, 8 mM MgSO4, 80 mM NaCl and 1.75 μL for both IsoAmp dNTP solution and IsoAmp Enzyme Mix) to the reaction. Finally, an enzyme denaturation step (95 °C for 10 min) was carried out. HDA products were later analyzed in a 2% agarose gel stained with Sybr Gold (Invitrogen, Oregon, USA). Primers used were as reported by ref. 18.Results and discussion
The protocol for the sodium phosphate buffer (Na2HPO4/NaH2PO4 buffer) based extraction was developed in different stages. The procedure was first optimized by using non GM-maize variety RWA38 and the ADH1 gene was used to assess the best extraction parameters. Lastly, the maize certified reference materials NK603, Bt11 and MON810 were used to screen for the 35S promoter by using helicase dependent amplification. A comparison of the method developed in this work with the more conventional protocol CTAB based extraction and the Promega Wizard® Genomic DNA purification kit was also carried out.Optimization of the best extraction conditions
In order to keep the extraction protocol simple and efficient, the influence of different conditions was investigated. Initially, the effect of the incubation time was defined, so as to know if a digestion of the enzymes and proteins present in the maize samples could increase the extracted amount of DNA. Results show (Fig. 1 from the ESI†) that no significant difference was seen when extracting at either of the times studied (10 min, 30 min, 45 min and 60 min). Therefore, an extraction time of 10 min was chosen. Furthermore, the amount of proteinase K for digesting the proteins in maize was also optimized. In this sense, extractions were carried out by adding 5, 10, 15 and 20 μL of a 20 mg mL−1 solution while following the same protocol as before. No dependency on the volume was observed when extracting gDNA from maize, suggesting a further use of only 5 μL of this enzyme to reduce costs of this protocol. In addition, the temperature at which the proteinase K during the extraction exerts its greatest activity was also optimized (Table 2, ESI†). A range of temperatures from 50 °C to 90 °C was evaluated, with 80 °C being the temperature at which the lowest Cq values were obtained and therefore the highest amount of DNA was extracted.With the aim of optimizing the concentration of the sodium phosphate buffer, experiments were performed at concentrations of 10, 50, 100 and 500 mM while extracting for 10 min at 80 °C in the presence of proteinase K (20 mg mL−1) (Fig. 1). Best results were obtained when working at 50 mM. A lower concentration of 10 mM considerably increased the Cq value, probably due to a lesser extraction effect (the greater the Cq value, the lower the concentration of extracted DNA). Concentrations higher than 50 mM decreased the Cq values, which might be caused by an inhibition effect of the polymerase during the PCR. Additionally, the combination of the sodium phosphate buffer with proteinase K (20 mg mL−1) considerably increased the amount of extracted DNA when compared to an extraction carried out with just buffer.
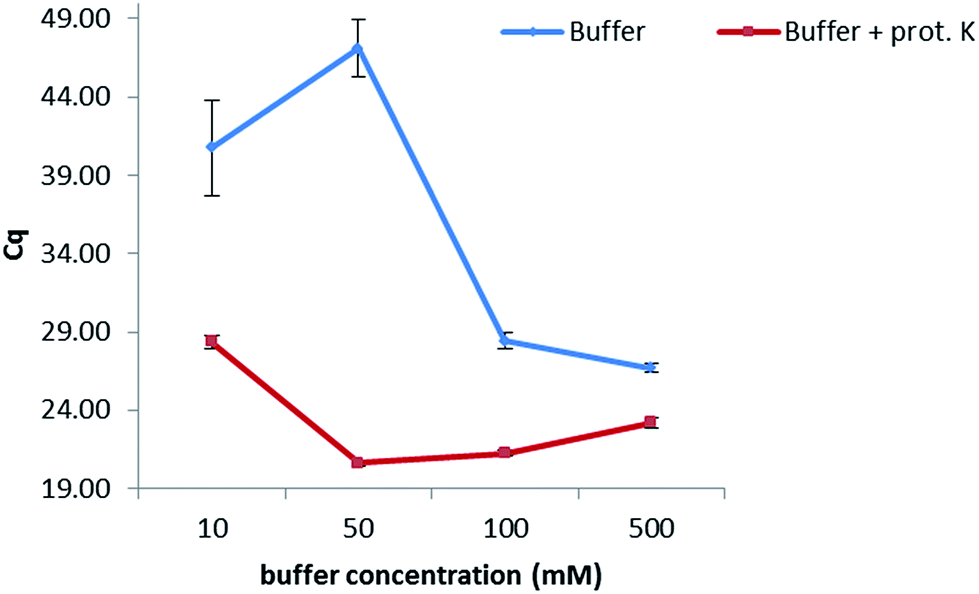 | ||
| Fig. 1 Optimization of the concentration of the extraction buffer. A difference in one Cq value corresponds to double the concentration of extracted DNA with optimal PCR efficiency. | ||
In addition, a treatment with alpha amylase was also performed to investigate if a digestion of the maize polysaccharides could increase the amount of gDNA released in the extract. In this sense, 10 or 100 μL of this enzyme (1 mg mL−1) were added before (pretreatment) and after (post-treatment) digesting with proteinase K (20 mg mL−1) while incubating for 5, 10, 20 and 30 min (Fig. 2). Values obtained were kept almost constant when extracting up to 20 min in all cases. After 20 min, the Cq values decreased when performing a pretreatment with the addition of 10 and 100 μL of alpha amylase as well as a post-treatment with 100 μL of alpha amylase, not so when adding 10 μL of this enzyme (1 mg mL−1) after the digestion with proteinase K, where the Cq values increased, reducing the amount of DNA extracted that could later be amplified in the PCR. Best results were obtained when incubating for 5 min with 10 μL of alpha amylase (1 mg mL−1) after digesting first with proteinase K (20 mg mL−1). However, as the Cq values did not considerably improve the results, the alpha amylase was not further used.
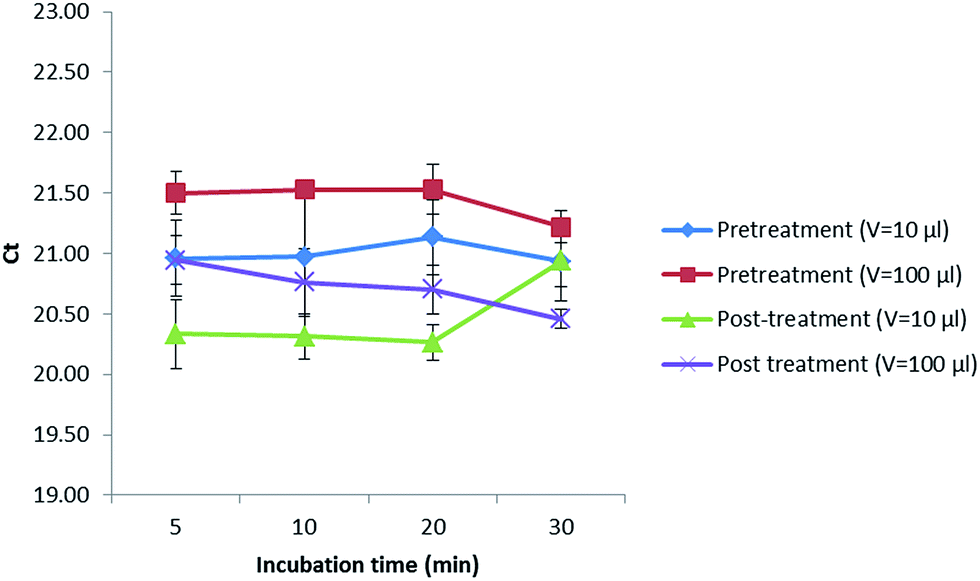 | ||
| Fig. 2 Treatment with alpha amylase at different incubation times and volumes before and after digesting with proteinase K. | ||
Furthermore, different chemicals, including sodium chloride (NaCl), potassium chloride (KCl), sodium sulphite (Na2SO3), ethylenediaminetetraacetic acid (EDTA), guanidine hydrochloride (GuHCl), sodium dodecyl sulfate (SDS) Tween 20 and Tween 80, were also added to the sodium phosphate buffer to enhance the results from the method (see ESI Fig. 3†). The salts NaCl and KCl were used for their salting out effect, while Na2SO3 prevents the oxidation of nucleic acid upon the release of sap from the cells.26,27 GuHCl and SDS were employed as they can remove proteins and polysaccharides by complexations28 and EDTA chelates metals in solution. Tween 20 and Tween 80 were used for their detergent effects. Despite all of their potential effects, no improvement in the Cq values was seen with any of the added compounds when compared to the extraction carried out with the phosphate buffer in combination with proteinase K (20 mg mL−1). Hence, they were not further employed. Maize powder samples were also pretreated before extraction with chloroform (CH3Cl), ethyl acetate (EtoAc) and a mixture 1:1 of CH3Cl:EtoAc to remove fats in the sample. However, no influence of this pretreatment was observed (Table 1 from the ESI†).
To simplify the method in order to make it applicable for on-site testing, a syringe filtration over a polypropylene syringe filter (pore size = 0.45 μm), instead of centrifugation, was applied. The obtained results showed that the simple filtration step (Cq of 20.68 ± 0.21) performed equally well as the more tedious centrifugation step (Cq 20.62 ± 0.20). Hence, filtration was chosen for further works due to its simplicity and applicability for on-site testing.
Applicability of the method developed to other maize varieties
The suitability of the extraction method developed in this work was expanded to other maize varieties, such as the NK603, Bt-11 and MON810. Two target sequences were studied; the ADH1 encoding for the alcohol dehydrogenase was used for the total amount of maize and the cauliflower mosaic virus (CaMV) P35S promoter for the detection of genetically modified sequences in maize. In this case, the DNA yield was calculated and calibration curves were used to check for the presence of inhibitors coextracted with the gDNA. Two other conventional methods, the CTAB method and the Promega Wizard® Genomic DNA purification kit were also used for comparing our new rapid method to well established protocols (Table 1). The DNA yield when using the here developed buffer based extraction system was lower than the ones obtained when extracting with the CTAB method, but was still approximately three times higher than the ones obtained with the Wizard Promega kit. All PCR assays were found to have high linearity (R2 = 0.99) and efficiencies (>0.90), except for the NK603 maize variety when analyzing the ADH1 gene (0.77). In addition, the linearity and PCR efficiency of the method developed in this work were comparable to those obtained with the CTAB method and the Promega commercial kit. These data indicated that there were few inhibitors coextracted with our method and the quality of DNA was good enough for PCR amplification.| Method of extraction | Maize variety | Target | Yield (μg DNA/g sample) ± STD | Slope | Efficiency | R 2 |
|---|---|---|---|---|---|---|
| CTAB | NK603 | ADH1 | 310.6 ± 2.8 | −3.37 | 0.98 | 0.99 |
| Bt-11 | 299.3 ± 4.8 | −3.39 | 0.97 | 0.99 | ||
| MON810 | 235.2 ± 4.7 | −3.22 | 1.04 | 0.99 | ||
| NK603 | P35S | 310.6 ± 2.8 | −3.53 | 0.92 | 1.00 | |
| Bt-11 | 299.3 ± 4.8 | −3.49 | 0.93 | 0.99 | ||
| MON810 | 235.2 ± 4.7 | −3.30 | 1.01 | 0.99 | ||
| Promega Wizard® Genomic DNA purification kit | NK603 | ADH1 | 47.5 ± 0.4 | −3.44 | 0.95 | 0.99 |
| Bt-11 | 41.42 ± 2 | −3.36 | 0.99 | 1.00 | ||
| MON810 | 55.7 ± 1.2 | −3.49 | 0.94 | 1.00 | ||
| NK603 | P35S | 47.5 ± 0.4 | −3.40 | 0.97 | 1.00 | |
| Bt-11 | 41.42 ± 2 | −3.37 | 0.98 | 0.99 | ||
| MON810 | 55.7 ± 1.2 | −3.33 | 1.00 | 0.99 | ||
| Phosphate buffer method | NK603 | ADH1 | 138.7 ± 0.6 | −4.02 | 0.77 | 1.00 |
| Bt-11 | 156 ± 1.2 | −3.53 | 0.92 | 0.99 | ||
| MON810 | 133 ± 1.1 | −3.54 | 0.92 | 0.99 | ||
| NK603 | P35S | 138.7 ± 0.6 | −3.42 | 0.96 | 0.99 | |
| Bt-11 | 156 ± 1.2 | −3.50 | 0.93 | 1.00 | ||
| MON810 | 133 ± 1.1 | −3.29 | 1.01 | 0.99 |
Moreover, the method of extraction developed here was combined with HDA to screen for the P35S promoter in the maize varieties NK603, Bt-11 and MON810 (Table 2). For comparison purposes, gDNA extracted from these varieties with the commercial kit from Promega and the CTAB protocol were used. As it can be seen, all maize lines analyzed were reliably detected down to a concentration of 0.5%, which is below the European threshold of 0.9%. Results obtained with the method developed in this work were comparable with those from the CTAB and the commercial Promega kit at this concentration. These data confirm that this method is suitable for detecting GM maize with isothermal DNA amplification methods as sufficient sensitivity was achieved to be under the lowest acceptance limit of GM in foods. The combination of this method with the HDA also contributes to enhance its value for on-site testing, as isothermal amplification methods require no PCR cyclers.
| c(maize) % | NK603 | Bt-11 | MON810 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| CTAB | Promega | Our method | CTAB | Promega | Our method | CTAB | Promega | Our method | |
| 2 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 |
| 1 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 |
| 0.5 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 |
| 0.1 | 4/5 | 3/5 | 2/5 | 4/5 | 5/5 | 0/5 | 3/5 | 4/5 | 3/5 |
| 0 | 0/5 | 0/5 | 0/5 | 0/5 | 0/5 | 0/5 | 0/5 | 0/5 | 0/5 |
Conclusions
In this work a simple, inexpensive and rapid method of DNA extraction from maize is proposed. Optimization of extraction parameters led to a simplified protocol where a high yield of gDNA could be extracted and the amount of coextracted inhibitors was kept low. The protocol developed showed comparable results to the conventional CTAB method and the commercial kit from Promega, while saving 3.5 h when comparing to the CTAB method and 5.5 h when comparing to the Promega kit while extracting 10 samples in parallel with these methods. Its combination with HDA for the detection of the P35S promoter in GM maize makes it suitable for reliable on-site detections of genetically modified sequences in maize containing this promoter with concentrations as low as 0.5%. In addition, as HDA can be carried out using an inexpensive heating block instead of a thermal cycler, the costs of the assays can be remarkably decreased.Acknowledgements
This work was supported by the state of Lower Austria in cooperation with the European Regional Development Fund.Notes and references
- Regulation (EC) No. 1829/2003 of the European Parliament and of the Council of 22 September 2003 on genetically modified food and feed, Official Journal of the European Union, L268.
- L. Asensio, I. González, T. García and R. Martín, Food Control, 2008, 19, 1–8 CrossRef CAS.
- E. Scaravelli, M. Brohée, R. Marchelli and A. J. van Hengel, Anal. Bioanal. Chem., 2009, 395, 127–137 CrossRef CAS PubMed.
- A. Holst-Jensen, Biotechnol. Adv., 2009, 27, 1071–1082 CrossRef PubMed.
- I. Mafra, S. A. Silva, E. J. M. O. Moreira, C. S. F. Silva, M. Beatriz and P. P. Olivera, Food Control, 2008, 19, 1183–1190 CrossRef CAS.
- I. Scholtens, E. Lauransee, B. Molenaar, S. Zaaijer, H. Gaballo, P. Boleij, A. Bak and E. Kok, J. Agric. Food Chem., 2013, 61, 9097–9109 CrossRef CAS PubMed.
- T. Demeke and G. R. Jenkins, Anal. Bioanal. Chem., 2010, 396, 1977–1990 CrossRef CAS PubMed.
- G. C. Allen, M. A. Flores-Vergara, S. Krasynanski, S. Kumar and W. F. Thompson, Nat. Protoc., 2006, 1, 2320–2325 CrossRef CAS PubMed.
- S. Santiago-Felipe, L. A. Tortajada-Genaro, R. Puchades and A. Maquiera, Anal. Chim. Acta, 2014, 811, 81–87 CrossRef CAS PubMed.
- R. M. Elsanhoty, M. F. Ramadan and K. D. Jany, Food Chem., 2011, 126, 1883–1889 CrossRef CAS PubMed.
- K. Cankar, D. Štebih, T. Dreo, J. Žel and K. Gruden, BMC Biotechnol., 2006, 6, 37 CrossRef PubMed.
- M. Chaouachi, R. El Malki, A. Berard, M. Romaniouk, V. Laval, D. Brunel and Y. Bertheau, J. Agric. Food Chem., 2008, 56, 1818–1828 CrossRef CAS PubMed.
- M. B. Gašparič, T. Tengs, J. L. La Paz, A. Holst-Jensen, M. Pla, T. Esteve, J. Žel and K. Gruden, Anal. Bioanal. Chem., 2010, 396, 2023–2029 CrossRef PubMed.
- G. Cottenet, C. Blancpain, V. Sonnard and P. F. Chuah, Anal. Bioanal. Chem., 2013, 405, 6831–6844 CrossRef CAS PubMed.
- F. Ahmad and S. A. Hashsham, Anal. Chim. Acta, 2012, 733, 1–15 CrossRef CAS PubMed.
- S. Fukuta, Y. Mizukami, A. Ishida, J. Ueda, M. Hasegawa, I. Hayashi, M. Hashimoto and M. Kanbe, Eur. Food Res. Technol., 2004, 218, 496–500 CrossRef CAS.
- C. Zahradnik, R. Martzy, R. L. Mach, R. Krska, A. H. Farnleitner and K. Brunner, Anal. Bioanal. Chem., 2014, 406, 6827–6833 CrossRef CAS PubMed.
- C. Zahradnik, C. Kolm, R. Martzy, R. L. Mach, R. Krska, A. H. Farnleitner and K. Brunner, Anal. Bioanal. Chem., 2014, 406, 6835–6842 CrossRef CAS PubMed.
- M. Vincent, Y. Xu and H. Kong, EMBO Rep., 2004, 5(8), 795–800 CrossRef CAS PubMed.
- P. M. Lizardi, X. Huang, Z. Zhu, P. Bray-Ward, D. C. Thomas and D. C. Ward, Nat. Genet., 1998, 19, 225–232 CrossRef CAS PubMed.
- B. A. Rohrman and R. R. Richards-Kortum, Lab Chip, 2012, 12, 3082–3088 RSC.
- Y. J. Jeong, K. Park and D. E. Kim, Cell. Mol. Life Sci., 2009, 66, 3325–3336 CrossRef CAS PubMed.
- J. Kim and C. J. Easley, Bioanalysis, 2011, 3(2), 227–239 CrossRef CAS PubMed.
- European Commission, Directorate General, Joint Research Centre, Community Reference Laboratory for GM Food and Feed, Ispra, 2007, http://gmo-crl.jrc.ec.europa.eu/summaries/3272_DNAExtrReport.pdf, accessed 8 November 2011.
- European Union Reference Laboratory for GM Food and Feed, European Network of GMO laboratories, 2011, http://ec.europa.eu/dgs/jrc/downloads/jrc_reference_report_2010_11_gmo_analysis_compendium.pdf, accessed on 3 April 2012.
- M. Byrne, B. Macdonald and M. Francki, BioTechniques, 2001, 30, 742–743 CAS.
- V. K. Baranwal, S. Majumder, Y. S. Ahlawat and R. P. Sing, J. Virol. Methods, 2003, 112, 153–156 CrossRef CAS PubMed.
- J. R. Mujico, M. Lombardía, M. C. Mena, E. Méndez and J. P. Albar, Food Chem., 2011, 128, 795–801 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Procedure for preparation of buffers, extraction procedures and additional results of the DNA extraction. See DOI: 10.1039/c5ay02628h |
| This journal is © The Royal Society of Chemistry 2016 |