
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular engineering and electrolyte optimization strategies for enhanced performance of Ru(II) polypyridyl-sensitized DSSCs
Islam M. Abdellah
 *ab
*ab
aDepartment of Chemistry, Faculty of Science, Aswan University, Aswan 81528, Egypt. E-mail: islamabdellah2@aswu.edu.eg; imabdell@ncsu.edu
bTECS Department, Wilson College of Textiles, North Carolina State University, Raleigh 27606, USA
First published on 31st March 2025
Abstract
Dye-sensitized solar cells (DSSCs) are a leading third-generation solar cell technology due to their low cost, ease of fabrication, and tunable photoelectrochemical properties. Among DSSC components, the photosensitizer plays a crucial role in light absorption and charge generation, with Ru(II)-polypyridyl complexes standing out due to their superior photostability, broad absorption spectra, and efficient charge injection. This review provides a comprehensive analysis of molecular engineering strategies for Ru(II)-polypyridyl photosensitizers, emphasizing ligand modifications to design and develop novel Ru(II) photosensitizers with prolonged excited-state lifetimes, reduced charge recombination, enhanced light-harvesting capabilities, and improved overall solar-to-power conversion efficiency (PCE). In addition, cyclometallated polypyridyl Ru(II) complexes are explored as promising alternatives to Ru(II) complexes incorporating labile thiocyanate (SCN) ligands for DSSCs, which offer improved stability. The relationship between the molecular structure of Ru(II) photosensitizers and their photovoltaic characteristics is analyzed by examining key factors that influence their photovoltaic performance, including light-harvesting efficiency, fine-tuning ground and excited state oxidation potentials (GSOP/ESOP), extending excited state lifetimes, and minimizing charge recombination. Additionally, the impact of co-adsorbents, electrolyte additives, and interfacial engineering on DSSC performance is explored. Emphasis is placed on optimizing redox electrolytes beyond conventional iodide/triiodide (I−/I−3) systems to minimize energy loss and enhance PCE. By carefully considering those challenges, this review lays the groundwork for the rational design of next-generation DSSCs that are more efficient, stable, and commercially viable.
 Islam M. Abdellah | Islam M. Abdellah is an Assistant Professor at Aswan University, Egypt, and a Postdoctoral Researcher at NC State University, USA. He earned his PhD through a joint program between both institutions. His research focuses on organic photovoltaic materials, molecular engineering, and machine learning for dye-sensitized and perovskite solar cells. He also works on sustainable textile dyeing and finishing, enhancing color fastness and antimicrobial properties. Additionally, his work extends to environmental sustainability through the development of innovative, cost-effective adsorbents for colorant removal, aiming to minimize environmental impact and operational costs. His interdisciplinary expertise integrates organic synthesis, polymers, energy materials, organic photovoltaics, computational chemistry, and sustainable materials science, advancing energy conversion, smart textiles, and environmental remediation. |
1. Introduction
The growing global reliance on electronic devices, combined with the rapid depletion of traditional fossil fuel reserves, emphasizes the importance of developing sustainable energy solutions.1–4 As energy demand grows, experts are actively studying renewable energy solutions that provide both efficiency and environmental benefits. Solar energy stands out as a particularly promising option, considering that the daily solar radiation reaching the Earth's surface greatly surpasses global energy consumption needs. The amount of solar radiation reaching the Earth's surface greatly exceeds global energy demand. On average, 173![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 terawatts (TW) of solar radiation continuously strikes the Earth,5 while global electricity demand is approximately 3.0 TW.6 A moderately efficient solar cell array (∼10% efficiency) covering a limited portion of the Earth's surface could generate substantial electricity, reducing dependence on fossil fuels and lowering greenhouse gas emissions.7 This led to the invention of new photovoltaic (PV) technologies, such as dye-sensitized solar cells (DSSCs), organic photovoltaics (OPVs), quantum dots solar cells (QDSCs), and as a third-generation alternative, perovskite solar cells (PSCs).8–11 DSSCs have drawn intensive attention and have the potential to replace silicon-based technology due to their low-cost, lightweight, facile solution processability, and superior photovoltaic performance.12 Since their inception by O'Regan and Grätzel in 1991,13 DSSCs have undergone significant advancements, particularly in their key components including photoanode (TiO2 or ZnO semiconductor coated FTO), photosensitizers (dyes), electrolyte (I−/I3− redox couple) and counter electrode (Pt coated FTO).14,15 Hence, DSSC researchers are still searching for novel and more efficient electrolytes,16–18 photosensitizers19,20 and/or semiconductors that would enhance the efficiency of DSSCs. The organic photovoltaics (OPVs), especially DSSCs, are a promising alternative to silicon-based solar cells with unique advantages such as low cost, ease of manufacturing, high surface to weight ratio, and flexibility.21–24
000 terawatts (TW) of solar radiation continuously strikes the Earth,5 while global electricity demand is approximately 3.0 TW.6 A moderately efficient solar cell array (∼10% efficiency) covering a limited portion of the Earth's surface could generate substantial electricity, reducing dependence on fossil fuels and lowering greenhouse gas emissions.7 This led to the invention of new photovoltaic (PV) technologies, such as dye-sensitized solar cells (DSSCs), organic photovoltaics (OPVs), quantum dots solar cells (QDSCs), and as a third-generation alternative, perovskite solar cells (PSCs).8–11 DSSCs have drawn intensive attention and have the potential to replace silicon-based technology due to their low-cost, lightweight, facile solution processability, and superior photovoltaic performance.12 Since their inception by O'Regan and Grätzel in 1991,13 DSSCs have undergone significant advancements, particularly in their key components including photoanode (TiO2 or ZnO semiconductor coated FTO), photosensitizers (dyes), electrolyte (I−/I3− redox couple) and counter electrode (Pt coated FTO).14,15 Hence, DSSC researchers are still searching for novel and more efficient electrolytes,16–18 photosensitizers19,20 and/or semiconductors that would enhance the efficiency of DSSCs. The organic photovoltaics (OPVs), especially DSSCs, are a promising alternative to silicon-based solar cells with unique advantages such as low cost, ease of manufacturing, high surface to weight ratio, and flexibility.21–24
Photosensitizers play a pivotal role in DSSCs, directly influencing their efficiency. Scientists have developed numerous highly efficient photosensitizing dyes, achieving a power conversion efficiency (PCE) stabilized at approximately 13% under one sun illumination (AM 1.5G sunlight, 100 mW cm−2).25 Since the discovery of DSSCs by O'Regan and Grätzel in 1991,13 several photosensitizers have been used throughout the last 34 years, including both metal-free and metal-based photosensitizers.26,27 Among these classes metal-based photosensitizers specially, Ru(II)-polypyridyl complexes have established themselves as the benchmark due to their exceptional photophysical and electrochemical properties. These complexes have broad absorbance spectra that extend into the near-infrared region, high molar extinction coefficients, and long-lived excited states, which all contribute to effective electron injection into the semiconductor.28 Moreover, Ru(II)-based photosensitizers exhibit outstanding photostability and redox reversibility, resulting in longer device lifetimes and improved stability under operational conditions. While metal-free photosensitizers, such as porphyrins, indoline, carbazole, triphenylamine derivatives, thiophene, indoline, BODIPY, and phthalocyanine dyes, have attained competitive PCEs, they frequently suffer from limitations such as dye aggregation, a shorter absorption range, and stability concerns.29 In contrast, Ru(II) complexes, which include highly investigated photosensitizers such as N3, N719, C101, and C106, have consistently exhibited superior performance, producing high PCEs while maintaining strong long-term stability. Although Ru-based photosensitizers are more expensive, their effectiveness and endurance make them vital for high-performance DSSCs and a preferred choice for commercial applications.28
Herein, we present a comprehensive analysis of molecular engineering options for improving the overall performance of DSSCs, with a special emphasis on Ru(II)-polypyridyl photosensitizers. It highlights significant changes in ligand design that improve light absorption, excited-state durations, and charge transfer qualities. Beyond Ru(II)-polypyridyl photosensitizer modifications, the review investigates critical DSSC components such as electrolytes, co-adsorbents, and counter electrodes, as shown in Fig. 1. In addition, alternative redox mediators beyond the traditional iodide/triiodide (I−/I3−) system are examined, with a focus on charge recombination and device stability. Similarly, the utilization of diverse co-adsorbent architectures to avoid dye aggregation, improve dye adsorption, and reduce charge recombination is investigated. The study also assesses different counter-electrode materials to replace the standard platinum-based material, providing cost-effective and efficient solutions. By addressing these combined molecular engineering and material optimization strategies, this work aims to offer valuable insights into the rational design of next-generation DSSCs with superior efficiency, long-term stability, and commercial viability.
 | ||
| Fig. 1 The key optimization strategies of the DSSCs components including modifications to electrolytes, Ru(II)-polypyridyl dyes, co-adsorbents, counter electrodes, and additives. | ||
2. Composition of DSSCs
A DSSC comprises several key components that work in unison to efficiently convert sunlight into electrical energy. These essential elements include photoanode, a sensitizing dye, a redox electrolyte, and a counter electrode, each playing a vital role in the device's photovoltaic performance. As shown in Fig. 2, the working and counter electrodes are commonly composed of transparent conductive glass covered with fluorine-doped tin oxide (FTO).30,31 The photoanode is made up of an FTO substrate coated with a layer of mesoporous titanium dioxide (TiO2), which acts as an effective surface for dye adsorption. Meanwhile, the counter electrode is often coated with a platinum catalyst. A redox electrolyte, typically comprising the I−/I3− redox pair, is injected between the two electrodes to enhance charge transport.32 When the DSSC is exposed to sunlight, the photosensitizer absorbs photons (E = hν), resulting in electron excitation and subsequent injection into TiO2's conduction band. A schematic diagram showing the main components of DSSCs as well as the flow of electrons inside the cell has been illustrated in Fig. 2. | ||
| Fig. 2 A schematic diagram showing the main components of DSSCs and the flow of the current inside the cell. | ||
2.1. Working electrode (photoanode)
The photoanode, serving as the working electrode in a typical dye-sensitized solar cell (DSSC), consists of three main components: a transparent conductive glass (TCG), a mesoporous semiconductor layer, and an adsorbed dye. TCGs are typically composed of thin films of doped metal oxide semiconductors, supported on either glass or flexible substrates. Among these, fluorine-doped tin oxide (FTO) is the most used due to its high thermal stability, excellent electrical conductivity, and optical transparency.33,34 More recently, alternative materials such as graphene-based electrodes have been explored for better compatibility with flexible substrates.35,36The semiconductor layer, typically an n-type metal oxide, is deposited on top of the TCG. The primary role of the semiconductor oxide layer is to provide a strong bond with the photosensitizer and facilitate efficient electron transport. The ideal characteristics of a semiconductor oxide layer for DSSCs include: (1) high surface area for enhanced dye adsorption and efficient light harvesting (2) minimal photon loss and high transparency in the visible spectrum (3) a conduction band (CB) lower than the lowest unoccupied molecular orbitals (LUMOs) of the dye, allowing effective electron injection (4) high electron mobility for efficient charge transport (5) chemical stability against redox electrolytes to reduce electron recombination (6) presence of hydroxyl groups or surface defects to enable strong dye bonding (7) ease of fabrication, stability, affordability, and environmental friendliness.
Titanium dioxide (TiO2) in its anatase crystalline form remains the most widely used semiconductor due to its superior photovoltaic efficiency, abundance, electron affinity, dye loading, surface area and cost-effective synthesis. Compared with other transition metal oxides, which makes it the most suitable choice as photoanode for DSSCs. This material was first employed in DSSCs by O'Regan and Grätzel (1991),13 and despite extensive research on alternatives such as zinc oxide (ZnO),37,38 Nb2O5,39 SrTiO3,40 Zn2SnO4,41 tin oxide (SnO2),42 and TiO2 continues to deliver the highest efficiencies.43 Beyond conventional TiO2, researchers have developed modified semiconductor oxides by doping them with transition metals, alkaline earth metals, rare earth metals, and non-metals to improve performance.44,45 Selenium nanoparticle films have also been applied over TiO2 layers to create cost-effective solar cells.46 Despite these advancements, TiO2 continues to outperform other materials due to its ideal electronic properties for DSSCs.
A variety of TiO2 nanostructures have been investigated for DSSCs, including one-dimensional (1D) nanostructures such as nanotubes,47 nanorods,48 nanowires,49 and nanosheets.50 These 1D nano structures are characterized by their excellent light-scattering ability and efficient electron transport, however their limitation lies in their relatively low surface area, which reduces dye loading.51 Furthermore, mesoporous TiO2 is widely used to optimize the balance between dye loading and electron mobility due its small pore sizes and large surface area.52 Three common crystalline phases of TiO2 have been studied: anatase (tetragonal), rutile (tetragonal), and brookite (orthorhombic). Anatase is preferred for DSSC applications due to its superior electron transport properties, while rutile is valued for its stability. Brookite is less common but may offer benefits in charge separation under specific conditions.53 Mixed-phase structures, particularly anatase–rutile combinations, can optimize performance in DSSCs by leveraging the strengths of each phase.54
High-efficiency DSSCs typically incorporate two layers of mesoporous TiO2 deposited on the FTO glass to enhance light absorption and scattering that help to improve overall light harvesting. The first layer, responsible for light absorption, is made of anatase TiO2 nanoparticles ranging from 15 to 20 nm in diameter and has a thickness of ∼10 μm. The second layer, designed to improve light scattering, is formed of bigger anatase particles (200–400 nm) with a thickness of ∼3 μm.55 These porous layers not only provide a huge surface area for dye adsorption, but they also promote electrolyte diffusion, resulting in efficient charge transport. To further enhance device performance, a thin TiO2 coating (∼1 nm) is deposited via aqueous TiCl4 treatment that help to facilitate charge injection and enhance the electron lifetime by minimizing surface impurities on TiO2 and improving dye adsorption.32 This process, often achieved through screen printing or doctor blading,56 forms a highly porous layer with an extremely large surface area, approximately 1000 times larger than the electrode's actual area, facilitating sufficient dye-sensitizer loading. The electrode is immersed in the photosensitizer solution to enable covalent bonding between the TiO2 and the dye through an anchoring group attached to the photosensitizer molecule.
2.2. Counter electrode
The counter electrode (CE) is a crucial component in DSSCs. Its primary role is to facilitate the transfer of electrons from the external circuit to the electrolyte, thereby regenerating the redox couple. For optimal performance, CE should possess excellent catalytic properties for the reduction of triiodide ions, alongside high electrical conductivity. Platinum is commonly used for the construction of CEs, typically deposited on FTO substrate using techniques such as screen printing or doctor blading. However, to lower costs and address the issue of platinum corrosion due to the iodide/triiodide electrolyte, researchers are investigating alternative carbon-based materials. Materials such as graphite, graphene, carbon nanotubes, and carbon black are gaining attention for their low cost, high surface area, superior catalytic activity, excellent electrical conductivity, and resistance to corrosion from the iodide/triiodide electrolyte, making them effective for charge transport.572.3. Photosensitizers
Photosensitizers are one of the critical components in DSSCs. Typically, a photosensitizer is an organometallic58 or organic molecule59 that harvests solar photons and converts them into electrical current. Its photochemical, photophysical, and molecular electronic characteristics significantly influence the electron transfer processes and solar spectrum absorption at the semiconductor interface. One of the most widely used organometallic photosensitizers is Ru(II)-polypyridyl complexes.60 These consist of a central Ru(II) ion coordinated with ancillary ligands that possess at least one anchoring group and a light-harvesting group. When the photosensitizer is photoexcited, an electron moves from the metal center to the π*-orbital, which is often found on the ligand with the anchoring group. This ensues by electron injection from the excited photosensitizer state into TiO2's conduction band (CB) across timescales ranging from femtoseconds to picoseconds.61 Ru(II)-polypyridyl complexes have demonstrated superior performance in DSSCs in terms of both overall conversion efficiency and stability.62,63 DSSCs based on Ru(II)-polypyridyl photosensitizers have achieved a certified power conversion efficiency (PCE) of 12.3% under standard illumination (AM 1.5G, 100 mW cm−2). In lab-scale devices, a record PCE of 14.3% has been reported under similar conditions.64 The current commercially relevant photosensitizers, such as N3, N719, and black dye (N749), are prone to degradation over time,63 Fig. 3. This degradation is at least partly due to the loss of monodentate, labile NCS-ligands.65 | ||
| Fig. 3 Chemical structures of benchmark N3, N719, and black dye (N749) photosensitizers. | ||
Most Ru(II)-polypyridyl complexes exhibit strong MLCT characteristics within the visible region, typically ranging from 400 nm to 600 nm. These complexes possess thermodynamically favorable ground and exciting state potentials, along with extended excited state lifetimes. All these criteria make Ru(II)-polypyridyl complexes versatile and powerful compounds used in a variety of high-tech applications. Their unique properties allow them to play crucial roles in medical treatments,66,67 catalysis,68 sensing,69 electronics,70 imaging,71 and energy conversion.65
3. Working principles of DSSCs
A DSSC operates through the following steps as shown in Fig. 4. First, when the photosensitizer absorbs the energy of the incident light, an electron is excited from the GSOP to ESOP, as illustrated in step (1). If the thermodynamic driving force is sufficient, the electron is injected from the ESOP of the photosensitizer into the CB of the TiO2, as shown in step (2). The injected electron to the CB may move into the external circuit if no recombination happened. The electron travel through the external circuit (e.g. light bulb) to the platinum counter electrode and enters the cell via reduction of the I3−/I− couple at the Pt/electrolyte interface, as shown in step (3). The reduction of the I3−/I− couple subsequently helps to regenerate the dye by reducing it (step 4). All the four steps (1–4) are the recommended main steps that should happen in the DSSC to enhance the PCE of the cell. While steps 5–7 represent electron recombination and are not recommended to happen in the cell because it helps to decrease the cell efficiency. One of the undesired steps is step 5 that represents recombination of electrons from the CB to the photosensitizer to regenerate the dye ground state. Also, relaxation may happen via non-radiative decay processes through electron transfer from the ESOP to the GSOP, as illustrated in step 6. Finally one of the common recombination is the reduction of the redox mediator (I3−/I−) by electrons in the conduction band of TiO2,74 as illustrated in step 7. | ||
| Fig. 4 Working mechanism of DSSCs. The diagram shows the electron flow and energy transfer processes, where blue arrows indicate the desired pathways for efficient energy conversion, and purple arrows highlight charge recombination losses. | ||
The solar spectrum at air mass 1.5, which represents hemispherical solar spectral irradiance, is distributed as follows: approximately 5% in the ultraviolet range (300–400 nm), 43% in the visible spectrum (400–700 nm), and 52% in the near-IR region (700–2500 nm).75 Consequently, an optimal photosensitizer for single-junction photovoltaic cells should exhibit broad absorption across the entire solar spectrum, particularly between 400 nm and 920 nm, to maximize light-to-electricity conversion. Effective light harvesting in the visible and near-IR regions requires that the photosensitizer's ESOP remains above the CB edge of TiO2, enabling efficient electron injection. Additionally, the photosensitizer's GSOP must be sufficiently negative compared to the electrolyte to ensure proper regeneration of the oxidized photosensitizer.76
The schematic diagram in Fig. 4 illustrates the thermodynamically favorable interfacial electron transfer process in a DSSC. When the photosensitizer absorbs a photon, it is excited to a higher energy state (step 1). This is followed by electron injection into the conduction band (CB) of TiO2 (step 2), while the oxidized photosensitizer is simultaneously reduced by the I3−/I− redox couple. An ideal Ru(II)-polypyridyl photosensitizer should have an ESOP higher than that of the TiO2-CB to ensure efficient electron injection. Additionally, its GSOP should be lower than the redox potential of the electrolyte to enable rapid regeneration of the photosensitizer by electron donation from I3−/I−. A lower ESOP value for the dye can decrease electron injection efficiency and enhance recombination reactions, which can adversely affect the performance of the DSSC.
4. Electronic transitions in Ru(II) complexes
Ruthenium(II)-polypyridyl complexes, are widely used as photosensitizers in DSSCs due to their strong light absorption, efficient charge transfer, and long excited-state lifetimes. Ru(II)–polypyridine complexes exhibit unique absorption characteristics due to their d6 electronic configuration and the nature of their polypyridine ligands. The nitrogen atoms in these ligands provide strong σ-donor capabilities, while the aromatic rings contribute π-donor and π*-acceptor orbitals, leading to complex electronic interactions.77 The absorption of light in these complexes results in various electronic transitions, which are crucial for their photovoltaic performance. As illustrated in Fig. 5, the molecular orbital (MO) diagram for a typical octahedral Ru(II) complex consists of σL orbitals (strong bonding, ligand-centered), πL orbitals (bonding, ligand-centered), πM orbitals with t2 t2g symmetry (nonbonding, metal-centered), orbitals with eg symmetry (antibonding, metal-centered),
orbitals with eg symmetry (antibonding, metal-centered),  orbitals (antibonding, ligand-centered), and strongly antibonding
orbitals (antibonding, ligand-centered), and strongly antibonding  orbitals (metal-centered).
orbitals (metal-centered).
 | ||
| Fig. 5 Molecular orbital diagram for the octahedral metal complex showing the main four types of transitions. | ||
Four primary electronic transitions define the absorption properties of Ru(II)-polypyridyl complexes.77 The first transition called metal-centered (MC) which occurs between different d orbitals of the Ru(II), typically transitions from the lower-energy t2g orbitals to the higher-energy eg orbitals.78 The second transition is Metal-to-Ligand Charge Transfer (MLCT) which involve the excitation of an electron from a metal-centered orbital (typically Ru d-orbitals) to a ligand-based π* orbital (usually polypyridyl ligands like bipyridine or phenanthroline). MLCT transitions occur in the visible to near-IR region (400–700 nm), making them essential for light absorption in DSSCs.64,77 The third is Ligand-to-Metal Charge Transfer (LMCT) transitions that occur when an electron is transferred from ligand-based orbitals to the Ru-based orbitals. Typically occurs when the metal is in a high oxidation state and can appear as intense bands in the UV (below 400 nm) but are less common in Ru(II)–polypyridine complexes.79,80 The fourth transition is Ligand-Centered (LC), also known as π–π* transitions which occur within the ligand orbitals. Usually, they appear as intense bands in the UV region.81 Additionally, an uncommon transition, Ligand-to-Ligand Charge Transfer (LLCT), can occur when electrons are transferred between different ligands within the same complex.82
5. Molecular engineering for the optimization of DSSCs
5.1. Evolution of Ru(II) photosensitizers
In 1993, Nazeeruddin and Grätzel introduced a photosensitizer known as cis-dithiocyanato-bis(2,2′-bipyridyl-4,4′-dicarboxylate)ruthenium(II), commercially named N3. DSSCs of N3 photosensitizer achieved a notable solar-to-electric power conversion efficiency of 10%.83,84 Further, the deprotonation of carboxylic protons in the N3 photosensitizer to form N719 resulted in a considerable shift in oxidation and reduction potentials towards more negative values and a significant increase in overall conversion efficiency to be 11.18%.83,85Studies suggest that the fully protonated N3 photosensitizer transfers most of its protons to the TiO2 surface, increasing its positive charge. This enhances electron injection efficiency, resulting in a higher short-circuit photocurrent density (JSC) of 18.20 mA cm−2, compared to 17.73 mA cm−2 for N719.86 However, surface protonation causes a positive shift in the TiO2 conduction band, leading to a reduction in open circuit photovoltage (VOC). As a result, N3 exhibits a lower VOC (0.72 V) than the deprotonated N719 (0.846 V). Extensive research on the impact of protonation on photovoltaic performance has shown that a singly protonated photosensitizer is the most effective in maximizing DSSC efficiency compared to its multiple protonated counterparts.63 Furthermore, in 2001, M. Nazeeruddin introduced a Ru(II)-based N749 photosensitizer known as (black dye) which achieved solar-to-power conversion efficiency of 10.4%, JSC of 20.53 mA cm−2, and VOC of 0.72 V under standard one sun illumination.87 It was suggested that the presence of three thiocyanato (S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CN–) and terpyridine tricarboxylic acid ligands caused a significant red shift in the metal-to-ligand charge transfer (MLCT) band by decreasing the π* energy levels of the terpyridine ligands. This shift enabled the photosensitizer to harvest photons across the entire visible spectrum and extend into the near-infrared region of the solar spectrum, thus creating a panchromatic photosensitizer.87
CN–) and terpyridine tricarboxylic acid ligands caused a significant red shift in the metal-to-ligand charge transfer (MLCT) band by decreasing the π* energy levels of the terpyridine ligands. This shift enabled the photosensitizer to harvest photons across the entire visible spectrum and extend into the near-infrared region of the solar spectrum, thus creating a panchromatic photosensitizer.87
An alternative strategy for improving the light-harvesting capabilities of Ru(II)-polypyridyl photosensitizers involves extending the conjugation of the bipyridine ligand. This modification adjusts the GSOP of the photosensitizer, thereby enhancing its absorption characteristics. In this regard, photosensitizers such as Z-910 and K-19 have been designed by substituting the carboxylic anchoring groups on one of the bipyridyl ligands with extended conjugated moieties such as methoxy-3-vinylbenzene and hexoxy-3-vinylbenzene, respectively (Fig. 6). These photosensitizers exhibit broader MLCT bands and significantly higher molar extinction coefficients than the widely used N719. The presence of methoxy-3-vinylbenzene and hexoxy-3-vinylbenzene moieties help to increase the conjugation in the complex that significantly enhances their ability to absorb light and causes red shift in their absorption spectrum due to the improved electron-donating ability, ultimately boosting the photovoltaic performance of the cell-sensitized with Z-910 and K-19. The π-conjugated photosensitizers Z-910 and K-19 achieved remarkable PCEs (light-to-power conversion efficiencies) of 10.2% and 7.1%,88,89 respectively. Furthermore, K-19 demonstrates exceptional thermal stability, attributed to its long alkyl chain.89 In 2006, K. Jiang introduced HRS-1, a ruthenium(II) complex featuring a hydrophobic moiety (2-hexyl-5-vinylthiophene) that helps to extend the π-conjugation inside the complex. The absorption spectrum of the photosensitizer (HRS-1) demonstrated a notable increase (33%) in the molar extinction coefficient and MLCT red shifted by 10 nm relative to N719.90 Comparative results under identical conditions demonstrated that HRS-1 achieved an overall conversion efficiency of 9.5% compared to 8.9% for N719. The high efficiency and stability of HRS-1 are attributed to the strong electron-donating thienyl groups in the 4,4′,9-di(hexylthienylvinyl)-2,2′-bipyridyl ligand.90 Furthermore, HRS-1 had a greater short-circuit current density (JSC) than N719-sensitized DSCs. This improvement is most likely attributable in part to the enhanced molecular extinction coefficient and red-shifted absorption. In addition, the photosensitizer C101, a homolog of HRS-1 with the ethylene groups between the EDOT and bipyridyl units deleted (as shown in Fig. 6), displayed a 10.33% increase in solar-to-power conversion efficiency. This enhancement is related to the enhanced molar extinction coefficients produced by extending the π-conjugation of the bipyridyl ligands. This boosts the optical absorptivity of the mesoporous titania film and the charge collection yield in dye-sensitized solar cells (DSSCs).91,92 Furthermore, replacing the hexyl chain substituent in C101 with thiohexyl chains resulted in C106, a novel heteroleptic Ru(II)-polypyridyl photosensitizer with a higher power conversion efficiency of 10.57% to 11.4%. The advantage of thioalkyl chains over simple alkyl chains is not yet explained.92
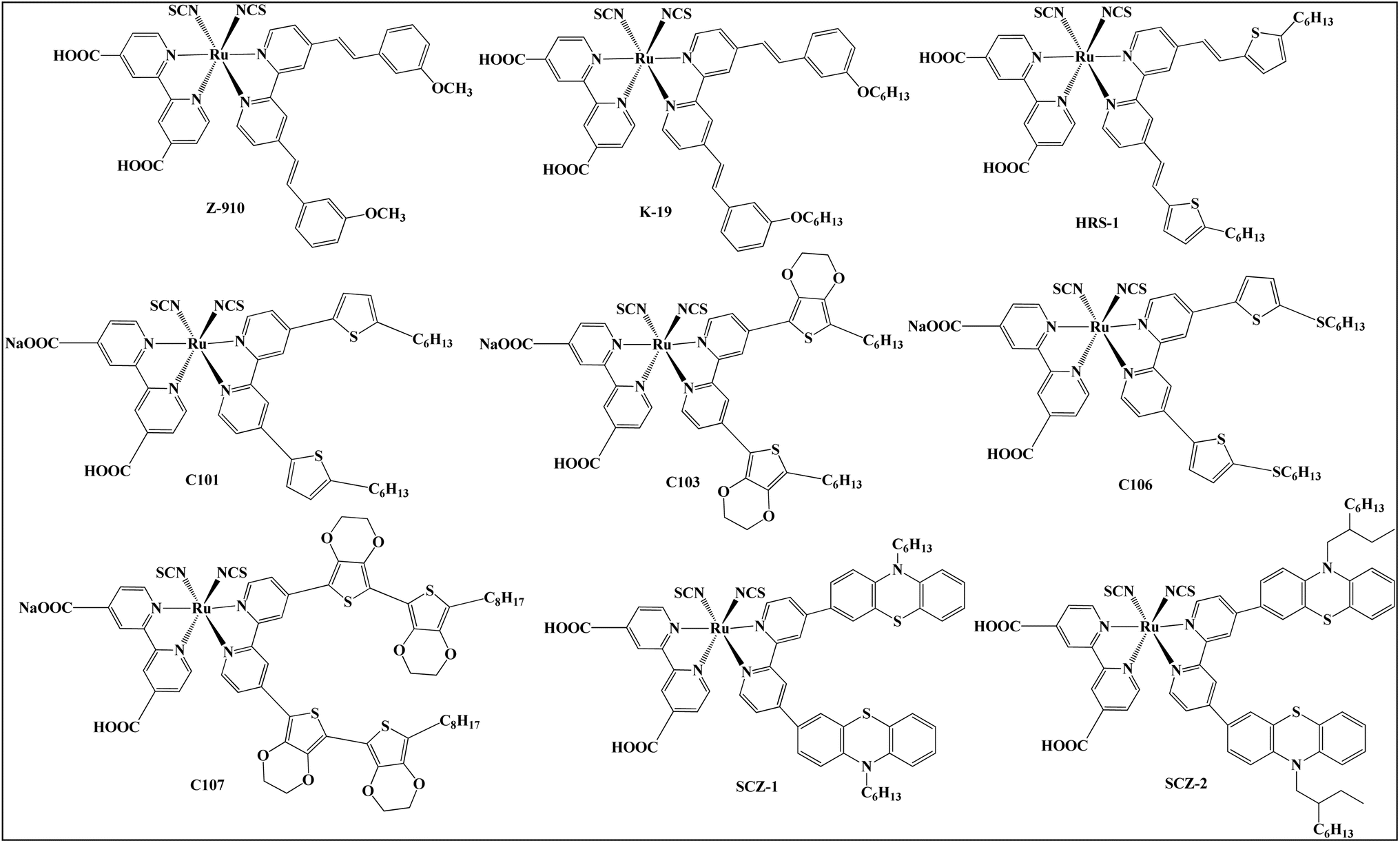 | ||
| Fig. 6 The molecular structures of the Ru(II) complexes photosensitizers. | ||
Additionally, Q. Yu et al. reported two heteroleptic Ru(II) complexes with electron-rich ligands, C103 and C107. The study found that the ruthenium complex C107 had a similar absorption profile to its analogue C103, but with a red shift and a greater molar extinction coefficient (27.4 × 103 M−1 cm−1 at 559 nm) than C103 (20.5 × 103 M−1 cm−1 at 550 nm). As a result, C107 had a higher PCE of 10.7% compared to 10.4% for C103.93 The red shift and enhanced molar extinction coefficient of C107 are due to its longer and bigger ligand, which increases electron-donating capacity. This powerful electron-donating ligand destabilizes the metal's GSOP by transferring electron density to the metal center. Consequently, the overall gap between the GSOP-metal and the ESOP-ligand decreases, leading to a red shift in the absorption spectrum.91 Finally, Z. She et al. reported two Ru(II) complexes, SCZ-1 and SCZ-2, featuring a phenothiazine-modified bipyridine as an ancillary ligand. These photosensitizers (SCZ-1 and SCZ-2) exhibit enhanced light-harvesting capacity due to higher molar extinction coefficients of 1.77 × 104 M−1 cm−1 and 1.66 × 104 M−1 cm−1, respectively, compared to 1.27 × 104 M−1 cm−1 for the reference (N719). Under AM 1.5G irradiation, SCZ-1 and SCZ-2 DSSCs achieved PCEs of 10.4% and 10.2%, respectively, surpassing N719's efficiency of 9.9%. The higher PCEs of SCZ-1 and SCZ-2 are primarily attributed to the increased JSC resulting from the improved absorption coefficient. The photovoltaic characteristics of SCZ-1 and SCZ-2 is comparable, suggesting that the difference in alkyl chains (hexyl or 2-ethyloctyl) on the phenothiazine moiety does not significantly impact their light-absorption ability. Both SCZ-1 and SCZ-2 photosensitizers showed similar VOC values to that of N719, but with higher JSC values. The dye loading amounts on TiO2 are lower for SCZ-1 and SCZ-2 compared to N719, yet their higher extinction coefficients and light-harvesting efficiency contribute to the enhanced JSC.94 The molecular structures of Z-910, K-19, HRS-1, C101, C106, C103, C107, SCZ-1 and SCZ-2 are depicted in Fig. 6 as well as its photovoltaic characteristics is tabulated in Table 1.
| No. | Photosensitizer code | JSC (mA cm−2) | VOC (V) | FF (%) | PCE (%) | Reference |
|---|---|---|---|---|---|---|
| 1 | N3 | 18.20 | 0.720 | 73.00 | 10.00 | 84 |
| 2 | N719 | 17.73 | 0.846 | 72.00 | 11.18 | 85 |
| 3 | N749 | 20.53 | 0.720 | 70.40 | 10.40 | 87 |
| 4 | Z-910 | 7.20 | 0.777 | 76.40 | 10.20 | 88 |
| 5 | K-19 | 13.20 | 0.718 | 74.60 | 7.10 | 89 |
| 6 | HRS-1 | 20.00 | 0.680 | 69.00 | 9.50 | 90 |
| 7 | C101 | 17.75 | 0.749 | 77.70 | 10.33 | 92 |
| 8 | C103 | 18.35 | 0.760 | 74.80 | 10.40 | 93 |
| 9 | C106 | 18.28 | 0.749 | 77.20 | 10.57 | 92 |
| 10 | C107 | 19.18 | 0.739 | 75.10 | 10.70 | 93 |
| 11 | SCZ-1 | 19.85 | 0.761 | 68.80 | 10.40 | 94 |
| 12 | SCZ-2 | 19.88 | 0.761 | 67.70 | 10.20 | 94 |
5.2. Thiocyanate free photosensitizers
Thiocyanate-free Ru(II) complexes for DSSCs are ruthenium-based photosensitizers that do not include thiocyanate (SCN) ligands in their molecular structure as illustrated in Fig. 7. These complexes typically employ other ligands to achieve the desired photophysical and electrochemical properties necessary for efficient light absorption and electron transfer processes in DSSCs. | ||
| Fig. 7 Thiocyanates free Ru(II) polypyridyl heteroleptic complexes. | ||
Wang et al. designed and synthesized thiocyanate-free Ru(II) complexes comprises from 4,4′-dicarboxylic acid-2,2′-bipyridine together with two functionalized pyridyl azolate ancillary ligands consisting of pyrazolate groups namely TFRS-4 provides VOC = 0.75 V, and ahigh PCE of 10.2%.95 Wu et al., modifying TFRS-4 by introducing two trans-oriented quinolinyl pyrazolate ancillaries afforded TFRS-52. The introduced tert-butyl group having quinoline in 6th position of TFRS-52 achieved improved VOC = 860 mV, and power conversion efficiency of 10.1%. The improved high open-circuit voltage is based on the increase of tert-butyl group the upward shift in conduction band edge and higher recombination resistances were noticed.96
Chou et al. designed and synthesized a heteroleptic tridentate ancillary ligand to replace traditional thiocyanates (SCN). This ancillary ligand is used to synthesize thiocyanate free Ru(II) complexes to replace SCN-based photosensitizers such as N3 and N719, which have been widely used in DSSCs. This approach helps to enhance light-harvesting capabilities and stability by replacing (SCN) ligands, which can lead to isomerization issues and reduce the stability of the solar cells. The synthesized TF complexes (TF1-4) exhibit improved photophysical and electrochemical properties compared to N749. They show intense visible absorption bands around 510 nm and broad absorption in the longer-wavelength region at approximately 650 nm and 720 nm, attributed to MLCT and LLCT transitions.97 The TF photosensitizers demonstrate higher PCEs compared to N749, with the highest PCE of 10.7% for TF-3. The VOC of TF photosensitizers range between 0.76–0.79 V, which is significantly higher than that of N749 (SCN-based) that achieved VOC of 0.72 V. The improved performance of TF photosensitizers is attributed to better packing on the TiO2 surface, which reduces charge recombination and enhances VOC. Additionally, the SCN-free design improves the stability of the solar cells, as evidenced by long-term aging experiments.97 Additionally, the negative pole of the dipole moment in complexes (TF1-4) is expected to be located closer to the TiO2 surface, resulting in the uplift of the TiO2 conduction band level and consequently enhanced VOC. Overall, the TF photosensitizers offer a promising alternative to traditional ruthenium-based photosensitizers for DSSCs.
H. Cheema et al. reported thiocyante-free ruthenium dye (HD-11) featuring a mono-dentate ligand of 3-(trifluoromethyl)pyrazole to replace the labile isothiocyanate ligand. HD-11 revealed 50 nm red shift in the low energy metal-to-ligand charge transfer (MLCT) absorption peak compared to N719. However, despite this spectral shift, HD-11 demonstrated a lower PCE (5.2%) than N719 under similar experimental conditions.98
S. Ashraf et al. synthesized thiocyanate-free Ru(II) photosensitizers, SD-15 and SD-16, utilizing 4,4′,5,5′-tetramethyl-1H,1′H-2,2′-bis-imidazole as an ancillary ligand for chelation. These complexes exhibited a red shift in the low-energy metal-to-ligand charge transfer (MLCT) absorption peak. However, under identical experimental conditions, SD-15 and SD-16 exhibited lower power conversion efficiencies (PCEs) of 3.32% and 1.06%, respectively. This decline in performance was attributed to a significantly reduced electron lifetime at the dye/TiO2 interface (0.71 ms), compared to 8.8 ms for N719, as determined by impedance measurements.99
On the other hand, cyclometallating ligands are commonly used in Ru(II) complexes for DSSCs applications. In 2009, Bessho, Grätzel, and colleagues introduced an efficient Ru(II) dye (1) for DSSCs featuring a cyclometallated 2-phenylpyridine ligand, with power conversion efficiencies comparable to N719.100 The complex (1) shows a red-shifted spectral response and higher external quantum efficiency, reaching over 80% at 600 nm and extending to 800 nm. This shift and enhanced absorption are due to the cyclometallated ligand, which destabilizes the HOMO more than the LUMO, leading to a conversion efficiency of 10.1% under standard sunlight. Moreover, Berlinguette reported a series of Ru photosensitizers with a substituted 3-(2′-pyridyl)-1,8-naphthalimide cyclometallated ligand, achieving high power conversion efficiencies (PCEs) in DSSCs. Substituting the pyridine ring with conjugated groups enhances molar absorption extinction coefficients, while the 1,8-naphthalimide fragment reduces electron density on the metal, keeping the Ru(III)/Ru(II) redox couple more positive than 0.8 V versus NHE. This maintains effective dye regeneration by the iodide-based redox mediator. The dye platform allows for modifications that improve light absorption and reduce recombination, achieving PCEs of 7% for complex (2) and 7.3% for complex (3).101 The molecular structures of all thiocyanate free Ru(II) complexes are illustrated in Fig. 7 and Table 2.
| No. | Photosensitizer code | JSC (mA cm−2) | VOC (V) | FF (%) | PCE (%) | Reference |
|---|---|---|---|---|---|---|
| 1 | TFRS-4 | 18.7 | 0.750 | 72.9 | 10.2 | 95 |
| 2 | TFRS-52 | 16.3–16.8 | 0.832–0.860 | 72–78 | 10.1–10.88 | 96 |
| 3 | TF1 | 18.22 | 0.740 | 0.676 | 9.11 | 97 |
| 4 | TF2 | 20.00 | 0.790 | 0.665 | 10.50 | 97 |
| 5 | TF3 | 21.39 | 0.760 | 0.660 | 10.70 | 97 |
| 6 | TF4 | 20.27 | 0.770 | 0.675 | 10.50 | 97 |
| 7 | HD-11 | 12.89 | 0.57 | 0.71 | 5.2 | 98 |
| 8 | SD-15 | 10.20 | 0.58 | 0.56 | 3.32 | 99 |
| 9 | SD-16 | 3.52 | 0.52 | 0.58 | 1.06 | 99 |
| 10 | 1 | 17.00 | 0.800 | 0.74 | 10.10 | 100 |
| 11 | 2 | 10.1 | 0.68 | 0.67 | 7.00 | 101 |
| 12 | 3 | 10.3 | 0.66 | 0.68 | 7.30 | 101 |
5.3. Excited state lifetime optimization
An efficient photosensitizer must have suitable excited state properties, as an electron injection from the photosensitizer's excited state to the semiconductor oxide is essential for the dye-sensitization process. The photosensitizer's excited state should last long enough to allow for proper energy transfer and electron injection. To increase the excited state lifetime of Ru(II)-polypyridyl complexes, specific ligand structural changes are necessary, such as adding electron-donating or withdrawing groups, extending π-conjugation, and utilizing stiff ligands. These changes improve the electronic properties of the complex, stabilize the excited state, and reduce non-radiative decay processes.102 To achieve efficient electron injections, the excited state lifetime should be long enough to allow for considerable electron transfer processes before the photosensitizer returns to the ground state. The photosensitizer's molecular structure affects its lifetime, which can be increased by adding suitable ligands.103 One effective strategy for increasing the excited state lifetime of Ru(II) complexes, particularly the terpyridine photosensitizers [Ru(tpy)2]2+, is to modify the ligands structure. Attaching strong electron-drawing substituents such as SO2Me to the 4′-position of terpyridine ligands can boost room temperature excited state durations to 25 ns, as illustrated in complex (4) in Fig. 8. Also, attaching electron-donating groups can destabilize metal-based GSOPs, leading to non-radiative decay, but they are less effective than electron-withdrawing groups.102 | ||
| Fig. 8 Tridentate heteroleptic Ru(II) complexes (4–9). | ||
A key strategy for enhancing the excited-state lifetime in tridentate-based Ru(II) photosensitizers involves ligand modifications to reduce non-radiative decay pathways.104,105 One effective approach is incorporating tridentate ligands with additional nitrogen atoms, which help stabilize the acceptor orbitals of the metal-to-ligand charge transfer (MLCT) excited state. For instance, complexes featuring the 2-aryl-4,6-bis(2-pyridyl)-s-triazine have demonstrated longer excited-state lifetimes at room temperature compared to conventional terpyridine-based systems. This improvement is attributed to the incorporation of triazine rings and the near-planar conformation of the non-coordinating ring, which lowers the MLCT state's energy and create a substantial energy gap to the metal-centered (MC) state, thereby suppressing non-radiative decay.105 Another promising strategy involves the design of dinuclear complexes incorporating planar bridging ligands. These structures enable extensive electron delocalization within the acceptor ligand upon MLCT excitation, reducing Franck–Condon factors associated with non-radiative decay and significantly prolonging the excited-state lifetime. For example, photosensitizer (5) is a binuclear Ru(II) complex featuring a large, planar bistridentate bridging ligand exhibits exceptionally long-lived emission and a high quantum yield in the near-infrared region, positioning it among the most stable near-IR-emitting Ru(II) complexes.104 The extension of excited-state lifetimes in these complexes is further influenced by electronic interactions between the metal centers and ligands. In dinuclear systems, strong electronic coupling between the two metal centers enhances the stability of the monooxidized species, contributing to prolonged emission lifetimes. A notable example is a tridentate Ru(II) complex incorporating N-heterocyclic carbene (NHC) ligands, designated as (6), which has demonstrated promising photophysical properties which is attributed to the electron-rich nature of NHC ligands.106 The chemical structures of complexes (5 and 6) are illustrated in Fig. 8.
Another approach is to use strong σ-donating ligands, such as carbenes, which are effective electron donors and form strong bonds with the ruthenium metal center. This destabilizes the lowest unoccupied molecular orbital (LUMO) and increases the energy of metal-centered (MC) states. Duati and colleagues reported heteroleptic photosensitizers (7–9) incorporating mixed ligands, with one being 2,2′;6′,2′′-terpyridine (tpy) and the other being one of the following: 2,6-bis([1,2,4]triazol-3-yl)pyridine or 2,6-bis(5-phenyl-[1,2,4]triazol-3-yl)pyridine, or 2,6-bis([1,2,3,4]tetrazol-5-yl)pyridine. These ligands are powerful σ-donors, as illustrated in Fig. 8, and they destabilize the ground state, lowering the energy gap between the metal LUMO and ligand HOMO. Consequently, the photosensitizer emits at a lower energy around 700 nm, with a prolonged excited state lifetime of 77 ns. However, protonation of these triazole rings quenches the excited state by decreasing the electron-donating ability, thereby reversing the process.107 Cyclometalated ligands, being strong σ-donors, destabilize the HOMO of the metal, reducing the gap between the metal HOMO and ligand LUMO, which significantly decreases the excited state lifetime.108,109
5.4. Strategies to minimize charge recombination reactions
Electron recombination at the semiconductor/dye/electrolyte interface is harmful to the photovoltages in DSSCs, leading to a significant reduction in the device's solar-to-power conversion efficiency.110 Electron recombination occurs when electrons deviate from the normal electron injection process (eqn (1)) and can recombine with oxidized dye molecules (eqn (2)) or with the electrolyte (I3−) (eqn (3)).
 | (1) |
| eTiO2− + dye+ → dye | (2) |
| eTiO2− + I3− → 3I− | (3) |
Therefore, minimizing charge recombination between TiO2 and oxidized photosensitizer molecules and/or redox couples in DSSCs is crucial for maximizing device solar-to-power conversion efficiency. Fig. 4 provides an overview of the kinetics, including electron injections, recombination reactions, and dye regeneration for each process.
In traditional DSSCs, the rate of electron recombination is significantly slower than the electron injection process from the excited photosensitizer into the CB of the semiconductor (TiO2). However, due to the relatively sluggish diffusion through the nanoporous TiO2, electrons tend to accumulate in the semiconductor/electrolyte interface. This proximity increases the likelihood of recombination with oxidized dye molecules or redox species within the electrolyte.111 Dye regeneration efficiency is primarily governed by two key factors. The first is the molecular structure of the photosensitizer, which plays a crucial role in determining its ground-state oxidation potential (GSOP), directly impacting the regeneration process. The second factor is the redox potential of the electrolyte, which must be appropriately aligned to facilitate efficient electron donation for regenerating the oxidized dye. To sustain a strong photocurrent, the regeneration process must outpace recombination reactions.112 Therefore, minimizing charge recombination between TiO2 and oxidized photosensitizer molecules and/or redox couples in DSSCs is crucial for maximizing device solar-to-power conversion efficiency. Fig. 4 provides an overview of the kinetics, including electron injections, recombination reactions, and dye regeneration for each process.
The effect of photosensitizer molecular structure on recombination reactions was studied. Research indicates that adding electron-donating groups to photosensitizers and separating dye cation centers from the TiO2 surface will greatly reduce charge recombination,113 resulting in long-lasting charge separation at the dye/TiO2 contact.114 Haque et al. confirm this by using transient absorption spectroscopy to investigate the charge-recombination dynamics of three Ru-based multifunctional photosensitizers (10–12) with varying triphenylamine antennas (Fig. 9). Photosensitizers (10 and 11) comprise electron-donating units with low molecular weight: triphenylamine (TPA) and N,N′-diphenyl-N,N′-bis(3-methylphenyl)-[1,1′-biphenyl]-4,4′-diamine (TPD), respectively. In contrast, photosensitizer (12) consists of poly(vinyl triphenylamine) chains (polyTPA) with around 100 repeat units linked to one of the Ru(II) core's bipyridine units. The recombination kinetics of these complexes vary significantly depending on the physical separation between the dye cation core and the TiO2 semiconductor oxide surface. The photosensitizer with the larger antenna had a substantially longer recombination half-time. Complexes 10, 11, and 12 exhibited recombination half-times of 350 μs, 5 ms, and 4 s, respectively. This shows that the spatial separation of the dye cation from the TiO2 surface, accomplished through the multistep migration of the dye moiety away from the TiO2 interface, is the key factor controlling charge transfer at the dye/TiO2 interface.114
 | ||
| Fig. 9 Ru(II) bipyridyl photosensitizer (10–12) incorporating different triphenylamine antenna and phenanthroline-based Ru(II) complexes (RuHDA1–3). | ||
Another strategy is to use monosubstituted ancillary ligands, which have been shown to effectively suppress recombination processes. Abdellah et al. synthesized two high molar extinction coefficient monosubstituted-bipy Ru(II) complexes, IA-5 and IA-6,115 as illustrated in Fig. 9. These complexes use an electron acceptor ancillary ligand in IA-5 and an electron donor ancillary ligand in IA-6 to study how these ligands and the quantity of anchoring groups (COOH) affect the photovoltaic characteristics of DSSCs. The results showed that the highly conjugated and strong donor auxiliary ligand in IA-6 reduced charge recombination and increased overall power conversion efficiency (PCE) to 7.81% at a VOC of 0.69 V. In contrast, the electron-accepting auxiliary ligand in IA-5 produced a lower PCE of 6.20% and a VOC of 0.62 V, outperforming the reference N719.115
Another way is to incorporate long aliphatic chains and bulky groups into the photosensitizer's auxiliary ligand to reduce recombination losses. Ashraf et al. created two heteroleptic polypyridyl Ru(II) complexes, SD-5 and SD-6, with electron-donating N-alkyl-2-phenylindole moieties in the auxiliary ligand, as illustrated in Fig. 9. In DSSCs, SD-6 had an overall efficiency of 8.14% with a VOC of 0.675 V, beating SD-5, which had a PCE of 4.99% and a VOC of 0.59 V. SD-6 outperforms SD-5 because of its long alkyl chains (–C18H37), which limit dye aggregation and suppress charge recombination, resulting in greater PCE and VOC values.116 The addition of indole moieties with extended alkyl chains to SD-6 increased performance compared to N719.116
On the other hand, Pashaei et al. conducted a similar investigation, synthesizing three phenanthroline-based Ru(II) complexes (RuHDA1–3) with long-chain auxiliary ligands and varied types and numbers of ancillary and anchoring ligands as illustrated in Fig. 9.117 The study revealed that RuHDA3, which has one auxiliary ligand and one anchoring ligand with two carboxylic acid groups and two NCS groups, is the most effective photosensitizer. RuHDA3 achieved a VOC of 0.65 V, compared to 0.58 V for RuHDA1 and 0.55 V for RuHDA2, and a PCE of 6.11%, vs. 4.41% and 3.40% for RuHDA1 and RuHDA2, respectively. Time-Resolved Absorption Spectroscopy (TAS) was utilized to investigate electron transfer kinetics in DSSCs and to link molecular structure to regeneration and recombination lifetimes. The findings demonstrated that balancing auxiliary and anchoring ligands is critical for reducing recombination losses and increasing DSSC efficiency.117 The final strategy involves treating photosensitizer-coated TiO2 electrodes with pyridine derivatives, providing a simple and effective method for increasing DSSC performance. This technique lowers recombination rates and raises open-circuit voltage, resulting in a considerable boost in overall DSSC efficiency. Huang et al. found that treating N3 photosensitizer-coated TiO2 electrodes with pyridine derivatives like 4-tert-butylpyridine, 2-vinylpyridine, and poly(2-vinylpyridine) considerably improved photovoltaic performance. The VOC exhibited an approximate 28% increase, reaching 0.73 V, while the PCE improved by 29.3%, achieving 7.5% compared to untreated electrodes. This enhancement in photovoltaic performance is ascribed to the pyridine derivatives, which may reduce the recombination rate.118
6. The role of co-adsorbents
The utilization of co-adsorbents plays a crucial role in enhancing the performance of DSSCs by mitigating recombination processes, preventing aggregation of the photosensitizer, and forming a dense protective layer on TiO2.119 The co-adsorbent enhances device efficiency in two ways. First, due to its small size, it acts as a filler, occupying the interspaces between the photosensitizer molecules to form a compact layer on the TiO2 surface. Second, it reduces aggregation among photosensitizer molecules, which can lower electron injection efficiency and increase electron recombination reactions at the TiO2/electrolyte interface. Co-adsorbents used with the photosensitizer significantly improve the device's open-circuit voltage (VOC).120 The selection of appropriate co-adsorbents is critical for improving the efficiency of DSSCs. An ideal co-adsorbent should possess a molecular structure that prevents competitive adsorption between dye molecules and minimizes dye aggregation on the TiO2 surface. Compounds such as chenodeoxycholic acid (CDCA) and hexadecylmalonic acid (HDMA) have demonstrated effectiveness in achieving uniform dye distribution, thereby maximizing light absorption. Additionally, co-adsorbents should form a compact monolayer on the TiO2 surface to mitigate electron recombination with I−/I3−.121 Anchoring groups, including phosphonate and carboxylate functional groups, enhance adsorption stability by strongly binding to the TiO2 surface.122 Furthermore, co-adsorbents with hydrophobic chains can reduce dye aggregation through repellent interactions, contributing to improved efficiency.123 Optimizing the concentration of co-adsorbents is essential to achieving optimal DSSC performance.124 Although a high molar extinction coefficient is not a primary requirement, co-adsorbents indirectly enhance light harvesting by facilitating optimal dye distribution. By fine-tuning these properties, co-adsorbents play a key role in maximizing the overall efficiency of DSSCs.Various co-adsorbents have been utilized to enhance the performance of DSSCs. Lim et al. demonstrated that incorporating stearic acid as a co-adsorbent with the N719 photosensitizer markedly enhances photovoltaic performance. Solar devices showed a notable 25% increase in short circuit current (JSC) and solar-to-power conversion efficiencies compared to control devices. This improvement is attributed to stearic acid's low dipole moment and high solubility, which slows down the photosensitizer anchoring rate during competitive adsorption. Consequently, a dense and strongly bonded dye layer form on the TiO2 surface, reducing photosensitizer aggregation and leading to enhanced device performance.125 Song et al. introduced triaryl amine-based co-adsorbent, 4-(bis(9,9-dimethyl-9H-fluoren-2-yl)amino)benzoic acid (HC-acid), designed to enhance the efficiency of DSSCs. HC-acid serves as an alternative to deoxycholic acid (DCA), offering dual functions: preventing π–π stacking of organic dye molecules and enhancing light harvesting at shorter wavelengths. When used with the organic dye NKX2677, HC-acid significantly improves the solar cell's efficiency, achieving a PCE of 9.09% under AM 1.5G conditions. This represents a 38% increase in efficiency compared to NKX2677 alone, attributed to a 20% increase in JSC and an 11% increase in VOC.126 Han et al. introduced innovative donor–acceptor type co-adsorbents (Y1 and Y2). These co-adsorbents consist of three units: an electron-donating group, a π-spacer, and an electron-accepting group. They effectively address challenges such as competitive light absorption by I−/I3−, prevent dye aggregation, and minimize charge recombination. The co-adsorbent Y1, with strong absorption around 390 nm, effectively restored the dip in the IPCE spectrum caused by I−/I3− in black dye-sensitized solar cell. In addition, the use of Y1 as co-adsorbent led to an increase of approximately 20 mV in open-circuit voltage (VOC) and an enhancement in short-circuit current density (JSC), resulting in a power conversion efficiency (PCE) of 11.28%, compared to 10.70% in the absence of the co-adsorbent. Meanwhile, Y2 provided a comparatively lower performance boost. The butyloxyl chains in Y1 helped prevent dye aggregation and reduce recombination, contributing to improved performance.127 Finally, Wang et al. discovered that incorporating 1-decylphosphonic acid as a co-adsorbent alongside the Ru(II)-bipyridyl complex (Z907) significantly enhanced the stability, short-circuit photocurrent density (JSC), and overall efficiency of the device. DPA helps to improve conversion efficiency from 6.8% without DPA to 7.3% as well as helps to improve the cells stability under thermal stress because DPA strongly binds to the oxide surface through P–O–metal bonds, reducing hydrophilic sites available for water adsorption.128 The chemical structure of the Ru(II)-bipyridyl complex (Z907) is depicted in Fig. 10.
 | ||
| Fig. 10 The molecular structures of the well-known co-adsorbents used in DSSCs, and Ru(II)-bipyridyl complex (Z907). | ||
The molecular structures of some co-adsorbents employed in DSSCs to enhance their performance such as stearic acid, 4-(bis(9,9-dimethyl-9H-fluoren-2-yl)amino)benzoic acid, Y1, Y2, 1-decylphosphonic acid, deoxycholic acid,129 chenodeoxycholic acid,130 3-phenylpropionic acid,131 4-guanidinobutyric acid,132 hexadecylmalonic acid,133 and 4-aminobutyric acid134 are listed in Fig. 10. In conclusion, although co-adsorbents play a beneficial role in DSSCs, their improper application can disrupt protective layers and enhance recombination processes, ultimately compromising the performance of the solar cell, especially in the absence of dye aggregation.135
7. Redox electrolyte function in DSSCs
Redox electrolytes are crucial components of DSSCs, significantly impacting both the device's performance and long-term stability. In DSSCs, the redox couple acts as a redox mediator in liquid or gel electrolytes, while in solid electrolytes, the hole transport material (HTM) serves as the hole mediator.136 The role of electrolytes becomes crucial following photon absorption, as the dye, upon excitement, swiftly injects electrons into the CB of TiO2, leading to its oxidation. The oxidized dye is then regenerated by receiving electrons from the reduced species in the electrolyte. The oxidized species of the redox mediator migrate towards the counter electrode, whereas the reduced species migrate from the counter electrode to the oxidized dye, primarily by diffusion. In DSSCs, the electrolyte plays a dual role: regenerating the oxidized dye and replenishing the reduced species of the redox couple at the CE.7.1. Electrolyte (I−/I3−) drawbacks
The interaction between photosensitizers and electrolytes can enhance charge recombination reactions, thereby diminishing the performance of DSSCs. Reynal et al. investigated a series of heteroleptic Ru(II)-polypyridyl complexes (12a–d) with various substituents, including –H, –CH3, –NH2, and –NO2, as illustrated in Fig. 11. These photosensitizers were found to significantly reduce the open-circuit voltage (VOC) when used with the I−/I3− electrolyte, particularly in complexes 12c and 12d, which incorporate –NH2 and –NO2 groups on the phenanthroline ligand. These groups caused significant changes in the interfacial charge transfer processes, thereby limiting device performance. The VOC was notably decreased for complexes 12c and 12d, with values of 0.48 V and 0.44 V, respectively, compared to 0.67 V for complexes 12a and 12b. The decline in VOC was ascribed to ineffective electron regeneration from I−/I3−, primarily due to charge recombination caused by the quenching the quenching of the excited-state photosensitizers (12a–d) by the I−/I3−.137 This highlights the strong correlation between open-circuit voltage (VOC) and the molecular structure of photosensitizers. The molecular structure greatly determines the strength of recombination processes between the photosensitizer and the I−/I3− electrolyte.138,139 Therefore, to prevent the redox pair from interacting with the adsorbed photosensitizer on the TiO2 surface, it's important to reduce complex formation between the photosensitizer and the electrolyte through careful molecular engineering of the photosensitizers. | ||
| Fig. 11 Molecular structure of Ru(II)-polypyridyl photosensitizers (12a–d). Where: R1 = H, R2 = H (12a); R1 = CH3, R2 = CH3 (12b); R1 = H, R2 = NH2 (12c); R1 = H, R2 = NO2 (12d). | ||
7.2. Developed strategy for efficient redox couples
In DSSCs, liquid electrolytes act as liquid redox systems, with the redox couple dissolved in aqueous or organic solvent media. The iodide/triiodide (I−/I3−) redox couple is the most often utilized electrolyte in DSSCs. However, it restricts the possible open-circuit voltage (VOC) to roughly 0.70 to 0.80 V due to high energy loss during the photosensitizer regeneration process, which is a key disadvantage of current DSSC technology. Replacing the I−/I3− electrolyte with more efficient redox couple can boost the device's photovoltage. To enhance the photovoltage, alternative redox couples with improved efficiency can replace the I−/I3− system. However, for optimal photocurrent generation, the regeneration of the photosensitizer by the redox mediator must occur at a faster rate than the back-transfer of electrons from TiO2 to the oxidized dye.140 Ensuring a sufficient driving force for dye regeneration is crucial to prevent recombination of injected electrons with the oxidized dye. If the driving force is inadequate, recombination can reduce photovoltaic performance by decreasing both JSC and VOC. Conversely, a strong driving force can limit VOC. Thus, efficient dye regeneration requires an ideal driving force of roughly 20–25 kJ mol−1. This can be achieved by selecting a redox couple matched to the specific photosensitizer.141 Therefore, to improve the open-circuit potential and photocurrent of DSSCs, innovative redox mediators with a higher redox potential while preserving the ideal driving force for dye regeneration are required. The general technique for raising VOC entails concurrently lowering the dye's ground state oxidation potential (GSOP) level and the redox shuttle's reduction potential to a higher potential (Fig. 12). This technique optimizes the driving force for dye regeneration, which varies depending on the redox couple used. | ||
| Fig. 12 Shows a strategy for boosting the VOC by concurrently moving the redox couple potential and the dye's ground-state oxidation potential (GSOP) to higher positive values, resulting in an ideal driving force for rapid dye regeneration. | ||
Since the discovery of DSSCs, various redox couples have been explored and used as potential redox shuttles in DSSCs, including: (1) halogenated redox couples such as I−/I3−142 and Br3−/Br−;143 (2) electrolytes incorporating transition-metal complexes, such as Co3+/Co2+,144 Cu2+/Cu+,145 Fe3+/Fe2+,146 and ferrocenium/ferrocene (Fc+/Fc);147 and (3) organic radical-based redox couples like TEMPO/TEMPO+148 and T2/T−.149 The chemical structures and the standard redox potentials of some developed redox shuttles used in DSSCs are illustrated in Fig. 13.
 | ||
| Fig. 13 Chemical structures of the most common redox couples used in DSSCs along with their standard redox potential. Where: TEMPO is 2,2,6,6-tetramethyl-1-piperidinyloxy & T2 (dimer of 5-mercapto-1-methyltetrazole ion) & T− (5-mercapto-1-methyltetrazole ion). | ||
7.3. Electrolyte working principle
To better understand the working principles of electrolytes in DSSCs, the most effective redox couples, including I−/I3−, cobalt complex electrolytes (Co2+/Co3+), and copper complex electrolytes (Cu2+/Cu+) were explored in depth, as shown in Fig. 14. Among these, the I−/I3− redox couple is recognized for its outstanding efficiency and remains the most widely used electrolyte since its inception, primarily due to its favorable standard redox potential of approximately 0.35 V relative to the normal hydrogen electrode (NHE). This optimal potential of I−/I3− enhances the driving force for dye regeneration across most photosensitizers. For example, the oxidation potential of I−/I3− relative to the oxidation potential of Ru(dcbpy)2(NCS)2 photosensitizer such as N3 or N719, which is around 1.1 V vs. NHE makes the driving force of regeneration of 0.75 V.150 This substantial driving force (0.75 V) promotes efficient dye regeneration while mitigating the recombination of injected electrons in TiO2 with the electrolyte, ultimately enhancing the JSC of the device. | ||
| Fig. 14 Redox potential values and the corresponding VOC values of the most common and efficient redox couples used in DSSCs, including I3−/I−, [Co(bpy)3]3+/2+, and [Cu(tmpy)3]2+/1+. | ||
Additionally, I−/I3− redox electrolytes absorb light in the blue area of the visible spectrum, competing with dye absorption and resulting in decreased photocurrent.151 The corrosive behavior of I−/I3− redox electrolytes, especially towards metal current collectors like silver (Ag), provides considerable hurdles for scaling up DSSCs to commercial modules.148 Studies on metal thin films in the presence of I−/I3− redox electrolytes demonstrate that metals like Ag, Au, and Al are extremely vulnerable to corrosion, while Pt, Ti, and Ni exhibit lesser corrosiveness.152 The I−/I3− redox pair has substantial problems such as competing light absorption, metal corrosion, and limited photovoltage. To solve these difficulties, an appropriate alternative to the I−/I3− redox pair is required.
To replace the I−/I3− electrolyte, researchers have explored and utilized several redox couples that are less corrosive and possess suitable redox potentials to achieve high VOC with promising solar-to-power conversion efficiencies.153,154 Saap et al. achieved a 12.3% efficiency in lab-scale DSSCs using the N3 sensitizer under 1 sun illumination by employing cobalt complexes with substituted polypyridine ligands as potential alternatives to the volatile and corrosive iodide/triiodide (I−/I3−) redox couple, the chemical structures of the developed polypyridyl cobalt(II/III) redox mediators incorporating are illustrated in Fig. 15. These cobalt complexes exhibit extinction coefficients around 102 M−1 cm−1 in the visible spectrum, minimizing interference with the light-harvesting capability of the photosensitizer.155 Cobalt complexes offer distinct advantages over the I−/I3− system, as the redox potential of the cobalt mediator can be fine-tuned by the ligands surrounding cobalt center. Additionally, bulky groups can be introduced as insulating spacers between the ligands, reducing recombination reactions between the I3− and titania, which ultimately leads to an increase in the VOC. However, the best-performing mediator, based on tris(4,4′-di-tert-butyl-2,2′-dipyridyl)cobalt(II/III), achieved efficiency of up to 80% when compared to the standard iodide/triiodide mediator.
 | ||
| Fig. 15 Molecular structure of the terpyridine, bipyridine, and phenanthroline complexes of cobalt(II) mediator used in DSSCs with N3 photosensitizer. | ||
In conclusion, cobalt (Co2+/Co3+) mediator is useful since it is commercially available, easy to synthesize, and has nonvolatile and noncorrosive electron-transfer properties in DSSCs. Furthermore, cobalt polypyridine complexes are attractive as redox shuttles due to their low visible light absorption, low metal corrosion, outer-sphere one-electron redox chemistry, and higher positive redox potential.156 Cobalt (Co2+/Co3+) electrolytes with bulky groups have much lower ion mobility during diffusion than iodine ions.157
7.4. Electrolyte additives
In DSSCs, various nitrogen-containing heterocyclic derivatives such as imidazole, triazole, benzimidazole, and pyridine are commonly used as electrolyte additives to enhance photovoltaic performance.158 The effects of these additives are well-documented and include modifying the redox couple potential, shifting the conduction band (CB) level of TiO2, forming a compact layer on the TiO2 surface, reducing dye aggregation, and blocking the TiO2 surface from the electrolyte. These additives adsorb onto the TiO2 surface, elevating the conduction band level and increasing VOC. The extent of this shift largely depends on the electron-donating power, charge density, and donating capability of the additives. However, while raising the TiO2 conduction band level increases VOC, it also reduces electron injection efficiency from the photosensitizers to TiO2, resulting in a decrease in the short-circuit current density (JSC).159 Kusama et al. investigated the impact of benzimidazole additives on DSSCs performance based on N3 photosensitizer and an I−/I3− redox electrolyte in acetonitrile. Their findings revealed that the addition of benzimidazoles enhances the VOC and fill factor (ff) but reduces the JSC. For example, 2-amino-1-methylbenzimidazole yielded the highest VOC of 0.83 V but the lowest JSC of 10.3 mA cm−2. Computational calculations show that higher partial charges of nitrogen atoms in benzimidazoles increase VOC but decrease JSC. Smaller benzimidazole molecules and those with a larger dipole moment difference from acetonitrile improves VOC and JSC, respectively. The highest efficiency of 7.6% was achieved with 5-chloro-1-ethyl-2-methylbenzimidazole and 2-(1-hydroxyethyl)benzimidazole under 100 mW cm−2 illumination. These effects are attributed to interactions between benzimidazoles and the TiO2, altering dark currents and the flatband potential of TiO2.160 The molecular structures of the benzimidazole-based additives used in electrolytes are listed in Fig. 16. | ||
| Fig. 16 Molecular structures of benzimidazole derivatives as additives in electrolyte. | ||
Kopidakis et al. explores the effect of guanidinium as an adsorbent on the performance of DSSCs. Specifically, it examines how guanidinium influences recombination and band-edge movement in these cells. Guanidinium added to the electrolyte was found to slow down recombination by a factor of about 20, which would typically increase the VOC. However, guanidinium also causes the TiO2 band edges to shift downward by 100 mV, which would decrease VOC. Despite this opposing effect, the overall impact of guanidinium results in a net improvement in VOC of about 20 mV due to the dominant effect of reduced recombination. This improvement is attributed to the dominant effect of reduced recombination over the unfavorable band-edge shift. The downward band-edge shift is likely due to the positive charge of the guanidinium cation interacting with the TiO2 surface, leading to a buildup of positive surface charges.161 Furthermore, Zhang et al. investigated the impact of 4-guanidinobutyric acid (GBA) on the performance of DSSCs fabricated with an amphiphilic ruthenium photosensitizer (K-19) and GBA. Their study demonstrated a notable increase in the VOC by ∼50 mV compared to cells without GBA. The inclusion of GBA caused a shift in the CB of TiO2 to a more negative potential, thereby reducing the back-reaction between electrons in the TiO2 conduction band and I3− in the electrolyte. This modification resulted in enhanced stability and efficiency, achieving a PCE of ∼8% under simulated sunlight.162 The chemical structures of the guanidinium cation and 4-guanidinobutyric acid are presented on Fig. 17.
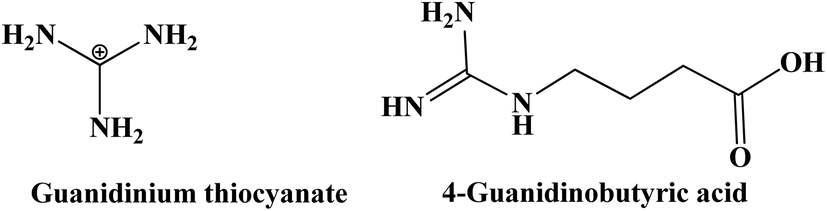 | ||
| Fig. 17 Chemical structure of guanidinium cation and 4-guanidinobutyric acid additives. | ||
Furthermore, Kusama et al. reported that using 10 different aminotriazole derivatives as additives improved the VOC and solar-to-power conversion efficiency (η) of DSSCs, although it reduced the JSC. The highest efficiency (η) of 7.6% was achieved by adding 3-amino-1H-1,2,4-triazole to the electrolyte mixture.159 The molecular structures of the ten aminotriazole derivatives additives used as electrolyte to additives to improve the VOC of DSSCs are illustrated in Fig. 18.
 | ||
| Fig. 18 Molecular structures of aminotriazole derivatives used as additives in electrolyte. | ||
Nazeeruddin et al. explored the addition of 4-tert-butylpyridine and 3-methyl-2-oxazolidinone in conjunction to enhance the performance of DSSCs based on N3 photosensitizer. Initially, without treating the N3-covered film with 4-tert-butylpyridine, the VOC was 0.38 V, and the PCE was approximately 3.7%. After treating the dye-coated TiO2 film with 4-tert-butylpyridine, VOC increases to 0.66 V, and the PCE improved to about 8.5%. The high VOC is attributed to the dark current suppressing at the semiconductor/electrolyte junction. This suppression occurs because 4-tert-butylpyridine adsorbs onto the TiO2 surface, blocking surface states that facilitate the reduction of I3− by conduction band electrons, thus reducing recombination and enhancing efficiency. Further improvement is achieved by using a mixture of acetonitrile and 3-methyl-2-oxazolidinone (90/10, v/v) as the electrolyte solvent, which increases VOC to 0.72 V and maintains a high PCE of ∼10%.163 The addition of 3-methyl-2-oxazolidinone to the electrolyte improves the overall stability and efficiency of the cell by optimizing the electrolyte's properties, such as viscosity and polarity, which can enhance ion mobility and reduce recombination losses. Finally, Afrooz et al. investigated the use of diethyl oxalate (DEOX) as an effective additive in dye-sensitized solar cells (DSSCs) with iodide/triiodide (I−/I−3) electrolytes. They examined the impact of DEOX and 4-tert-butylpyridine (TBP) additives on DSSC performance by comparing various electrolyte compositions. Their findings revealed that electrolytes containing both DEOX and TBP additives exhibited the best photovoltaic performance. The addition of DEOX led to increased short-circuit current density and open-circuit voltage, resulting in improved overall efficiency. They proposed that DEOX forms a complex with iodine molecules, influencing the electron transfer processes in the cell.164
8. Conclusion
This paper offers a comprehensive review of optimization strategies for dye-sensitized solar cells (DSSCs), highlighting recent advancements across key components, including Ru(II) photosensitizers, electrolytes, co-adsorbents, and additives. The review underscores the critical role of modifying Ru(II) photosensitizers to enhance excited state lifetimes and minimize charge recombination. By incorporating electron-donating or withdrawing groups and utilizing ligands with extended π-systems, significant improvements in electron injection efficiency and reduced non-radiative decay have been achieved, leading to enhanced photogenerated current and overall cell performance. Structural modifications of Ru(II) photosensitizers concluded that the most effective photosensitizer is one with an architecture consisting of one auxiliary ligand and one anchoring ligand, incorporating two carboxylic acid groups and two thiocyanate (SCN) groups. Additionally, the development of thiocyanate-free Ru(II) complexes and cyclometallated polypyridyl Ru(II) complexes has shown promising potential in enhancing the long-term stability of DSSCs. Furthermore, charge recombination is addressed not only through structural modifications of Ru(II) photosensitizers and careful engineering of the semiconductor/electrolyte interface but also through the exploration of efficient electrolytes. These electrolytes are designed to outperform conventional I−/I3− electrolytes, which can cause corrosion, light absorption competition with the photosensitizer, suppress the open-circuit voltage (VOC) to about 0.70 to 0.80 V, and decrease overall power conversion efficiency. Various alternative redox couples have been encouraged such as transition-metal complexes (e.g., Co3+/Co2+, Cu2+/Cu+). These alternatives aim to optimize the redox potential and improve overall DSSC performance. Finally, the development of convenient co-adsorbents has been instrumental in preventing photosensitizer aggregation, thereby improving device stability and efficiency. In summary, these advancements represent a significant and forward-thinking approach to optimizing DSSCs. Continued refinement of molecular designs, reduction of charge recombination, and innovation in co-adsorbents and redox electrolytes are essential for achieving higher efficiencies and enhancing commercial viability. This review not only highlights the substantial progress made but also sets the stage for future breakthroughs that will further advance DSSC technology and its application in the renewable energy sector.Data availability
No new data were created or analyzed in this study. Data sharing is not applicable to this article.Conflicts of interest
The author has no conflict to declare.References
- R. Munir, A. F. Zahoor, M. N. Anjum, U. Nazeer, A. U. Haq, A. Mansha, A. R. Chaudhry and A. Irfan, Top. Curr. Chem., 2024, 383, 1–54, DOI:10.1007/S41061-024-00488-3.
- I. M. Abdellah and A. El-Shafei, J. Photochem. Photobiol., A, 2020, 387, 112133, DOI:10.1016/j.jphotochem.2019.112133.
- A. Dutta, M. Nayak, A. J. Akhtar and S. K. Saha, J. Phys. Chem. Solids, 2025, 199, 112569, DOI:10.1016/J.JPCS.2025.112569.
- I. M. Abdellah and A. El-Shafei, Sol. Energy, 2020, 198, 25–35, DOI:10.1016/j.solener.2020.01.040.
- National Oceanic and Atmospheric Administration, Energy on a Sphere, 2017, https://sos.noaa.gov/catalog/live-programs/energy-on-a-sphere/ Search PubMed.
- U.S. EIA, International Energy Statistics, 2024, https://www.eia.gov/international/data/world Search PubMed.
- M. Bilgili, S. Tumse and S. Nar, Arabian J. Sci. Eng., 2024, 49, 14503–14531, DOI:10.1007/S13369-024-09390-Y.
- M. A. Fazal and S. Rubaiee, Sol. Energy, 2023, 258, 203–219, DOI:10.1016/J.SOLENER.2023.04.066.
- I. M. Abdellah, T. H. Chowdhury, J.-J. Lee, A. Islam, M. K. Nazeeruddin, M. Gräetzel and A. El-Shafei, Sustainable Energy Fuels, 2021, 5(1), 199–211, 10.1039/d0se01323d.
- R. Kesavan, I. M. Abdellah, S. P. Singh, A. El-Shafei and A. V. Adhikari, Phys. Chem. Chem. Phys., 2019, 21, 10603–10613, 10.1039/c9cp01032g.
- (a) E. Danladi, E. E. Oguzie and F. I. Ezema, Multiscale Multidiscip. Model. Exp. Des., 2024, 8, 1–31, DOI:10.1007/S41939-024-00699-7; (b) I. M. Abdellah and A. El-Shafei, New J. Chem., 2024, 48, 18666–18682, 10.1039/D4NJ03777D; (c) A. I. Abdellah, A. I. Koraiem and A. El-Shafei, Dyes Pigm., 2019, 164, 244–256, DOI:10.1016/J.DYEPIG.2019.01.035.
- R. Sasikumar, S. Thirumalaisamy, B. Kim and B. Hwang, Renewable Sustainable Energy Rev., 2024, 199, 114549, DOI:10.1016/J.RSER.2024.114549.
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740, DOI:10.1038/353737a0.
- R. Munir, A. F. Zahoor, M. N. Anjum, U. Nazeer, A. U. Haq, A. Mansha, A. R. Chaudhry and A. Irfan, Top. Curr. Chem., 2024, 383, 1–54, DOI:10.1007/S41061-024-00488-3.
- S. S. Singh and B. Shougaijam, Lect. Notes Electr. Eng., 2022, vol. 850, pp. 85–109, DOI:10.1007/978-981-16-9124-9_5.
- V. Armel, M. Forsyth, D. R. MacFarlane and J. M. Pringle, Energy Environ. Sci., 2011, 4, 2234–2239, 10.1039/C1EE01062J.
- C. Aumaitre, D. Joly, D. Aldakov and R. Demadrille, in The Future of Semiconductor Oxides in Next-Generation Solar Cells, Elsevier, 2018, pp. 85–115, DOI:10.1016/B978-0-12-811165-9.00003-X.
- C. Law, S. C. Pathirana, X. Li, A. Y. Anderson, P. R. F. Barnes, A. Listorti, T. H. Ghaddar and B. C. O'Regan, Adv. Mater., 2010, 22, 4505–4509, DOI:10.1002/adma.201001703.
- C.-Y. Chen, S.-J. Wu, C.-G. Wu, J.-G. Chen and K.-C. Ho, Angew. Chem., 2006, 118, 5954–5957, DOI:10.1002/ange.200601463.
- M. Hussain, A. Islam, I. Bedja, R. K. Gupta, L. Han and A. El-Shafei, Phys. Chem. Chem. Phys., 2014, 16, 14874–14881, 10.1039/C4CP00907J.
- S. B. Darling and F. You, RSC Adv., 2013, 3(39), 17633–17648, 10.1039/c3ra42989j.
- K. Karuppasamy, K. Prasanna, V. R. Jothi, D. Vikraman, S. Hussain, J. H. Hwang and H. S. Kim, Nanomaterials, 2020, 10(11), 2106, DOI:10.3390/nano10112106.
- T. Jayaraman, A. P. Murthy, V. Elakkiya, S. Chandrasekaran, P. Nithyadharseni, Z. Khan, R. A. Senthil, R. Shanker, M. Raghavender, P. Kuppusami, M. Jagannathan and M. Ashokkumar, J. Ind. Eng. Chem., 2018, 64, 16–59, DOI:10.1016/j.jiec.2018.02.029.
- P. Arunachalam, in Polymer-based Nanocomposites for Energy and Environmental Applications, Elsevier, 2018, pp. 185–203 Search PubMed.
- (a) Z. Yao, M. Zhang, H. Wu, L. Yang, R. Li and P. Wang, J. Am. Chem. Soc., 2015, 137, 3799–3802, DOI:10.1021/jacs.5b01537; (b) Z. Yao, H. Wu, Y. Li, J. Wang, J. Zhang, M. Zhang, Y. Guo and P. Wang, Energy Environ. Sci., 2015, 8, 3192–3197, 10.1039/c5ee02822a.
- J. Ji, H. Zhou, Y. K. Eom, C. H. Kim and H. K. Kim, Adv. Energy Mater., 2020, 10, 2000124, DOI:10.1002/aenm.202000124.
- T. F. Lu, K. Wang, X. Han, Y. Cui, H. Tao, M. He, Z. Zhang and B. He, Struct. Chem., 2025, 1–15, DOI:10.1007/S11224-024-02447-6.
- G. C. Vougioukalakis, A. I. Philippopoulos, T. Stergiopoulos and P. Falaras, Coord. Chem. Rev., 2011, 255, 2602–2621, DOI:10.1016/J.CCR.2010.11.006.
- A. Kumar, I. Kathuria and S. Kumar, Next Mater., 2025, 7, 100352, DOI:10.1016/J.NXMATE.2024.100352.
- J. Zhang, J. Han, Z. Shi, Y. Ju, Z. Zhang and M. Gu, Appl. Surf. Sci., 2019, 465, 357–361, DOI:10.1016/J.APSUSC.2018.09.191.
- C. G. Granqvist, Sol. Energy Mater. Sol. Cells, 2007, 91, 1529–1598, DOI:10.1016/J.SOLMAT.2007.04.031.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663, DOI:10.1021/CR900356P.
- X. L. Pinheiro, A. Vilanova, D. Mesquita, M. Monteiro, J. A. M. Eriksson, J. R. S. Barbosa, C. Matos, A. J. N. Oliveira, K. Oliveira, J. Capitão, E. Loureiro, P. A. Fernandes, A. Mendes and P. M. P. Salomé, Ceram. Int., 2023, 49, 13019–13030, DOI:10.1016/J.CERAMINT.2022.12.175.
- J. Kim, Ceram. Int., 2024, 50, 38342–38349, DOI:10.1016/J.CERAMINT.2024.07.198.
- D. B. Ferry, T. Rasheed, M. T. Anwar and M. Imran, ChemistrySelect, 2024, 9, e202301442, DOI:10.1002/SLCT.202301442.
- E. Guo, G. Lu and J. Chen, Front. Energy Res., 2015, 3, 171508, DOI:10.3389/FENRG.2015.00050.
- M. Ramya, T. K. Nideep, V. P. N. Nampoori and M. Kailasnath, J. Mater. Sci. Mater. Electron., 2021, 32, 3167–3179, DOI:10.1007/S10854-020-05065-0.
- C. Pozzi, S. Ferrari, R. Luciani, G. Tassone, M. P. Costi and S. Mangani, Molecules, 2019, 24, 1257, DOI:10.3390/MOLECULES24071257.
- N. Tomar, A. Agrawal, V. S. Dhaka and P. K. Surolia, Sol. Energy, 2020, 207, 59–76, DOI:10.1016/J.SOLENER.2020.06.060.
- E. Guo and L. Yin, J. Mater. Chem. A, 2015, 3, 13390–13401, 10.1039/C5TA02556G.
- D. Hwang, J. S. Jin, H. Lee, H. J. Kim, H. Chung, D. Y. Kim, S. Y. Jang and D. Kim, Sci. Rep., 2014, 4, 7353, DOI:10.1038/srep07353.
- I. M. Hung and R. Bhattacharjee, Energies, 2016, 9, 641, DOI:10.3390/EN9080641.
- Y. Yan, Y. Zhang, Y. Zhao, F. Ding, Y. Lei, Y. Wang, J. Zhou and W. Kang, J. Mater. Sci., 2025, 60(11), 4975–5005, DOI:10.1007/S10853-025-10734-8.
- C. B. Ong, L. Y. Ng and A. W. Mohammad, Renewable Sustainable Energy Rev., 2018, 81, 536–551, DOI:10.1016/j.rser.2017.08.020.
- N. Tomar, A. Agrawal, V. S. Dhaka and P. K. Surolia, Sol. Energy, 2020, 207, 59–76, DOI:10.1016/j.solener.2020.06.060.
- M. Panahi-Kalamuei, M. Salavati-Niasari and S. M. Hosseinpour-Mashkani, J. Alloys Compd., 2014, 617, 627–632, DOI:10.1016/j.jallcom.2014.07.174.
- M. Yavarzadeh, F. Nasirpouri, L. J. Foruzin and A. Pourandarjani, Heliyon, 2024, 10, e24247, DOI:10.1016/j.heliyon.2024.e24247.
- M. Meena, A. Kavitha, S. Karthick, S. Pavithra and S. Shanmugan, Bull. Mater. Sci., 2022, 45, 1–9, DOI:10.1007/s12034-022-02828-9.
- D. Maheswari and D. Sreenivasan, Appl. Sol. Energy, 2015, 51, 112–116, DOI:10.3103/S0003701X15020085.
- R. Yang, J. Cai, K. Lv, X. Wu, W. Wang, Z. Xu, M. Li, Q. Li and W. Xu, Appl. Catal., B, 2017, 210, 184–193, DOI:10.1016/j.apcatb.2017.03.064.
- M. E. Yeoh and K. Y. Chan, Int. J. Energy Res., 2017, 41, 2446–2467, DOI:10.1002/er.3764.
- J. T. Park, W. S. Chi, S. J. Kim, D. Lee and J. H. Kim, Sci. Rep., 2014, 4, 1–7, DOI:10.1038/srep05505.
- D. R. Eddy, M. D. Permana, L. K. Sakti, G. A. N. Sheha, G. A. N. Solihudin, S. Hidayat, T. Takei, N. Kumada and I. Rahayu, Nanomaterials, 2023, 13, 704, DOI:10.3390/nano13040704.
- Y. H. Fan, C. Y. Ho and Y. J. Chang, Scanning, 2017, 2017, 9152973, DOI:10.1155/2017/9152973.
- A. Carella, F. Borbone and R. Centore, Front. Chem., 2018, 6, 416256, DOI:10.3389/fchem.2018.00481.
- (a) I. M. Abdellah and A. El-Shafei, RSC Adv., 2020, 10, 610–619, 10.1039/c9ra06150a; (b) L. K. Bommineedi, N. Upadhyay and R. Minnes, Simple Chemical Methods for Thin Film Deposition, 2023, pp. 449–507, DOI:10.1007/978-981-99-0961-2_11.
- S. Ding, C. Yang, J. Yuan, H. Li, X. Yuan and M. Li, RSC Adv., 2023, 13, 12309, 10.1039/d3ra00926b.
- D. Kim, V. Q. Dang and T. S. Teets, Chem. Sci., 2023, 15, 77–94, 10.1039/d3sc04580c.
- X. L. Wang, J. F. Huang, J. M. Liu and P. Tsiakaras, Coord. Chem. Rev., 2025, 522, 216143, DOI:10.1016/j.ccr.2024.216143.
- J. L. Fillaut, Coord. Chem. Rev., 2024, 518, 216050, DOI:10.1016/j.ccr.2024.216050.
- M. Grätzel, J. Photochem. Photobiol., C, 2003, 4, 145–153, DOI:10.1016/S1389-5567(03)00026-1.
- Y. Qin, Q. Peng, N. Martins and B. Neto, Int. J. Photoenergy, 2012, 2012, 291579, DOI:10.1155/2012/291579.
- P. T. Nguyen, R. Degn, H. T. Nguyen and T. Lund, Sol. Energy Mater. Sol. Cells, 2009, 93, 1939–1945, DOI:10.1016/J.SOLMAT.2009.07.008.
- H. A. Fetouh, A. E. Dissouky, H. A. Salem, M. Fathy, B. Anis and A. E. H. Kashyout, Sci. Rep., 2024, 14, 1–13, DOI:10.1038/s41598-024-66808-1.
- A. O. Adeloye and P. A. Ajibade, Molecules, 2014, 19, 12421–12460, DOI:10.3390/MOLECULES190812421.
- A. Gul, M. Ahmad, R. Ullah, R. Ullah, Y. Kang and W. Liao, J. Inorg. Biochem., 2024, 255, 112523, DOI:10.1016/J.JINORGBIO.2024.112523.
- R. Youf, A. Nasir, M. Müller, F. Thétiot, T. Haute, R. Ghanem, U. Jonas, H. Schönherr, G. Lemercier, T. Montier and T. le Gall, Pharmaceutics, 2022, 14, 1664, DOI:10.3390/PHARMACEUTICS14081664.
- A. M. Peyrot, M. P. Zelaya and F. Fagalde, Eur. J. Inorg. Chem., 2024, 27, e202300652, DOI:10.1002/EJIC.202300652.
- H. Yin, Z. Liu, S. Yu, Y. Yang, J. Dong, X. Yang, F. Wang, C. He and F. Cheng, Transit. Met. Chem., 2021, 46, 49–56, DOI:10.1007/S11243-020-00420-W.
- R. Kaur, B. Singh, V. Singh, M. Zharnikov and P. C. Mondal, Coord. Chem. Rev., 2024, 514, 215872, DOI:10.1016/J.CCR.2024.215872.
- L. Conti, G. E. Giacomazzo, B. Valtancoli, M. Perfetti, A. Privitera, C. Giorgi, P. S. Sfragano, I. Palchetti, S. Pecchioli, P. Bruni and F. Cencetti, Int. J. Mol. Sci., 2022, 23, 13302, DOI:10.3390/IJMS232113302.
- R. Singh and R. R. Singh, Mater. Horiz.: From Nature to Nanomater., 2023, vol. F1208, pp. 91–120, DOI:10.1007/978-981-99-3866-7_4.
- J. Albero, P. Atienzar, A. Corma and H. Garcia, Chem. Rec., 2015, 15, 803–828, DOI:10.1002/TCR.201500007.
- M. Kurucz, I. Nikolinakos, J. Soueiti, T. Baron, F. Grifoni, W. Naim, Y. Pellegrin, F. Sauvage, F. Odobel and S. Haacke, ChemPhotoChem, 2024, 8, e202300175, DOI:10.1002/CPTC.202300175.
- Y. A. Eltbaakh, M. H. Ruslan, M. A. Alghoul, M. Y. Othman, K. Sopian and M. I. Fadhel, Renewable Sustainable Energy Rev., 2011, 15, 1403–1426, DOI:10.1016/J.RSER.2010.10.018.
- R. Singh, Green Energy Technol., 2018, 221, 221–250, DOI:10.1007/978-981-10-7326-7_12.
- S. Campagna, F. Puntoriero, F. Nastasi, G. Bergamini and V. Balzani, Top. Curr. Chem., 2007, 280, 117–214, DOI:10.1007/128_2007_133.
- M. T. Rupp, N. Shevchenko, G. S. Hanan and D. G. Kurth, Coord. Chem. Rev., 2021, 446, 214127, DOI:10.1016/J.CCR.2021.214127.
- Y. Yang, X. Huang and Y. Jin, Chem. Commun., 2025, 61, 1944–1961, 10.1039/D4CC06099G.
- G. V. Loukova, Springer Handb., 2022, vol. 459, pp. 459–492, DOI:10.1007/978-3-030-63713-2_19.
- M. Bharath, G. Roy, A. Ghosh, M. Ghosh and D. Asthana, ACS Omega, 2025, 10, 3511, DOI:10.1021/ACSOMEGA.4C07748.
- C. Pan, P. Y. Ho, W. Q. Huang, G. F. Huang, L. H. Zhang, D. N. Tritton, S. M. Yiu, W. L. Man, C. C. Ko, C. F. Leung and W. X. Ni, Inorg. Chem. Front., 2025, 12(5), 1969–1978, 10.1039/D4QI02665A.
- A. Sen, M. H. Putra, A. K. Biswas, A. K. Behera and A. Groβ, Dyes Pigm., 2023, 213, 111087, DOI:10.1016/J.DYEPIG.2023.111087.
- M. K. Nazeeruddin, A. Kay, I. Rodicio, R. Humphry-Baker, E. Müller, P. Liska, N. Vlachopoulos and M. Grätzel, J. Am. Chem. Soc., 1993, 115, 6382, DOI:10.1021/JA00067A063.
- M. K. Nazeeruddin, F. de Angelis, S. Fantacci, A. Selloni, G. Viscardi, P. Liska, S. Ito, B. Takeru and M. Grätzel, J. Am. Chem. Soc., 2005, 127, 16835, DOI:10.1021/JA052467L.
- M. K. Nazeeruddin, R. Humphry-Baker, P. Liska and M. Grätzel, J. Phys. Chem. B, 2003, 107, 8981, DOI:10.1021/JP022656F.
- M. K. Nazeeruddin, P. Péchy, T. Renouard, S. M. Zakeeruddin, R. Humphry-Baker, P. Cointe, P. Liska, L. Cevey, E. Costa, V. Shklover, L. Spiccia, G. B. Deacon, C. A. Bignozzi and M. Grätzel, J. Am. Chem. Soc., 2001, 123, 1613, DOI:10.1021/JA003299U.
- P. Wang, S. M. Zakeeruddin, J. E. Moser, R. Humphry-Baker, P. Comte, V. Aranyos, A. Hagfeldt, M. K. Nazeeruddin and M. Grätzel, Adv. Mater., 2004, 16, 1806, DOI:10.1002/ADMA.200400039.
- D. Kuang, S. Ito, B. Wenger, C. Klein, J. E. Moser, R. Humphry-Baker, S. M. Zakeeruddin and M. Grätzel, J. Am. Chem. Soc., 2006, 128, 4146, DOI:10.1021/JA058540P.
- K. J. Jiang, N. Masaki, J. b. Xia, S. Noda and S. Yanagida, Chem. Commun., 2006, 23, 2460, 10.1039/B602989B.
- F. Gao, Y. Wang, D. Shi, J. Zhang, M. Wang, X. Jing, R. Humphry-Baker, P. Wang, S. M. Zakeeruddin and M. Grätzel, J. Am. Chem. Soc., 2008, 130, 10720, DOI:10.1021/JA801942J.
- Y. Cao, Y. Bai, Q. Yu, Y. Cheng, S. Liu, D. Shi, F. Gao and P. Wang, J. Phys. Chem. C, 2009, 113, 6290, DOI:10.1021/JP9006872.
- Q. Yu, S. Liu, M. Zhang, N. Cai, Y. Wang and P. Wang, J. Phys. Chem. C, 2009, 113, 14559, DOI:10.1021/JP904096G.
- Z. She, Y. Cheng, L. Zhang, X. Li, D. Wu, Q. Guo, J. Lan, R. Wang and J. You, ACS Appl. Mater. Interfaces, 2015, 7, 27831, DOI:10.1021/ACSAMI.5B09160.
- S. W. Wang, K. L. Wu, E. Ghadiri, M. G. Lobello, S. te Ho, Y. Chi, J. E. Moser, F. de Angelis, M. Grätzel and M. K. Nazeeruddin, Chem. Sci., 2013, 4, 2423, 10.1039/C3SC50399B.
- K. L. Wu, W. P. Ku, J. N. Clifford, E. Palomares, S. te Ho, Y. Chi, S. H. Liu, P. T. Chou, M. K. Nazeeruddin and M. Grätzel, Energy Environ. Sci., 2013, 6, 859, 10.1039/C2EE23988D.
- C.-C. Chou, K.-L. Wu, Y. Chi, W.-P. Hu, S. J. Yu, G.-H. Lee, C.-L. Lin and P.-T. Chou, Angew. Chem., 2011, 123, 2102, DOI:10.1002/ANGE.201006629.
- H. Cheema, A. Islam, L. Han and A. El-Shafei, Dyes Pigm., 2015, 120, 93, DOI:10.1016/J.DYEPIG.2015.04.005.
- S. Ashraf, J. Akhtar, H. M. Siddiqi and A. El-Shafei, New J. Chem., 2017, 41, 6272–6277, 10.1039/C7NJ01363A.
- T. Bessho, E. Yoneda, J. H. Yum, M. Guglielmi, L. Tavernelli, H. Imai, U. Rothlisberger, M. K. Nazeeruddin and M. Grätzel, J. Am. Chem. Soc., 2009, 131, 5930–5934, DOI:10.1021/JA9002684.
- D. V. Pogozhev, M. J. Bezdek, P. A. Schauer and C. P. Berlinguette, Inorg. Chem., 2013, 52, 3001–3006, DOI:10.1021/IC3024524.
- M. Maestri, N. Armaroli, V. Balzani, E. C. Constable and A. M. C. Thompson, Inorg. Chem., 1995, 34, 2759–2767 CrossRef CAS.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. von Zelewsky, Coord. Chem. Rev., 1988, 84, 85–277, DOI:10.1016/0010-8545(88)80032-8.
- M. I. J. Polson, F. Loiseau, S. Campagna and G. S. Hanan, Chem. Commun., 2006, 1301–1303, 10.1039/B515493F.
- M. I. J. Polson, E. A. Medlycott, G. S. Hanan, L. Mikelsons, N. J. Taylor, M. Watanabe, Y. Tanaka, F. Loiseau, R. Passalacqua and S. Campagna, Chem.–Eur. J., 2004, 10, 3640–3648, DOI:10.1002/CHEM.200400032.
- S. U. Son, K. H. Park, Y. S. Lee, B. Y. Kim, C. H. Choi, M. S. Lan, Y. H. Jang, D. J. Jang and Y. K. Chung, Inorg. Chem., 2004, 43, 6896–6898, DOI:10.1021/IC049514F.
- M. Duati, S. Tasca, F. C. Lynch, H. Bohlen, J. G. Vos, S. Stagni and M. D. Ward, Inorg. Chem., 2003, 42, 8377–8384, DOI:10.1021/IC034691M.
- H. Amouri, Chem. Rev., 2023, 123, 230–270, DOI:10.1021/ACS.CHEMREV.2C00206.
- J. Steube, L. Burkhardt, A. Päpcke, J. Moll, P. Zimmer, R. Schoch, C. Wölper, K. Heinze, S. Lochbrunner and M. Bauer, Chem.–Eur. J., 2019, 25, 11826–11830, DOI:10.1002/CHEM.201902488.
- P. Sánchez-Fernández, C. A. Aranda, R. Escalante, A. J. Riquelme, R. Demadrille, P. Pistor, G. Oskam and J. A. Anta, Sol. RRL, 2024, 8, 2400149, DOI:10.1002/SOLR.202400149.
- N. Masud and H. K. Kim, ACS Omega, 2023, 8, 6139, DOI:10.1021/ACSOMEGA.2C06843.
- Y. Xie, J. Baillargeon and T. W. Hamann, J. Phys. Chem. C, 2015, 119, 28155–28166, DOI:10.1021/ACS.JPCC.5B08244.
- J. N. Clifford, E. Martínez-Martínez-Ferrero, A. Viterisi and E. Palomares, Chem. Soc. Rev., 2011, 40, 1635, 10.1039/b920664g.
- S. A. Haque, S. Handa, K. Peter, E. Palomares, M. Thelakkat and J. R. Durrant, Angew. Chem., Int. Ed., 2005, 44, 5740–5744, DOI:10.1002/ANIE.200500363.
- I. M. Abdellah, A. I. Koraiem and A. El-Shafei, Sol. Energy, 2019, 177, 642–651, DOI:10.1016/J.SOLENER.2018.11.047.
- S. Ashraf, R. Su, J. Akhtar, A. Shuja, H. M. Siddiqi and A. El-Shafei, New J. Chem., 2022, 46, 2739–2746, 10.1039/D1NJ04362E.
- B. Pashaei and H. Shahroosvand, Inorg. Chem. Commun., 2020, 112, 107737, DOI:10.1016/J.INOCHE.2019.107737.
- S. Y. Huang, G. Schlichthörl, A. J. Nozik, M. Grätzel and A. J. Frank, J. Phys. Chem. B, 1997, 101, 2576–2582, DOI:10.1021/JP962377Q.
- S. Rahman, A. Haleem, M. Siddiq, M. K. Hussain, S. Qamar, S. Hameed and M. Waris, RSC Adv., 2023, 13, 19508, 10.1039/D3RA00903C.
- D. Zhang, M. Stojanovic, Y. Ren, Y. Cao, F. T. Eickemeyer, E. Socie, N. Vlachopoulos, J. E. Moser, S. M. Zakeeruddin, A. Hagfeldt and M. Grätzel, Nat. Commun., 2021, 12, 1, DOI:10.1038/S41467-021-21945-3.
- W. Vallejo, M. Lerma and C. Díaz-Uribe, Heliyon, 2024, 11, e41092, DOI:10.1016/J.HELIYON.2024.E41092.
- V. S. Manthou, E. K. Pefkianakis, P. Falaras and G. C. Vougioukalakis, ChemSusChem, 2015, 8, 588–599, DOI:10.1002/CSSC.201403211.
- A. Saha and B. Ganguly, J. Mol. Graph. Model., 2024, 127, 108678, DOI:10.1016/J.JMGM.2023.108678.
- M. Mazloum-Ardakani and R. Arazi, Heliyon, 2019, 5, e01444, DOI:10.1016/J.HELIYON.2019.E01444.
- J. Lim, Y. S. Kwon and T. Park, Chem. Commun., 2011, 47, 4147–4149, 10.1039/c0cc04999a.
- H. M. Song, K. D. Seo, M. S. Kang, I. T. Choi, S. K. Kim, Y. K. Eom, J. H. Ryu, M. J. Ju and H. K. Kim, J. Mater. Chem., 2012, 22, 3786–3794, 10.1039/C2JM16021H.
- L. Han, A. Islam, H. Chen, C. Malapaka, B. Chiranjeevi, S. Zhang, X. Yang and M. Yanagida, Energy Environ. Sci., 2012, 5, 6057–6060, 10.1039/C2EE03418B.
- P. Wang, S. M. Zakeeruddin, R. Humphry-Baker, J. E. Moser and M. Grätzel, Adv. Mater., 2003, 15, 2101–2104, DOI:10.1002/ADMA.200306084.
- P. R. Kumar and V. Ramasubbu, Solid-State Electron., 2022, 198, 108480, DOI:10.1016/J.SSE.2022.108480.
- V. Kumar, R. Gupta and A. Bansal, Sol. Energy, 2020, 196, 589–596, DOI:10.1016/J.SOLENER.2019.12.034.
- H. Matsuyoshi, H. Tomita, H. Nishino, H. Sakamoto and K. Manabe, Int. J. Photoenergy, 2013, 2013, 439717, DOI:10.1155/2013/439717.
- Z. Zhang, N. Evans, S. M. Zakeeruddin, R. Humphry-Baker and M. Grätzel, J. Phys. Chem. C, 2007, 111, 398–403, DOI:10.1021/JP0648745.
- P. Wang, S. M. Zakeeruddin, P. Comte, R. Charvet, R. Humphry-Baker and M. Grätzel, J. Phys. Chem. B, 2003, 107, 14336–14341, DOI:10.1021/JP0365965.
- M. Wang, C. Grätzel, S. J. Moon, R. Humphry-Baker, N. Rossier-Iten, S. M. Zakeeruddin and M. Grätzel, Adv. Funct. Mater., 2009, 19, 2163–2172, DOI:10.1002/ADFM.200900246.
- Z. Ning, Q. Zhang, W. Wu and H. Tian, J. Organomet. Chem., 2009, 694, 2705–2711, DOI:10.1016/J.JORGANCHEM.2009.02.016.
- N. Masud and H. K. Kim, ACS Omega, 2023, 8, 6139–6163, DOI:10.1021/ACSOMEGA.2C06843.
- A. Reynal, A. Forneli, E. Martínez-Ferrero, A. Sánchez-Díaz, A. Vidal-Ferran, B. C. O'Regan and E. Palomares, J. Am. Chem. Soc., 2008, 130, 13558–13567, DOI:10.1021/JA800513M.
- A. Carella, F. Borbone and R. Centore, Front. Chem., 2018, 6, 416256, DOI:10.3389/FCHEM.2018.00481/BIBTEX.
- T. Marinado, K. Nonomura, J. Nissfolk, M. K. Karlsson, D. P. Hagberg, L. Sun, S. Mori and A. Hagfeldt, Langmuir, 2010, 26, 2592–2598, DOI:10.1021/LA902897Z.
- Y. Saygili, M. Söderberg, N. Pellet, F. Giordano, Y. Cao, A. B. Munoz-García, S. M. Zakeeruddin, N. Vlachopoulos, M. Pavone, G. Boschloo, L. Kavan, J. E. Moser, M. Grätzel, A. Hagfeldt and M. Freitag, J. Am. Chem. Soc., 2016, 138, 15087–15096, DOI:10.1021/JACS.6B10721.
- T. Daeneke, A. J. Mozer, Y. Uemura, S. Makuta, M. Fekete, Y. Tachibana, N. Koumura, U. Bach and L. Spiccia, J. Am. Chem. Soc., 2012, 134, 16925–16928, DOI:10.1021/JA3054578.
- D. Zhou, Z. Xia, H. Shang, H. Xiao, Z. Jiang, H. Li, L. Zheng, J. Dong and W. Chen, Mater. Chem. Front., 2021, 5, 3085–3092, 10.1039/D0QM00806K.
- C. Teng, X. Yang, C. Yuan, C. Li, R. Chen, H. Tian, S. Li, A. Hagfeldt and L. Sun, Org. Lett., 2009, 11, 5542–5545, DOI:10.1021/OL9022936.
- S. H. Kang, M. J. Jeong, Y. K. Eom, I. T. Choi, S. M. Kwon, Y. Yoo, J. Kim, J. Kwon, J. H. Park and H. K. Kim, Adv. Energy Mater., 2017, 7, 1602117, DOI:10.1002/AENM.201602117.
- H. Michaels, M. Rinderle, R. Freitag, I. Benesperi, T. Edvinsson, R. Socher, A. Gagliardi and M. Freitag, Chem. Sci., 2020, 11, 2895–2906, 10.1039/C9SC06145B.
- C. Curiac, R. R. Rodrigues, J. Watson, L. A. Hunt, A. Devdass, J. W. Jurss, N. I. Hammer, R. C. Fortenberry and J. H. Delcamp, ChemSusChem, 2021, 14, 3084–3096, DOI:10.1002/CSSC.202100884.
- T. Daeneke, T. H. Kwon, A. B. Holmes, N. W. Duffy, U. Bach and L. Spiccia, Nat. Chem., 2011, 3, 211–215, DOI:10.1038/nchem.966.
- Z. Zhang, P. Chen, T. N. Murakami, S. M. Zakeeruddin and M. Grätzel, Adv. Funct. Mater., 2008, 18, 341–346, DOI:10.1002/ADFM.200701041.
- M. Wang, N. Chamberland, L. Breau, J. E. Moser, R. Humphry-Baker, B. Marsan, S. M. Zakeeruddin and M. Grätzel, Nat. Chem., 2010, 2, 385–389, DOI:10.1038/nchem.610.
- G. Boschloo and A. Hagfeldt, Acc. Chem. Res., 2009, 42, 1819–1826, DOI:10.1021/AR900138M.
- H. Tian, X. Jiang, Z. Yu, L. Kloo, A. Hagfeldt, L. Sun, H. Tian, L. Sun, X. Jiang, Z. Yu, L. Kloo and A. Hagfeldt, Angew. Chem., 2010, 122, 7486–7489, DOI:10.1002/ANGE.201003740.
- K. Okada, H. Matsui, T. Kawashima, T. Ezure and N. Tanabe, J. Photochem. Photobiol., A, 2004, 164, 193–198, DOI:10.1016/J.JPHOTOCHEM.2004.01.028.
- H. Iftikhar, G. G. Sonai, S. G. Hashmi, A. F. Nogueira and P. D. Lund, Materials, 2019, 12, 1998, DOI:10.3390/MA12121998.
- R. A. A. Talip, W. Z. N. Yahya and M. A. Bustam, Sustainability, 2020, 12, 7598, DOI:10.3390/SU12187598.
- S. A. Sapp, C. M. Elliott, C. Contado, S. Caramori and C. A. Bignozzi, J. Am. Chem. Soc., 2002, 124, 11215–11222, DOI:10.1021/JA027355Y.
- S. M. Feldt, E. A. Gibson, E. Gabrielsson, L. Sun, G. Boschloo and A. Hagfeldt, J. Am. Chem. Soc., 2010, 132, 16714–16724, DOI:10.1021/JA1088869.
- J. J. Nelson, T. J. Amick and C. M. Elliott, J. Phys. Chem. C, 2008, 112, 18255–18263, DOI:10.1021/JP806479K.
- Q. Liu and J. Wang, Sol. Energy, 2019, 184, 454–465, DOI:10.1016/J.SOLENER.2019.04.032.
- H. Kusama and H. Arakawa, J. Photochem. Photobiol., A, 2004, 164, 103–110, DOI:10.1016/J.JPHOTOCHEM.2003.11.013.
- H. Kusama and H. Arakawa, J. Photochem. Photobiol., A, 2004, 162, 441–448, DOI:10.1016/S1010-6030(03)00414-3.
- N. Kopidakis, N. R. Neale and A. J. Frank, J. Phys. Chem. B, 2006, 110, 12485–12489, DOI:10.1021/JP0607364.
- Z. Zhang, S. M. Zakeeruddin, B. C. O'Regan, R. Humphry-Baker and M. Grätzel, J. Phys. Chem. B, 2005, 109, 21818–21824, DOI:10.1021/JP054305H.
- M. K. Nazeeruddin, A. Kay, I. Rodicio, R. Humphry-Baker, E. Müller, P. Liska, N. Vlachopoulos and M. Grätzel, J. Am. Chem. Soc., 1993, 115, 6382–6390, DOI:10.1021/JA00067A063.
- M. Afrooz and H. Dehghani, Electrochim. Acta, 2015, 174, 521–531, DOI:10.1016/J.ELECTACTA.2015.06.024.
| This journal is © The Royal Society of Chemistry 2025 |