
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCofacial porphyrin organic cages. Metals regulating excitation electron transfer and CO2 reduction electrocatalytic properties†
Xiaolin
Liu
a,
Chenxi
Liu
a,
Xiaojuan
Song
b,
Xu
Ding
a,
Hailong
Wang
 *a,
Baoqiu
Yu
a,
Heyuan
Liu
*b,
Bin
Han
a,
Xiyou
Li
*b and
Jianzhuang
Jiang
*a
*a,
Baoqiu
Yu
a,
Heyuan
Liu
*b,
Bin
Han
a,
Xiyou
Li
*b and
Jianzhuang
Jiang
*a
aBeijing Advanced Innovation Center for Materials Genome Engineering, Beijing Key Laboratory for Science and Application of Functional Molecular and Crystalline Materials, Department of Chemistry and Chemical Engineering, School of Chemistry and Biological Engineering University of Science and Technology Beijing, Beijing 100083, China. E-mail: hlwang@ustb.edu.cn; jianzhuang@ustb.edu.cn
bSchool of Materials Science and Engineering China University of Petroleum (East China), Qingdao 266580, China. E-mail: 20170053@upc.edu.cn; xiyouli@upc.edu.cn
First published on 10th August 2023
Abstract
Herein, we introduce a comprehensive study of the photophysical behaviors and CO2 reduction electrocatalytic properties of a series of cofacial porphyrin organic cages (CPOC-M, M = H2, Co(II), Ni(II), Cu(II), Zn(II)), which are constructed by the covalent-bonded self-assembly of 5,10,15,20-tetrakis(4-formylphenyl)porphyrin (TFPP) and chiral (2-aminocyclohexyl)-1,4,5,8-naphthalenetetraformyl diimide (ANDI), followed by post-synthetic metalation. Electronic coupling between the TFPP donor and naphthalene-1,4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5,8-bis(dicarboximide) (NDI) acceptor in the metal-free cage is revealed to be very weak by UV-vis spectroscopic, electrochemical, and theoretical investigations. Photoexcitation of CPOC-H2, as well as its post-synthetic Zn and Co counterparts, leads to fast energy transfer from the triplet state porphyrin to the NDI unit according to the femtosecond transient absorption spectroscopic results. In addition, CPOC-Co enables much better electrocatalytic activity for CO2 reduction reaction than the other metallic CPOC-M (M = Ni(II), Cu(II), Zn(II)) and monomeric porphyrin cobalt compartment, supplying a partial current density of 18.0 mA cm−2 at −0.90 V with 90% faradaic efficiency of CO.
:5,8-bis(dicarboximide) (NDI) acceptor in the metal-free cage is revealed to be very weak by UV-vis spectroscopic, electrochemical, and theoretical investigations. Photoexcitation of CPOC-H2, as well as its post-synthetic Zn and Co counterparts, leads to fast energy transfer from the triplet state porphyrin to the NDI unit according to the femtosecond transient absorption spectroscopic results. In addition, CPOC-Co enables much better electrocatalytic activity for CO2 reduction reaction than the other metallic CPOC-M (M = Ni(II), Cu(II), Zn(II)) and monomeric porphyrin cobalt compartment, supplying a partial current density of 18.0 mA cm−2 at −0.90 V with 90% faradaic efficiency of CO.
Introduction
The great development of molecular cages, which are termed as void molecular structures with confined nano-sized cavities, has been witnessed in the field of supramolecular chemistry, spanning from initial synthetic chemistry to interdisciplinary applications covering supramolecular chemistry and materials science.1–16 Their categories are diversified due to the various construction processes based mainly on coordination interactions,17–34 covalent bonds,35–45 and hydrogen bonds.46–50 Since 2009, porous organic cages (POCs) have emerged as a new class of functional organic materials with unique porous architectures through crystal engineering of dynamic covalent-bonded molecular cages.6,44,51–57 The progress of dynamic covalent chemistry (DCvC) has broadened the versatile cages with predesigned structures and functionalities by efficiently assembling discrete molecular modules.8,51 The open intrinsic and inter-cage porosities in solid state POCs ensure the diffusion and penetration of special ions and small molecules for applications in heterogeneous catalysis,58–62 separation,63–66 and storage.67–69 Unlike covalent- and coordination-bonded reticular frameworks, POCs have excellent solution processability because of the weak interactions between the neighboring void molecular building blocks, enabling the further generation of reformative cages3,54,70 as well as porous alloys,71 core–shell crystals72 and liquids73,74 through smart chemical and physical methods. In addition, POCs have been demonstrated as a new kind of synthon to transform cages into framework structures with enlarged external cage porosities.54,75,76 The intrinsic receptor nature of POCs allows the immobilization of various active species in a confined environment for catalysis and energy storage.58–62,77,78 Nevertheless, the photophysical behaviors of POCs with multiple chromophores or redox-active components in a three-dimensional spatial distribution have been rarely studied, possibly due to the synthetic difficulty in incorporating those different building blocks into a cage.79–82Porphyrins, as chlorophyll derivatives, have been widely explored in relation to understanding natural processes involving light harvesting, electron and energy transfer, and catalysis.83–88 The diversification of porphyrins has become prominent via the functionalization of the aromatic core and insertion of metals into the molecular cavity, greatly tuning their chemical, electronic, and photophysical properties.83–88 Thus far, porphyrins have been incorporated into a large number of not only molecules and oligomers, but also polymeric and framework architectures, showing outstanding functionality in the field of photosynthesis89,90 molecular electronics,91,92 molecular machines,93,94 catalysis,95–102 and therapy.103,104 In this regard, the preparation of molecular cages based on redox and photoactive porphyrins has been an attractive research subject due to the accessible active sites, favorable guest diffusion and confinement inside the cage, and reduced aggregation of catalytic centers in this system.41,105–107
Taking inspiration from porphyrin and POC chemistry, we started this research to obtain new porphyrin POCs towards biomimetic models and selective heterocatalysts. Herein, a functional cofacial porphyrin organic cage, CPOC-H2, has been constructed through the self-assembly of a 5,10,15,20-tetrakis(4-formylphenyl)porphyrin (TFPP) donor (D) and chiral (2-aminocyclohexyl)-1,4,5,8-naphthalenetetraformyl diimide (ANDI) acceptor (A) derived from naphthalene-1,4:5,8-bis(dicarboximide) (NDI), Scheme 1. The following post-synthetic metalation of the metal-free cage with different metal ions leads to the generation of four other D–A-type cages, CPOC-M (M = Co(II), Ni(II), Cu(II), Zn(II)). In CPOC-H2, there exists a weak electronic coupling between the porphyrin donor and NDI acceptor according to the UV-vis spectroscopy, electrochemistry, and theoretical calculation results. In addition, only the metal-free cage together with the Zn/Co-metalated samples shows the excitation electron transfer based on the femtosecond transient absorption spectroscopic results. In particular, CPOC-Co possesses much superior electrocatalytic activity for the CO2 reduction reaction compared to the other metallic CPOC-M (M = Ni(II), Cu(II), Zn(II)) and monomeric cobalt porphyrin compartment, supplying a partial current density of 18.0 mA cm−2 at −0.9 V with 90% faradaic efficiency of CO due to the high surface concentration of active sites.
 | ||
| Scheme 1 Schematic synthesis of CPOC-M (M = H2, Co(II), Ni(II), Cu(II), Zn(II)). | ||
Results and discussion
Synthesis and simulated structure of CPOC-M
Cofacial porphyrin dimers and cages, with two porphyrin components in a face-to-face arrangement, have been demonstrated to allow multiple interactions between metal centers and substrates, enabling efficient promotion of oxygen and carbon dioxide reduction reactions.108–112 The step-wise synthetic method to chiral POCs based on an NDI unit pioneered by Wasielewski's group enables the fixation of another chromophore or redox-active component in a cofacial arrangement.79 For the purpose of building multiple functional cofacial POCs, the two functional building blocks, including TFPP and enantiomeric ANDI, have been assembled using a dynamic imine reaction, Scheme 1 and Fig. S1–S5,† leading to a D–A-type metal-free cage, CPOC-H2. It is worth noting that an excessive amount of enantiomeric ANDI (5 eq. TFPP) is necessary for the formation of the chiral metal-free cages in a good yield of 80%. The chemical components of CPOC-H2 were determined by MALDI-TOF mass spectrometry. The observed molecular ion peak for CPOC-H2 reveals the presence of two porphyrin units and four NDI species, consistent with the predesigned cofacial porphyrin organic square prism. The symmetric structure of CPOC-H2 is confirmed by the 1H NMR spectrum for a ((R)-2-aminocyclohexyl)-1,4,5,8-naphthalenetetraformyl diimide derived cage (R)-CPOC-H2, showing seven sets of aromatic proton signals in Fig. S6.† According to the integrals, the signals at δ 8.8, 8.2, and 7.9–8.0 are assigned to the corresponding protons in the D-A-type porphyrin-NDI organic square prism with the help of 13C and 1H–1H COSY spectra, Scheme 1 and Fig. S7 and S8.† In addition, the Fourier transform infrared (FT-IR) data for CPOC-H2 exhibit the appearance of a vibration band at 1630 cm−1, corresponding to the newly formed C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond, Fig. S9.† The complete metalation of the cofacial porphyrin units in the presence of different acetates is monitored using UV-vis spectroscopy in combination with MALDI-TOF mass spectrometry, Fig. S10.†
N bond, Fig. S9.† The complete metalation of the cofacial porphyrin units in the presence of different acetates is monitored using UV-vis spectroscopy in combination with MALDI-TOF mass spectrometry, Fig. S10.†
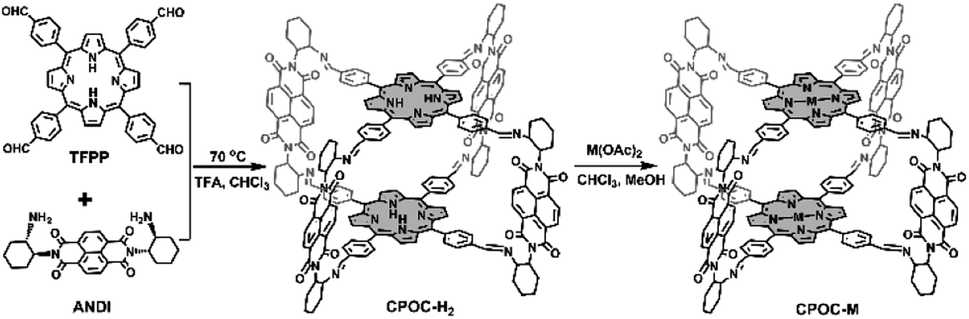
The molecular structure of empty cage CPOC-H2 was optimized based on density functional theory (DFT). The cofacial porphyrin units are separated with the distance of 3.6 Å between two parallel porphyrin planes, Fig. 1a and b. Four NDI units are connected to two porphyrin building blocks, forming four intramolecular microporous cavities. This allows the easy diffusion of suitable substrates to approach these two kinds of active units. The rotation angles for the cofacial porphyrin units are 29.73–30.98°, revealing the distorted quadrangular prism structure for this cage. The homogeneous chiral cyclohexanediamine induces a helical arrangement of the four NDI units in the CPOC-H2 structure model. The chirality of the CPOC-H2 enantiomers was confirmed by observation of a positive and negative Cotton effect in their circular dichroism spectra, Fig. 1c. The dihedral angles of NDI and the porphyrin N4 planes are 84.0–89.6°. As a consequence, this new D–A-type cage with two chromophores in a nearly orthogonal arrangement seems to be an interesting molecular model to understand their physicochemical properties. Solid CPOC-H2 is able to exhibit 4.0 wt% and 5.6 wt% CO2 uptakes under 760 mmHg pressure at 298 and 273 K, Fig. 1d.
 | ||
| Fig. 1 Molecular structure of (R)-CPOC-H2 optimized by the DFT method in top (a) and side (b) view, respectively (carbon (except carbon from NDI: green): grey, nitrogen: blue, oxygen: red, central yellow ball represents void). (c) Circular dichroism spectra of (R)-CPOC-H2 (blue) and (S)-CPOC-H2 (red) in CHCl3. (d) CO2 adsorption (solid) and desorption (hollow) curves of POC-H2 at 298 K (blue) and 273 K (red). (e) Comparison of the UV-vis spectra of CPOC-H2 and the corresponding monomeric references in CHCl3 (inset: the enlarged spectrum from 460 to 700 nm). | ||
Photophysical characterization of CPOC-M
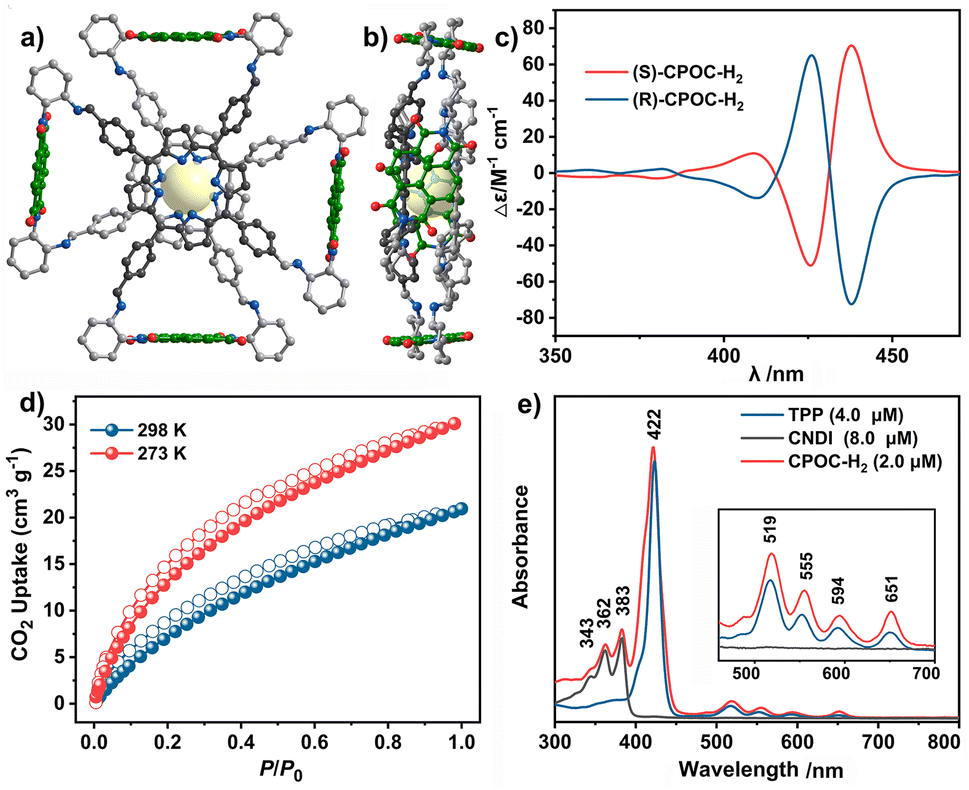
As shown in Fig. 1e and S11,† 5,10,15,20-tetrakisphenylporphyrin (TPP) has a characteristic absorption made up of one S-band and four Q-bands in the UV-vis absorption spectrum at 422, 519, 555, 594, and 651 nm, respectively. In contrast, the electronic adsorption of the monomeric NDI derivative reference (CNDI) appears at 343, 362, and 383 nm, Fig. 1e and S12.† After the dynamic covalent chemistry self-assembly, CPOC-H2 still obtains similar adsorption bands to individual monomers, indicating the weak electronic coupling between porphyrin and the NDI units, Fig. 1e. This is further supported by the observation of similar redox potentials of porphyrin and NDI units in the cyclic voltammograms of the cage and monomers, Fig. S13.† It is worth noting that the molar absorption coefficient for the electronic absorption band at 422 nm for (R)-CPOC-H2 is much less than twice the value for the porphyrin monomer, possibly due to the spatially coupled transition dipole moments,79 Fig. S14.† In contrast, the molar absorption coefficients for the 362 and 383 nm electronic absorption bands of (R)-CPOC-H2 are bigger than quadruple the value for the CNDI monomer due to the long distance separation. After the immobilization of different metals into CPOC-H2, the metalated cages have similar S-band and molar absorption coefficients to those for the metal-free cage, Fig. S15–S18 and Table S1.†To reveal the excited-state dynamics of these D–A-type cages, their femtosecond transient absorption (TA) spectra were comparatively investigated with the help of monomeric chromophores ANDI and TPP. Upon photoexcitation at 380 nm, a broad excited-state absorption (ESA) band at ∼421 nm associated with the singlet (S1) state of ANDI is observed at 0.53 ps, Fig. 2a. The increase in response time to 1.00 ps induces a serious intensity decrease for the S1 absorptions, accompanied by the rise of a broad ESA band from 450 to 600 nm. The newly observed band is caused by the formation of the NDI radical anion (NDI˙−).78,81,113 As for the metal free TPP with excitation at 514 nm, its TA spectra collected at the initial low response time show a main ESA band at 442 nm as well as four weak bands at 533, 567, 621, and 691 nm, belonging to the corresponding S1 state, Fig. 2b. Subsequently, the S1 absorption bands at 442 and 691 nm show a serious decrease within 5.06 ps; meanwhile, the peak of the first band slightly blue-shifts to 440 nm. These changes are ascribed to the intersystem crossing (ISC) to generate the triplet state of TPP based on a previous report.114
 | ||
| Fig. 2 TA spectra of ANDI (a), TPP (b), CPOC-H2 (c), and CPOC-Co (d) in degassed toluene excited at 380, 514, 530, and 530 nm, respectively. | ||
As for CPOC-H2, a broad ESA band of 444–800 nm with only one main peak at 455 nm was observed at 0.87 ps, originating from the S1 state. Similar to the ESA spectrum of TPP, a fast decay accompanied by a slight blue shift is observed for the main band within 2.25 ps due to the formation of the triplet state via ISC, Fig. 2c. Then, as the triplet signal decreased, a new ESA band at 475 nm associated with NDI˙− emerged at a high response time, suggesting the presence of electron transfer from TFPP to NDI in CPOC-H2. After metalation of the cofacial porphyrin units of CPOC-H2 with Co and Zn ions, similar spectral evolution was also monitored, Fig. 2d and S19.† This indicates that similar electron transfer from the triplet state porphyrin to the NDI unit is also presented in CPOC-Co and CPOC-Zn. However, for CPOC-Ni and CPOC-Cu, only their spectral changes associated with the ISC process are distinguished, Fig. S20–S22.† The lack of observation of the NDI˙− spectral feature implies the absence of electron transfer for these two cages, possibly due to the different metallic nature.
To get the rate of the corresponding photophysical process in these compounds, we conducted global analysis for their fs-TA data, and the corresponding time constants are shown in Table S2.† For ANDI, a two-species decay model was employed (S1 → CS), and the formation rate of the NDI˙− is ∼0.3 ps from the singlet state (S1) in ANDI, which is close to the charge transfer rate in similar compounds.79 This suggests that our deconvolution method is reliable. For TPP, the triplet state (T1) generation rate was determined to be ∼0.2 ps with a two-component decay model (S1 → T1). Similarly, the same two-component decay model (S1 → T1) was also employed for CPOC-Ni and CPOC-Cu, and the time constant of T1 formation is 1.3 and 2.5 ps for CPOC-Ni and CPOC-Cu, respectively. In CPOC-H2, CPOC-Co, and CPOC-Zn, except for the ISC process, the charge separation (CS) process is also presented. Therefore, a sequential three-species decay model (S1 → T1 → CS) was adopted in CPOC-H2, CPOC-Co, and CPOC-Zn. The ISC rate is 0.2, 1.6, and 2.0 ps for CPOC-H2, CPOC-Co, and CPOC-Zn, respectively. The corresponding charge separation rate is 28.0, 9.6, and 21.0 ps.
Electrochemical CO2 reduction activity
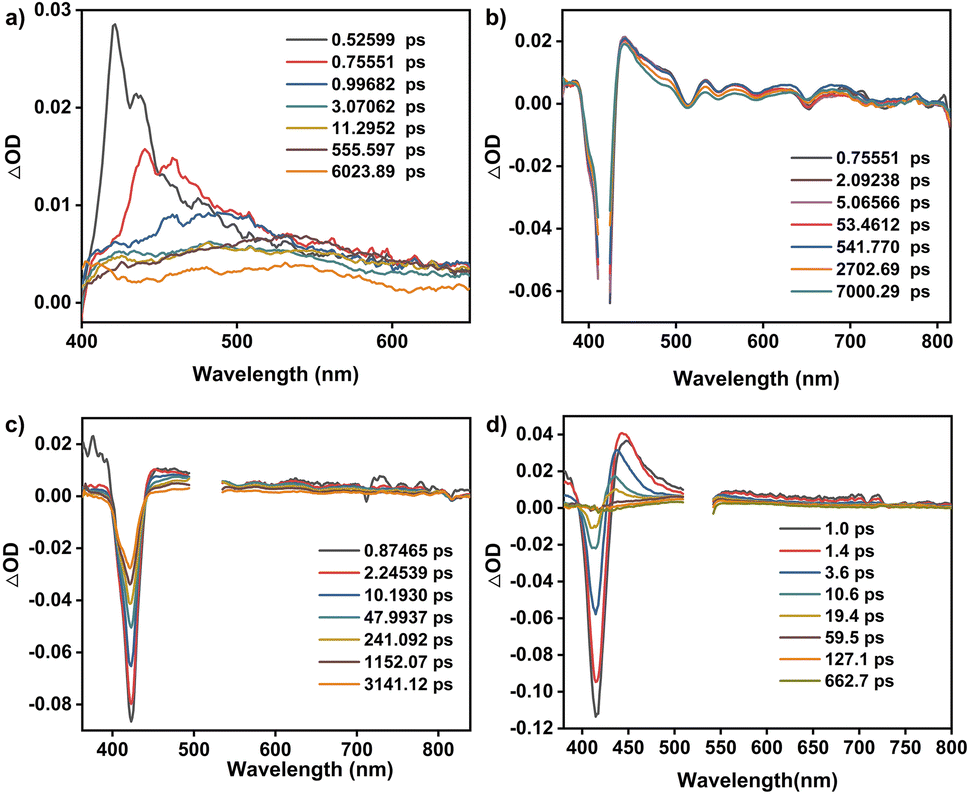
The cofacial cobalt porphyrin111 and metallomacrocycle-based POCs115,116 have been demonstrated to possess active electrocatalytic capability towards the CO2 reduction reaction (CO2RR). Prior to the electrocatalysis, the permanent porosities of amorphous CPOC-Co were determined with a carbon dioxide sorption test at ambient temperatures. Similar to CPOC-H2, CPOC-Co is able to hold moderate 3.3 wt% and 4.4 wt% CO2 uptakes under 760 mmHg pressure at 298 and 273 K, Fig. S23,† respectively. It is worth noting that the porosities made up of the external and internal cage voids are useful for CO2 diffusion to the catalytic active sites within CPOC-Co. The CO2RR electrocatalytic activities for CPOC-Co and cobalt 5,10,15,20-tetrakisphenylporphyrin (TPP-Co) were comparatively evaluated using a two-compartment H-type cell using an electrolyte of a CO2-saturated 0.5 M KHCO3 aqueous solution (pH = 7.2), Fig. S24.† These active molecular materials were mixed with single-walled carbon nanotubes (CNTs) and subsequently deposited on a piece of carbon fiber paper as a working electrode (for details, please see ESI†). Preliminary linear sweep voltammetry (LSV) curves were firstly screened in Ar and CO2-saturated 0.5 M KHCO3 electrolytes, respectively. As shown in Fig. 3a, the LSV curve for the CPOC-Co electrode exhibits a rapid increase upon the current density at −0.50 to −1.00 V (vs. RHE) in Ar-saturated electrolyte. The change of the electrolyte saturated gas to CO2 leads to a much more quickly increased current density in this potential range, indicating the electrocatalytic CO2RR capability for this cage. The constant total geometric current of CPOC-Co is collected at the potential range between −0.40 and −0.90 V with each potential for 20 min, Fig. 3b. Gas chromatography (GC) and 1H NMR spectroscopic analyses double identify the binary CO and H2 gaseous products obtained from the CO2RR electrocatalysis upon the CPOC-Co electrode at −0.40 to −0.90 V, instead of any liquid product, Fig. S25–S27.† In good contrast, almost no CO product is detected in the control electrocatalysis under an Ar-saturated electrolyte or sole CNT electrode, confirming the carbon source in the CO product from CO2 and electrocatalytic activity of CPOC-Co, Fig. S28 and S29.† The increase of the potentials applied upon the CPOC-Co electrode from −0.50 to −0.70 V results in an increase in the faradaic efficiency of CO (FECO) from 74% to 92%. A tiny decrease for FECO to 90% is detected from −0.70 to −0.90 V. Such a tendency for FECO is similar to that for the TPP-Co electrode, Fig. 3c. However, the partial current density (jCO) for the CPOC-Co electrode is much bigger than that of the monomeric TPP-Co counterpart under the potential range of −0.60 V to −0.90 V, Fig. 3d. In particular, jCO (18.0 mA cm−2) for the CPOC-Co electrode at −0.90 V is 34% higher than that of TPP-Co (13.4 mA cm−2) as well as the reported metalloporphyrin cage Fe-PB (∼1.6 mA cm−2 at −0.8 V)115 and metallophthalocyanine cage NiPc-cage (∼4.8 mA cm−2 at −0.9 V)116 and even those COFs including CoPc-PI-COF-2 (16.6 mA cm−2 at −0.9 V),117 Co-TTCOF (4.5 mA cm−2),118 and TTF-Por(Co)-COF (6.9 mA cm−2 at −0.9 V)119 under the same conditions, suggesting the cofacial and porous structure superiority of the porphyrin cage in CO2RR electrocatalysis. | ||
| Fig. 3 (a) LSV curves for CPOC-Co. (b) Chronoamperometric responses at different potentials (vs. RHE) for CPOC-Co. (c) Comparison of the faradaic efficiency between CPOC-Co and TPP-Co. (d) Comparison of the partial CO current density between CPOC-Co and TPP-Co. (e) Tafel plots of CPOC-Co and TPP-Co. (f) Comparison of the performance to other electrocatalytic materials. | ||
Electrochemical impedance spectroscopy (EIS) measurements disclose a much smaller charge transfer resistance for CPOC-Co in comparison with TPP-Co, Fig. S30,† allowing the better charge transfer capability of the cage-based electrode in CO2RR electrocatalysis. Furthermore, the surface concentrations of electrochemically active metal sites on the CPOC-Co and TPP-Co electrodes were compared by the integration of the corresponding CV anodic waves at different scan rates, Fig. S31 and S32.† The deduced average values for the CPOC-Co and TPP-Co electrodes are 8.1 × 10−8 and 5.4 × 10−8 mol cm−2, respectively, representing 13.4% and 8.8% of the total cobalt(II) porphyrin moieties as active sites in electrocatalysis. The higher percentage of active molecules for the CPOC-Co electrode than the TPP-Co electrode, as well as some good catalysts such as CoPc-PI-COF-1 (3.5%),117 COF-367-Co (4%),101 Co-TTCOF (0.9%),118 CoPcPDQ-COF (4.7%),120 and COP-SA (0.9%),121 might be associated with the unique porous structure of the cage molecule and good dispersion on the carbon substrate through solution processing, as indicated by the TEM photos and energy dispersive X-ray spectroscopy element mapping in Fig. S33 and S34.† In order to compare the CO2RR electrocatalysis dynamics of the CPOC-Co and TPP-Co electrodes, Tafel plots with the overpotential (η) vs. the logarithm of current density [log(jCO)] were recorded, Fig. 3e. The Tafel slope shows a linear relationship in the overpotential range of −0.19 and −0.36 V, providing fitting data of 84 and 136 mV dec−1 for CPOC-Co and TPP-Co, respectively. These small Tafel slopes suggest their faster CO2RR kinetics, possibly due to the effective electron transfer and large active surface area. Moreover, the smaller Tafel slope for CPOC-Co further implies the important role of the special structure of the porphyrin cage in CO2RR electrocatalysis.
Toward practical applications, the long-term durability of CPOC-Co for the electrocatalytic CO2RR was tested at −0.70 V (vs. RHE). After 10 h, the FECO for the CPOC-Co electrode is still kept above 89% with slight decay in comparison with the fresh catalyst (92%), illustrating its excellent durability, Fig. S35.† During the 10 h electrocatalytic test at −0.70 V (vs. RHE), the turnover number of CPOC-Co accumulated to 35128, comparable to the excellent reticular framework electrocatalysts COF-367-Co (3901)101 and MOF-1992/CB (5800)122 under similar conditions, Fig. 3f and Table S3.† After a 10 h test at −0.70 V, the XPS peaks of Co 2p1/2 and Co 2p3/2 observed at 795.80 and 780.60 eV for divalent cobalt ions in the used electrode are in good agreement with those found for CPOC-Co, Fig. S36.† Furthermore, FT-IR spectroscopic data disclose the lack of any significant change between the fresh CPOC-Co and used catalyst, Fig. S37.† These characterization results for the used electrode support the good chemical stability of this cage during the present electrocatalysis conditions. It is worth noting that no CO product was detected in the control electrocatalysis for the CPOC-Zn and CPOC-Cu analogues. In contrast, a little CO product is determined for the CPOC-Ni control cathode under the large potential of −0.70 V. However, the FECO for the CPOC-Ni cathode is lower than 58% even at the applied potential of −0.90 V, Fig. S38–S41.† These data verify the best electrochemical catalytic activities of CPOC-Co among these metallic cages.
To better understand the good catalytic performance of the cobalt cage rather than the other metal species, DFT calculations were performed to study the mechanism of the active metallic porphyrin models (Por-M, M = Co, Ni, Cu, Zn) for the CO2RR, Fig. 4a. Their CO2RR processes contain four classic steps, namely CO2 adsorption with Por-M, the formation of one electron-mediated *COOH and *CO products around the metal sites, and the CO desorption process. According to the energy barriers (ΔG) for each step that occurred on the metal site in Fig. 4, the potential-determining step for Por-Co was assigned to the reduction of *CO2 to *COOH. The value of ΔG for Por-Co is 1.15 eV, much smaller than that (about 2.00 eV) for Por-Ni, Por-Cu, and Por-Zn, suggesting the higher catalytic performance of the cobalt cage towards the CO2RR from a thermodynamic perspective.
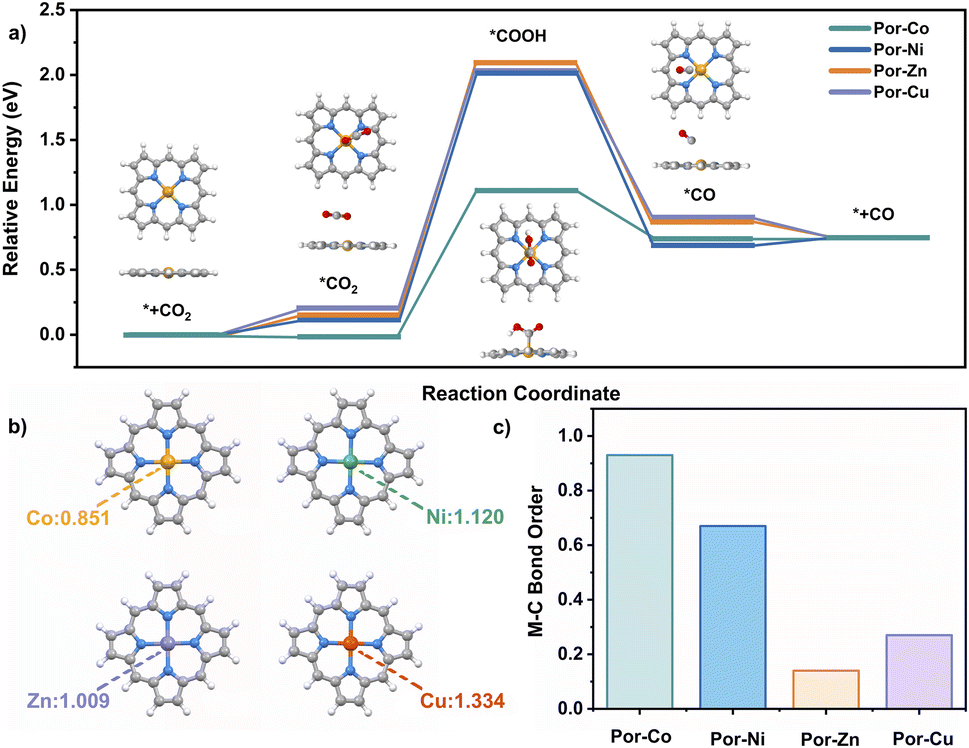 | ||
| Fig. 4 (a) Free energy diagrams for CO2 reduction to CO on Por-M. (b) NPA charge of Co, Ni, Cu and Zn atoms in the porphyrin without introduction of CO2. (c) Mayer bond order of M-C in the *COOH intermediate. | ||
The different metallic nature of these cages inspired us to find the origin of the different activity in the CO2RR from their electronic structures. As a result, charge distribution, adsorption capacity and intermediate stability upon Por-M were investigated. The natural population analysis (NPA) charge of the four kinds of metal atoms illustrates the more electron-rich environment for Por-Co (0.851) compared with the other three metallic species Por-M (1.009–1.334 for M = Ni, Zn or Cu) sites, Fig. 4b. As a result, Por-Co possesses stronger physical adsorption capability for CO2 molecules.123 This is supported by the lower adsorption energy of CO2 on the Co site (−0.02 eV) than on the Ni, Zn, and Cu center metal sites (0.13–0.19 eV). In addition, the Co–C Mayer bond order (0.93) of the *COOH intermediate on Por-Co is much higher than that in other Por-M (M = Ni, Cu, Zn), indicating the better intermediate stability for the cobalt molecule. These results well rationalize the excellent electrocatalytic activity of the cobalt porphyrin and certainly for the cofacial cobalt cage towards the CO2RR, Fig. 4c.
In order to gain an insight into the high selectivity for the cobalt cage, theoretical calculations for the hydrogen evolution reaction (HER) were performed over the Por-Co model as well as the other three Por-M (M = Ni, Zn, Cu) models, Fig. S42.† Por-Co has a much lower energy barrier of 1.16 eV relative to that of 2.29–2.99 eV for the Por-M (M = Ni, Zn, Cu) models, indicating the possible excellent electrocatalytic behavior toward the HER for the former catalyst. The value of CO2RR for Por-Co is similar to that of the HER, indicating that CO2RR happened preferentially on cobalt sites. However, it is worth noting that Por-Co possesses stronger physical adsorption capability for CO2 molecules, supported by the lower adsorption energy of CO2 on the Co site (−0.02 eV) than that of the Ni, Cu, and Zn sites (0.13–0.19 eV). The preferred adsorption of CO2 on the active metal site in thermodynamics level may be favorable for the CO2RR with high selectivity for Por-Co and thus the cobalt cage. For other metallic units, the high energy for both the CO2RR and HER may introduce poor electrochemical catalytic activity and the corresponding cages.
Conclusions
In summary, we have designed and prepared a series of D–A-type functional cofacial organic cages based on the stepwise assembly of a porphyrin donor and naphthalene-1,4:5,8-bis(dicarboximide) acceptor followed by post-metalation. Their excitation energy transfer and electrocatalytic carbon dioxide behaviors have been carefully studied, revealing the important role of metals in the photophysical and electrocatalytic mechanism. In particular, the cofacial and porous structure for the cobalt cage ensures efficient and selective electrocatalytic carbon dioxide reduction. This work not only provides multiple functional organic cages though the synergistic predesign and post-synthesis methods, but also gives a clear structure-related photophysical and electrocatalytic study.Data availability
All the data supporting this article have been included in themain text and the ESI.†Author contributions
X. L., C. L. H. W., H. L., X. L., and J. J. conceived the subject and designed the experiments. X. L. and C. L. synthesized the materials and performed most of the characterization experiments with the help of B. Y. and B. H. X. S., H. L., and X. L. collected the femtosecond transient absorption data. X. D. performed the theoretical simulation. X. L., H. W., H. L., X. L., and J. J. interpreted the results and wrote the paper. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Natural Science Foundation of China (No. 22235001, 22175020, 22133006, 22261132512, and 22131005), the Xiaomi Young Scholar Program, the Fundamental Research Funds for the Central Universities and the University of Science and Technology Beijing.Notes and references
- M. Yamashina, Y. Tanaka, R. Lavendomme, T. K. Ronson, M. Pittelkow and J. R. Nitschke, Nature, 2019, 574, 511–515 CrossRef CAS PubMed.
- J. Dong, Y. Liu and Y. Cui, Acc. Chem. Res., 2021, 54, 194–206 CrossRef CAS PubMed.
- M. Liu, L. Zhang, M. A. Little, V. Kapil, M. Ceriotti, S. Yang, L. Ding, D. L. Holden, R. Balderas-Xicohténcatl, D. He, R. Clowes, S. Y. Chong, G. Schütz, L. Chen, M. Hirscher and A. I. Cooper, Science, 2019, 366, 613–620 CrossRef CAS PubMed.
- J. Liu, Z. Wang, P. Cheng, M. J. Zaworotko, Y. Chen and Z. Zhang, Nat. Rev. Chem., 2022, 6, 339–356 CrossRef PubMed.
- M. Yoshizawa, M. Tamura and M. Fujita, Science, 2006, 312, 251–254 CrossRef CAS PubMed.
- T. Tozawa, J. T. A. Jones, S. I. Swamy, S. Jiang, D. J. Adams, S. Shakespeare, R. Clowes, D. Bradshaw, T. Hasell, S. Y. Chong, C. Tang, S. Thompson, J. Parker, A. Trewin, J. Bacsa, A. M. Z. Slawin, A. Steiner and A. I. Cooper, Nat. Mater., 2009, 8, 973–978 CrossRef CAS PubMed.
- Y. Ni, T. Y. Gopalakrishna, H. Phan, T. Kim, T. S. Herng, Y. Han, T. Tao, J. Ding, D. Kim and J. Wu, Nat. Chem., 2020, 12, 242–248 CrossRef CAS PubMed.
- R. Saha, B. Mondal and P. S. Mukherjee, Chem. Rev., 2022, 122, 12244–12307 CrossRef CAS PubMed.
- N. Ahmad, H. A. Younus, A. H. Chughtai and F. Verpoort, Chem. Soc. Rev., 2015, 44, 9–25 RSC.
- M. Han, D. M. Engelhard and G. H. Clever, Chem. Soc. Rev., 2014, 43, 1848–1860 RSC.
- T. Jiao, G. Wu, Y. Zhang, L. Shen, Y. Lei, C.-Y. Wang, A. C. Fahrenbach and H. Li, Angew. Chem., Int. Ed., 2020, 59, 18350–18367 CrossRef CAS PubMed.
- X. Jing, C. He, L. Zhao and C. Duan, Acc. Chem. Res., 2019, 52, 100–109 CrossRef CAS PubMed.
- L. Liang, W. Zhao, X.-J. Yang and B. Wu, Acc. Chem. Res., 2022, 55, 3218–3229 CrossRef CAS PubMed.
- A. J. Gosselin, C. A. Rowland and E. D. Bloch, Chem. Rev., 2020, 120, 8987–9014 CrossRef CAS PubMed.
- N. Hosono and S. Kitagawa, Acc. Chem. Res., 2018, 51, 2437–2446 CrossRef CAS PubMed.
- Y. Wang, J.-P. Chang, R. Xu, S. Bai, D. Wang, G.-P. Yang, L.-Y. Sun, P. Li and Y.-F. Han, Chem. Soc. Rev., 2021, 50, 13559–13586 RSC.
- W. Cullen, H. Takezawa and M. Fujita, Angew. Chem., Int. Ed., 2019, 58, 9171–9173 CrossRef CAS PubMed.
- E. Sánchez-González, M. Y. Tsang, J. Troyano, G. A. Craig and S. Furukawa, Chem. Soc. Rev., 2022, 51, 4876–4889 RSC.
- R. Ham, C. J. Nielsen, S. Pullen and J. N. H. Reek, Chem. Rev., 2023, 123, 5225–5261 CrossRef CAS PubMed.
- Y. Fang, J. A. Powell, E. Li, Q. Wang, Z. Perry, A. Kirchon, X. Yang, Z. Xiao, C. Zhu, L. Zhang, F. Huang and H.-C. Zhou, Chem. Soc. Rev., 2019, 48, 4707–4730 RSC.
- S.-C. Li, L.-X. Cai, M. Hong, Q. Chen and Q.-F. Sun, Angew. Chem., Int. Ed., 2022, 61, e202204732 CAS.
- E.-S. M. El-Sayed, Y. D. Yuan, D. Zhao and D. Yuan, Acc. Chem. Res., 2022, 55, 1546–1560 CrossRef CAS PubMed.
- X. Tang, C. Meng, N. Rampal, A. Li, X. Chen, W. Gong, H. Jiang, D. Fairen-Jimenez, Y. Cui and Y. Liu, J. Am. Chem. Soc., 2023, 145, 2561–2571 CrossRef CAS PubMed.
- C. Tan, J. Jiao, Z. Li, Y. Liu, X. Han and Y. Cui, Angew. Chem., Int. Ed., 2018, 57, 2085–2090 CrossRef CAS PubMed.
- K. Li, K. Wu, Y.-Z. Fan, J. Guo, Y.-L. Lu, Y.-F. Wang, G. Maurin and C.-Y. Su, Natl. Sci. Rev., 2021, 9, nwab155 CrossRef PubMed.
- D. Li, X. Liu, L. Yang, H. Li, G. Guo, X. Li and C. He, Chem. Sci., 2023, 14, 2237–2244 RSC.
- S. Pullen and G. H. Clever, Acc. Chem. Res., 2018, 51, 3052–3064 CrossRef CAS PubMed.
- S. Lee, H. Jeong, D. Nam, M. S. Lah and W. Choe, Chem. Soc. Rev., 2021, 50, 528–555 RSC.
- T. Nakama, A. Rossen, R. Ebihara, M. Yagi-Utsumi, D. Fujita, K. Kato, S. Sato and M. Fujita, Chem. Sci., 2023, 14, 2910–2914 RSC.
- C. Xu, Q. Lin, C. Shan, X. Han, H. Wang, H. Wang, W. Zhang, Z. Chen, C. Guo, Y. Xie, X. Yu, B. Song, H. Song, L. Wojtas and X. Li, Angew. Chem., Int. Ed., 2022, 61, e202203099 CAS.
- M. D. Pluth, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2009, 42, 1650–1659 CrossRef CAS PubMed.
- H. Amouri, C. Desmarets and J. Moussa, Chem. Rev., 2012, 112, 2015–2041 CrossRef CAS PubMed.
- R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810–6918 CrossRef CAS PubMed.
- P.-F. Cui, X.-R. Liu, Y.-J. Lin, Z.-H. Li and G.-X. Jin, J. Am. Chem. Soc., 2022, 144, 6558–6565 CrossRef CAS PubMed.
- G. Zhang and M. Mastalerz, Chem. Soc. Rev., 2014, 43, 1934–1947 RSC.
- G. Montà-González, F. Sancenón, R. Martínez-Máñez and V. Martí-Centelles, Chem. Rev., 2022, 122, 13636–13708 CrossRef PubMed.
- Y. Chen, G. Wu, B. Chen, H. Qu, T. Jiao, Y. Li, C. Ge, C. Zhang, L. Liang, X. Zeng, X. Cao, Q. Wang and H. Li, Angew. Chem., Int. Ed., 2021, 60, 18815–18820 CrossRef CAS PubMed.
- X. Liu, Z. Shi, M. Xie, J. Xu, Z. Zhou, S. Jung, G. Cui, Y. Zuo, T. Li, C. Yu, Z. Liu and S. Zhang, Angew. Chem., Int. Ed., 2021, 60, 15080–15086 CrossRef CAS PubMed.
- H. Qu, Z. Huang, X. Dong, X. Wang, X. Tang, Z. Li, W. Gao, H. Liu, R. Huang, Z. Zhao, H. Zhang, L. Yang, Z. Tian and X. Cao, J. Am. Chem. Soc., 2020, 142, 16223–16228 CrossRef CAS PubMed.
- Z. Wang, X. He, T. Yong, Y. Miao, C. Zhang and B. Zhong Tang, J. Am. Chem. Soc., 2020, 142, 512–519 CrossRef CAS PubMed.
- P. Ballester, Chem. Soc. Rev., 2010, 39, 3810–3830 RSC.
- R. D. Mukhopadhyay, Y. Kim, J. Koo and K. Kim, Acc. Chem. Res., 2018, 51, 2730–2738 CrossRef CAS PubMed.
- S. Bera, A. Basu, S. Tothadi, B. Garai, S. Banerjee, K. Vanka and R. Banerjee, Angew. Chem., Int. Ed., 2017, 56, 2123–2126 CrossRef CAS PubMed.
- X. Zhao, Y. Liu, Z.-Y. Zhang, Y. Wang, X. Jia and C. Li, Angew. Chem., Int. Ed., 2021, 60, 17904–17909 CrossRef CAS PubMed.
- D. Luo, Y. He, J. Tian, J. L. Sessler and X. Chi, J. Am. Chem. Soc., 2022, 144, 113–117 CrossRef CAS PubMed.
- L. R. MacGillivray and J. L. Atwood, Nature, 1997, 389, 469–472 CrossRef CAS.
- Y. Liu, C. Hu, A. Comotti and M. D. Ward, Science, 2011, 333, 436–440 CrossRef CAS PubMed.
- J. Rebek Jr, Acc. Chem. Res., 2009, 42, 1660–1668 CrossRef PubMed.
- X. Zhao, H. Wang, B. Li, W. Zhang, X. Li, W. Zhao, C. Janiak, A. W. Heard, X.-J. Yang and B. Wu, Angew. Chem., Int. Ed., 2022, 61, e202115042 CrossRef CAS PubMed.
- D. Beaudoin, F. Rominger and M. Mastalerz, Angew. Chem., Int. Ed., 2016, 55, 15599–15603 CrossRef CAS PubMed.
- Y. Jin, Q. Wang, P. Taynton and W. Zhang, Acc. Chem. Res., 2014, 47, 1575–1586 CrossRef CAS PubMed.
- T. Hasell and A. I. Cooper, Nat. Rev. Mater., 2016, 1, 16053 CrossRef CAS.
- M. Mastalerz, Acc. Chem. Res., 2018, 51, 2411–2422 CrossRef CAS PubMed.
- H. Wang, Y. Jin, N. Sun, W. Zhang and J. Jiang, Chem. Soc. Rev., 2021, 50, 8874–8886 RSC.
- C. Halliwell, J. F. Soria and A. Fernandez, Angew. Chem., Int. Ed., 2023, 62, e202217729 CrossRef CAS PubMed.
- F. Beuerle and B. Gole, Angew. Chem., Int. Ed., 2018, 57, 4850–4878 CrossRef CAS PubMed.
- Z. Shan, X. Wu, B. Xu, Y.-l. Hong, M. Wu, Y. Wang, Y. Nishiyama, J. Zhu, S. Horike, S. Kitagawa and G. Zhang, J. Am. Chem. Soc., 2020, 142, 21279–21284 CrossRef CAS PubMed.
- X. Yang, J.-K. Sun, M. Kitta, H. Pang and Q. Xu, Nat. Catal., 2018, 1, 214–220 CrossRef CAS.
- J.-K. Sun, W.-W. Zhan, T. Akita and Q. Xu, J. Am. Chem. Soc., 2015, 137, 7063–7066 CrossRef CAS PubMed.
- M. Hua, S. Wang, Y. Gong, J. Wei, Z. Yang and J.-K. Sun, Angew. Chem., Int. Ed., 2021, 60, 12490–12497 CrossRef CAS PubMed.
- N. Sun, C. Wang, H. Wang, L. Yang, P. Jin, W. Zhang and J. Jiang, Angew. Chem., Int. Ed., 2019, 58, 18011–18016 CrossRef CAS PubMed.
- K. Acharyya and P. S. Mukherjee, Angew. Chem., Int. Ed., 2019, 58, 8640–8653 CrossRef CAS PubMed.
- K. Su, W. Wang, S. Du, C. Ji, M. Zhou and D. Yuan, J. Am. Chem. Soc., 2020, 142, 18060–18072 CrossRef CAS PubMed.
- C. Liu, Y. Jin, Z. Yu, L. Gong, H. Wang, B. Yu, W. Zhang and J. Jiang, J. Am. Chem. Soc., 2022, 144, 12390–12399 CrossRef CAS PubMed.
- E. Martínez-Ahumada, D. He, V. Berryman, A. López-Olvera, M. Hernandez, V. Jancik, V. Martis, M. A. Vera, E. Lima, D. J. Parker, A. I. Cooper, I. A. Ibarra and M. Liu, Angew. Chem., Int. Ed., 2021, 60, 17556–17563 CrossRef PubMed.
- L. Cheng, P. Tian, H. Duan, Q. Li, X. Song, A. Li and L. Cao, Chem. Sci., 2023, 14, 833–842 RSC.
- L.-J. Wang, S. Bai and Y.-F. Han, J. Am. Chem. Soc., 2022, 144, 16191–16198 CrossRef CAS PubMed.
- Z. Wang, N. Sikdar, S.-Q. Wang, X. Li, M. Yu, X.-H. Bu, Z. Chang, X. Zou, Y. Chen, P. Cheng, K. Yu, M. J. Zaworotko and Z. Zhang, J. Am. Chem. Soc., 2019, 141, 9408–9414 CrossRef CAS PubMed.
- T. Hasell, M. Miklitz, A. Stephenson, M. A. Little, S. Y. Chong, R. Clowes, L. Chen, D. Holden, G. A. Tribello, K. E. Jelfs and A. I. Cooper, J. Am. Chem. Soc., 2016, 138, 1653–1659 CrossRef CAS PubMed.
- M. W. Schneider, I. M. Oppel, A. Griffin and M. Mastalerz, Angew. Chem., Int. Ed., 2013, 52, 3611–3615 CrossRef CAS PubMed.
- S. Jiang, Y. Du, M. Marcello, E. W. Corcoran Jr, D. C. Calabro, S. Y. Chong, L. Chen, R. Clowes, T. Hasell and A. I. Cooper, Angew. Chem., Int. Ed., 2018, 57, 11228–11232 CrossRef CAS PubMed.
- T. Hasell, S. Y. Chong, M. Schmidtmann, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2012, 51, 7154–7157 CrossRef CAS PubMed.
- N. Giri, M. G. Del Pópolo, G. Melaugh, R. L. Greenaway, K. Rätzke, T. Koschine, L. Pison, M. F. C. Gomes, A. I. Cooper and S. L. James, Nature, 2015, 527, 216–220 CrossRef CAS PubMed.
- K. Jie, N. Onishi, J. A. Schott, I. Popovs, D.-e. Jiang, S. Mahurin and S. Dai, Angew. Chem., Int. Ed., 2020, 59, 2268–2272 CrossRef CAS PubMed.
- J.-X. Ma, J. Li, Y.-F. Chen, R. Ning, Y.-F. Ao, J.-M. Liu, J. Sun, D.-X. Wang and Q.-Q. Wang, J. Am. Chem. Soc., 2019, 141, 3843–3848 CrossRef CAS PubMed.
- B. Han, H. Wang, C. Wang, H. Wu, W. Zhou, B. Chen and J. Jiang, J. Am. Chem. Soc., 2019, 141, 8737–8740 CrossRef CAS PubMed.
- R. McCaffrey, H. Long, Y. Jin, A. Sanders, W. Park and W. Zhang, J. Am. Chem. Soc., 2014, 136, 1782–1785 CrossRef CAS PubMed.
- X. Zhang, K. Su, A. G. A. Mohamed, C. Liu, Q. Sun, D. Yuan, Y. Wang, W. Xue and Y. Wang, Energy Environ. Sci., 2022, 15, 780–785 RSC.
- T. Šolomek, N. E. Powers-Riggs, Y.-L. Wu, R. M. Young, M. D. Krzyaniak, N. E. Horwitz and M. R. Wasielewski, J. Am. Chem. Soc., 2017, 139, 3348–3351 CrossRef PubMed.
- C. Liu, K. Liu, C. Wang, H. Liu, H. Wang, H. Su, X. Li, B. Chen and J. Jiang, Nat. Commun., 2020, 11, 1047 CrossRef CAS PubMed.
- A. Aster, C. Rumble, A.-B. Bornhof, H.-H. Huang, N. Sakai, T. Šolomek, S. Matile and E. Vauthey, Chem. Sci., 2021, 12, 4908–4915 RSC.
- T. D. Lash, Chem. Rev., 2022, 122, 7987–7989 CrossRef CAS PubMed.
- Y. Ding, W.-H. Zhu and Y. Xie, Chem. Rev., 2017, 117, 2203–2256 CrossRef CAS PubMed.
- J. L. Sessler, Z. Gross and H. Furuta, Chem. Rev., 2017, 117, 2201–2202 CrossRef CAS PubMed.
- T. Tanaka and A. Osuka, Chem. Rev., 2017, 117, 2584–2640 CrossRef CAS PubMed.
- W. Zhang, W. Lai and R. Cao, Chem. Rev., 2017, 117, 3717–3797 CrossRef CAS PubMed.
- J. Jiang and D. K. P. Ng, Acc. Chem. Res., 2009, 42, 79–88 CrossRef CAS PubMed.
- P. S. Bols and H. L. Anderson, Acc. Chem. Res., 2018, 51, 2083–2092 CrossRef CAS PubMed.
- R. L. Milot and C. A. Schmuttenmaer, Acc. Chem. Res., 2015, 48, 1423–1431 CrossRef CAS PubMed.
- E. Nikoloudakis, I. López-Duarte, G. Charalambidis, K. Ladomenou, M. Ince and A. G. Coutsolelos, Chem. Soc. Rev., 2022, 51, 6965–7045 RSC.
- M. Gilbert and B. Albinsson, Chem. Soc. Rev., 2015, 44, 845–862 RSC.
- J. S. Lindsey and D. F. Bocian, Acc. Chem. Res., 2011, 44, 638–650 CrossRef CAS PubMed.
- Y. Oka, H. Masai and J. Terao, Angew. Chem., Int. Ed., 2023, 62, e202217002 CrossRef CAS PubMed.
- W. Danowski, F. Castiglioni, A. S. Sardjan, S. Krause, L. Pfeifer, D. Roke, A. Comotti, W. R. Browne and B. L. Feringa, J. Am. Chem. Soc., 2020, 142, 9048–9056 CrossRef CAS PubMed.
- C. Costentin, S. Drouet, M. Robert and J.-M. Savéant, Science, 2012, 338, 90–94 CrossRef CAS PubMed.
- X.-L. Lv, K. Wang, B. Wang, J. Su, X. Zou, Y. Xie, J.-R. Li and H.-C. Zhou, J. Am. Chem. Soc., 2017, 139, 211–217 CrossRef CAS PubMed.
- T. He, S. Chen, B. Ni, Y. Gong, Z. Wu, L. Song, L. Gu, W. Hu and X. Wang, Angew. Chem., Int. Ed., 2018, 57, 3493–3498 CrossRef CAS PubMed.
- K. Sun, Y. Qian and H.-L. Jiang, Angew. Chem., Int. Ed., 2023, 62, e202217565 CrossRef CAS PubMed.
- Y.-R. Wang, H.-M. Ding, X.-Y. Ma, M. Liu, Y.-L. Yang, Y. Chen, S.-L. Li and Y.-Q. Lan, Angew. Chem., Int. Ed., 2022, 61, e202114648 CAS.
- E.-X. Chen, M. Qiu, Y.-F. Zhang, L. He, Y.-Y. Sun, H.-L. Zheng, X. Wu, J. Zhang and Q. Lin, Angew. Chem., Int. Ed., 2022, 61, e202111622 CAS.
- S. Lin, C. S. Diercks, Y.-B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang, O. M. Yaghi and C. J. Chang, Science, 2015, 349, 1208–1213 CrossRef CAS PubMed.
- L. An, P. De La Torre, P. T. Smith, M. R. Narouz and C. J. Chang, Angew. Chem., Int. Ed., 2023, 62, e202209396 CAS.
- C. F. G. C. Geraldes, M. M. C. A. Castro and J. A. Peters, Coord. Chem. Rev., 2021, 445, 214069 CrossRef CAS.
- G. Yu, T.-Y. Cen, Z. He, S.-P. Wang, Z. Wang, X.-W. Ying, S. Li, O. Jacobson, S. Wang, L. Wang, L.-S. Lin, R. Tian, Z. Zhou, Q. Ni, X. Li and X. Chen, Angew. Chem., Int. Ed., 2019, 58, 8799–8803 CrossRef CAS PubMed.
- S. Durot, J. Taesch and V. Heitz, Chem. Rev., 2014, 114, 8542–8578 CrossRef CAS PubMed.
- F. J. Rizzuto, L. K. S. von Krbek and J. R. Nitschke, Nat. Rev. Chem., 2019, 3, 204–222 CrossRef.
- A. Dhamija, C. K. Das, Y. H. Ko, Y. Kim, R. D. Mukhopadhyay, A. Gunnam, X. Yu, I.-C. Hwang, L. V. Schäfer and K. Kim, Chem, 2022, 8, 543–556 CAS.
- C. J. Chang, Z.-H. Loh, C. Shi, F. C. Anson and D. G. Nocera, J. Am. Chem. Soc., 2004, 126, 10013–10020 CrossRef CAS PubMed.
- S. Fukuzumi, K. Okamoto, C. P. Gros and R. Guilard, J. Am. Chem. Soc., 2004, 126, 10441–10449 CrossRef CAS PubMed.
- M. R. Crawley, D. Zhang, A. N. Oldacre, C. M. Beavers, A. E. Friedman and T. R. Cook, J. Am. Chem. Soc., 2021, 143, 1098–1106 CrossRef CAS PubMed.
- E. A. Mohamed, Z. N. Zahran and Y. Naruta, Chem. Mater., 2017, 29, 7140–7150 CrossRef CAS.
- C. G. Oliveri, P. A. Ulmann, M. J. Wiester and C. A. Mirkin, Acc. Chem. Res., 2008, 41, 1618–1629 CrossRef CAS PubMed.
- D. Gosztola, M. P. Niemczyk, W. Svec, A. S. Lukas and M. R. Wasielewski, J. Phys. Chem. A, 2000, 104, 6545–6551 CrossRef CAS.
- Y. Venkatesh, M. Venkatesan, B. Ramakrishna and P. R. Bangal, J. Phys. Chem. B, 2016, 120, 9410–9421 CrossRef CAS PubMed.
- P. T. Smith, B. P. Benke, Z. Cao, Y. Kim, E. M. Nichols, K. Kim and C. J. Chang, Angew. Chem., Int. Ed., 2018, 57, 9684–9688 CrossRef CAS PubMed.
- Y. Hu, S. Huang, L. J. Wayment, J. Wu, Q. Xu, T. Chang, Y.-P. Chen, X. Li, B. Andi, H. Chen, Y. Jin, H. Zhu, M. Du, S. Lu and W. Zhang, Cell Rep. Phys. Sci., 2023, 4, 101285 CrossRef CAS.
- B. Han, X. Ding, B. Yu, H. Wu, W. Zhou, W. Liu, C. Wei, B. Chen, D. Qi, H. Wang, K. Wang, Y. Chen, B. Chen and J. Jiang, J. Am. Chem. Soc., 2021, 143, 7104–7113 CrossRef CAS PubMed.
- H.-J. Zhu, M. Lu, Y.-R. Wang, S.-J. Yao, M. Zhang, Y.-H. Kan, J. Liu, Y. Chen, S.-L. Li and Y.-Q. Lan, Nat. Commun., 2020, 11, 497 CrossRef CAS PubMed.
- Q. Wu, R.-K. Xie, M.-J. Mao, G.-L. Chai, J.-D. Yi, S.-S. Zhao, Y.-B. Huang and R. Cao, ACS Energy Lett., 2020, 5, 1005–1012 CrossRef CAS.
- N. Huang, K. H. Lee, Y. Yue, X. Xu, S. Irle, Q. Jiang and D. Jiang, Angew. Chem., Int. Ed., 2020, 59, 16587–16593 CrossRef CAS PubMed.
- Y. Song, J.-J. Zhang, Z. Zhu, X. Chen, L. Huang, J. Su, Z. Xu, T. H. Ly, C.-S. Lee, B. I. Yakobson, B. Z. Tang and R. Ye, Appl. Catal. B, 2021, 284, 119750 CrossRef CAS.
- R. Matheu, E. Gutierrez-Puebla, M. Á. Monge, C. S. Diercks, J. Kang, M. S. Prévot, X. Pei, N. Hanikel, B. Zhang, P. Yang and O. M. Yaghi, J. Am. Chem. Soc., 2019, 141, 17081–17085 CrossRef CAS PubMed.
- X. Zhang, Y. Wang, M. Gu, M. Wang, Z. Zhang, W. Pan, Z. Jiang, H. Zheng, M. Lucero, H. Wang, G. E. Sterbinsky, Q. Ma, Y.-G. Wang, Z. Feng, J. Li, H. Dai and Y. Liang, Nat. Energy, 2020, 5, 684–769 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc01816d |
| This journal is © The Royal Society of Chemistry 2023 |