
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Two-dimensional self-assembly and co-assembly of two tetracarboxylic acid derivatives investigated by STM†
Xuan
Peng‡
ad,
Linlin
Gan‡
ac,
Wenchao
Zhai
d,
Xiaoling
Chen
d,
Ke
Deng
 *a,
Wubiao
Duan
*c,
Wei
Li
*d and
Qingdao
Zeng
*ab
*a,
Wubiao
Duan
*c,
Wei
Li
*d and
Qingdao
Zeng
*ab
aCAS Key Laboratory of Standardization and Measurement for Nanotechnology, CAS Center for Excellence in Nanoscience, National Center for Nanoscience and Technology (NCNST), Beijing 100190, China. E-mail: kdeng@nanoctr.cn; zengqd@nanoctr.cn
bCenter of Materials Science and Optoelectronics Engineering, University of Chinese Academy of Sciences, Beijing 100049, China
cDepartment of Chemistry, School of Science, Beijing Jiaotong University, Beijing 100044, China. E-mail: wbduan@bjtu.edu.cn
dSchool of Science, Nanchang Institute of Technology, Nanchang 330099, China. E-mail: liweidting@nit.edu.cn
First published on 3rd August 2023
Abstract
In this work, the two-dimensional self-assembly and co-assembly behaviors of two tetracarboxylic acid derivatives (H4BDETP and H4BTB) were investigated by scanning tunneling microscopy (STM). H4BDETP molecules self-assembled into linear nanostructures, and H4BTB molecules formed lamellar and tetragonal nanostructures. The formation of a H4BDETP/H4BTB co-assembly nanostructure was closely related to the deposition sequence of H4BDETP and H4BTB on highly oriented pyrolytic graphite (HOPG). The introduction of H4BTB into the self-assembly system of H4BDETP resulted in the emergence of the H4BDETP/H4BTB nanostructure, while the addition of H4BDETP had no effect on the self-assembly system of H4BTB and a H4BDETP/H4BTB co-assembly nanostructure was not obtained.
1. Introduction
Supramolecular self-assembly has been a wide concern in many fields such as nanopattern fabrication and heterogeneous nanomaterials ascribed to its ability to guide simple molecular components to spontaneously form complicated and well-organized molecular aggregation states via molecular non-covalent interactions.1–4 Supramolecular self-assembly is promising in the preparation of diverse functional nanostructures through designing the chemical structures of functional building blocks at the molecular level and controlling the connection of building blocks. The precise construction of multi-component self-assembly systems anticipated to possess functions that cannot be achieved using mono-component systems has attracted attention and yet remains challenging.5,6 The fabrication of multi-component nanostructures is affected not only by the complementarity of the chemically distinct molecular building blocks in spatial size and shape, but also by the uncontrollable connection between building blocks attributed to their abundant binding sites.7,8 The research on two-dimensional assembly systems at the molecular level is conducive to explore the characteristics and mechanism of molecular assembly and provides guidance for the controllable preparation of complex hybrid nanostructures. Scanning tunneling microscopy (STM) can provide information about self-assembly morphology at the molecular level, which is helpful for investigating the intermolecular interaction mode and the laws of molecular self-assembly.9–11As one of the non-covalent interactions, a hydrogen bond is supposed to be a powerful driving force for inducing molecular self-assembly and the formed self-assembly nanostructures are more predictable attributed to its directivity and saturation. Functional groups including carboxyl groups, amino groups, cyan groups etc. are modified in the molecular structure to provide hydrogen bonding sites for intermolecular recognition.12–17 Among them, the modification of carboxyl groups at the ends of benzene rings is a commonly used method for constructing hydrogen-bonded supramolecular assembly systems.18–20 Molecular interaction sites are closely related to the number and substitution position of carboxyl groups, which influences the molecular symmetry and the intermolecular interaction mode and further the final self-assembly nanostructure.21 Earlier research studies mostly focused on the construction of binary nanostructures composed of carboxylic acid derivatives and pyridine derivatives, and it was found that the introduced pyridine derivatives could disrupt the O–H⋯O hydrogen bonds between carboxylic acid derivatives and formed stronger O–H⋯N hydrogen bonds with carboxylic acid derivatives.22–26 However, in addition to pyridine derivatives, external aromatic acid derivatives can also regulate the self-assembly structure of carboxylic acid derivatives. Initially, the interface co-assembly behavior of two C3-symmetric triacid derivatives, 1,3,5-tris(4-carboxyphenyl)benzene (BTB) and trimesic acid (TMA), in two solvents has been revealed by STM.27 Three binary hydrogen-bonded networks were obtained by adjusting the mixed solution concentration of BTB and TMA. However, BTB and TMA separately self-assembled into mono-component nanostructures and phase separation was observed when mixing them in a ultra-high vacuum on Au(111).28 Later, the regulatory effects of BTB and TMA on the self-assembly of other carboxylic acid derivatives were investigated. For instance, TMA can act as a bridging molecule to connect with low-symmetry carboxylic acid derivative H4OBDB (or H4ADDI molecules) via hydrogen bonds, thus forming two-component nanostructures with alternating arrangement between H4OBDB (or H4ADDI) and TMA molecules.29 Terephthalic acid (TPA) and TMA co-assembled into a rectangular nanostructure or compact nanostructure, depending on the substrate bias.30 Tetracarboxylic acid (PBPTTBA) and BTB formed a novel co-crystallized network driven by homomeric and heteromeric R22(8) hydrogen bonds.31 In this work, C2-symmetric 5′-(4-carboxyphenyl)-[1,1′:3′,1′′-terphenyl]-3,4′′,5-tricarboxylic acid (H4BTB) which possesses a terminal isophthalic acid group different from BTB was introduced to regulate the self-assembly nanostructure of 5′,5′′′′-([2,2′-bithiophene]-5,5′-diyl)bis(([1,1′:3′,1′′-terphenyl]-4,4′′-dicarboxylic acid)) (H4BDETP). And their chemical structures are displayed in Scheme 1.
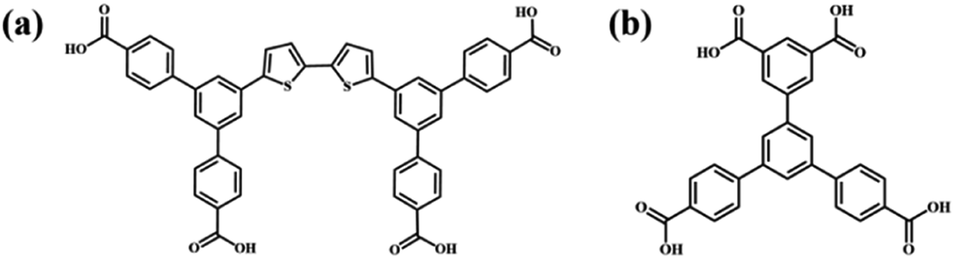 | ||
| Scheme 1 Chemical structures of (a) H4BDETP and (b) H4BTB. | ||
In this article, the mono-component and bi-component assembly systems of the two C2-symmetric tetracarboxylic acid derivatives (H4BDETP and H4BTB) at the 1-heptanoic acid/HOPG interface were studied by STM. H4BDETP molecules constituted dimeric building blocks and further aggregated into an ordered linear nanostructure based on hydrogen bonds. The addition of H4BTB destroyed the H4BDETP's linear structure and gave rise to the formation of a H4BDETP/H4BTB co-assembly nanostructure. H4BDETP's dimer building blocks were unaffected and H4BTB functioned as a bridging molecule to connect with H4BDETP's dimers through hydrogen bonds. Interestingly, the deposition sequence is crucial for the formation of the H4BDETP/H4BTB co-assembly nanostructure.
2. Experimental section
2.1 STM detection
H4BDETP and H4BTB were bought from Jilin Chinese Academy of Sciences – Yanshen Technology Co., Ltd and 1-heptanoic acid from J&K company. All samples were directly used without further purification here. H4BDETP and H4BTB were dissolved in 1-heptanoic acid solvent with a solution concentration around 10−4 mol L−1. Their solutions were respectively dropped on the freshly cleaved HOPG (grade ZYB, NTMDT, Russia) surface treated with Scotch tape and then probed using a Nanoscope IIIa scanning probe microscope (Bruker, USA). And a Pt/Ir (80/20) wire after mechanically cutting was employed as the STM tip. After observing the linear assembly nanostructure of H4BDETP, a droplet of H4BTB solution was dropped on HOPG to obtain the bi-component nanostructure and then detected by STM. To investigate the effect of the molecular deposition sequence on the co-assembly result, H4BDETP was deposited on the HOPG substrate covered by the self-assembly nanostructures of H4BTB followed by STM characterization. Detailed imaging conditions are provided in the titles of all figures.2.2 Theoretical calculations
Theoretical analysis in this work was fulfilled using density functional theory (DFT) calculations offered by the DMol3 code.32 Periodic boundary conditions (PBCs) were adopted to present the 2D periodic alignment on graphite. In order to give a depiction of exchange and correlation, the Perdew and Wang parametrization of the local exchange correlation energy was employed in the local spin density approximation (LSDA).33,34 All-electron spin-unrestricted Kohn–Sham wave functions were expanded in a local atomic orbital basis. A numerical basis set was applied for the large system. The calculations were equipped with medium mesh and were all-electron ones. A self-consistent field procedure was carried out by applying a convergence criterion of 10−5 au to the energy and electron density. Integrating with the experimental results, the lattice constants and the conformation of adsorbed molecules were ulteriorly optimized. With the achievement of the density convergence criterion and energy requirements, we attained the optimized lattice constants and the intermolecular interaction energy between adsorbed molecules.The model system presented the interactions between adsorbed molecules and the HOPG substrate. Owing to the similarity in the adsorption of adsorbed molecules containing a π-conjugated benzene ring on graphite and graphene, calculations were carried out on infinite graphene monolayers by utilizing PBC here. Along the normal direction, the interlayer spacing of graphene in the superlattice was 40 Å. As we built models for the adsorbates on graphene, graphene supercells were adopted, and the Brillouin zone was sampled using a 1 × 1 × 1 k-point mesh. The interaction energy (Einter) of the adsorbed molecules on graphite was given by Einter = Etot(adsorbates/graphene) − Etot(isolated adsorbates in vacuum) − Etot(graphene).
3. Results and discussion
3.1 Self-assembly nanostructure of H4BDETP
In the self-assembly system of H4BDETP, a well-ordered linear nanostructure (Fig. S1†) was formed at the liquid/solid interface. Fig. 1(a) provides more details about the linear structure. It shows that arched bright spots represented H4BDETP molecules which adsorbed on the substrate in two different orientations. Two H4BDETP molecules marked by yellow lines were arranged in a face-to-face manner and constituted a dimer. In a dimer, the distance between the branch chain ends of the two molecules was about 0.2 ± 0.1 nm, which implied that there may be hydrogen bonds between their terminal carboxyl groups. The measured distance between the branch chain ends of the yellow dimer and the neighboring blue dimer was 0.3 ± 0.1 nm, and this indicated that hydrogen bonds may also have existed between them. The measured lattice parameters in Fig. 1(a) were: a = 3.5 ± 0.1 nm, b = 3.2 ± 0.1 nm, and α = 108 ± 1°. According to the optimized structural model (Fig. 1(b)), the two molecules in a dimer interacted with each other through two pairs of O–H⋯O hydrogen bonds labeled with red circles. At the same time, the H4BDETP dimer formed a pair of O–H⋯O hydrogen bonds with two neighboring dimers respectively, which were marked by green circles. And the two remaining carboxyl groups of the dimer were unsaturated. Table 1 shows that the calculated lattice parameters were in good agreement with the experimental data, indicating that the molecular packing model was reasonable. | ||
| Fig. 1 (a) High-resolution STM image of H4BDETP's linear structure at the 1-heptanoic acid/HOPG interface, and the tunneling conditions were: Iset = 216.7 pA, Vbias = 707.4 mV (32.0 nm × 32.0 nm). (b) The calculated molecular model for the linear structure. | ||
| a (nm) | b (nm) | α (deg) | ||
|---|---|---|---|---|
| H4BDETP | Exptl | 3.5 ± 0.1 | 3.2 ± 0.1 | 108 ± 1 |
| Calcd | 3.42 | 3.1 | 108.4 | |
| H4BTB lamellar structure | Exptl | 1.7 ± 0.1 | 3.6 ± 0.1 | 118 ± 1 |
| Calcd | 1.65 | 3.7 | 118.5 | |
| H4BTB tetragonal structure | Exptl | 1.6 ± 0.1 | 1.6 ± 0.1 | 95 ± 1 |
| Calcd | 1.63 | 1.63 | 94.5 | |
| H4BDETP/H4BTB co-assembly structure | Exptl | 7.8 ± 0.1 | 5.3 ± 0.1 | 45 ± 1 |
| Calcd | 7.77 | 5.36 | 44.5 |
3.2 Self-assembly nanostructures of H4BTB
When a droplet of H4BTB solution was dropped on the HOPG surface, H4BTB molecules aggregated into a lamellar structure (domain I) and tetragonal structure (domain II) as shown in Fig. S2(a) and (b).† In the high-resolution image of the lamellar structure (Fig. 2(a)), H4BTB molecules obviously appeared as triangle bright spots due to the high electron cloud density of the π-conjugated structure and were adsorbed on HOPG in two opposite orientations. One vertex of the H4BTB molecule labeled with a white triangle was adjacent to the vertices of the two H4BTB molecules, which implied that the meta carboxyl groups of the H4BTB molecule may form hydrogen bonds with the carboxyl groups of the adjacent molecules. Simultaneously, the two remaining vertices were also close to the vertices of another two molecules respectively, and this suggested that the two remaining terminal carboxyl groups also possibly formed hydrogen bonds with the carboxyl groups of other molecules. The unit cell was labeled with a white parallelogram in Fig. 2(a) and the lattice parameters were measured to be: a = 1.7 ± 0.1 nm, b = 3.6 ± 0.1 nm, and α = 118 ± 1°. Based on the STM results, the optimized model (Fig. 2(b)) of H4BTB's lamellar structure was provided by theoretical calculations. Fig. 2(b) shows that all carboxyl groups of H4BTB molecules interacted with the carboxyl groups of adjacent molecules via O–H⋯O hydrogen bonds. Every four H4BTB molecules with two orientations constituted a cavity, and two kinds of cavities were observed in the lamellar structure. As shown in Table 1, the calculated lattice parameters agreed well with the experimental parameters.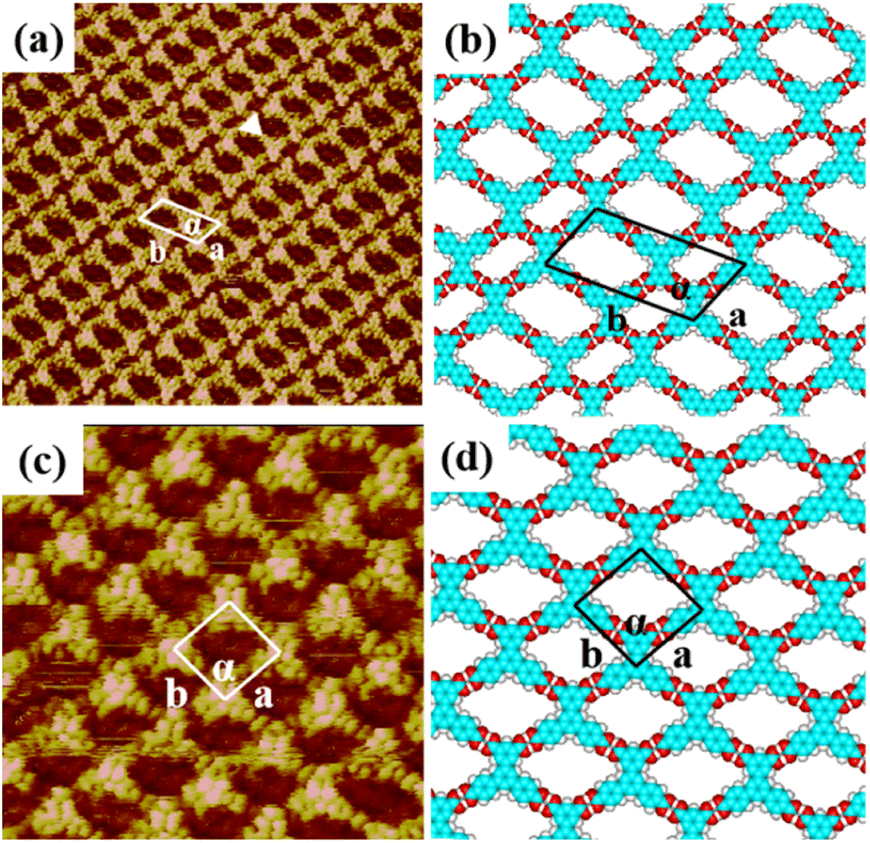 | ||
| Fig. 2 High-resolution STM image of H4BTB's (a) lamellar structure (25 nm × 25 nm) and (c) tetragonal structure (25 nm × 25 nm) at the 1-heptanoic acid/HOPG interface both with the tunneling conditions of Iset = 362.2 pA and Vbias = 668.0 mV. (b) and (d) The corresponding optimized molecular packing models for H4BTB's lamellar nanostructure in (a) and tetragonal nanostructure in (c) respectively. | ||
In the tetragonal structure of H4BTB (Fig. 2(c)), each triangle bright spot corresponded to one H4BTB molecule. Similar to the lamellar structure, the vertices of one H4BTB molecule in the tetragonal structure were adjacent to those of four H4BTB molecules, suggesting the existence of hydrogen bonds between their carboxyl groups. However, the difference from the lamellar structure was that all H4BTB molecules in the tetragonal structure were arranged in the same orientation and just formed one type of cavity. The measured parameters of the unit cell marked by white lines in Fig. 2(c) were: a = 1.6 ± 0.1 nm, b = 1.6 ± 0.1 nm, and α = 95 ± 1°. As shown in the optimized assembly model (Fig. 2(d)), all carboxyl groups of H4BTB molecules were saturated by forming O–H⋯O hydrogen bonds with the adjacent ones, which contributed to the generation of a tetragonal supramolecular network.
3.3 Co-assembly nanostructure of H4BDETP/H4BTB
To expand H4BDETP's packing structure, H4BTB as a bridging molecule was introduced into the pre-assembled structure of H4BDETP, and then characterized by STM. As displayed in (Fig. S3(a)–(c)†), a novel porous supramolecular network (domain I) different from the self-assembly nanostructures of H4BDETP or H4BTB was formed on the substrate. The formation of the H4BDETP/H4BTB co-assembly nanostructure was affected by the molar ratio of H4BDETP to H4BTB and the concentration of H4BTB to some extent. When the molar ratio of H4BDETP to H4BTB exceeded 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Fig. S3(a)†), H4BTB molecules disturbed a part of H4BDETP's self-assembly nanostructure and almost all H4BTB molecules formed a co-assembly nanostructure with H4BDETP. With the molar ratio reaching 2:1 (Fig. S3(b)†), the HOPG surface was nearly completely covered by the H4BDETP/H4BTB co-assembled nanostructure. As the concentration of H4BTB was increased and the molar ratio was controlled below 2:1 (Fig. S3(c)†), excessive H4BTB molecules formed a self-assembly structure that co-existed with the H4BDETP/H4BTB co-assembly nanostructure on HOPG.
:1 (Fig. S3(a)†), H4BTB molecules disturbed a part of H4BDETP's self-assembly nanostructure and almost all H4BTB molecules formed a co-assembly nanostructure with H4BDETP. With the molar ratio reaching 2:1 (Fig. S3(b)†), the HOPG surface was nearly completely covered by the H4BDETP/H4BTB co-assembled nanostructure. As the concentration of H4BTB was increased and the molar ratio was controlled below 2:1 (Fig. S3(c)†), excessive H4BTB molecules formed a self-assembly structure that co-existed with the H4BDETP/H4BTB co-assembly nanostructure on HOPG.
More details about the H4BDETP/H4BTB co-assembly nanostructure could be obtained from the high-resolution STM image (Fig. 3(a)). It showed that each arched bright spot represented one H4BDETP molecule, and every two H4BDETP molecules tended to constitute a dimer labeled with blue lines. Triangle bright spots representing H4BTB molecules isolated the H4BDETP dimers and increased the experimental unit cell parameters a = 7.8 ± 0.1 nm and b = 5.3 ± 0.1 nm respectively (Table 1). The optimized molecular model in Fig. 3(b) revealed that each H4BTB molecule was combined with four H4BDETP dimers via O–H⋯O hydrogen bonds between carboxyl groups. Every two H4BTB molecules with two different orientations and a pair of H4BDETP dimers constituted a hexagonal cavity. And the size of the hexagonal cavity depended on the position of H4BTB's carboxyl groups that formed hydrogen bonds with H4BDETP molecules.
 | ||
| Fig. 3 (a) High-resolution STM image of the H4BDETP/H4BTB co-assembly structure at the 1-heptanoic acid/HOPG interface, Iset = 265.5 pA and Vbias = 754.4 mV (46.3 nm × 46.3 nm). (b) The calculated molecular model for the H4BDETP/H4BTB co-assembly structure. | ||
To investigate the forming mechanism of the H4BDETP/H4BTB co-assembly nanostructure, DFT calculations were performed and energy values for the self-assembly nanostructures of H4BTB, H4BDETP and the co-assembly nanostructure of H4BDETP/H4BTB are presented in Table 2. The total energies included the energies of the interactions between molecules and the interactions between molecules and substrates. The thermodynamic stability can be evaluated by comparing the total energy of the distinct systems with the same lattice parameters. Due to the differences in the lattice parameters of systems, the total energy per unit area was adopted to compare the thermodynamic stability of distinct systems. The lower the total energy per unit area, the more stable the system was. As the total energies per unit area for the self-assembly nanostructures of H4BTB were lower than that for the self-assembly nanostructure of H4BDETP (Table 2), H4BTB could affect H4BDETP's linear structure, giving rise to the generation of the H4BDETP/H4BTB co-assembly nanostructure. The introduced H4BTB interacted with H4BDETP dimers through hydrogen bonds and saturated all carboxyl groups of H4BDETP dimers, which helped to reduce the energy of intermolecular interactions and led to a lower total energy per unit area of the H4BDETP/H4BTB co-assembly nanostructure (−0.276 kcal mol−1 Å−2) than that of H4BDETP's self-assembly nanostructure (−0.260 kcal mol−1 Å−2).
| Interactions between molecules (kcal mol−1) | Interactions between molecules and substrate (kcal mol−1) | Total energy (kcal mol−1) | Total energy per unit area (kcal mol−1 Å−2) | |
|---|---|---|---|---|
| H4BDETP | −79.737 | −181.944 | −261.681 | −0.260 |
| H4BTB lamellar structure | −118.258 | −109.912 | −228.170 | −0.425 |
| H4BTB tetragonal structure | −60.359 | −52.624 | −112.983 | −0.426 |
| H4BDETP/H4BTB co-assembly structure | −331.934 | −473.123 | −805.057 | −0.276 |
It was interesting that the formation of the H4BDETP/H4BTB co-assembly nanostructure was associated with the deposition sequence. We adopted the opposite deposition sequence. When H4BDETP was introduced in the pre-assembled system of H4BTB, H4BTB's self-assembly nanostructure remained unchanged and the H4BDETP/H4BTB co-assembly nanostructure was not observed in Fig. S4(a)–(c),† despite the molar ratio of the two components being adjusted. The introduced H4BDETP was unable to break the pre-assembled nanostructure of H4BTB. Although the calculated energies of the intermolecular interactions and the molecules–substrate interactions for H4BTB's self-assembly nanostructure were higher than those for the H4BDETP/H4BTB co-assembly structure (Table 2), the smaller lattice parameters of H4BTB's self-assembly nanostructure (Table 1) lowered the total energies per unit area, which was thermodynamically unfavorable for the formation of the H4BDETP/H4BTB co-assembly nanostructure. The theoretical calculations were consistent with the experimental results showing that the sturdy pre-assembled nanostructure of H4BTB was unaffected by the introduced H4BDETP. We have also investigated the self-assembly behavior of the pre-mixed bi-component solution. Fig. S5(a) and (b)† show that no H4BDETP/H4BTB co-assembly nanostructure was formed except for the individual self-assembly nanostructures of H4BDETP and H4BTB. The above experimental observations indicated that the formation of the H4BDETP/H4BTB co-assembly nanostructure depended on the deposition sequence of the two components.
4. Conclusions
In conclusion, the self-assembly nanostructures of H4BDETP and H4BTB and their co-assembly nanostructure at the liquid/solid interface were investigated by STM. H4BDETP molecules aggregated into a linear nanostructure in the form of O–H⋯O hydrogen bonded dimers. And H4BTB molecules formed lamellar and tetragonal nanostructures. The introduced H4BTB molecules disturbed the intermolecular O–H⋯O hydrogen bonds between H4BDETP dimers and formed new O–H⋯O hydrogen bonds with H4BDETP dimers, contributing to the generation of a H4BDETP/H4BTB co-assembly nanostructure. Notably, the H4BDETP/H4BTB co-assembly nanostructure was not observed when the deposition sequence of H4BDETP and H4BTB on HOPG was altered. This work was interesting and may have an enlightening significance for selecting bridging molecules to expand the supramolecular nanostructure of carboxylic acid derivatives and help deepen the understanding of the formation of binary supramolecular nanostructures.Author contributions
Xuan Peng and Linlin Gan contributed to this work equally.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (no. XDB36000000), the National Natural Science Foundation of China (no. 21972031, 22272039 and 12064026), the Science and Technology Research Project of Jiangxi Provincial Department of Education (no. GJJ2201523 and GJJ2201526), and the Jilin Chinese Academy of Sciences-Yanshen Technology Co., Ltd.Notes and references
- K. Nickmans and A. P. Schenning, Adv. Mater., 2018, 30, 1703713 CrossRef PubMed
.
- S. K. Saraswathi and J. Joseph, ACS Appl. Nano Mater., 2022, 5, 3018–3027 CrossRef CAS
- D. O. Shin, J. H. Mun, G.-T. Hwang, J. M. Yoon, J. Y. Kim, J. M. Yun, Y.-B. Yang, Y. Oh, J. Y. Lee and J. Shin, ACS Nano, 2013, 7, 8899–8907 CrossRef CAS PubMed
- N. P. King, J. B. Bale, W. Sheffler, D. E. McNamara, S. Gonen, T. Gonen, T. O. Yeates and D. Baker, Nature, 2014, 510, 103–108 CrossRef CAS PubMed
- B. O. Okesola and A. Mata, Chem. Soc. Rev., 2018, 47, 3721–3736 RSC
- L. Li, R. Sun and R. Zheng, Mater. Des., 2021, 197, 109209 CrossRef CAS
- K. S. Mali, J. Teyssandier, N. Bilbao and S. De Feyter, Supramol. Chem., 2022, 43–79 Search PubMed
- G. Velpula, T. Takeda, J. Adisoejoso, K. Inukai, K. Tahara, K. S. Mali, Y. Tobe and S. De Feyter, Chem. Commun., 2017, 53, 1108–1111 RSC
- J. Fang, X. Zhu, W. Luo, J. Shi, L. Wang, B. Tu, Q. Zeng and X. Xiao, Chin. Chem. Lett., 2022, 33, 1100–1104 CrossRef CAS
- Y. Xiao, L. Cheng, X. Sui, Q. Wang, J. Chen, D. Deng, J. Zhang, X. Peng, X. Li and X. Xiao, Nano Res., 2022, 15, 8019–8027 CrossRef CAS
- Y. Xiao, F. Cai, X. Peng, X. Kang, P. Lei, X. Li, H. Xu, X. Xiao, B. Tu and Q. Zeng, Chin. Chem. Lett., 2021, 32, 3566–3569 CrossRef CAS
- X. Peng, T. Meng, L. Wang, L. Cheng, W. Zhai, K. Deng, C.-Q. Ma and Q. Zeng, Chin. Chem. Lett., 2023, 34, 107568 CrossRef CAS
- Z. Feng, C. C. Cudia, L. Floreano, A. Morgante, G. Comelli, C. Dri and A. Cossaro, Chem. Commun., 2015, 51, 5739–5742 RSC
- Y. Liu, Y. Wang, S. Zhang, H. Zou, X. Miao and W. Deng, J. Phys. Chem. C, 2023, 127, 7402–7409 CrossRef CAS
- X. Peng, Y. Xiao, B. Mu, K. Deng, W. Tian, X. Xiao, X. Li and Q. Zeng, Appl. Surf. Sci., 2021, 565, 150529 CrossRef CAS
- X. Zhang, N. Li, G.-C. Gu, H. Wang, D. Nieckarz, P. Szabelski, Y. He, Y. Wang, C. Xie and Z.-Y. Shen, ACS Nano, 2015, 9, 11909–11915 CrossRef CAS PubMed
- X. Zhang, H. Ding, S. Yang, H. Yang, X. Yang, B. Li, X. Xing, Y. Sun, G. Gu and X. Chen, Small, 2023, 2207111 CrossRef CAS PubMed
- Y. Xie, C. Liu, L. Cheng, Y. Fan, H. Li, W. Liu, L. Zhu, X. Li, K. Deng and Q. Zeng, Chin. Chem. Lett., 2022, 33, 4649–4654 CrossRef CAS
- X. Li, J. Li, C. Ma, C. Chen, S. Zhang, B. Tu, W. Duan and Q. Zeng, Chin. Chem. Lett., 2021, 32, 1077–1080 CrossRef CAS
- J. Li, W. Luo, S. Zhang, C. Ma, X. Xiao, W. Duan and Q. Zeng, Nano Res., 2022, 15, 1691–1697 CrossRef CAS
- L. Cai, L. Wang, S. Kang, Y. Geng, K. Deng, Q. Zheng and Q. Zeng, J. Phys. Chem. C, 2016, 120, 27259–27267 CrossRef CAS
- F. Cometto, K. Frank, B. Stel, N. Arisnabarreta, K. Kern and M. Lingenfelder, Chem. Commun., 2017, 53, 11430–11432 RSC
- T. Meng, P. Lei, Y. Zhang, K. Deng, X. Xiao and Q. Zeng, Chin. J. Chem., 2022, 40, 2727–2733 CrossRef CAS
- H. Pinfold, C. Greenland, G. Pattison and G. Costantini, Chem. Commun., 2020, 56, 125–128 RSC
- P. Lei, L. Ma, S. Zhang, J. Li, L. Gan, K. Deng, W. Duan, W. Li and Q. Zeng, Chin. Chem. Lett., 2022, 108005 Search PubMed
- X. Peng, L. Cheng, X. Zhu, Y. Geng, F. Zhao, K. Hu, X. Guo, K. Deng and Q. Zeng, Nano Res., 2018, 11, 5823–5834 CrossRef CAS
- L. Kampschulte, T. L. Werblowsky, R. S. Kishore, M. Schmittel, W. M. Heckl and M. Lackinger, J. Am. Chem. Soc., 2008, 130, 8502–8507 CrossRef CAS PubMed
- L. Yan, G. Kuang and N. Lin, Chem. Commun., 2018, 54, 10570–10573 RSC
- S. Zhang, J. Li, L. Gan, L. Ma, W. Ma, M. Zhang, F. Cheng, K. Deng and Q. Zeng, Nanoscale, 2023, 15, 4353–4360 RSC
- W. Li, S. Xu, X. Chen and C. Xu, Chin. Chem. Lett., 2021, 32, 480–484 CrossRef CAS
- Q. Liang, Y. Yu, G. Feng, Y. Shen, L. Yang and S. Lei, Chem. Commun., 2020, 56, 12182–12185 RSC
- B. Delley, J. Chem. Phys., 2000, 113, 7756–7764 CrossRef CAS
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244–13249 CrossRef PubMed
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary large-scale STM images of self-assembly nanostructures and the co-assembly nanostructure of H4BDETP and H4BTB. See DOI: https://doi.org/10.1039/d3na00389b |
| ‡ Xuan Peng and Linlin Gan contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |