
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring the molecular landscape of multicomponent crystals formed by naproxen drug and acridines†
Artur
Mirocki
 a,
Mattia
Lopresti
b,
Luca
Palin
bc,
Eleonora
Conterosito
b,
Artur
Sikorski
a and
Marco
Milanesio
*b
a,
Mattia
Lopresti
b,
Luca
Palin
bc,
Eleonora
Conterosito
b,
Artur
Sikorski
a and
Marco
Milanesio
*b
aFaculty of Chemistry of the University of Gdansk, ul. Wita Stwosza 63, 80-308 Gdansk, Poland
bUniversità del Piemonte Orientale, Dipartimento di Scienze e Innovazione Tecnologica, Viale T. Michel 11, 15121 Alessandria, Italy. E-mail: marco.milanesio@uniupo.it
cNova Res s.r.l., Via D. Bello 3, 28100 Novara, Italy
First published on 6th September 2022
Abstract
The cocrystallization of active pharmaceutical ingredient naproxen with some acridines (acridine, 9-aminoacridine, 6,9-diamino-2-ethoxyacridine) has been explored and the conditions under which the crystallization can be carried out have been investigated. While the crystallization of acridine-based molecular crystals was widely studied under solution conditions, solvent-free and/or mechanochemical method potentialities are still unknown. To fill this gap, the cocrystallization of naproxen with the above-mentioned acridines was attempted using different approaches, e.g., by heat treatment of the dry mechanical mixture and by liquid-assisted grinding (LAG), as alternatives to the traditional precipitation by a proper solution. In the first case, the reaction is driven under dry conditions by the temperature and gave no results independently of the temperature used, below or above the melting point of the reactants. In the second case, the reaction is driven by the mechanical action of grinding assisted by a few drops of solvent to facilitate and improve the reaction. This screening allowed obtaining three new molecular crystals for naproxen coupled to acridine and a mono-aminoacridine and solved by single-crystal and powder X-ray diffraction (PXRD). Two host–guest structures were obtained by solution crystallization, while a layered structure was obtained under LAG conditions. Interconversion between molecular crystals formed by the same chemical species was hindered once a molecular crystal was obtained by a specific technique. Hirshfeld and energy framework calculations confirmed the remarkable structural differences between 1α and 1β packing and suggested that 1β is kinetically more stable. Variable-temperature PXRD, DSC and TGA were used to explore the stability of the compounds. 6,9-Diamino-2-ethoxyacridine proved to be too polar and/or too bulky to form crystals with naproxen regardless of the preparation method and the different stoichiometric ratios used. It is noteworthy that LAG allowed the preparation of the naproxen/acridine molecular crystal with a yield higher than 99% under almost solvent-free conditions. DSC indicated the formation of a eutectic between naproxen and acridine, with the possibility of recrystallizing the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex also from the melt solution.
:1 complex also from the melt solution.
1. Introduction
Crystal engineering is an intensively developing field, and one of its main assumptions is to identify and understand intermolecular interactions in crystals.1–4 It allows the design and preparation of new materials based on a repeatable structural motif (synthons) arising through intermolecular interactions between different functional groups in the molecules. The knowledge on how the synthons assemble is fundamental to obtain multicomponent crystals with the desired, predictable structure as well as the expected properties. However, a change of the stoichiometry of the coformers and an increase in the number of functional groups in the molecules used may lead to unexpected changes in the structural landscape of multicomponent crystals. All these issues are even more important when the synthons are active pharmaceutical ingredients (APIs). In this case, small changes in the molecular formula and/or in the crystal packing might affect the solubility, bioavailability and other properties related to their biological activity. The formulation of APIs is thus one among the key steps when transforming a bioactive molecule into a drug. A common way to modify, tailor and control such properties is exploiting the possible polymorphs of an API. A more tackling but intriguing route is obtaining multicomponent crystals (cocrystals, salts, salt cocrystals or their solvates) involving the API. In this way, not only can the properties be modified but new properties can be obtained by properly selecting the coformer molecules and also modifying the stoichiometric ratio.5–7 Combining different APIs can also have synergic effects with cocrystals, showing improved performance with respect to the sum of the separate properties. For instance, the cocrystal of tramadol–celecoxib proved to have a much better biopharmacological profile than their free combination or the two APIs alone.8 With these premises, the structural properties of cocrystals involving naproxen and acridines were explored to understand when cocrystals can be obtained, if different stoichiometric ratios are possible and which are the interactions driving their crystal packing. Naproxen (IUPAC name: (2S)-2-(6-methoxy-2-naphthyl) propanoic acid) is a commonly used nonsteroidal anti-inflammatory drug (NSAID) because it has a wide range of applications. It is used as an anti-inflammatory and analgesic agent in pain conditions, such as migraine, tension headaches or postoperative pain, and also for painful rheumatic conditions (e.g. osteoarthritis diseases).9–11 Currently naproxen is the subject of a research topic under investigation as an anti-COVID 19 compound.12–14 To explore the crystal structures of molecular crystals involving naproxen, we have performed a search of the Cambridge Structural Database (CSD).15 A search of the CSD (ver. 5.43, update November 2021) shows that there are 73 crystal structures of organic compounds containing naproxen, including 37 structures of salts, 35 structures of cocrystals and 1 structure of salt cocrystal. Molecular crystals and materials can be obtained by several approaches from solution to solid-state approaches.16 However, in the CSD there are only a few examples showing the possibility of forming crystalline multicomponent systems involving naproxen and cyclic nitrogen containing bases. For example, Zaworotko and co-workers (2009) described the synthesis and structural characterization of cocrystals of naproxen and trans-1,2-bis(4-pyridyl)ethylene.17 Castro et al. (2011) reported the structural characterization of naproxen cocrystals with pyridinecarboxamide isomers (picolinamide, nicotinamide, isonicotinamide, and pyrazinamide).18 Tilborg et al. (2013)19 and Tumanova et al. (2018)20 extensively studied the polymorphism and stoichiometric diversity of cocrystals formed from naproxen and proline. Manoj et al. (2014) described the crystal structures of cocrystals of racemic and (S)-naproxen with bipyridine/piperazine and determined their physicochemical properties using DSC, PXRD, hot-stage microscopy, and FT-IR spectroscopy.21 Neurohr et al. (2015) investigated the cocrystallization of naproxen racemic mixture and nicotinamide using compressed CO2 as an antisolvent.22 Nechipadappu and Trivedi (2017) reported the structural and physicochemical characterization of pyridine derivative (4-aminopyridine, 4-dimethylaminopyridine, and 2-aminopyridine) salts of naproxen.23 Among cyclic nitrogen-containing bases, acridines are an interesting object of research. This group of compounds is recognized as APIs because they exhibit anti-cancer, antibacterial, and antiviral properties. Acridines are used as bacteriostatic antiseptic drugs and for various infections and inflammations.24–27 The most important property of acridines is their ability to interact with DNA through intercalations.28,29 Recently we have presented results regarding the capabilities of different solvents to induce cocrystallization in acridine-based molecular crystals of APIs.30 Some of us demonstrated that, in similar cases, cocrystallization31 and solid-state reactions32 can be carried out directly in the solid state under dry conditions with high conversion rates and with the additional advantage of eliminating solvents.33 In this work, the cocrystallization of naproxen with some acridines is thus attempted by three different approaches: (i) solution methods, (ii) liquid-assisted grinding (LAG), and (iii) solid-state thermal approach, heating the sample just below or above the melting point of the reactants to explore the molecular landscape of the target compounds. While the first case requires large quantities of solvents, in the LAG case, the reaction is driven by the mechanical action of the grinding assisted by a few drops of solvent to facilitate and improve the reaction, as described in ref. 34. In the last case, the reaction is driven by the temperature and by the conditions of autogenous pressure that develops inside the reaction chamber, if a sealed tube is used for the preparation and the temperature is below the reactants' melting points and no eutectic is present. As naproxen counterparts, acridine, 9-aminoacridine, 6,9-diamino-2-ethoxyacridine, depicted in Scheme 1, were chosen because of their complementary biological activities, i.e., NSAID for naproxen and antibacterial and antiviral for acridines. Their combination in a molecular crystal can give synergic powering of their complementary properties. The aim is to obtain new coupled compounds with high yields while exploring the effects of the different numbers of acridine lateral chains under the different crystallization conditions. All couples were tested to shed light on the more stable molecular crystal, depending on the synthesis method. Single-crystal X-ray and powder X-ray diffraction (PXRD) were performed to determine the crystal structures of the title compounds. Hirshfeld surface analysis and energy framework calculations were utilized to further explore the packing differences among the solved structures. Variable-temperature PXRD, differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) were carried out to assess the thermal behaviours and stability of the reactant and products. | ||
| Scheme 1 Molecular structures of naproxen and acridines reported in the paper (the specific chemical forms used are detailed in the Experimental section). | ||
2. Experimental
2.1. Crystallization from solution, LAG and dry synthesis
All chemicals (acridine, 9-aminoacridine hydrochloride monohydrate, 6,9-diamino-2-ethoxyacridine-DL-lactate monohydrate, naproxen) were purchased from Sigma-Aldrich. The pKa values of acridine, 9-aminoacridine and 6,9-diamino-2-ethoxyacridine are 5.60, 9.90 and 11.22, respectively, while that of naproxen is 4.15. Since for 9-aminoacridine and 6,9-diamino-2-ethoxyacridine the pKa difference is larger than 3 and the salt or salt cocrystal is expected, their salt form is used as the reactant.The crystals of the investigated compounds were obtained after many attempts of solution crystallization, varying the molar ratio of reagents: naproxen:acridine: 1:1, 1:1.5, 1.5:1, 1:2 and 2:1, naproxen:9-aminoacridine: 1:1, 1:2 and 2:1. For naproxen:6,9-diamino-2-ethoxyacridine no cocrystals were obtained after trying the ratios: 1:1, 1:1.5, 1.5:1, 1:2, 2:1, 1:3 and 3:1. The successful recipes are the following. Acridine (0.024 g, 0.134 mmol) and naproxen (0.015 g, 0.065 mmol) were dissolved in 4 mL of an ethanol/water mixture (1:1 v/v) and heated. The solution was allowed to evaporate for a few days to give light yellow crystals of 1α. 9-Aminoacridine hydrochloride monohydrate (0.026 g, 0.105 mmol) and naproxen (0.012 g, 0.052 mmol) were dissolved in 4 mL of an ethanol/water mixture (3:1 v/v) and heated. The solution was allowed to evaporate for a few days to give yellow crystals of 2β.
The preparation was carried out under almost solvent-free conditions by the mechanical action of manual grinding of the reactant mixtures, assisted by a few drops of solvent, as in Conterosito et al.35 The procedure was optimized by carrying out the preparation many times, increasing the number of solvent drops and repeating the LAG with the same number of drops (between 5 and 10). After all preliminary attempts, an PXRD measurement was carried out to assess the yield. Repeated LAG with a few drops of solvent was found to be more efficient than a single LAG with more drops to increase the yield. The procedures were applied to the three couples of naproxen with the three acridines in Scheme 1. These preliminary tests allowed us to obtain a complete conversion only for 1β with the following recipe. Acridine (0.087 g, 0.485 mmol) and naproxen (0.11 g, 0.478 mmol) were ground together with 10 drops (about 0.4 ml) of ethanol three times to obtain complete conversion, then treated in an oven at 93 °C for 3 hours. At this temperature, the reactants are below their melting points, and the cocrystal (when formed in the case of 1β) is solid and stable according to DSC (Fig. S5†) and in situ XRD data (Fig. S7†). It is worth noting that no washing of the product is needed, and no waste is produced, so that all the reactants were transformed into the product 1β: with a traditional formula of the yield using the ratio between the product and reactant weight, we could write a 100% yield. However, we are aware that the purity of 1β was checked by PXRD whose sensitivity is close to 0.1–1 wt%: we can thus prudently conclude that the yield is at least larger than 99%. According to Friščić et al.,34 the solvent/mass ratio is between that in the LAG and slurry conditions considering the total amount of solvent (30 drops) but in the LAG range considering each single grinding (10 drops). Therefore, from hereon, the preparation is referred to as “LAG” procedure. All similar attempts for 9-aminoacridine hydrochloride monohydrate (0.084 g, 0.338 mmol) and naproxen (0.078 g, 0.338 mmol) and 6,9-diamino-2-ethoxyacridine DL-lactate monohydrate (ethacridine) (0.122 g, 0.338 mmol) and naproxen (0.078 g, 0.339 mmol) failed to obtain a cocrystal.
Dry synthesis was carried out by a thermal method, inserting an equimolar amount of reactant in a sealed capillary and then treating in an oven at a temperature just below the melting point of one of the counterparts, exploiting the approach by Palin et al.31 to calculate the best reaction temperature range depending on the lowest melting point between the two reactants. The experiments were carried out by treating the dry mechanical mixture at 85 °C, 93 °C and 101 °C (for acridine and naproxen) and 106 °C, 121 °C and 136 °C (for 9-aminoacridine/6,9-diamino-2-ethoxyacridine and naproxen) for 3 hours. For the acridine/naproxen couple, crystallization was also attempted, with no results, to form the melt mixture as a comparison in both a DSC crucible and under in situ XRD conditions.
Concerning physical and chemical properties, all molecular complexes were obtained in water-based solutions with a solubility similar to that of naproxen (15.9 mg L−1), an important feature envisaging biopharmacological applications.
2.2. Single-crystal X-ray diffraction
Single-crystal X-ray diffraction data were collected on an Oxford Diffraction Gemini R ULTRA Ruby CCD diffractometer with Mo Kα (λ = 0.71073 Å) radiation at T = 295(2) K (Table S1†). The lattice parameters were obtained by least-squares fit to the optimized setting angles of the reflections collected by means of CrysAlis CCD software.36 Data were reduced using CrysAlis RED software36 and applying multiscan absorption corrections. The structural resolution procedure was carried out using the SHELX package.37 The structures were solved with direct methods that carried out refinements by full-matrix least-squares on F2 using the SHELXL-2017/1 program.37 All H atoms bound to O/N atoms were located on a difference Fourier map and refined freely with Uiso(H) = 1.5/1.2Ueq(O/N). All H atoms bound to C atoms were placed geometrically and refined using a riding model with d(C–H) = 0.93–0.98 Å and Uiso(H) = 1.2Ueq(C) (d(C–H) = 0.96 Å and Uiso(H) = 1.5Ueq(C) for the methyl groups). All interactions were calculated using the PLATON program.38 The following programs were used to prepare the molecular graphics: ORTEPII,39 PLUTO-78,40 and Mercury.41 Full crystallographic details of the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre (deposition no. CCDC 2168756, CCDC 2169121 and CCDC 2168757 for 1α, 1β and 2α, respectively).2.3. Powder X-ray diffraction
PXRD analysis was performed on a Bruker D8 Advance diffractometer with a Lynx-Eye XE-T detector and Cu Kα (λ = 1.5418 Å) radiation (Table 2). The goniometer radius is 280 mm. The tube was set at 40 mA current and 40 kV electric potential. The instrument was at first used as an analytical qualitative tool to evaluate if the reaction occurred and to assess the purity of the product. Then it was used to understand if the single-crystal structure is representative of the complete batch or if just the mechanical mixture of the reactant was present after synthesis attempts and thus to identify the best (or not) preparation procedures. When enough sample was available, an auto-sampler with nine positions, rotating sample holders and an air scatter knife was used. On both the primary and secondary optics, Soller slits of 2.5° opening were positioned. The patterns were collected in Bragg–Brentano geometry from 3° to 70° 2θ with a step size of 0.01° and exposure time of 0.05 s; automatic divergence slits were set to obtain constant sample illumination of 17 mm. The dry synthesis was carried out in sealed 0.7 mm glass capillaries placed in an oven and directly used for the measurements (0.6 mm planar slit used with a step size of 0.0204° and exposure time of 0.5 s). The capillary setup was also utilized to obtain an optimal angle resolution with a FWHM of about 0.01° suitable for indexing of 1β, obtained only by LAG in polycrystalline form. The measurement was carried out using a planar 0.6 mm slit, a step-size of 0.005° and exposure time of 5 s, and this 18 hour measurement resulted in a 1β crystal structure suitable for solving and refinement. Structure solution was carried out by simulated annealing in real space by EXPO42 from capillary data, while final structure refinement was carried out by Topas Academic43,44 on capillary and flat sample data. In situ X-ray diffraction study of the LAG procedure was applied to single crystals of 1α using a focusing optic (Goebel mirror with 1 mm focusing hole) after positioning the sample by the xyz stage. A variable-temperature powder X-ray diffraction experiment was performed by installing a Linkam THMS600 variable-temperature stage within the motorized xyz stage (UMC compact stage) of a Bruker D8 Advance diffractometer to obtain a PXRD setup able to manage liquids. The standard Linkam cover was removed to allow X-rays to reach the sample and a Kapton foil was used to avoid evaporation. The sample was positioned in a 10 mm diameter sample holder for liquids and patterns were collected on a same length illuminated area every 40 seconds. PXRD patterns were measured in the 2θ range of 8–31° with an increment of 0.025° and a step time of 0.03 s. A USB microscope was used to obtain the image of the sample before each PXRD measurement to check if the sample appears liquid or solid.2.4. Thermal analysis
DSC was carried out using a TA Q200 and a METTLER TOLEDO DSC 3 analyzer. For both instruments a closed aluminium pan was used with a ramp rate of 5 °C min−1. TGA was carried out using a TA Q600 STD analyzer and an alumina crucible at a ramp rate of 10 °C min−1 in an oxidizing environment.2.5. Hirshfeld surfaces and energy framework calculations
Hirshfeld surfaces with electron density, fingerprint plots and energy frameworks were calculated using CrystalExplorer 17.5 (ref. 45) and analysed for the three solved structures. Hirshfeld surfaces were calculated with a high-resolution setting. The wavefunctions for each molecule and pairwise interaction for the calculation of energy frameworks were calculated using Tonto with the B3LYP DFT method by employing the 6-31G(d,p) basis set, as implemented in CrystalExplorer. The scale for the tube size employed for energy framework pictures is 80 and the cut-off value for energies was set to 0 kJ mol−1. Interaction energies between each independent molecule and its neighbours were calculated and from this the sum of the lattice energy for each unique molecule was obtained as one-half the product of the number of symmetry equivalent molecules in the cluster and the total energy.463. Results and discussion
3.1. Crystal structure solution
The preparation of the three possible couples between naproxen and the three acridines in Scheme 1 was carried out by solution, LAG and a thermal dry procedure and the results are summarized in Table 1. A cocrystal of naproxen with acridine (2:3 stoichiometry) (1α) and a monohydrate salt cocrystal of naproxen with 9-aminoacridine (2:1 stoichiometry) (2α) were obtained through the reactions carried out in solution. Conversely, powdered cocrystals of naproxen with acridine (1:1 stoichiometry) (1β) were obtained by the reactions carried out under LAG conditions, as reported in Table 1. These forms are those expected from the pKa difference of the reactants, which is larger than 3 only for the 2α couple.
| API | Coformer | Compound | Solution | Dry | LAG |
|---|---|---|---|---|---|
| Naproxen | Acridine | 1 | Single crystals 1α | Mechanical mixture of reactants | Powder 1β |
| 9-Aminoacridine | 2 | Single crystals 2α | Mechanical mixture of reactants | ||
| 6,9-Diamino-2-ethoxyacridine | — | Mechanical mixture of reactants | Mechanical mixture of reactants |
Crystallization from solution produced single crystals by coupling naproxen to acridine and 9-aminoacridine, while no results were obtained for naproxen/6,9-diamino-2-ethoxyacridine. Single-crystal X-ray diffraction measurements show that compound 1α crystallizes from solution in the monoclinic P21 space group as a cocrystal with two naproxen and three acridine molecules in the asymmetric unit (Fig. S1† and Table 2). Compound 2α crystallizes in the monoclinic P21 space group as a monohydrate salt cocrystal with one naproxen molecule and anion, one 9-aminoacridinium cation, and one water molecule in the asymmetric unit (Fig. S2† and Table 2). Both 1α (Fig. 1, top) and 2α (Fig. 2) show a host–guest-like packing. LAG and dry thermal synthesis were thus performed as described in the Experimental section for all the three acridines and the results are summarized in Table 1. While dry synthesis was unsuccessful in all cases, the LAG approach allowed obtaining a cocrystal of naproxen with acridine (1:1 stoichiometry) (1β). In fact, analysis by powder X-ray diffraction (Fig. 3) confirmed that the patterns of the products obtained by LAG (the reactant with a 3:2 naproxen/acridine ratio as in 1α) did not coincide with those obtained by solution or with the patterns corresponding to the mechanical mixture of the reagents. The new molecular cocrystal appeared with a residual amount of acridine reactant. The LAG procedure was thus repeated with a 1:1 naproxen/acridine reactant ratio and pure 1β was obtained, within the sensibility limitation of powder diffraction (Table 2). For further confirmation, the reagents were also subjected to the LAG procedure individually to verify that the products obtained were not mechanical mixtures of molecular crystals of the reagents alone due to the LAG process. In Fig. 3, the PXRD patterns of both molecular crystals 1α and 1β of naproxen/acridine cocrystals confirm that their crystal structures are different. To obtain an PXRD pattern suitable for structure solution, a capillary was filled, and a long measurement was carried out to obtain a good resolution. This pattern was used to index and obtain a cell. Then, the structure solution was carried out using the same data based on the available diffraction data; the obtained crystal structure is presented in Fig. 1, bottom. It is worth noting that the product is pure, within the sensitivity limitation of PXRD, as indicated by the Rietveld refinement reported in Fig. 3.
| Compound | 1α (SX) | 2α (SX) | 1β (PXRD) |
|---|---|---|---|
| Chemical formula | C67H55N3O6 | C41H40N2O7 | C27H23NO3 |
| Formula weight/g mol−1 | 998.14 | 672.75 | 409.48 |
| Crystal system | Monoclinic | Monoclinic | Orthorhombic |
| Space group | P21 | P21 | P212121 |
| a/Å | 17.3435(14) | 10.0768(11) | 12.6466(4) |
| b/Å | 5.9437(6) | 6.2918(5) | 29.6246(12) |
| c/Å | 25.672(2) | 27.347(3) | 5.63596(19) |
| α/° | 90 | 90 | 90 |
| β/° | 101.975(9) | 98.004(11) | 90 |
| γ/° | 90 | 90 | 90 |
| V/Å3 | 2588.8(4) | 1717.0(3) | 2111.52 |
| ρ calc/g cm−3 | 1.280 | 1.301 | 1.288 |
| Final R1 value | 0.0585 | 0.0677 | R p = 2.043 |
| Final wR2 value | 0.0927 | 0.0843 | R wp = 2.635 |
| CCDC number | 2168756 | 2168757 | 2169121 |
 | ||
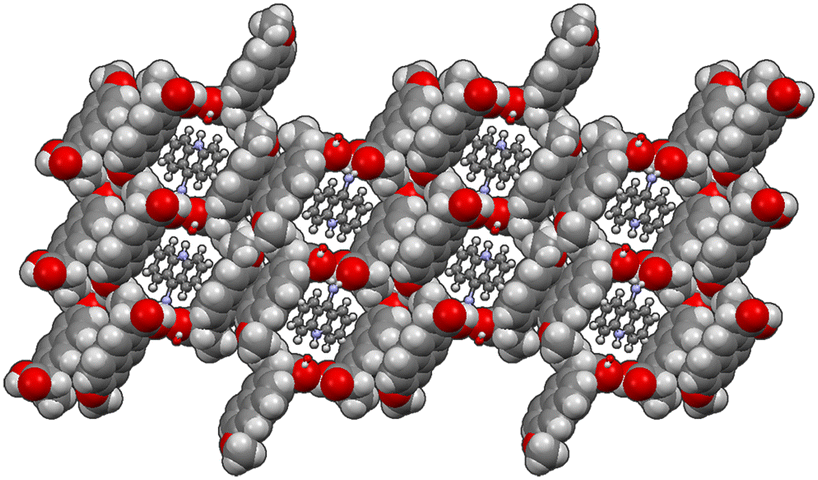
| Fig. 1 Crystal packing of 1α and 1β. | ||
 | ||
| Fig. 2 Crystal packing of 2α. | ||
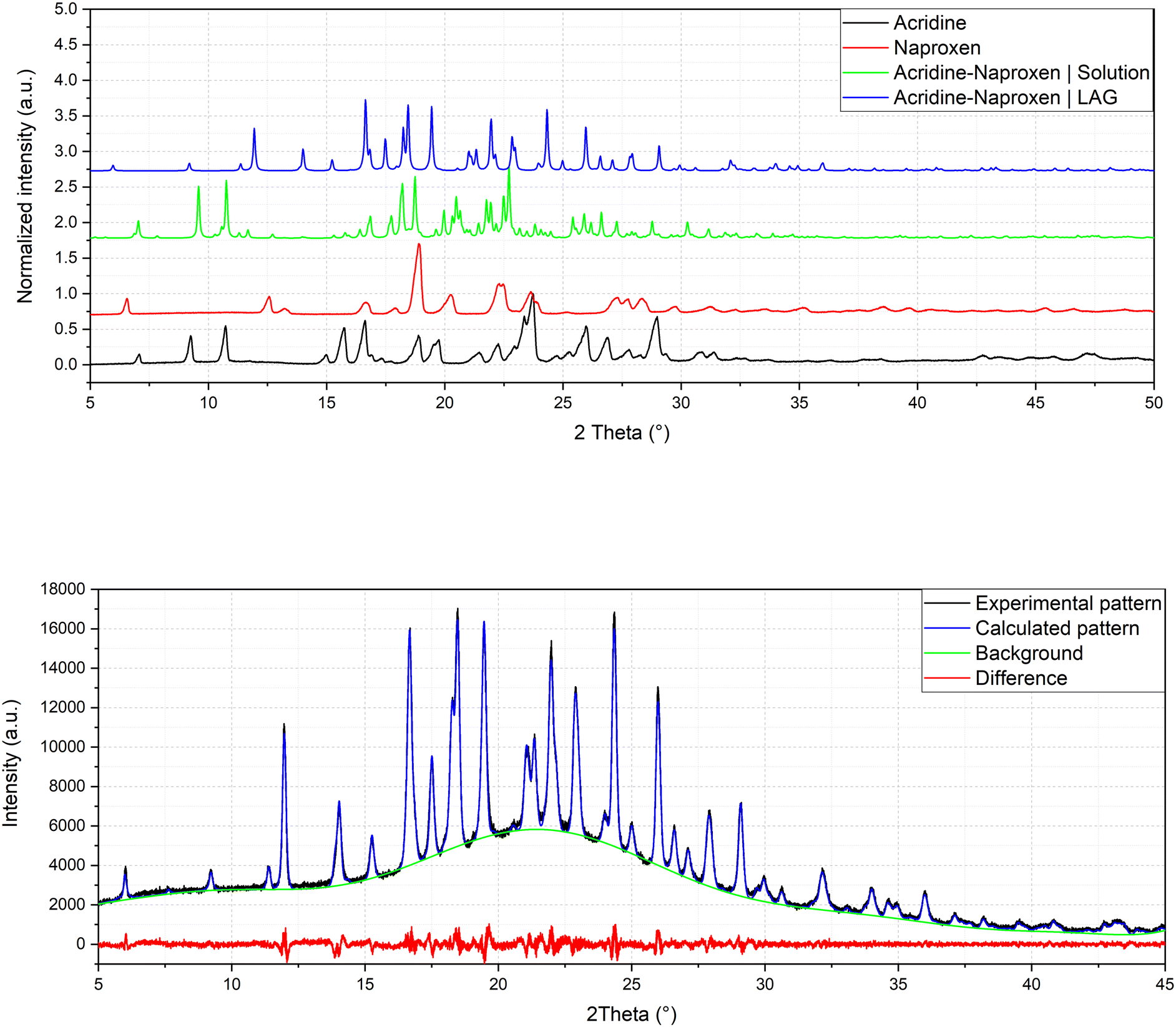 | ||
| Fig. 3 Comparison of powder patterns of 1α and 1β and refinement of crystal structure solved by powder diffraction of 1β (Rp = 2.043, Rwp = 2.635). | ||
In both molecular crystals 1α and 1β, the packing is due to two different driving forces: in the case of the 2:3 molecular crystal 1α, intermolecular COOH(naproxen)⋯N(acridine) and OH(naproxen)⋯N(acridine) hydrogen bonds and C–H(naproxen/acridine)⋯π(naproxen/acridine) interactions prevail (Tables S3 and S4†), while in the case of the 1:1 ratio molecular crystal 1β, there is an overall stabilization of the structure given by π-stacking and intermolecular and the above-mentioned O–H⋯N hydrogen bonds. In detail, each naproxen is bonded by one H-bond and CH⋯O interactions to two different acridine molecules (Table S2†). A T-like interaction and a parallel π–π interaction connect adjacent acridine layers as detailed in Table S3.† Several CH⋯C longer contacts (mainly involving naproxen methyl groups) complete the packing. Consequently, in the crystal of 1α, hydrogen-bonded pairs of naproxen and acridine molecules formed the 3D network, with voids filled by the non-hydrogen-bonded acridine molecules. Of course, the host–guest structure cannot be considered a MOF-like compound since the guest cannot be removed without destroying its crystal structure. Conversely, in 1β, π-stacked columns of acridine molecules and the layered association of naproxen molecules can be observed as previously observed in the molecular crystals of naproxen with proline (1:1) (described by authors as type II).20 A weak CH⋯O and a strong OH⋯N hydrogen bond interaction connect the layers of naproxen with those of acridine. This packing was obtained after carefully checking all the possibilities in placing the acridine molecule (that can be flipped by 180° without changing the packing) and selecting the correct location of the hydrogen of the hydroxyl.
The chosen solution minimizes the agreement factor and gives more reliable interactions and intermolecular contacts without geometrical warnings. On the other hand, the crystal structure of 2α may be considered as a 3D network formed by monoanionic dimers of the naproxen molecule and anion with π-stacked 9-aminoacridinium cations located in the cavities formed by the network, similarly to 1α. To date, no molecular crystals belonging to this family are available; however different structures of molecular crystals of naproxen and acridine with other molecules are present within the CCDC database. In this broad landscape of structures, molecular crystals can be found crystallizing with similar cell parameters and with equivalent space groups. For example, naproxen crystallizes according to space group P21 with (S)-1-phenylethylammonium, as reported by Rossi et al.,47 with a needle crystal habit, as in the case of the single crystal presented in this work. Naproxen also crystallizes in space group P212121 with (1S,2S)-trans-1-aminobenz[f]indan-2-ol,48 but with a very different arrangement of the asymmetric unit, i.e., without an alternation between the two molecules in the structure. Acridine also has similar molecular crystal structures reported in the CCDC, such as that of Bao et al.49 with space group P21, or with P212121, reported by Rajkumar et al.50
The same procedure used to obtain 1β was applied to compound 2 and (hypothetical) 3 but no results were obtained, as can be seen from the PXRD data reported in Fig. 4. It can be concluded that compound 2 (with a monoamino acridine) has only one stable molecular crystal with the host–guest hydrate structure, while hypothetical compound 3 (with a diamino acridine) cannot be obtained, probably because of the higher polarity of the diamino acridine and/or its steric hindrance is not suitable for a host–guest-like or layered structures such as 1α and 1β, respectively.
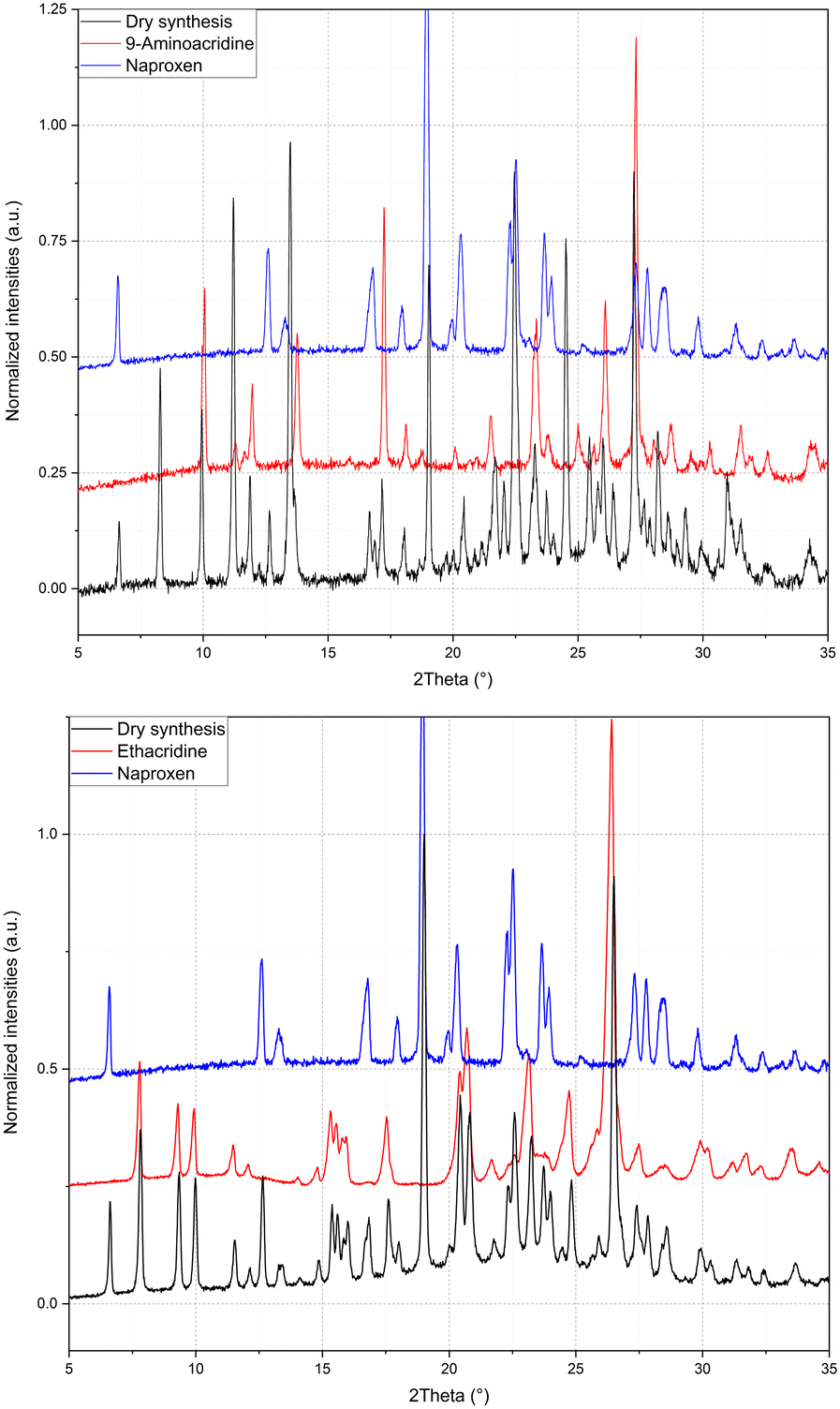 | ||
| Fig. 4 Refinement of LAG of 2 and 3 as mechanical mixture. | ||
3.2. Exploring the landscape of cocrystals of naproxen with acridines with different molecular ratios
The presence of two molecular crystals of naproxen with acridine raises questions on which is the more stable one and on their possible interconversion. To shed light on this point, single crystals of 1α obtained by solution precipitation were subjected to the LAG procedure under in situ PXRD conditions: surprisingly, after this procedure, 1α did not produce 1β (Fig. 5, top), suggesting that 1α should be the more stable compound, able to resist the harsh mechanochemical treatment. Only a degradation of the structure of 1α (obtained from solution crystallization) is visible after applying the LAG procedure. The main peak of compound 1α (8° in Fig. 5, top) remained after the treatments and the main peaks (indicated by asterisks) of 1β do not appear. After the failure of conversion of 1α to 1β by LAG, the opposite experiment was attempted. 1β formerly obtained by LAG was dissolved and crystallized from solution with the same procedure used to obtain 1α using water, ethanol and a 50:50 mixture as solvent. Even more surprisingly, solution crystallization of 1β did not produce 1α but still 1β as indicated by the refinement in Fig. 5, bottom. Clearly, the interconversion between 1α and 1β seems unfeasible, suggesting a similar stability between the two cocrystals and/or a very high energy barrier between the two. Also, the different stoichiometry might play a role in hindering their interconversion, which was not observed despite several attempts (Fig. 5 represents only one of several experiments).
 | ||
| Fig. 5 Refinement of 1α after LAG (top) and solution crystallization of 1β after full dissolution (bottom). | ||
To further understand the driving forces of the crystal packing and estimate which compound is more stable between 1α and 1β, Hirshfeld surface analysis and energy framework calculations were carried out. Fig. 6 presents a crystal structure model of 1α viewed along the b-axis, showing Hirshfeld surfaces with d-norm plotted, and the fingerprint plots for each individual molecule in 1α are reported. Acridine molecules are labelled with “a” and naproxen molecules with “n”.
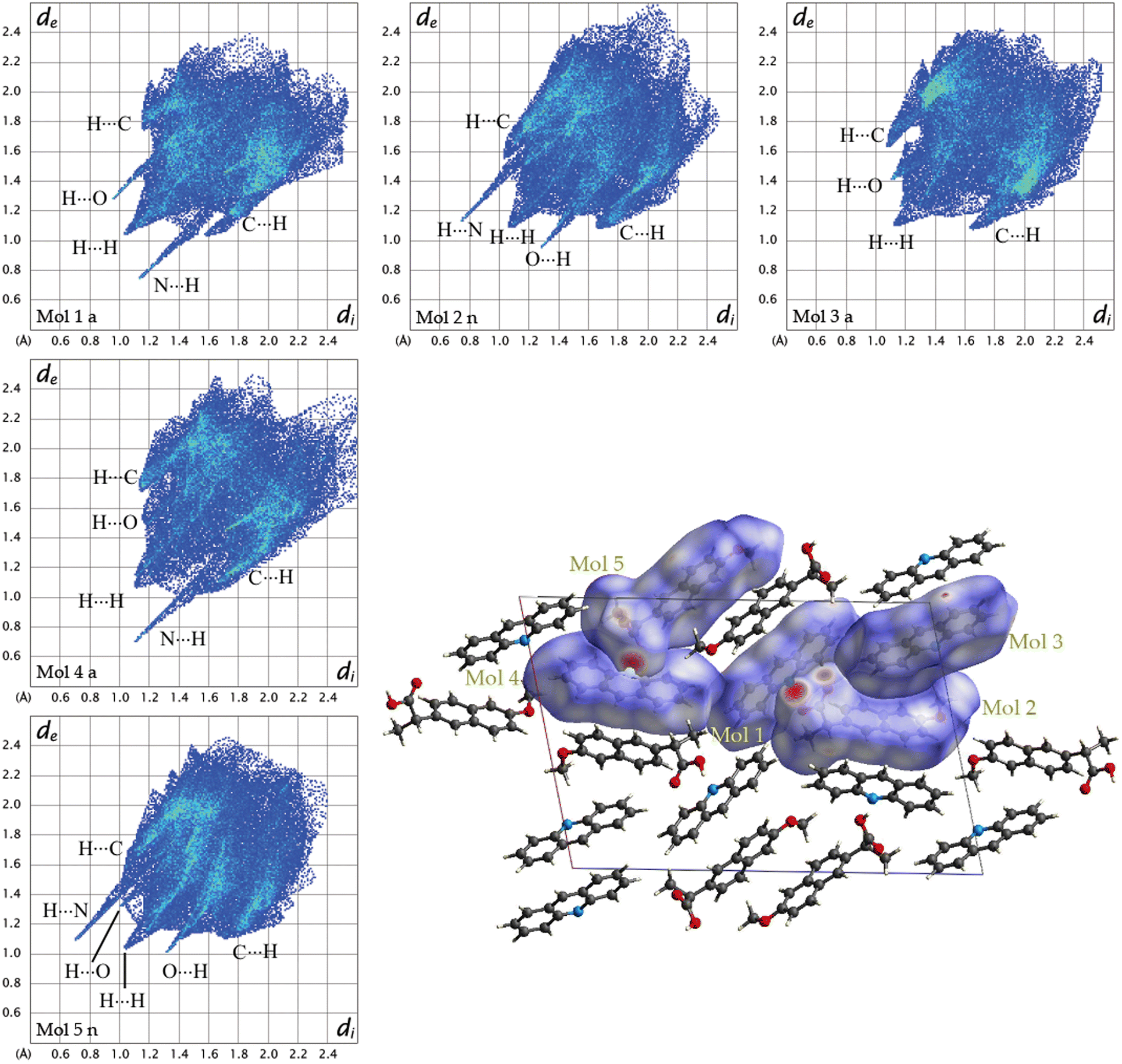 | ||
| Fig. 6 Fingerprint plots for each symmetry-independent molecule in 1α and crystal structure model of 1a viewed along the b-axis showing Hirshfeld surfaces with d-norm plotted. | ||
By looking at the Hirshfeld surfaces and fingerprint plots of 1α the presence of two short contact OH⋯N hydrogen bonds can be readily seen between Mol_1 and Mol_2_n and a slightly shorter one between Mol_4_a and Mol_5_n. Mol_1_a and Mol_2_n also share an O⋯H interaction, while Mol_5_n has an O⋯H interaction with a symmetry equivalent molecule The more symmetrical plot of Mol_3_a evidences a predominance of interactions with symmetry equivalent molecules. There is no evidence by looking at both the Hirshfeld surfaces and fingerprint plots of π–π interactions (C⋯C).
The most important interactions in 1β shown by Hirshfeld surface analysis (Fig. 7) are the OH⋯N hydrogen bond between acridine and naproxen, separated by a short contact distance (2.08 Å) clearly evidenced by plotting d-norm on the surfaces (Fig. 7, bottom), and the presence of π–π stacking between acridine molecules. The fingerprint plots are shown in the top part of Fig. 7 for acridine (Mol_1_a) and naproxen (Mol_2_n). The fingerprint plot features are labelled according to the interaction; the first atom is the one inside the surface. The hydrogen bond is clearly shown by a spike in both plots, indicating a short-range contact between the N atom in acridine and the OH group in naproxen, while the π–π interaction (C⋯C) is found at longer distances and occurs between acridine molecules. This feature is clearly visible only in the acridine plot, represented by a green symmetrical spot at 1.8 Å on both axes, indicating a contact distance of about 3.6 Å.
 | ||
| Fig. 7 (Top) Fingerprint plots for acridine (Mol_1_a) and naproxen (Mol_2_n) in 1β and (bottom) the crystal structure model of 1β viewed along the a-axis showing Hirshfeld surfaces with d-norm plotted. Some molecules were omitted for clarity. | ||
From the analysis of the Hirshfeld surfaces and fingerprint plots of 2α (Fig. S9 and S10, respectively, in the ESI†) it appears that the shorter interactions are hydrogen bonds, the one between the water molecule and Mol_1_a (9-aminoacridinium). Other hydrogen bonds are formed between the naproxen moieties and water, while N⋯H interactions are long range and can be seen in the fingerprint plot only after filtering (Fig. S10,† bottom). It is also worth noting the formation of π–π interactions between 9-acridinium molecules (Mol_1_a) evidenced by a green spot in the fingerprint plot as in 1β.
The energy frameworks were also calculated for all structures. In Fig. 8 the energy frameworks for the coulombic interactions are reported showing that in 1α the stronger interactions (thicker tubes in the picture) form a crossed pattern, while in 1β the interactions are almost parallel, forming a layered structure. Remarkable differences can be seen also in the dispersion interactions, forming a honeycomb-like motif in 1α viewed along the b-axis, while a square motif is observed for 1β. In this structure, along the b-axis the packing is stabilised mainly by coulombic forces, while dispersion forces are stronger in the perpendicular directions. Therefore, the layers are kept together by π–π interactions while the interaction between the layers is polar (i.e. hydrogen bonds).
 | ||
| Fig. 8 Energy framework plot of coulomb interactions in red (top) and dispersion interactions in green (bottom) for structure 1α and 1β. | ||
From the calculation of pairwise interactions, the lattice energy was calculated for each independent molecule. For structure 1β the sum of energies for the acridine molecule was found to be about −90 kJ mol−1, while for naproxen it was −108 kJ mol−1. For structure 1α the energies for the acridine molecules resulted to be −100, −82 and −97 kJ mol−1 for Mol_1_a, Mol_3_a and Mol_4_a, respectively (avg. −93 kJ mol−1), while the energies for the naproxen molecules are −122 kJ mol−1 for Mol_2_n and −125 kJ mol−1 for Mol_5_n (avg. 124 kJ mol−1). Therefore, the average energy for acridine molecules was found to be similar in the two structures, while naproxen appears to be more stable in 1α. These data, keeping in mind the different stoichiometry, allow estimating the lattice energies to be −263 and −198 kJ mol−1 for 1α and 1β, respectively, suggesting that 1α is the thermodynamically stable cocrystal.
The energy frameworks for 2α are shown in Fig. S11 and S12 in the ESI.† Coulombic energy is shown in Fig. S11;† the attractive interactions are shown in red while repulsive forces are in yellow. The water molecule is involved in the stronger attractive interaction involving the 9-aminoacridinium and naproxen molecules, while repulsion occurs between the aromatic rings and the deprotonated naproxen. The layered motif of the packing is driven by the presence of parallel dispersion forces like in 1β (see Fig. S12,† view along the a-axis). The key role of the water molecule in the crystal packing of 2α could be the reason why both thermal dry synthesis and LAG were unsuccessful in obtaining a second cocrystal, as in the case of compound 1.
Finally, to experimentally assess which compound is more stable between 1α and 1β and further explore the thermal behaviour of reactants and products, TGA and DSC analysis of the mechanical mixture of acridine and naproxen and of the cocrystal in their various forms was investigated. TGA was carried out from 40 to 600 °C and the profiles are in Fig. S3.† The maximum of the loss degradation slope profile is at 241 °C for 1α and at 283 °C for 1β, clearly suggesting that 1β is the more stable cocrystal. This indication is apparently contradictory with respect to energy framework calculations, and it could in principle be ascribed to the limitations of this theoretical approach. Another hypothesis could be that the stability of 1β is due to kinetic reasons. DSC was also measured from 40 to 160 (in the case of acridine when the samples melt at around 105°) or 180 °C (in the case of 9-aminoacridine to reach full melting) and the results are reported in the ESI.† The heating/cooling ramp was cycled to study the reversibility of the process (Fig. S4†). While acridine and naproxen melt at 107 and 153 °C, respectively, their mechanical mixture melts as a eutectic at about 100 °C. A double DSC peak is observed for both the mechanical mixture and 1β, indicating two steps in the eutectic formation. The high temperature and more intense peak is attributed to the melting, while the first DSC peak can be ascribed to a metastable phase. It must be noticed that a double DSC peak is not unusual in a molecular complex21 where a monotropic phase transformation of a metastable form is supposed to explain the low temperature DSC signal. The liquid mixture obtained by heating the mechanical mixture and 1β up to 160 °C was cooled and solidified without any DSC peak, suggesting the formation of amorphous solid materials. To confirm the DSC data, variable temperature PXRD was carried out on the 1β molecular adduct at the same temperature range of DSC (Fig. S7,† left). The crystalline compounds show peaks until 118 °C, and above this temperature their disappearance suggests the melting, confirmed by imaging data. On cooling to RT from 160 °C, the formation of an amorphous solid is confirmed by XRD (Fig. S7,† right). Surprisingly, after heating 1β just above the eutectic temperature (120°) and cooling to 40 °C, crystalline 1β is again formed, similarly to the solution crystallization after solubilizing 1β instead. At this temperature, couples of acridine and naproxen are probably still present in the melt, and they favour the crystallization instead of the amorphous solidification. In fact, the DSC profile of 1α (Fig. S4†) shows two broader peaks under heating, but at much lower temperatures, e.g., 50 and 70 °C This behaviour further confirmed that 1β appears to be the more stable compound. From a thermodynamic viewpoint, within the limitation of energy framework calculations, 1α seemed more stable than 1β, explaining why the LAG treatment of 1α is not able to transform this crystal in 1β. The unsuccessful conversion of 1β to 1α, together with the higher temperature signal of 1β in TGA and DSC, should be ascribed to kinetic reasons. Conversely, the DSC profile of the mechanical mixture of naproxen and 9-aminoacridine (the reactants of compound 2) suggests their separate melting (at the expected melting temperature) and a eutectic solidification at 110 °C (Fig. S6†) of the two crystalline reactants, confirming a weaker affinity with respect to the acridine/naproxen couple. The PXRD pattern of the analysed sample was collected after the cooling ramp of the DSC (Fig. S7†) and, as expected, represents the sum of the two pure phase PXRD profiles, confirming that no reactions occurred, with crystallization of the reactants. Compound 2 can be obtained only by solvent crystallization in the host/guest structure, in analogy to 1α and no 1:1 phase was obtained. Finally, these DSC experiments explain the unsuccessful thermal synthesis in the capillary because the crystallization of the melt, independently of the chosen temperature, if the eutectic is reached, gives an amorphous phase.
To summarize, the molecular and structural landscape of compound 1 appears as in Fig. 9. We must conclude that solubilizing 1β induces a different result (still 1β crystallizes) with respect to the crystallization after solubilizing the reactants separately (1α crystallizes). Moreover, it must be noted that 1β from solution shows a habit (long needles in the optical microscope) different from that of 1β obtained from LAG (powder made up of small crystallites) as can be seen by the preferred orientation evident in the pattern of recrystallized 1β in Fig. 5, bottom. Except for preferred orientations, this “needle form” showed the same powder pattern of the LAG-obtained sample, confirming the same crystal structure despite the different morphology. This seeding effect of a molecular crystal (1β) prevailing, although it does not appear to be the more thermodynamically stable one, is rare but not new and observed also in famous case studies as those depicted as “the disappearing polymorph” summarized in review some years ago.51 However, in the present case, the different stoichiometries and solvation degrees of 1α and 1β can also be other explanations of the impossibility of the mutual interconversion between the two forms.
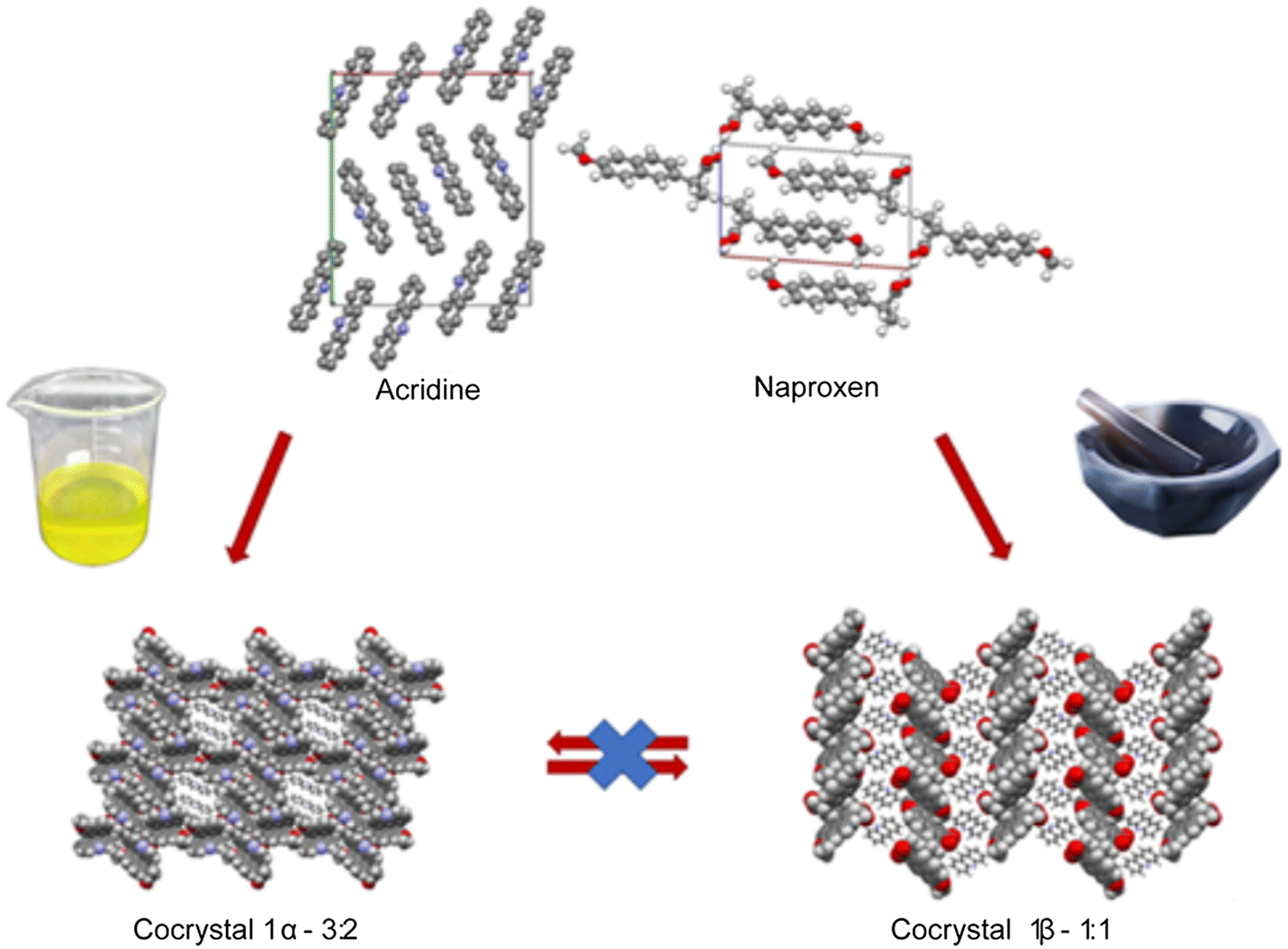 | ||
| Fig. 9 The molecular landscape of compound 1. | ||
Concerning the number of molecular crystals obtained using the reactant in Scheme 1, it can be concluded that a clear trend from acridine (two molecular crystals), 9-aminoacridine (one molecular crystal) and 6,9-diamino-2-ethoxyacridine (no cocrystal) is evident. Such results suggest that only compounds with a limited number of polar side chains can be used as cocrystals with naproxen. When the affinity is higher as in the case of 1, a eutectic is formed upon heating and the 1:1 compound (1β) can be obtained by the LAG method, besides the host/guest compound (1α). When the affinity is smaller, as in the case of 2, the eutectic is formed only upon cooling of the melt, but the cooling causes crystallization of the same reactant and the 1:1 layered phase equivalent to 1β cannot be obtained. Only the host/guest structure (2α) can be obtained by solution crystallization. With a bulkier and more polar counterpart (6,9-diamino-2-ethoxyacridine in Scheme 1) no crystallization is observed by any preparation technique. In this case the DSC profile of the mechanical mixture gave contradictory results: a eutectic partial melting is observed at 120 °C (6,9-diamino-2-ethoxyacridine lactate melts at 245°), then a large bump around 140° but a net sharp peak is observed at 175 °C, just some degrees above the naproxen MP (165 °C), followed by amorphous solidification under cooling. These results are not much different from the acridine/naproxen couple, but in this case not one among the three methods used allowed obtaining a crystalline molecular complex despite the affinity observed in the melt state by DSC. The unsuccessful attempt could be due to the use of its common and stable lactate form.
Conclusions
A total of three cocrystals of naproxen with acridines, comprising two cocrystals with a different stoichiometry, were obtained: two through the reaction carried out in solution and one carried out by the LAG procedure. The efficacy of LAG as a means of exploring stoichiometric diversity in molecular cocrystals was confirmed in this study.Three new crystal structures were obtained, one of them solved by powder diffraction data. The crystallization from solution allowed obtaining solvated 3:2 and 2:1 host–guest structures for 1α and 2. Conversely, LAG allowed obtaining a 1:1 layered head-to-tail crystal structure (1β) with a yield larger than 99% under almost dry conditions. Surprisingly, the two molecular cocrystals of naproxen with acridine cannot be converted from one to the other, probably because of the different stoichiometry and complex equilibria between reactants and products, governed by contrasting thermodynamic and kinetic effects, favouring 1α and 1β, respectively. Interestingly, during the review of the present article, a very recent publication reported that mechanochemistry resulted in a unique way to expand the crystal landscape of dexamethasone to obtain kinetically stable cocrystals with benzenediols (catechol and resorcinol).52
The 1β form was experimentally found to be more stable than 1α, as indicated by TGA and DSC measurements, in contrast to energy framework calculations. DSC also allowed identification of eutectic melting and amorphous solidification in the case of acridine/naproxen mechanical mixtures and cocrystals. Conversely, aminoacridine and naproxen melt separately and then crystallize as a mixture of the reactants with a eutectic solidification. As a final consideration, increasingly substituted acridines are less prone to cocrystallize with naproxen, with acridine showing two molecular cocrystals, 9-aminoacridine showing one molecular cocrystal, and 6,9-diamino-2-ethoxyacridine giving no cocrystals, independently of the adopted preparation procedure.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors acknowledge the Research of Young Scientists grant (BMN) no. 539-T080-B027-22 (University of Gdansk), DS no. 531-T080-D738-22 (University of Gdansk), Project 288–105 by FINPIEMONTE within the Programma Pluriennale Attività Produttive 2015/2017 Misura `3.1 “Contratto d'insediamento” (Università del Piemonte Orientale).Notes and references
- G. R. Desiraju, Supramolecular synthons in crystal engineering—a new organic synthesis, Angew. Chem., Int. Ed. Engl., 1995, 34(21), 2311–2327 CrossRef CAS
.
- B. Moulton and M. J. Zaworotko, From molecules to crystal engineering: supramolecular isomerism and polymorphism in network solids, Chem. Rev., 2001, 101(6), 1629–1658 CrossRef CAS
- G. R. Desiraju, Hydrogen bridges in crystal engineering: interactions without borders, Acc. Chem. Res., 2002, 35(7), 565–573 CrossRef CAS PubMed
- G. R. Desiraju, Crystal engineering: a holistic view, Angew. Chem., Int. Ed., 2007, 46(44), 8342–8356 CrossRef CAS PubMed
- N. Shan and M. J. Zaworotko, The role of cocrystals in pharmaceutical science, Drug Discov. Today, 2008, 13(9–10), 440–446 CrossRef CAS
- D. J. Good and N. Rodriguez-Hornedo, Solubility advantage of pharmaceutical cocrystals, Cryst. Growth Des., 2009, 9(5), 2252–2264 CrossRef CAS
- N. K. Duggirala, M. L. Perry, Ö. Almarsson and M. J. Zaworotko, Pharmaceutical cocrystals: Along the path to improved medicines, Chem. Commun., 2016, 52(4), 640–655 RSC
- A. Port, C. Almansa, R. Enrech, M. Bordas and C. R. Plata-Salamán, Differential Solution Behavior of the New API–API Co-Crystal of Tramadol–Celecoxib (CTC) versus Its Constituents and Their Combination, Cryst. Growth Des., 2019, 19(6), 3172–3182 CrossRef CAS
- M. Isidori, M. Lavorgna, A. Nardelli, A. Parrella, L. Previtera and M. Rubino, Ecotoxicity of naproxen and its phototransformation products, Sci. Total Environ., 2005, 348(1–3), 93–101 CrossRef CAS
- P. A. Todd and S. P. Clissold, Naproxen: A Reappraisal of its Pharmacology, and Therapeutic Use in Rheumatic Diseases and Pain States, Drugs, 1990, 40(1), 91–137 CrossRef CAS PubMed
- C. Bombardier, L. Laine, A. Reicin, D. Shapiro, R. Burgos-Vargas, B. Davis, R. Day, M. B. Ferraz, C. J. Hawkey, M. C. Hochberg, T. K. Kvien and T. J. Schnitzer, Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis, N. Engl. J. Med., 2000, 343(21), 1520–1528 CrossRef CAS PubMed
- M. Asadi, S. Sayar, E. Radmanesh, S. Naghshi, S. Mousaviasl, S. Jelvay, M. Ebrahimzadeh, A. Mohammadi, S. Abbasi, S. Mobarak, S. Bitaraf, F. Zardehmehri and A. Cheldavi, Efficacy of naproxen in the management of patients hospitalized with COVID-19 infection: A randomized, double-blind, placebo-controlled, clinical trial, Diabetes Metab. Syndr.: Clin. Res. Rev., 2021, 15(6), 102319 CrossRef PubMed
- M. Yousefifard, A. Zali, A. Zarghi, A. Madani Neishaboori, M. Hosseini and S. Safari, Non-steroidal anti-inflammatory drugs in management of COVID-19; a systematic review on current evidence, Int. J. Clin. Pract., 2020, 74(9), e13557 CAS
- O. Terrier, S. Dilly, A. Pizzorno, J. Henri, F. Berenbaum, B. Lina, B. Fève, F. Adnet, M. Sabbah, M. Rosa-Calatrava, V. Maréchal and A. Slama Schwok, Broad-spectrum antiviral activity of naproxen: from Influenza A to SARS-CoV-2 Coronavirus, 2020, hal-02988352.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, The Cambridge Structural Database, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS
- E. Conterosito, V. Gianotti, L. Palin, E. Boccaleri, D. Viterbo and M. Milanesio, Facile preparation methods of hydrotalcite layered materials and their structural characterization by combined techniques, Inorg. Chim. Acta, 2018, 470, 36–50 CrossRef CAS
- D. R. Weyna, T. Shattock, P. Vishweshwar and M. J. Zaworotko, Synthesis and structural characterization of cocrystals and pharmaceutical cocrystals: mechanochemistry vs slow evaporation from solution, Cryst. Growth Des., 2009, 9(2), 1106–1123 CrossRef CAS
- R. A. E. Castro, J. D. Ribeiro, T. M. Maria, M. Ramos Silva, C. Yuste-Vivas, J. Canotilho and M. E. S. Eusébio, Naproxen cocrystals with pyridinecarboxamide isomers, Cryst. Growth Des., 2011, 11(12), 5396–5404 CrossRef CAS
- A. Tilborg, G. Springuel, B. Norberg, J. Wouters and T. Leyssens, On the influence of using a zwitterionic coformer for cocrystallization: structural focus on naproxen–proline cocrystals, CrystEngComm, 2013, 15(17), 3341–3350 RSC
- N. Tumanova, N. Tumanov, F. Fischer, F. Morelle, V. Ban, K. Robeyns, Y. Filinchuk, J. Wouters, F. Emmerling and T. Leyssens, Exploring polymorphism and stoichiometric diversity in naproxen/proline cocrystals, CrystEngComm, 2018, 20(45), 7308–7321 RSC
- K. Manoj, R. Tamura, H. Takahashi and H. Tsue, Crystal engineering of homochiral molecular organization of naproxen in cocrystals and their thermal phase transformation studies, CrystEngComm, 2014, 16(26), 5811–5819 RSC
- C. Neurohr, M. Marchivie, S. Lecomte, Y. Cartigny, N. Couvrat, M. Sanselme and P. Subra-Paternault, Naproxen–nicotinamide cocrystals: Racemic and conglomerate structures generated by CO2 antisolvent crystallization, Cryst. Growth Des., 2015, 15(9), 4616–4626 CrossRef CAS
- S. K. Nechipadappu and D. R. Trivedi, Structural and physicochemical characterization of pyridine derivative salts of anti-inflammatory drugs, J. Mol. Struct., 2017, 1141, 64–74 CrossRef CAS
- M. M. Patel, M. D. Mali and S. K. Patel, Bernthsen synthesis, antimicrobial activities and cytotoxicity of acridine derivatives, Bioorg. Med. Chem. Lett., 2010, 20(21), 6324–6326 CrossRef CAS PubMed
- T. S. Huang, J. J. Lee, Y. S. Li and S. P. Cheng, Ethacridine induces apoptosis and differentiation in thyroid cancer cells in vitro, Anticancer Res., 2019, 39(8), 4095–4100 CrossRef CAS
- M. Tonelli, G. Vettoretti, B. Tasso, F. Novelli, V. Boido, F. Sparatore, B. Busonera, A. Ouhtit, P. Farci, S. Blois, G. Giliberti and P. La Colla, Acridine derivatives as anti-BVDV agents, Antiviral Res., 2011, 91(2), 133–141 CrossRef CAS PubMed
- S. Hassan, D. Laryea, H. Mahteme, J. Felth, M. Fryknäs, W. Fayad, S. Linder, L. Rickardson, J. Gullbo, W. Graf, L. Påhlman, B. Glimelius, P. Larsson and P. Nygren, Novel activity of acriflavine against colorectal cancer tumor cells, Cancer Sci., 2011, 102(12), 2206–2213 CrossRef CAS PubMed
- S. Nafisi, A. A. Saboury, N. Keramat, J. F. Neault and H. A. Tajmir-Riahi, Stability and structural features of DNA intercalation with ethidium bromide, acridine orange and methylene blue, J. Mol. Struct., 2007, 827(1–3), 35–43 CrossRef CAS
- G. P. Moloney, D. P. Kelly and P. Mack, Synthesis of acridine-based DNA bis-intercalating agents, Molecules, 2001, 6(3), 230–243 CrossRef CAS
- A. Mirocki and A. Sikorski, The influence of solvent on the crystal packing of ethacridinium phthalate solvates, Materials, 2020, 13(22), 5073 CrossRef CAS
- L. Palin, E. Conterosito, R. Caliandro, E. Boccaleri, G. Croce, S. Kumar, W. van Beek and M. Milanesio, Rational design of the solid-state synthesis of materials based on poly-aromatic
molecular complexes, CrystEngComm, 2016, 18(31), 5930–5939 RSC
- R. Caliandro, V. Toson, L. Palin, E. Conterosito, M. Aceto, V. Gianotti, E. Boccaleri, E. Dooryhee and M. Milanesio, New Hints on the Maya Blue Formation Process by PCA-Assisted In Situ XRPD/PDF and Optical Spectroscopy, Chem. – Eur. J., 2019, 25(49), 11503–11511 CrossRef CAS PubMed
- L. Palin, M. Milanesio, W. van Beek and E. Conterosito, Understanding the ion exchange process in LDH nanomaterials by fast in situ XRPD and PCA-assisted kinetic analysis, J. Nanomater., 2019, 4612493 CAS
- T. Friščić, S. L. Childs, S. A. Rizvi and W. Jones, The role of solvent in mechanochemical and sonochemical cocrystal formation: a solubility-based approach for predicting cocrystallisation outcome, CrystEngComm, 2009, 11(3), 418–426 RSC
- E. Conterosito, M. Milanesio, L. Palin and V. Gianotti, Rationalization of liquid assisted grinding intercalation yields of organic molecules into layered double hydroxides by multivariate analysis, RSC Adv., 2016, 6(110), 108431–108439 RSC
- CrysAlis CCD and CrysAlis RED, Version 1.171.36.24, Oxford Diffraction Ltd., Yarnton, UK, 2012
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 CrossRef PubMed
- A. L. Spek, Structure validation in chemical crystallography, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed
- Johnson, C.K., ORTEP II, Report ORNL-5138, Oak Ridge National Laboratory, Oak Ridge, TN, USA, 1976.
- Motherwell, S.; Clegg, S. PLUTO-78, Program for Drawing and Molecular Structure, University of Cambridge, Cambridge, UK, 1978.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, Mercury CSD 2.0—New Features for the Visualization and Investigation of Crystal Structures, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS
- A. Altomare, C. Cuocci, C. Giacovazzo, A. Moliterni, R. Rizzi, N. Corriero and A. Falcicchio, EXPO2013: a kit of tools for phasing crystal structures from powder data, J. Appl. Crystallogr., 2013, 46(4), 1231–1235 CrossRef CAS
- A. A. Coelho, TOPAS and TOPAS-Academic: an optimization program integrating computer algebra and crystallographic objects written in C++, J. Appl. Crystallogr., 2018, 51(1), 210–218 CrossRef CAS
- URL: https://www.topas-academic.net/
- C. F. Mackenzie, P. R. Spackman, D. Jayatilaka and M. A. Spackman, CrystalExplorer model energies and energy frameworks: extension to metal coordination compounds, organic salts, solvates and open-shell systems, IUCrJ, 2017, 4(5), 575–587 CrossRef CAS
- S. P. Thomas, P. R. Spackman, D. Jayatilaka and M. A. Spackman, Accurate lattice energies for molecular crystals from experimental crystal structures, J. Chem. Theory Comput., 2018, 14(3), 1614–1623 CrossRef CAS PubMed
- P. Rossi, J. Ceccarelli, S. Milazzo, P. Paoli, J. Morais Missina, S. Ciattini, A. Ienco, G. Tuci, M. Valleri, M. P. Giovannoni, G. Guerrini and L. Conti, Nonsteroidal Anti-Inflammatory Drugs–1-Phenylethylamine Diastereomeric Salts: A Systematic Solid-State Investigation, Cryst. Growth Des., 2021, 21(12), 6947–6960 CrossRef CAS
- Y. Kobayashi, K. Kinbara, M. Sato and K. Saigo, Synthesis, absolute configuration, and application of enantiopure trans-1-aminobenz [f] indan-2-ol, Chirality, 2005, 17(2), 108–112 CrossRef CAS PubMed
- J. Bao, Z. Zhang, Z. Yan, J. R. Wang and X. Mei, Cocrystallization in vitamin B9 gels to construct stoichiometry-controlled isostructural materials, CrystEngComm, 2018, 20(12), 1644–1648 RSC
- M. Rajkumar, A non-centrosymmetric cocrystal assembled by OH…N and CH…π supramolecular synthons, J. Mol. Struct., 2021, 1245, 131105 CrossRef CAS
- D. K. Bučar, R. W. Lancaster and J. Bernstein, Disappearing polymorphs revisited, Angew. Chem., Int. Ed., 2015, 54(24), 6972–6993 CrossRef PubMed
- S. N. Wong, K. H. Low, J. Weng, H. W. Chan and S. F. Chow, Expanding the solid-state landscape of dexamethasone: a specific sandwich structure in facilitating the formation of kinetically stable cocrystals from mechanochemistry, CrystEngComm, 2022, 24, 5875–5879 RSC
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2168756, 2168757 and 2169121. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2ce00890d |
| This journal is © The Royal Society of Chemistry 2022 |