
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Identification of a selective DNA ligase for accurate recognition and ultrasensitive quantification of N6-methyladenosine in RNA at one-nucleotide resolution†
Weiliang
Liu
,
Jingli
Yan
*,
Zhenhao
Zhang
,
Hongru
Pian
,
Chenghui
Liu
 and
Zhengping
Li
*
and
Zhengping
Li
*
Key Laboratory of Applied Surface and Colloid Chemistry, Ministry of Education, Key Laboratory of Analytical Chemistry for Life Science of Shaanxi Province, School of Chemistry and Chemical Engineering, Shaanxi Normal University, Xi’an 710062, Shaanxi Province, P. R. China. E-mail: yanjingli@snnu.edu.cn; lzpbd@snnu.edu.cn
First published on 15th February 2018
Abstract
N 6-Methyladenosine (m6A) is the most frequent post-transcriptional modification in RNA, and it plays a critical role in biological processes. The functions of m6A remain largely unexplored due to a lack of highly sensitive methods to quantitatively determine the m6A modification fraction at a precise location. Here, we first reveal that T3 DNA ligase has significant selectivity towards the m6A modification. On the basis of the new finding, we establish an ultrasensitive quantitation assay for accurately determining m6A at one-nucleotide resolution in RNA. With the proposed assay, as low as 4 fM RNA containing m6A can be determined and the selectivity is up to 54.1-fold to discriminate m6A against unmodified adenosine (A). The sensitivity has been improved about 106-fold so the proposed method can be successfully employed to accurately determine m6A in real biological samples, even in low abundance RNA.
Introduction
N 6-Methyladenosine (m6A) is the most frequent form of post-transcriptional modification in RNA. Recent studies have shown that m6A occurs not only in eukaryotic and bacterial messenger RNA (mRNA)1,2 but also in viral RNA,3,4 ribosomal RNA (rRNA) and long non-coding RNA (lncRNA).5 The discovery of methyltransferase6–9 and demethylase10 for m6A suggests that m6A modification is revisable and dynamically regulated, and further indicates that m6A modification is associated with gene regulation, including mRNA translation efficiency,11 stability and splicing.12,13 Therefore, m6A modification plays a critical role in many biological processes and is linked to human health.14 However, the biology of m6A remains largely uncovered, due to the lack of sensitive and robust methods to quantitatively determine the degree of m6A modification at a precise location.In 2012, by combining m6A-specific methylated RNA immunoprecipitation with massively parallel sequencing (MeRIP-seq), S. R. Jafferey’s and D. Dominissini’s groups developed high-throughput sequencing methods to analyze m6A modification on a transcriptome-wide scale.15,16 These studies showed that more than 7000 mammalian mRNAs contain m6A and that the m6A sites preferentially appear near stop codons and in 3′ UTRs. However, the sequencing methods cannot quantitatively detect the m6A modification fraction at the precise location, which generally locate the m6A residues within approximately 200 nucleotides. As the MeRIP-seq methods offer the distribution of m6A at a transcriptome-wide scale, quantitative methods for the determination of site-specific m6A in RNA have become increasingly important, and are greatly significant for revealing the biological functions of m6A and the association between m6A and human disease and clinical diagnosis. Liu et al. first reported a RNase H cleavage-based method for quantifying m6A at a specific site, which required site-specific cleavage and radioactive labelling followed by ligation-assisted extraction and Thin Layer Chromatography (SCARLET).17 The multiple enzymatic steps and the separation lead to very laborious processes, expensive reagents and radioactive hazards. More recently, E. T. Kool et al. have identified a DNA polymerase (Tth DNA Pol) with selectivity for the incorporation of thymidine opposite unmodified A over m6A.18 Zhou et al. have also revealed that Bst DNA polymerase can significantly hinder RNA-directed DNA synthesis.19 On the basis of the selectivity of DNA polymerases, probe extension-based methods have been established for quantifying m6A at specific sites. Although these methods also need electrophoresis separation and radioactive or fluorescent labels, the procedures have been simplified. Of particular note is that the sensitivity of all methods mentioned above is too low to quantify m6A in low abundance mRNA or lncRNA due to the lack of an amplification step, in which about 10 fmole RNA containing m6A can be detected in a large amount of total RNA samples. The reason may be that the products of probe extension and enzymatic cleavage are difficult to specifically amplify.
Generally, DNA ligase shows high specificity compared to DNA polymerase and the ligated DNA products can be specifically amplified. Dai et al. have described the T4 DNA ligase-based method to discriminate between A and m6A in RNA.20 However, no attempt was made to detect m6A in RNA samples, possibly due to the low selectivity of T4 DNA ligase (see below). In this work, we first reveal that T3 DNA ligase has strong selectivity to discriminate m6A from A in RNA. By ligating the DNA probes with T3 DNA ligase using a template of RNA and amplifying the ligated DNA products with PCR, as low as 4 fM (corresponding to 40 zmol in 10 μL solution) lncRNA containing m6A can be accurately determined and the selectivity for discriminating m6A in RNA can be greatly improved up to 54.1-fold.
Experimental section
Materials and reagents
RNA2577-A, RNA2577-m6A and RNA2488-A segments were synthesized by Integrated DNA Technologies (America). DEPC-treated water, RNAiso (lysis buffer), dNTPs and the DNA probes were obtained from TaKaRa Biotechnology Co. Ltd. (Dalian, China). JumpStart™ Taq DNA Polymerase was purchased from Sigma-Aldrich (Shanghai, China). T3 DNA ligase, T7 DNA ligase, T4 DNA ligase and Taq DNA ligase as well as T4 RNA ligase 2 were purchased from New England Biolabs (Beijing, China). Super Green I was purchased from Fanbo Biochemicals Co. Ltd. (Beijing, China). Trichloromethane, isopropyl alcohol and ethanol were purchased from Sinopharm Chemical Reagent Co. Ltd. (Beijing, China). All the solutions for reactions were prepared with DEPC-treated water.Ligation of DNA probes using a template of RNA
The ligation reaction mixture A consisted of 20 nM probe L, 20 nM probe R, the ligation buffer (66 mM Tris–HCl, 10 mM MgCl2, 1 mM ATP, 1 mM DTT, and 7.5% polyethylene glycol (PEG 6000) at pH 7.6 and 25 °C) and an appropriate amount of the RNA target. The ligation reaction mixture B consisted of 0.9 U ligase and the ligation buffer. Mixture A was heated at 85 °C for 3 min and then incubated at 35 °C for 10 min; then the ligation reaction mixture B was added. The final volume of the ligation reaction mixture was 10 μL. Then the ligation reaction mixture was incubated at an appropriate temperature to carry out ligation. After ligation, the reaction mixture was put on ice immediately. The reaction conditions for T3 DNA ligase, T4 DNA ligase, T7 DNA ligase, T4 RNA ligase 2 and Taq DNA ligase were 35 °C for 15 min, 25 °C for 30 min, 35 °C for 15 min, 37 °C for 30 min, and 35 °C for 15 min, respectively.PCR amplification reaction and real-time fluorescence measurements
A volume of 2 μL of the ligation product was transferred to a PCR reaction mixture with a final volume of 10 μL. The PCR reaction mixture contained a forward primer, a reverse primer (the concentration of each was 200 nM), 250 μM dNTPs, 0.4× Super Green I, 0.5 U JumpStart™ Taq DNA Polymerase and the PCR buffer (10 mM Tris–HCl at pH 8.3, 50 mM KCl, 1.5 mM MgCl2, and 0.001% (w/v) gelatin). The PCR reaction was carried out with a StepOne Real-Time PCR System (Applied Biosystems, USA) using a hot start of 94 °C for 2 min, followed by 45 cycles of 94 °C for 20 s and 60 °C for 30 s. The real-time fluorescence intensity was monitored at 60 °C.Isolation of total RNA from HeLa cells
HeLa cells were cultured in DMEM (GBICO, Cat. 12100-046) supplemented with 10% (v/v) Fetal Bovine Serum (HyClone), 100 U mL−1 penicillin and 100 μg mL−1 streptomycin. The cells were maintained at 37 °C under a humidified atmosphere containing 5% CO2.The DMEM was removed from the Petri dish while the bottom of the Petri dish was filled with cells. After that the cells were washed three times with cold PBS buffer (10 mM sodium phosphate buffer, 0.1 M NaCl, at pH 7.4 and 25 °C). Next, 6 mL RNAiso solution was added into the Petri dish, and then the cells were incubated at room temperature for 2 min. After incubation, the cell lysates were collected in a centrifuge tube which contained 1.2 mL trichloromethane. The centrifuge tube was shaken slightly at room temperature for 2 min, and then the cell lysates were centrifuged for 15 min at 12000 rpm at 4 °C, and the aqueous phase was collected in a new tube. Next, 3 mL isopropyl alcohol was added to the aqueous phase and the mixture was incubated at room temperature for 10 min. Then the mixture was centrifuged for 20 min at 12000 rpm at 4 °C. The precipitate was washed once with 75% ethanol and dissolved in DEPC-treated water. Finally, the concentration of total RNA was determined with a Nano Drop 2000 (Thermo Scientific).
Extraction of poly A+ RNA from total RNA
The structure of most lncRNA is similar to that of mRNA containing a polyA tail.21,22 The m6A to be detected is located in MALAT1 lncRNA which has a polyA tail. Therefore, MALAT1 lncRNA can be obtained by the extraction of poly A+ RNA.Determination of melting temperatures (Tm) of the hybrids of the RNA segments and the probes
To determine the melting temperature (Tm) of the hybrids of RNA targets and the DNA probes in the ligation reaction, 200 nM RNA target (RNA2577-A or RNA2577-m6A) was mixed with 200 nM probe L1 and 200 nM probe R1 D in a 10 μL mixture solution which contained T3 DNA ligase reaction buffer (66 mM Tris–HCl, 2 mM ATP, 10 mM MgCl2, 1 mM DTT, and 3.75% PEG 6000) and 0.4× Super Green I. The mixture was put into a StepOne Real-Time PCR System (Applied Biosystems, USA) to determine the fluorescence signal changes when increasing the temperature from 25 °C to 95 °C. Fig. S1(a) and (b)† show the derivatives of the fluorescence intensity signal as a function of temperature, respectively, produced by RNA2577-A/(probe L1 – probe R1 D) and RNA2577-m6A/(probe L1 – probe R1 D). After that, probe R1 D was replaced by probe R1 and we conducted the same experiment. The derivatives of the fluorescence intensity signal as a function of temperature, produced by RNA2577-A/(probe L1 – probe R1) and RNA2577-m6A/(probe L1 – probe R1) are depicted in Fig. S1(c) and (d),†respectively.Results and discussion
Principle of the ligation-dependent PCR method for RNA m6A detection
The general outline of the proposed ligase-dependent PCR assay for m6A determination is schematically illustrated in Fig. 1. m6A at the 2577th site of MALAT1 lncRNA, which has been demonstrated to contain an m6A modification,17 is employed as the model target in this study. lncRNA containing m6A and A at the 2577th site are respectively defined as RNA2577-m6A and RNA2577-A. For detection of RNA2577-m6A, DNA probe L1 (left probe) and probe R1 (right probe) were designed (see the sequence in Table S1†). Each probe contains a universal primer-specific sequence used for PCR amplification (red and orange) and a target-specific sequence (blue and black), which is respectively complementary to the RNA target immediately downstream and upstream of the 2577th site. Probe L1 is modified with a phosphate group at its 5′-terminus and probe R1 is modified with two ribonucleotides at its 3′-terminus. As shown in Fig. 1(I), in the presence of RNA2577-A, probe L1 and probe R1 will adjacently hybridize with RNA around the 2577th A site, and thus they can be ligated by catalysis with T3 DNA ligase. The ligated products are subsequently amplified with PCR using the universal forward primer and reverse primer. When the 2577th A is methylated at the N6 position, the m6A at the 2577th site can significantly hinder the ligation of probe L1 and probe R1 (Fig. 1(II)). Therefore, in the presence of RNA2577-m6A, the ligated products and final PCR products will be greatly reduced. Thus, RNA2577-m6A can be detected. In order to quantitatively determine the m6A modification fraction at the 2577th site in MALAT1 lncRNA, the A at the 2488th site of the same MALAT1 lncRNA molecule is selected as the reference site (Fig. 1(III)), which is known to only contain A.15,16 Probe L2 and probe R2 are designed to be the same as probe L1 and probe R1, respectively, except for the target-specific sequences, which are respectively complementary to the RNA target immediately downstream and upstream of the 2488th site. In the presence of MALAT1 lncRNA, probe L2 and probe R2 will be ligated and the PCR signal of the ligated products can accurately indicate the lncRNA concentration. As demonstrated below, by comparing the real-time fluorescence PCR signals at the 2577th site and 2488th site, the m6A modification fraction can be precisely determined.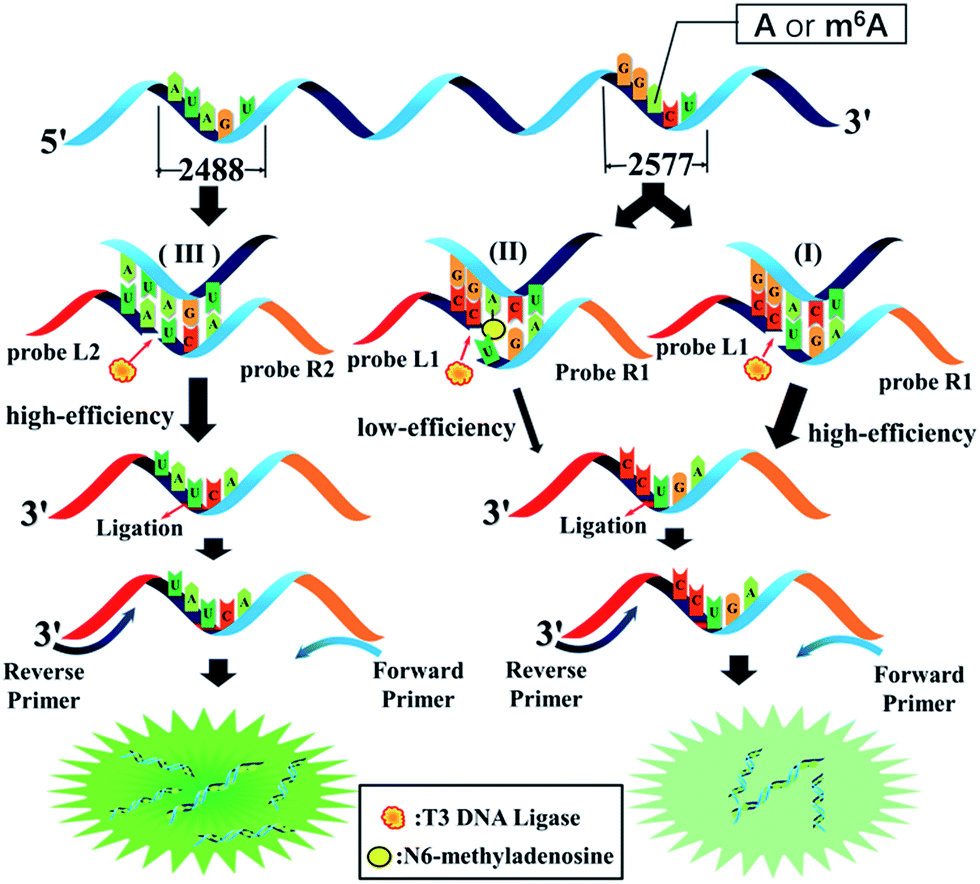 | ||
| Fig. 1 The general outline of the ligation-dependent PCR assay for m6A detection. | ||
Identification of a ligase with substantial selectivity towards m6A
The selectivity of the ligases for discriminating m6A from A is the key issue for the ligase-dependent PCR assay to detect m6A in RNA. So, we first identify the substantial selectivity of the ligases towards m6A. A pair of 69 mer synthetic MALAT1 lncRNA segments containing either m6A or A at the 2577th site are employed as the templates. Probe L1 and probe R1 D, which is the same as probe R1 but without modification at its 3′-terminus with two ribonucleotides, are first hybridized with the RNA2577-A or RNA2577-m6A segments, and are then ligated through catalysis by various ligases. The ligated products are amplified with PCR and the PCR products are detected with gel electrophoresis. As demonstrated in Fig. 2(a), when the pixel intensity of the PCR products produced from the RNA2577-A segment is defined as 100, the ratio of the pixel intensity produced from the RNA2577-m6A segment to that produced from the RNA2577-A segment using different ligases is 24.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 (T3 DNA ligase), 32.1:100 (T4 RNA ligase 2), 79.1:100 (T4 DNA ligase), 64.4:100 (T7 DNA ligase) and 77.7:100 (Taq DNA ligase), in which the T3 DNA ligase and T4 RNA ligase 2 show good selectivity for recognizing m6A from A in RNA. Using an RNA molecule as the template to ligate the DNA probes, it has been reported that modification with two ribonucleotides of one DNA probe at its 3′-terminus can greatly improve the specificity and efficiency of the ligase reaction.23 Therefore, probe L1 and probe R1 with the modification of two ribonucleotides at the 3′-terminus are used to further investigate the selectivity of T3 DNA ligase and T4 RNA ligase 2. As shown in Fig. 2(b), using probe L1 and R1, the ratio of the pixel intensity produced from the RNA2577-m6A segment to that produced from the RNA2577-A segment reaches 6.87:100 (T3 DNA ligase) and 30.5:100 (T4 RNA ligase 2). These results indicate that T3 DNA ligase has the strongest selectivity to discriminate m6A from A in RNA and modification of probe R1 with two ribonucleotides can greatly improve the selectivity.
:100 (T3 DNA ligase), 32.1:100 (T4 RNA ligase 2), 79.1:100 (T4 DNA ligase), 64.4:100 (T7 DNA ligase) and 77.7:100 (Taq DNA ligase), in which the T3 DNA ligase and T4 RNA ligase 2 show good selectivity for recognizing m6A from A in RNA. Using an RNA molecule as the template to ligate the DNA probes, it has been reported that modification with two ribonucleotides of one DNA probe at its 3′-terminus can greatly improve the specificity and efficiency of the ligase reaction.23 Therefore, probe L1 and probe R1 with the modification of two ribonucleotides at the 3′-terminus are used to further investigate the selectivity of T3 DNA ligase and T4 RNA ligase 2. As shown in Fig. 2(b), using probe L1 and R1, the ratio of the pixel intensity produced from the RNA2577-m6A segment to that produced from the RNA2577-A segment reaches 6.87:100 (T3 DNA ligase) and 30.5:100 (T4 RNA ligase 2). These results indicate that T3 DNA ligase has the strongest selectivity to discriminate m6A from A in RNA and modification of probe R1 with two ribonucleotides can greatly improve the selectivity.
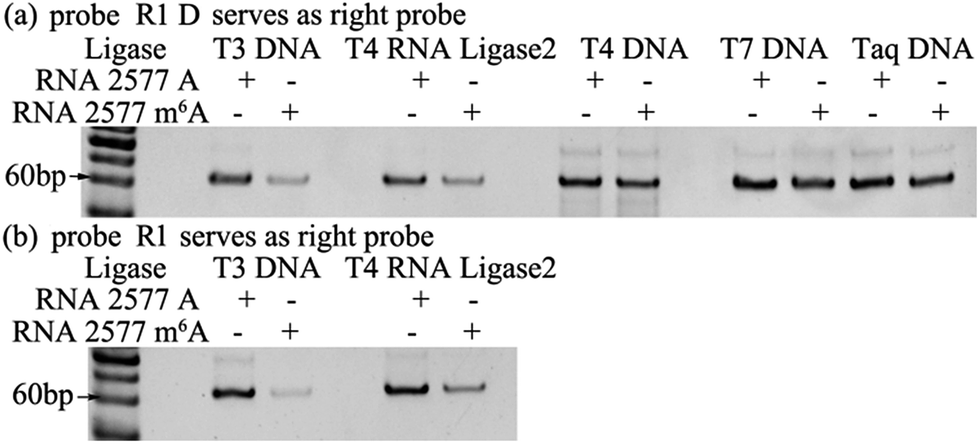 | ||
| Fig. 2 Gel electrophoresis for identifying a ligase with substantial selectivity towards m6A. The probes used in the reactions were probe R1 D (a) and probe R1 (b). | ||
The m6A modification is a single methylation of adenosine at the N6 position. The methyl group is small and chemically inert. More importantly, the methylated N6-position retains its ability to donate a hydrogen bond, so that m6A can still form Watson–Crick base pairs.17 Therefore, it is extremely difficult to specifically recognize m6A from A in RNA.18 Fortunately, as demonstrated above, we have revealed that different ligases have different identifying abilities towards m6A, and T3 DNA ligase shows the strongest selectivity, which is ascribed to its ability to recognize between m6A:T and A:T. We also demonstrate that T3 DNA ligase shows better selectivity to discriminate between m6A:U and A:U when probe R1 is modified with two ribonucleotides at its 3′-terminus. The melting temperatures (Tm) of the hybrids of the RNA segments and the probes were detected, and are 44.3 °C (RNA2577-A/probe L1 – probe R1D), 43.8 °C (RNA2577-m6A/probe L1 – probe R1D), 43.7 °C (RNA2577-A/probe L1 – probe R1), and 41.4 °C (RNA2577-m6A/probe L1 – probe R1) (see Fig. S1†). Accordingly, the m6A modification decreases the Tm value by only 0.5 °C in the presence of m6A:T and A:T pairs, and in the presence of m6A:U and A:U pairs, the Tm value can be decreased by 2.3 °C, which also makes T3 DNA ligase more selective for recognizing m6A and A in RNA.
Assessment of the effects of core sequences on specificity
In 2012, Dan Dominissini et al. found that all sequences with m6A conform to the degenerate consensus RRACH (A = m6A, R = A or G, H![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) U or A),15 and the commonest core sequences for m6A are GGACU, AAACU, AGACU and GGACA. The core sequence of RNA 2577 is GGACU. To assess the effects of the core sequences on the specificity of the proposed ligase-dependent PCR assay, the core sequence of RNA 2577 (GGACU) is replaced with AAACU, AGACU, and GGACA, respectively. The corresponding RNAs containing A or m6A are respectively defined as RNA AAACU-A or RNA AAACU-m6A, RNA AGACU-A or RNA AGACU-m6A, and RNA GGACA-A or RNA GGACA-m6A. These RNA molecules were used to perform the specificity experiments. The experiments were performed with the same procedures using T3 DNA ligase as demonstrated above. The results are shown in Fig. 3, and the ratio of the pixel intensity produced from the m6A segment to that produced from the A segment reaches 7.20:100 (RNA AAACU), 7.70:100 (RNA AGACU), and 5.46:100 (RNA GGACA). These results indicate that core sequences have little effect on the specificity of the proposed ligase-dependent PCR assay.
U or A),15 and the commonest core sequences for m6A are GGACU, AAACU, AGACU and GGACA. The core sequence of RNA 2577 is GGACU. To assess the effects of the core sequences on the specificity of the proposed ligase-dependent PCR assay, the core sequence of RNA 2577 (GGACU) is replaced with AAACU, AGACU, and GGACA, respectively. The corresponding RNAs containing A or m6A are respectively defined as RNA AAACU-A or RNA AAACU-m6A, RNA AGACU-A or RNA AGACU-m6A, and RNA GGACA-A or RNA GGACA-m6A. These RNA molecules were used to perform the specificity experiments. The experiments were performed with the same procedures using T3 DNA ligase as demonstrated above. The results are shown in Fig. 3, and the ratio of the pixel intensity produced from the m6A segment to that produced from the A segment reaches 7.20:100 (RNA AAACU), 7.70:100 (RNA AGACU), and 5.46:100 (RNA GGACA). These results indicate that core sequences have little effect on the specificity of the proposed ligase-dependent PCR assay.
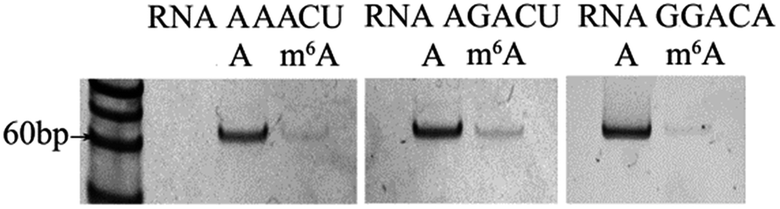 | ||
| Fig. 3 Gel electrophoresis for assessing the effects of core sequences on specificity. | ||
Analytical performance of the ligation-dependent PCR method for RNA m6A detection
With the proposed ligase-dependent PCR assay, we systematically investigate the influence of experimental parameters, including the ligation temperature, ligation time and amounts of T3 DNA ligase, on the selectivity and sensitivity for detecting m6A, and optimize the experimental conditions (see Fig. S2–S4†). Under the optimum experimental conditions, the analytical performance of the ligase-dependent PCR assay is evaluated using real-time fluorescence PCR measurements, in which Super Green I is utilized as the fluorescent dye for real-time detection of the PCR products. As shown in Fig. 4(a), well-defined real-time fluorescence curves can be obtained which are produced by the RNA2577-A segment in the concentration range from 4 fM to 4 nM. When the CT values (the number of cycles experienced by the fluorescence signal of each reaction up to the threshold value) are plotted against the logarithm (lg) of RNA2577-A concentration, as depicted in Fig. 4(b), an excellent linear relationship is obtained. The linear regression equation is CT = −8.59 − 3.14lgCRNA2577-A (M) with a corresponding correlation coefficient, R2, of 0.998. At the same time, as demonstrated in Fig. 4(c), the CT values produced by RNA2577-m6A at the same concentrations are much less than those produced by RNA2577-A. According to the calibration curve shown in Fig. 4(b), the CT values produced by 4 pM RNA2577-m6A correspond to the concentration of RNA2577-A at 73.9 fM. Therefore, the selectivity of the ligase-dependent PCR assay for detecting m6A in RNA is up to 54.1-fold. It is particularly worthwhile to note that the real-time fluorescence signal produced by 400 fM RNA2577-m6A is almost the same as the blank. That is to say, when the total concentration of the RNA target to be detected is less than 400 fM, RNA containing m6A produced a negligible signal, which will be of no contribution to the real-time fluorescence signal. The result means that the RNA-m6A concentration should be equal to that of the total RNA target concentration minus the RNA2577-A concentration when the total RNA concentration is less than 400 fM, which can be determined with the ligase-dependent PCR assay.
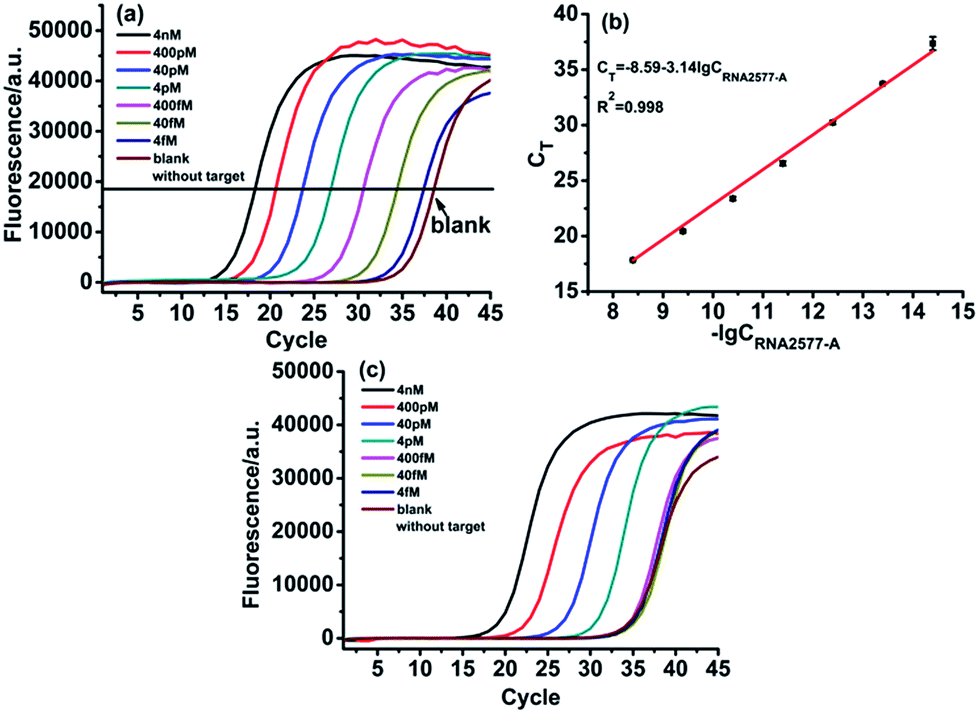 | ||
| Fig. 4 Specificity and sensitivity of the ligase-dependent PCR assay: (a) real-time fluorescence curves produced by the RNA2577-A segment in the concentration range from 4 fM to 4 nM under optimum experimental conditions. (b) The relationship between CT and lg of RNA2577-A concentration (M). (c) Real-time fluorescence curves produced by the RNA2577-m6A segment in the concentration range from 4 fM to 4 nM under optimum experimental conditions. | ||
Subsequently, we tested whether the ligase-dependent PCR assay can be employed for quantitative detection of m6A-containing RNA. The RNA2577-A segment and RNA2577-m6A segment were firstly mixed with a total concentration of 400 fM as the synthetic samples, in which the RNA 2577-m6A proportions were 0%, 10%, 30%, 50%, 80%, and 100%. Then, the RNA2577-A concentration was determined with the ligase-dependent PCR assay according to the calibration curve shown in Fig. 4(b). The RNA2577-m6A concentration was equal to 400 fM minus the RNA2577-A concentration. As shown in Table S2,† the detected RNA2577-m6A concentrations are in good agreement with the added ones, indicating that the proposed assay can be used for quantitative evaluation of the extent of RNA containing m6A in RNA samples at a specific site when the total RNA concentration is less than 400 fM.
Detection of m6A in real poly A+ RNA samples
For determination of RNA containing m6A in real biological samples, the vital issue is how to obtain the total concentration of the RNA target. To address this issue, as demonstrated before, we selected an adenosine (A) site in the same RNA target molecule as the reference site, which is known to only contain A, such as the 2488th site in the MALAT1 lncRNA. The ligase-dependent assay can easily determine the total concentration of the RNA target molecule at the reference site, which can also control the total RNA target concentration at less than 400 fM for determination of real biological samples.The designed RNA2488-A specific probe L2 and probe R2 are used to perform the ligation reaction by catalysis with T3 DNA ligase and then the ligated products are amplified with PCR. A synthetic RNA2488-A segment is employed to construct the calibration curve. As demonstrated in Fig. 5(a) and (b), there is an excellent linear relationship between the CT value and lg of RNA2488-A concentration in the range from 4 fM to 4 nM. The correlation equation is CT = −6.39 − 3.16lgCRNA2488-A (M) and the correlation coefficient, R2, is 0.990. Finally, we apply the ligase-dependent PCR assay to determine the m6A modification fraction at the 2577th site of the MALAT1 lncRNA in 85 ng poly A+ RNA extracted from HeLa cells. As shown in Fig. 5(b) and (c), the total concentration of MALAT1 lncRNA is determined to be 285.9 fM (CRNA2488-A) in the sample with the ligase-dependent PCR assay at the 2488th site. As shown in Fig. 5(d) and 4(b), the RNA2577-A concentration is determined to be 55.2 fM (CRNA2577-A) with the ligase-dependent PCR assay at the 2577th site. Therefore, the RNA2577-m6A concentration is calculated as 230.7 fM from the formula CRNA2488-A–CRNA2577-A, and the m6A modification fraction at the 2577th site is estimated to be 80.7%, which very much coincides with that obtained by the SCARLET assay (∼80%).17 The same method is applied to the measurement of the m6A modification fraction at the 2577th site of the MALAT1 lncRNA in poly A+ RNA extracted from HEK293T, and the m6A modification fraction at the 2577th site is estimated to be 51.3% (Fig. S5†), which also coincides well with that obtained by the SCARLET assay (∼51%).17 The above results indicate the good applicability of the ligase-dependent PCR assay for quantitation of m6A in real biological samples.
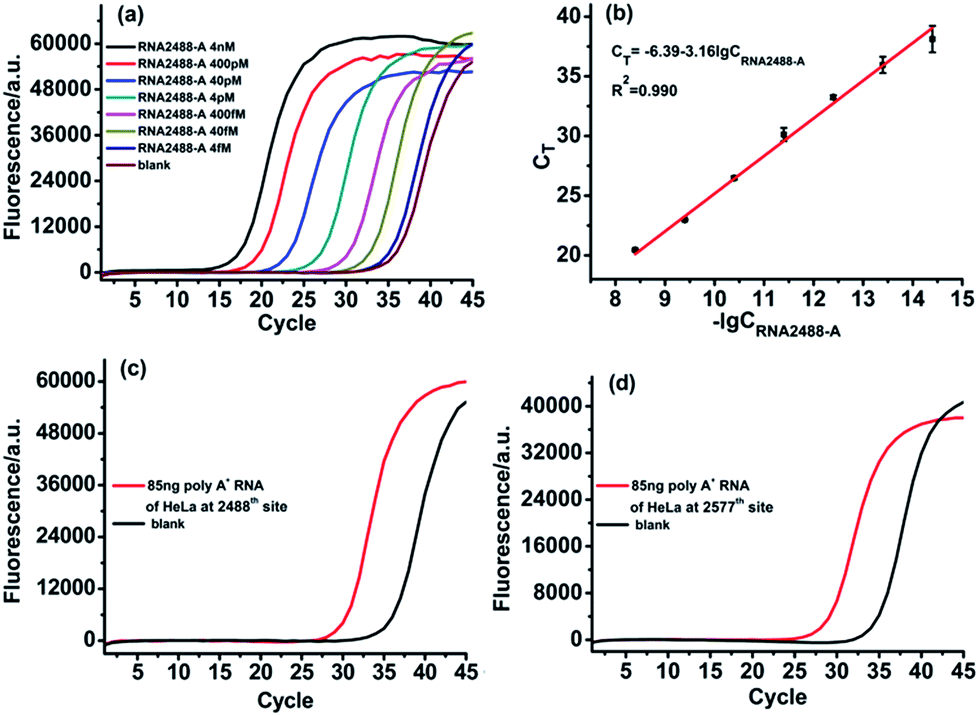 | ||
| Fig. 5 Determination of m6A in poly A+ RNA of HeLa cells. (a) Real-time fluorescence curves produced by the RNA2488-A segment in the concentration range from 4 fM to 4 nM. (b) The linear relationship between CT and lg of RNA2488-A concentration (M). (c) Real-time fluorescence curves produced by RNA2488-A in 85 ng poly A+ RNA of HeLa cells under optimum experimental conditions. (d) Real-time fluorescence curves produced by RNA2577-A in 85 ng poly A+ RNA of HeLa cells under optimum experimental conditions. | ||
Conclusions
In summary, we have first revealed that T3 DNA ligase, a commercially available ligase, has strong selectivity for the discrimination of m6A from A in RNA molecules. On this basis, a highly sensitive and selective ligase-dependent PCR assay has been established, which can accurately determine the m6A modification fraction at any specific site in RNA. Most importantly, owing to its ultrahigh sensitivity, the proposed assay can be used to quantify m6A in low abundance cellular RNA in real biological samples, which is currently not possible using existing techniques due to the lack of practical ability for nucleic acid amplification. In addition, the ligase-dependent PCR assay requires only common and available lab equipment and materials. Therefore, we believe that the proposed assay should be readily promising for applications in m6A related fundamental studies and clinical diagnosis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the National Natural Science Foundation of China (grant numbers 21505090 and 21335005), the Postdoctoral Sustentation Fund of China (2015M582604), the Program for Changjiang Scholars and innovative Research Team in University (IRT_15R43), the Fundamental Research Funds for the Central Universities (GK201501003) and the 111 Project (B14041).Notes and references
- R. Desrosiers, K. Friderici and F. Rottman, Proc. Natl. Acad. Sci. U. S. A., 1974, 71, 3971–3975 CrossRef CAS.
- X. Deng, K. Chen, G.-Z. Luo, X. Weng, Q. Ji, T. Zhou and C. He, Nucleic Acids Res., 2015, 43, 6557–6567 CrossRef CAS PubMed.
- K. Dimock and C. M. Stoltzfus, Biochemistry, 1977, 16, 471–478 CrossRef CAS PubMed.
- S. E. Kane and K. Beemon, Mol. Cell. Biol., 1985, 5, 2298–2306 CrossRef CAS PubMed.
- Y. C. Choi and H. Busch, Biochemistry, 1978, 17, 2551–2560 CrossRef CAS PubMed.
- J. A. Bokar, M. E. Shambaugh, D. Polayes, A. G. Matera and F. M. Rottman, RNA, 1997, 3, 1233–1247 CAS.
- J. Liu, Y. Yue, D. Han, X. Wang, Y. Fu, L. Zhang, G. Jia, M. Yu, Z. Lu, X. Deng, Q. Dai, W. Chen and C. He, Nat. Chem. Biol., 2014, 10, 93–95 CrossRef CAS PubMed.
- X.-L. Ping, B.-F. Sun, L. Wang, W. Xiao, X. Yang, W.-J. Wang, S. Adhikari, Y. Shi, Y. Lv, Y.-S. Chen, X. Zhao, A. Li, Y. Yang, U. Dahal, X.-M. Lou, X. Liu, J. Huang, W.-P. Yuan, X.-F. Zhu, T. Cheng, Y.-L. Zhao, X. Wang, J. M. R. Danielsen, F. Liu and Y.-G. Yang, Cell Res., 2014, 24, 177–189 CrossRef CAS PubMed.
- Y. Wang, Y. Li, J. I. Toth, M. D. Petroski, Z. Zhang and J. C. Zhao, Nat. Cell Biol., 2014, 16, 191–198 CrossRef CAS PubMed.
- G. Jia, Y. Fu, X. Zhao, Q. Dai, G. Zheng, Y. Yang, C. Yi, T. Lindahl, T. Pan, Y.-G. Yang and C. He, Nat. Chem. Biol., 2011, 7, 885–887 CrossRef CAS PubMed.
- X. Wang, B. S. Zhao, I. A. Roundtree, Z. Lu, D. Han, H. Ma, X. Weng, K. Chen, H. Shi and C. He, Cell, 2015, 161, 1388–1399 CrossRef CAS PubMed.
- X. Wang, Z. Lu, A. Gomez, G. C. Hon, Y. Yue, D. Han, Y. Fu, M. Parisien, Q. Dai, G. Jia, B. Ren, T. Pan and C. He, Nature, 2014, 505, 117–120 CrossRef PubMed.
- W. Xiao, S. Adhikari, U. Dahal, Y. S. Chen, Y. J. Hao, B. F. Sun, H. Y. Sun, A. Li, X. L. Ping, W. Y. Lai, X. Wang, H. L. Ma, C. M. Huang, Y. Yang, N. Huang, G. B. Jiang, H. L. Wang, Q. Zhou, X. J. Wang, Y. L. Zhao and Y. G. Yang, Mol. Cell, 2016, 61, 507–519 CrossRef CAS PubMed.
- A. Klungland and J. A. Dahl, Curr. Opin. Genet. Dev., 2014, 26, 47–52 CrossRef CAS PubMed.
- D. Dominissini, S. Moshitch-Moshkovitz, S. Schwartz, M. Salmon-Divon, L. Ungar, S. Osenberg, K. Cesarkas, J. Jacob-Hirsch, N. Amariglio, M. Kupiec, R. Sorek and G. Rechavi, Nature, 2012, 485, 201–206 CrossRef CAS PubMed.
- K. D. Meyer, Y. Saletore, P. Zumbo, O. Elemento, C. E. Mason and S. R. Jaffrey, Cell, 2012, 149, 1635–1646 CrossRef CAS PubMed.
- N. Liu, M. Parisien, Q. Dai, G. Zheng, C. He and T. Pan, RNA, 2013, 19, 1848–1856 CrossRef CAS PubMed.
- E. M. Harcourt, T. Ehrenschwender, P. J. Batista, H. Y. Chang and E. T. Kool, J. Am. Chem. Soc., 2013, 135, 19079–19082 CrossRef CAS PubMed.
- S. Wang, J. Wang, X. Zhang, B. Fu, Y. Song, P. Ma, K. Gu, X. Zhou, X. Zhang, T. Tian and X. Zhou, Chem. Sci., 2016, 7, 1440–1446 RSC.
- Q. Dai, R. Fong, M. Saikia, D. Stephenson, Y.-T. Yu, T. Pan and J. A. Piccirilli, Nucleic Acids Res., 2007, 35, 6322–6329 CrossRef CAS PubMed.
- M. Guttman, I. Amit, M. Garber, C. French, M. F. Lin, D. Feldser, M. Huarte, O. Zuk, B. W. Carey, J. P. Cassady, M. N. Cabili, R. Jaenisch, T. S. Mikkelsen, T. Jacks, N. Hacohen, B. E. Bernstein, M. Kellis, A. Regev, J. L. Rinn and E. S. Lander, Nature, 2009, 458, 223–227 CrossRef CAS PubMed.
- T. Hung and H. Y. Chang, RNA Biol., 2010, 7, 582–585 CrossRef CAS PubMed.
- J. Zhang, Z. Li, H. Wang, Y. Wang, H. Jia and J. Yan, Chem. Commun., 2011, 47, 9465–9467 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc05233b |
| This journal is © The Royal Society of Chemistry 2018 |