
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Environmentally benign and diastereoselective synthesis of 2,4,5-trisubstituted-2-imidazolines†
Hai Anh Le Phuong‡
 a,
Levente Cseri‡a,
George F. S. Whiteheadb,
Arthur Garfortha,
Peter Buddb and
Gyorgy Szekely*a
a,
Levente Cseri‡a,
George F. S. Whiteheadb,
Arthur Garfortha,
Peter Buddb and
Gyorgy Szekely*a
aSchool of Chemical Engineering & Analytical Science, The University of Manchester, The Mill, Sackville Street, Manchester, M1 3BB, UK. E-mail: gyorgy.szekely@manchester.ac.uk; Tel: +44 (0)161 306 4366
bSchool of Chemistry, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK
First published on 20th November 2017
Abstract
2-Imidazolines are heterocycles with a wide range of applications in biomedical science, pharmaceuticals, surfactants and catalysis. The 2,4,5-trisubstituted derivatives are conventionally prepared through the reaction of aromatic aldehydes and ammonia, requiring harsh conditions and toxic solvents. Herein, we report a sustainable, one-pot synthetic route to obtain cis or trans diastereomers of 2,4,5-trisubstituted-2-imidazolines. The reaction parameters, such as the ammonia source, the substituents of the aromatic aldehydes and the effect of temperature and solvent, were systematically explored. The synthesis was attempted in sixteen different green solvents. Reaction kinetics and quantum mechanical studies (DFT B3LYP/6-31G(d,p)) revealed the elementary steps of the cyclisation and refuted two common speculations. The limitations of the synthesis were explored through varying the p-, o-, and m-substituents on the aldehyde as well as employing heterocyclic and non-aromatic derivatives. Ten out of the twelve products synthesised in this work can be solely purified by crystallisation, without the need for extraction or chromatography. Owing to the bulky nature of the products, the applicability of membrane-based purification was demonstrated to further improve the sustainability of the synthesis.
Introduction
2-Imidazoline derivatives are an important class of heterocycles with a wide range of industrial applications (Fig. 1). They exhibit biological activity and can be found in natural products such as topsentines or spongotines, which have been attractive for biomedical purposes due to their antitumor, antiviral and anti-inflammatory characteristics.1 Several pharmaceutical compounds that contain a 2-imidazoline ring have been synthesised and studied for blood pressure control, hyperglycemia, depression and schizophrenia.2 Imidazolines and imidazolinium ions have been used as surfactants,3 ionic liquids,4 ionic polymers,5 and in the field of coordination chemistry and catalysis.6–10 Among the 2-imidazoline derivatives, 2,4,5-triaryl imidazolines (also called amarines) have been gaining increased interest because their hydrolysis provides 1,2-diaryl-1,2-diaminoethanes featuring a C2-symmetric structure, important in catalysis.11–13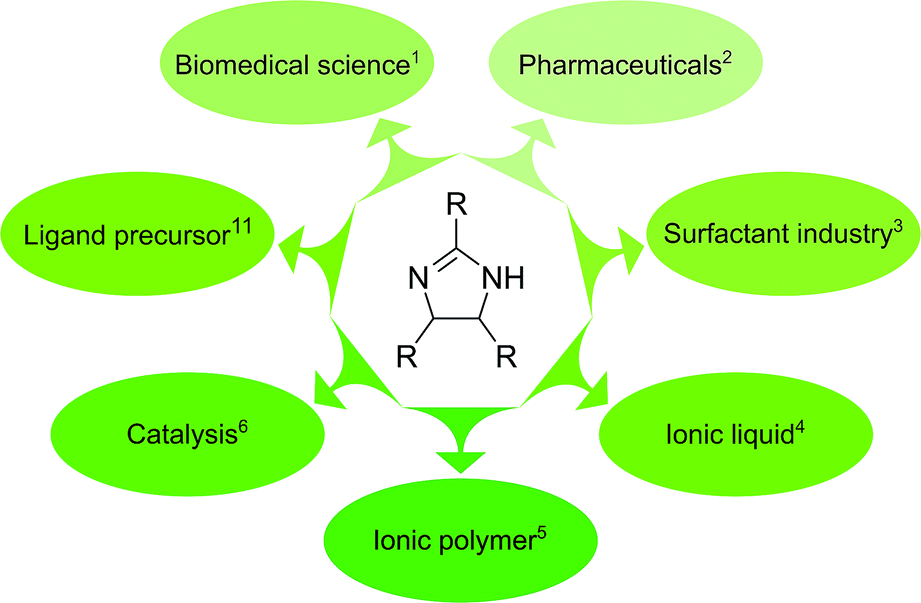 | ||
| Fig. 1 2-Imidazoline derivatives are versatile building blocks for various applications. | ||
The reported syntheses for obtaining 2,4,5-trisubstituted-2-imidazolines are summarised in Table 1. 2,4,5-Trisubstituted-2-imidazolines were successfully synthesised from benzylamine and carbon tetrachloride,14 and 1,2-cyclic sulphates and amidine.15 However, none of these syntheses were as prominent as the reaction from benzaldehyde and ammonia.16 Benzaldehyde reacts with liquid ammonia to produce hydrobenzamide (also known as 1,3,5-triaryl-2,4-diazapentadiene), an intermediate which upon heating or employing strong base deprotonates and subsequently forms amarine, supposedly via disrotatory pericyclic closure.11,17 The rate of pericyclisation usually depends on both the steric and electronic character of the substituents.18 However, the effect of substituents on the synthesis of 2-imidazolines and the reaction mechanism have yet to be investigated.
| Reactants | T (°C) | Solvent | Additives | Purification | Yield (%) | Ref. |
|---|---|---|---|---|---|---|
| a Two-step reaction. The 2,4-diazapentadiene intermediate was isolated via recrystallisation from cyclohexane. The 2-imidazoline product was purified by recrystallisation from petroleum ether. | ||||||
| Benzylamine & carbon tetrachloride | 125 | Carbon tetrachloride | Cu, Fe catalyst | Column chromatography | 1–24 | 14 |
| 1,2-Cyclic sulphates & amidine | 85–162 (reflux) | Dimethoxyethane/Methoxyethyl ether | — | Extraction | 22–64 | 15 |
| Benzaldehyde & ammoniaa | 130 | Benzene | — | Recrystallisation | 81 | 12 |
| Benzaldehyde & HDMS | 120 | — | — | Column chromatography | 47–89 | 19 |
| Benzaldehyde & urea | 120 | DMF | Cs2CO3 | Column chromatography | 40–79 | 20 |
Notwithstanding the moderate to good yields, most reactions require additives, extensive purification, or harsh conditions, i.e., high temperature, toxic or banned solvents. In addition, additives and catalysts were usually used for the preparation of the desired product, which results in additional waste that has to be managed. While attempts have been made towards a more sustainable synthesis of 2,4,5-trisubstituted-2-imidazolines, there is a need for exploring green solvents and purification processes.
The 21st century's sustainability grand challenge requires the chemical industry to employ mild reaction conditions, and non-toxic, non-flammable chemicals and solvents, with the aim of minimising environmental impact.21 Solvents for reaction and purification account for more than 80% of the total mass used for the production of pharmaceuticals,22 and consume 60% of the total energy.23 The solvent selection guides established by pharmaceutical companies provide practical advice to reduce the environmental burden associated with solvent usage.24,25 Following the guidelines, sixteen different green solvents were screened for the syntheses presented in this work. As an effective process intensification tool due to the mild conditions and low energy consumption,26 membrane technology is a sustainable alternative to conventional purification techniques such as crystallisation and chromatography,27 which can be more easily implemented in continuous processing.28 Consequently, the potential of membrane-based purification of the 2-imidazoline products was evaluated.
Herein, we report the development toward an environmentally benign, diastereoselective, one-pot synthesis of 2,4,5-trisubstituted-2-imidazolines employing non-toxic ammonium salts, green solvents and purification. The kinetics and reaction mechanism were explored to reveal the governing factors of the pericyclisation. To the best of our knowledge the kinetic studies and quantum mechanical calculations for the reaction of aldehyde and ammonia to produce 2,4,5-trisubstituted-2-imidazolines have not yet been reported.
Results and discussion
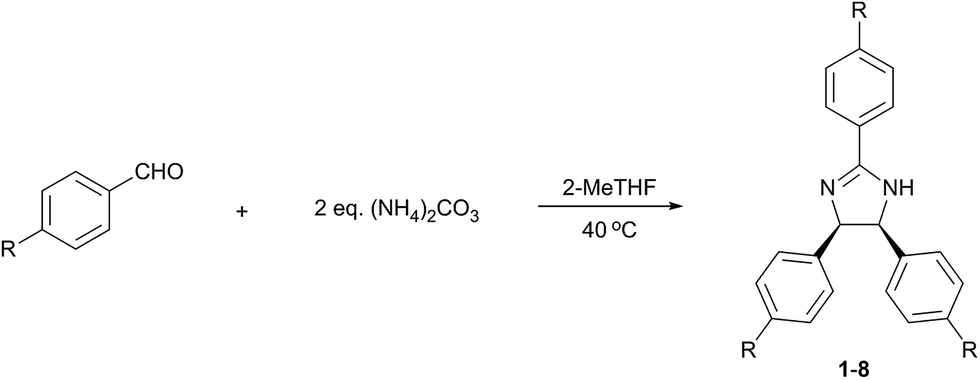
The reaction optimisation was initiated by investigating the effect of temperature and solvent on the reaction of 4-(trifluoromethyl)benzaldehyde and (NH4)2CO3 (Table 2). In line with the 12 principles of green chemistry,21 all reactions were performed at mild temperatures (20–50 °C) in solvents, which were chosen according to GlaxoSmithKline's solvent selection guide.25 Although, the isolated yield markedly increased with the temperature up to 40 °C (entries 1–3), a considerable decrease at 50 °C was observed (entry 4), which indicates the decomposition of (NH4)2CO3. The influence of solvent is presented in entries 3 and 5–16 in Table 2. While 1 was obtained in most of the solvents, good yield was only obtained in polar solvents (2-MeTHF, ethyl lactate, propylene carbonate, sulfolane, 2-propanol, methanol and DMSO), probably due to the stabilisation of the ionic intermediates. High solvent polarity favours the reaction, which resulted in reduced reaction time. For instance, the use of DMSO resulted in virtually 100% conversion of the starting material within 24 h, while achieving the same conversion in 2-MeTHF required 96 h. However, the higher reaction rate facilitated by-product formation, which subsequently decreased selectivity and isolated yield of 1. The product was not formed in Cyrene, choline chloride![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :urea (1:2 n/n) deep eutectic solvent (DES) and [bmim][OAc] ionic liquid. Cyrene, a relatively new green alternative for polar aprotic solvents,29 resulted in quasi-complete substrate consumption within 24 h. Nonetheless, a multicomponent reaction mixture was obtained without any trace of 1, which reiterates the labile and reactive nature of this new solvent.30 Considering the trade-off between selectivity and reaction rate, 2-MeTHF, ethyl lactate, propylene carbonate and sulfolane provide a suitable solvent for the reaction as they produced 1 with high selectivity in a manageable reaction time. However, since the product isolation from the last three is more solvent and energy intensive (extraction and evaporation), 2-MeTHF was used for further reaction optimisation.
:urea (1:2 n/n) deep eutectic solvent (DES) and [bmim][OAc] ionic liquid. Cyrene, a relatively new green alternative for polar aprotic solvents,29 resulted in quasi-complete substrate consumption within 24 h. Nonetheless, a multicomponent reaction mixture was obtained without any trace of 1, which reiterates the labile and reactive nature of this new solvent.30 Considering the trade-off between selectivity and reaction rate, 2-MeTHF, ethyl lactate, propylene carbonate and sulfolane provide a suitable solvent for the reaction as they produced 1 with high selectivity in a manageable reaction time. However, since the product isolation from the last three is more solvent and energy intensive (extraction and evaporation), 2-MeTHF was used for further reaction optimisation.
|
|||
|---|---|---|---|
| Entry | Solvent | T (°C) | Yieldb (%) |
| a The reactions were carried out with 4-(trifluoromethyl)benzaldehyde (3 mmol), (NH4)2CO3 (6 mmol), and solvent (6 mL) for 96 h.b Isolated yield.c The reaction was performed for 24 h at which point product formation stopped according to HPLC monitoring. | |||
| 1 | 2-MeTHF | 20 | 21 |
| 2 | 2-MeTHF | 30 | 40 |
| 3 | 2-MeTHF | 40 | 67 |
| 4 | 2-MeTHF | 50 | 48 |
| 5 | Ethyl lactate | 40 | 66 |
| 6 | Propylene carbonate | 40 | 66 |
| 7 | Sulfolane | 40 | 65 |
| 8 | 2-Propanol | 40 | 55 |
| 9 | Methanol | 40 | 41 |
| 10 | DMSOc | 40 | 38 |
| 11 | MTBE | 40 | 22 |
| 12 | CPME | 40 | 11 |
| 13 | 1-Butanol | 40 | 10 |
| 14 | Butyl acetate | 40 | 6 |
| 15 | Dimethyl carbonate | 40 | 2 |
| 16 | Water | 40 | 1 |
To study the effect of ammonia sources (2 eq.) on the reaction, non-toxic ammonium salts of acids covering a wide range of pKa values (−6.30 to 10.32) were selected (Table 3). The highest conversion was achieved using (NH4)2CO3 (entry 1), while moderate and poor yield was obtained with NH4HCO3 and NH4OAc (entries 2 and 3), respectively. NH4HCOO (entry 4) did not provide the desired product at 40 °C, but a moderate yield was obtained at the reflux temperature of 81 °C. Moderate yield was also achieved using NH4CF3COO at reflux (entry 5). In contrast, no product was formed with NH4Cl (entry 6) under reflux conditions. These results suggest that employing the ammonium salt of a weaker acid (therefore with higher pKa value) leads to increased reaction rate due to the more favoured dissociation of the salt and the more basic environment. In addition, the amount of ammonia source was also investigated (entry 1). The increase of ammonium carbonate from 1 to 2 eq. led to 23% yield enhancement. However, further increase to 4 eq. did not result in significant improvement.
|
||||
|---|---|---|---|---|
| Entry | NH3 source | pKa of acid31 | T (°C) | Yieldb (%) |
| a The reactions were carried out with 4-(trifluoromethyl)benzaldehyde (3 mmol), ammonium salt (6 mmol), and 2-MeTHF (6 mL) for 96 h.b Isolated yield.c 1 eq. ammonium salt (3 mmol) was used.d 4 eq. ammonium salt (12 mmol) was used.e Instead of 40 °C the reaction was performed at 35 °C due to the decomposition of NH4HCO3 at 40 °C.32 | ||||
| 1 | (NH4)2CO3 | 10.32 | 40 | 44c/67/71d |
| 2 | NH4HCO3 | 6.37 | 35e | 42 |
| 3 | NH4OAc | 4.75 | 40 | 11 |
| 4 | NH4HCOO | 3.75 | 40/reflux | —/48 |
| 5 | NH4CF3COO | 0.23 | reflux | 44 |
| 6 | NH4Cl | −6.30 | reflux | — |
The effect of both electron withdrawing and electron donating p-substituents on the reaction were systematically investigated (Table 4). Substrates with strongly or moderately electron-withdrawing groups (EWG) (entries 1–7), i.e., moderate to high Hammett sigma constants (σ = 0.45–0.82), provided the desired product in good to excellent yield. The substrate with weakly electron-withdrawing bromide group (entry 8; σ = 0.23) only afforded the 2-imidazoline derivative in poor yield. Reactions with non-EWGs (–H, –OH) were attempted but did not yield the imidazoline products.
|
|||||
|---|---|---|---|---|---|
| Entry | R | σb (−) | t (h) | Yieldc (%) | Product |
| a Unless otherwise noted, the reaction was performed with p-substituted benzaldehyde (3 mmol), (NH4)2CO3 (6 mmol), and 2-MeTHF (6 mL) at 40 °C.b Hammett sigma constant.33c Isolated yield.d Due to the poor substrate solubility in pure 2-MeTHF, the reaction required the use of 2-MeTHF (5 mL) and DMSO (1 mL) solvent mixture.e The lower 58% isolated yield is a result of excessive by-product formation when (NH4)2CO3 (6 mmol) was used, while the higher 85% isolated yield was obtained with the use of NH4OAc (6 mmol) ammonia source. This result implies that 3 is sensitive to oxidation under basic conditions. | |||||
| 1 | –NMe3+I−d | 0.82 | 24 | 83 | 2 |
| 2 | –NO2e | 0.78 | 24 | 58/85 | 3 |
| 3 | –SO2Med | 0.72 | 48 | 69 | 4 |
| 4 | –SF5 | 0.68 | 36 | 87 | 5 |
| 5 | –CN | 0.66 | 96 | 68 | 6 |
| 6 | –CF3 | 0.54 | 96 | 67 | 1 |
| 7 | –COOMe | 0.45 | 96 | 65 | 7 |
| 8 | –Br | 0.23 | 168 | 8 | 8 |
| 9 | –H | 0.00 | 168 | — | — |
In order to further explore the effect of EWGs on the reaction rate, the kinetics of the reaction was studied. The reactions of (NH4)2CO3 with 4-nitrobenzaldehyde, 4-(pentafluorosulfanyl)-benzaldehyde, 4-(trifluoromethyl)benzaldehyde, and methyl 4-formyl benzoate were chosen as model systems because they provided the desired product under the same conditions with a considerable yield, and they cover a wide range of σ (0.45–0.78). A consecutive reaction, 3A → B → C, was suggested for the reaction kinetics based on the formation of stable intermediate B identified as hydrobenzamide in the literature.11 In order to obtain the reaction rate constants k1 and k2, the rate eqn (1)–(3) have to be solved.
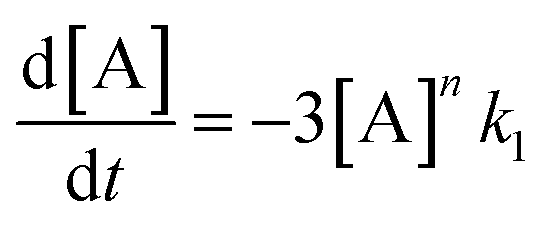 | (1) |
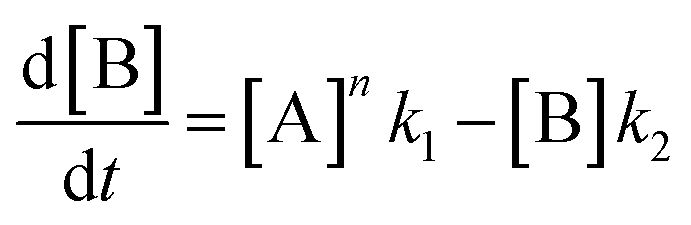 | (2) |
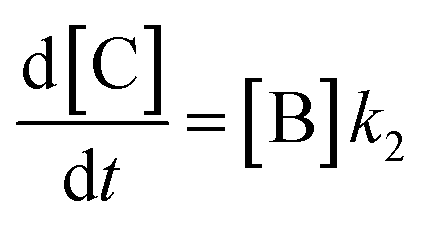 | (3) |
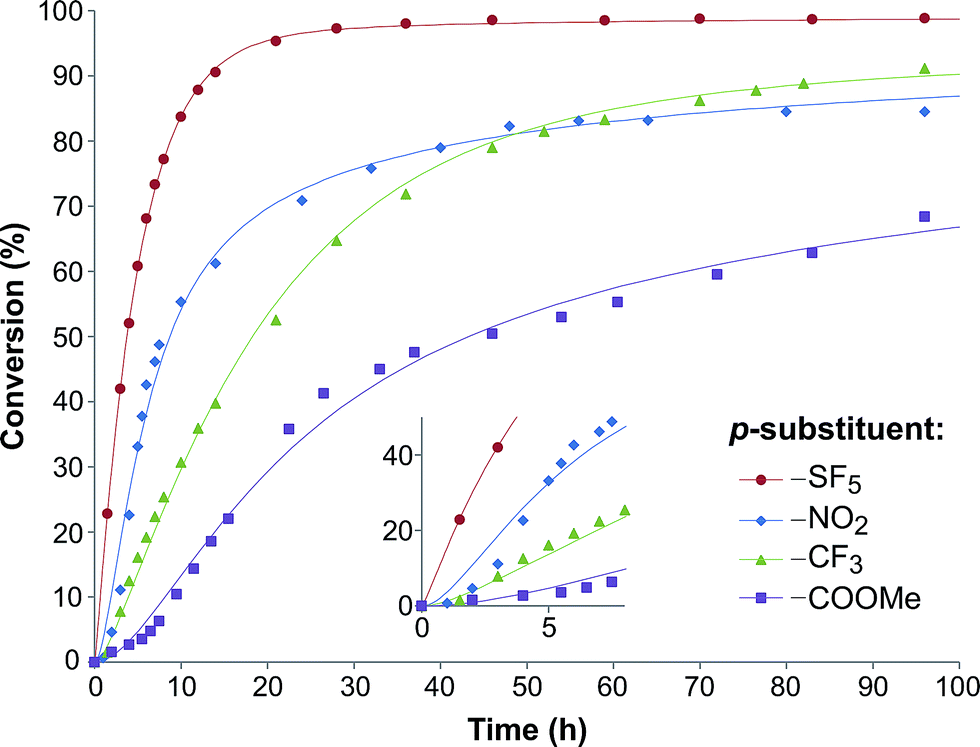 | ||
| Fig. 2 Substrate conversion profile as a function of reaction time. Symbols denote the experimental data, while the lines signify the calculated plots. | ||
In order to investigate the relationship between the reaction rates and the electron-withdrawing substituents, the calculated k1 and k2 values are displayed as a function of the σ in Fig. 3a. Although increasing rate constants for both k1 and k2 were expected with increasing σ no direct relationship between the three constants was found. The rate of hydrobenzamide formation increases from σ = 0.45 (–COOMe) until σ = 0.68 (–SF5), however, k1 significantly drops at σ = 0.78. The rate constant of the intramolecular ring closure decreases between σ = 0.45 (–COOMe) and σ = 0.54 (–CF3) and then grows with increasing electron-withdrawing property. These results could be the consequence of other factors, such as low solubility of the intermediate.
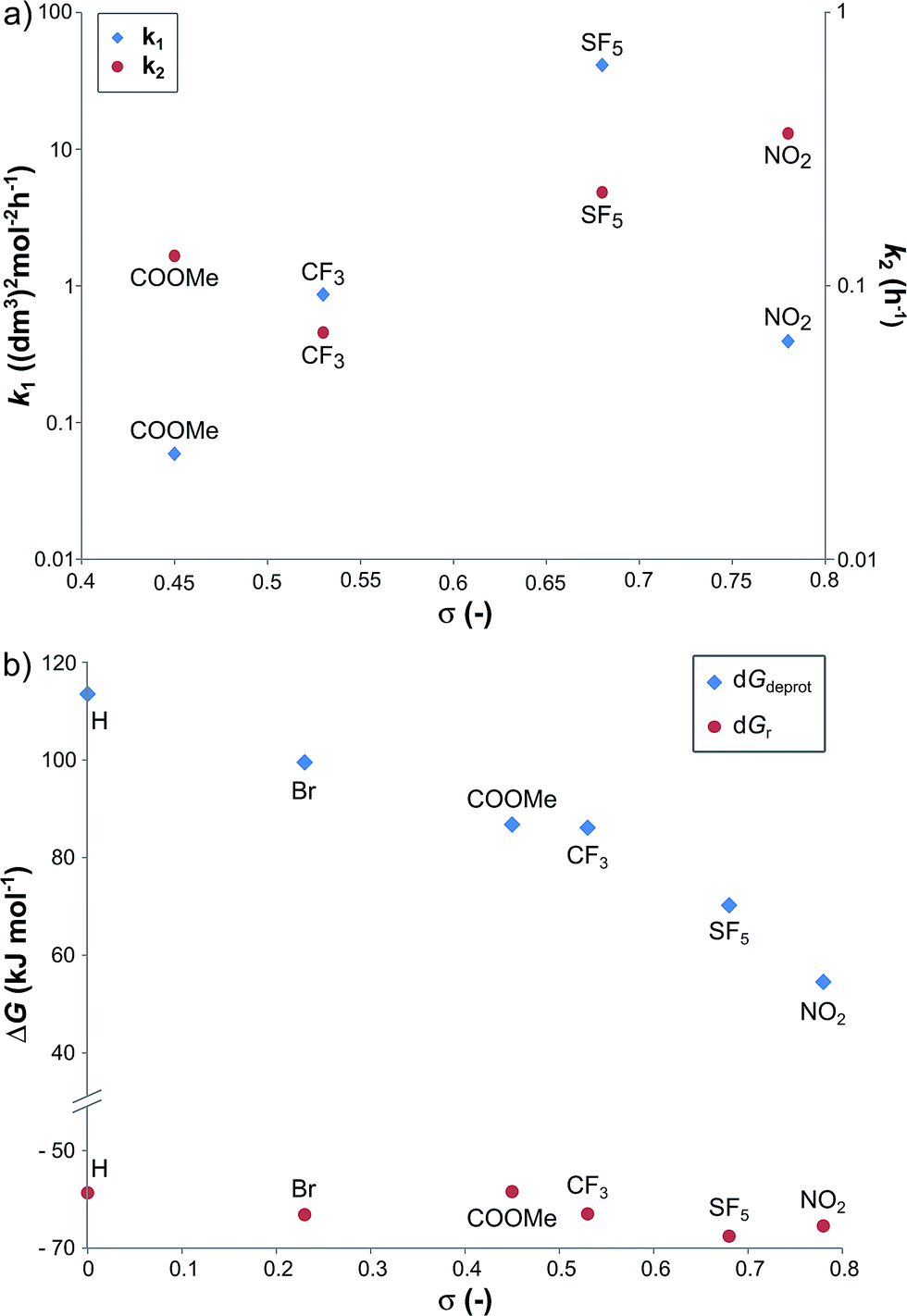 | ||
| Fig. 3 The k1 and k2 reaction rate constants (a), and the relative Gibbs free energy of the B → C reaction (ΔGr) and the deprotonation step (ΔGdeprot) (b) as a function of Hammett sigma constant (σ). | ||
Density functional theory computational analysis (DFT B3LYP/6-31G(d,p)) revealed the cyclisation mechanism of 1 from the hydrobenzamide intermediate (1hb) (Fig. 4). The deprotonation of 1hb forms a high energy carbanion intermediate (1cU), which stabilises by the translation of the ammonium counter ion. Subsequent protonation of 1cU on its nitrogen atom results in 1zw zwitterionic intermediate, which provides 1 via ring closure. The Gibbs free energy of both the cyclisation (ΔGr) and the deprotonation (ΔGdeprot) was derived for substituents with different electron-withdrawing properties (different σ values). The dependence of ΔGr on σ is negligible; however, the ΔGdeprot values significantly decrease with higher σ values (Fig. 3b). Consequently, the reaction favours EWGs, which is in line with the experimental observations (see Table 4).
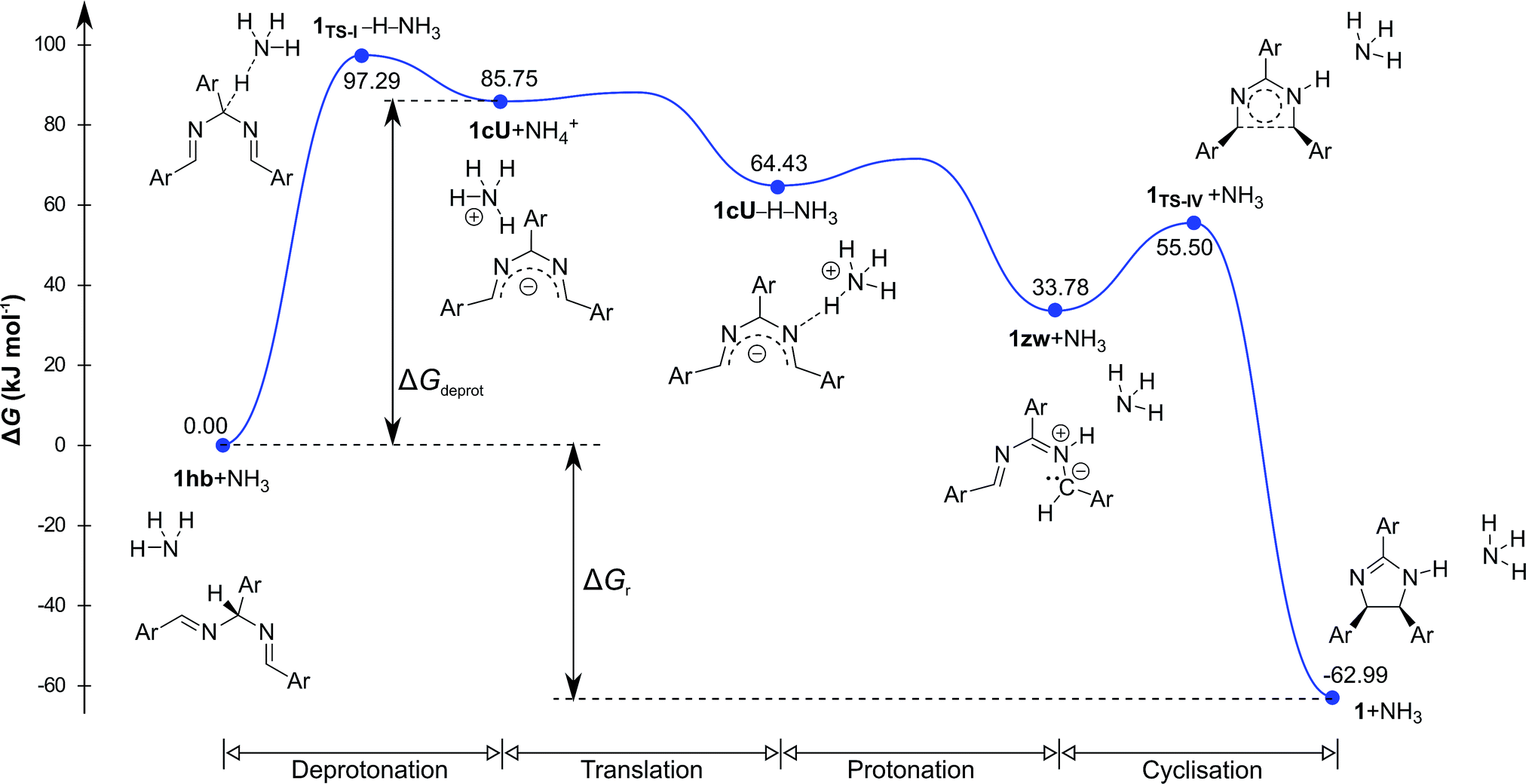 | ||
| Fig. 4 Computed relative Gibbs free energy profile of hydrobenzamide cyclisation in the presence of ammonia. | ||
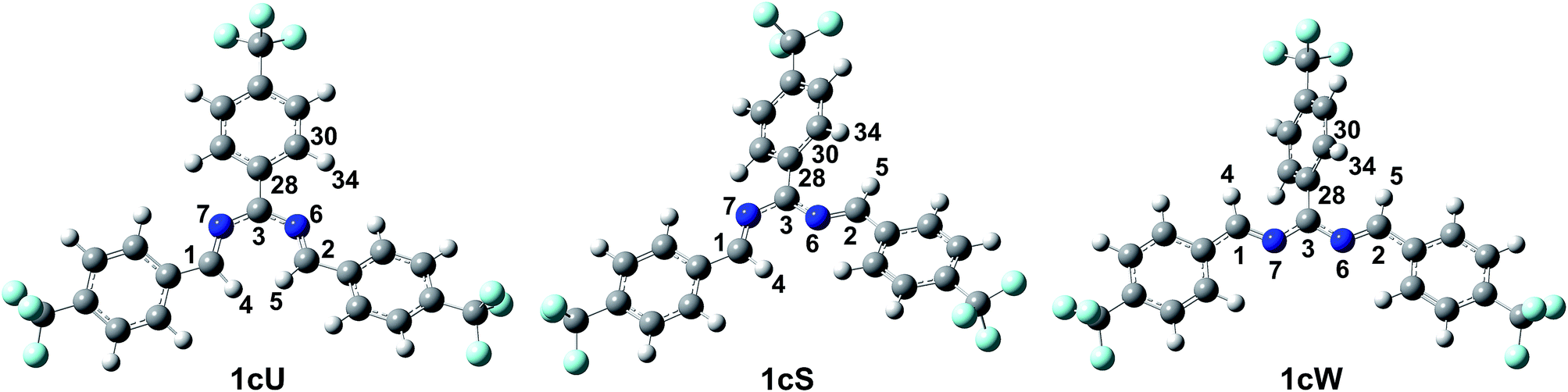
The DFT results refuted two speculations about the mechanism of the cyclisation. First, the cyclisation from the carbanion species with electron-withdrawing substituents is not favourable thermodynamically (Table S2, ESI†), despite speculations that the reaction proceeds with the ring closure of the carbanion intermediate leading to the negatively charged imidazoline which is the deprotonated form of 2-imidazoline (Scheme S1a, ESI†).11 Second, 1cU is the energetically most favoured carbanion conformer (Table 5). Despite speculations that 1cW has the lowest energy due to the lack of steric hindrance between H(4) and H(5)16 1cU has a quasi-planar structure, which allows the conjugation of the π system to be extended to the whole molecule. However, the steric repulsion between the H(5) and H(34) atoms in 1cS and 1cW forces the ring to turn out of the plane, which decreases the conjugation. The cyclisation was further studied using nuclear independent chemical shift (NICS) calculations.34 NICS(0) values of −1.94 and −3.55 ppm were obtained for the five-membered rings in 1zw and 1, respectively. These relatively small negative NICS(0) values revealed the non-aromatic nature of the rings. In contrast, the large negative NICS(0) value of −11.34 ppm for 1TS-IV proved the aromaticity of the transition state, and consequently, the pericyclic nature of the reaction. The lobes in the highest occupied molecular orbital (HOMO) of 1zw indicate that the cyclisation is allowed progress only in disrotatory mode leading to the cis product (1) according to the Woodward–Hoffman rule.35 The experimental results are in good agreement with the calculations because for all substrates, the formation of solely the cis product was observed. The stereochemistry of 1 (Fig. 5a), 2, and 5 (Fig. S115 and S116, ESI†) was confirmed by single-crystal X-ray diffraction. The stereochemistry of 3, 4 and 6–8 was deduced to be cis as the synthetic methods were identical. This deduction was further supported by the high coupling constants (10.9–11.2 Hz) for the imidazoline hydrogens in the NMR spectra.
|
|||
|---|---|---|---|
| Carbanion conformer | ΔG (ΔH) (kJ mol−1) | Boltzman population at 40 °C (%) | Dihedral angle N(6)–C(3)–C(28)–C(30) (deg) |
| 1cU | 0.0 (0.0) | 67.2 | −1.4 |
| 1cS | +2.02 (+2.20) | 30.9 | −27.4 |
| 1cW | +9.33 (+13.69) | 1.9 | −61.8 |
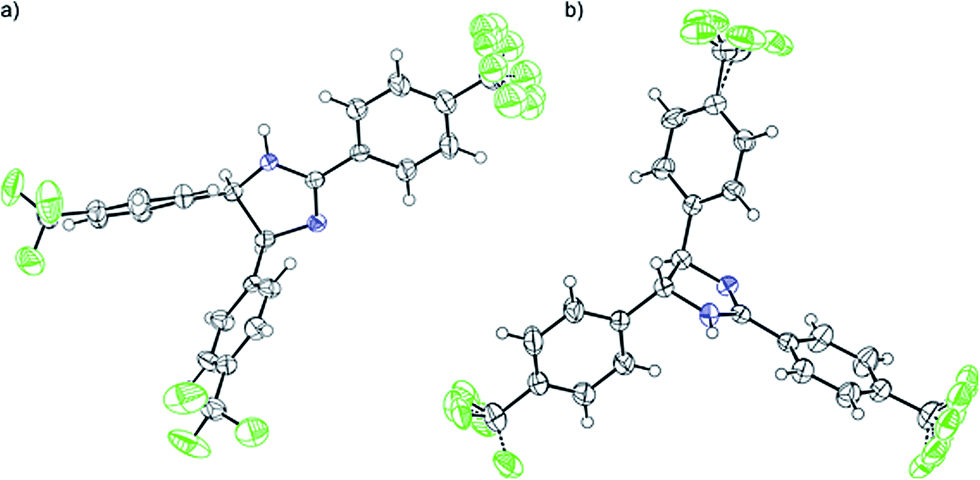 | ||
| Fig. 5 ORTEP representation of the molecular structure of cis-1 (a) and trans-9 (b) at 50% probability as determined by single-crystal X-ray diffraction, which revealed that some of the –CF3 groups can have more than one arrangement in the crystal. | ||
As a consequence of the electrocyclic nature of the reaction, the diastereoselectivity can be influenced by the method of reaction initiation. Photoirradiation at 254 nm and 365 nm for 96 h resulted in 39% and 13% conversion with trans:cis product ratio of 1.42 and 1.61, respectively (Scheme 1).
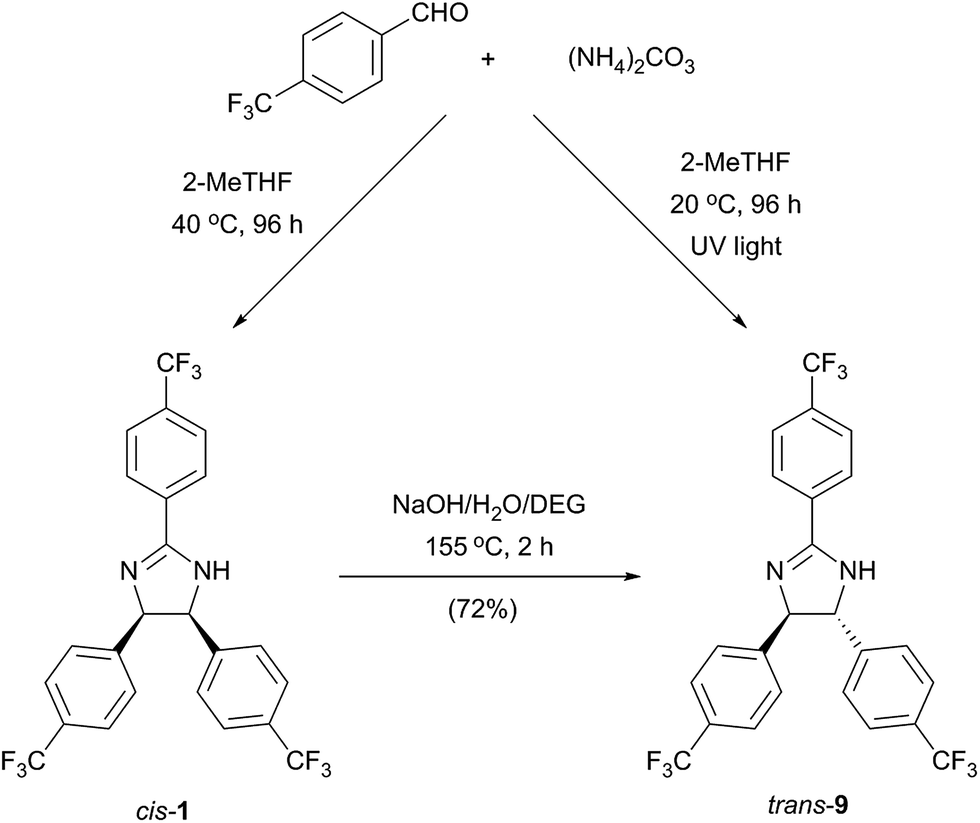 | ||
| Scheme 1 Synthetic methods yielding cis or trans stereoisomers of a 2-imidazoline. | ||
Furthermore, the thermodynamically favoured trans 9 can be selectively obtained by alkali treatment of the cis isomer, similarly to a previously described procedure.13 The trans stereochemistry of 9 (Fig. 5b) was indicated by the lower coupling constant of imidazoline hydrogens (J = 8.8 Hz) compared to the case of 1 (J = 10.9 Hz).
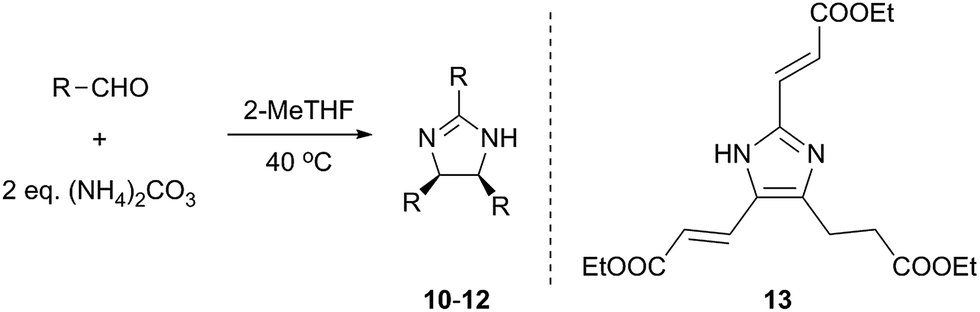
Besides p-substituted aldehydes, the synthesis was also performed from o-, and m-substituted aldehydes, in order to investigate the effect of the substituent position on the reaction (Table 6). Moreover, attempts were made to expand the substrate scope to heterocyclic and non-aromatic aldehydes as well. Comparing the position of substituents (entries 1–2), the m-substitution provided the desired product in good yield, but the reaction was slower compared to the p-substituted analogue. The o-substitution did not afford the imidazoline product as a consequence of steric effects, i.e., the close proximity of the bulky –CF3 substituent to the formyl group sterically hinders the reaction. When it was attempted to produce the product from 4-pyridinecarboxaldehyde and (NH4)2CO3 with the optimised conditions, a product mixture of imidazoline and imidazole (1:1) was obtained. However, when NH4OAc was used as ammonia source, the desired 2-imidazoline product was isolated in a good yield of 72%. This result suggests that, similarly to 3, product 11 is also sensitive to oxidation under basic conditions. The synthesis of 12 was performed using trans-cinnamaldehyde and (NH4)2CO3 as shown in entry 4 in Table 6. The reaction provided the product in good yield within 24 h, which indicates that highly conjugated aldehydes also afford the desired 2-imidazoline product. Therefore, the reaction was also attempted using ethyl 4-oxobut-2-enoate as a non-aromatic starting material. Similarly to trans-cinnamaldehyde, the consumption of starting material was virtually 100% within 24 h (entry 5). The reaction, however, did not stop at the 2-imidazoline product, but through an intramolecular double bond migration a trisubstituted imidazole product was formed instead. Consequently, it could be concluded that a conjugated system which is sterically unhindered is required for the presented synthetic methodology to yield 2-imidazolines.
|
||||
|---|---|---|---|---|
| Entry | R–CHO | Time (h) | Yieldb (%) | Product |
| a The reactions were performed with aldehyde (3 mmol), (NH4)2CO3 (6 mmol), and 2-MeTHF (6 mL) at 40 °C.b Isolated yield.c The ammonia source was NH4OAc (6 mmol).d Formation of the desired 2-imidazoline was not observed, but 13 was obtained instead. | ||||
| 1 | 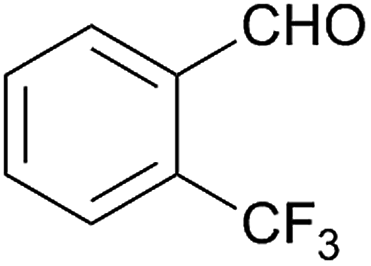 |
128 | — | — |
| 2 | 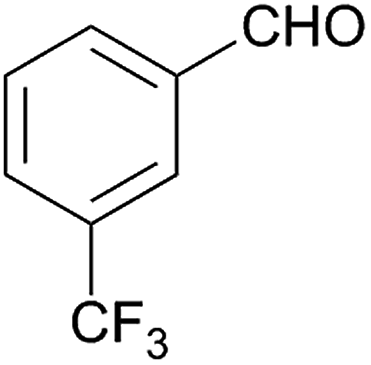 |
128 | 61 | 10 |
| 3 | 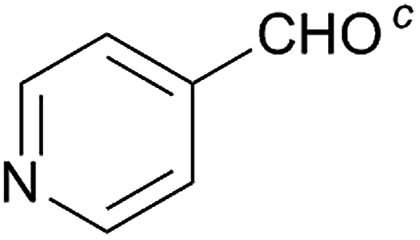 |
36 | 72 | 11 |
| 4 | 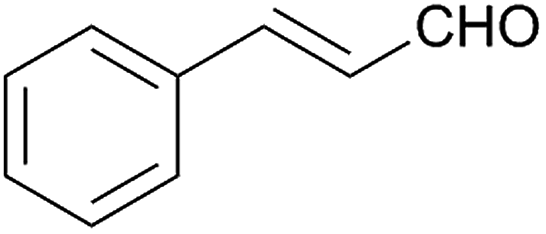 |
24 | 84 | 12 |
| 5 | 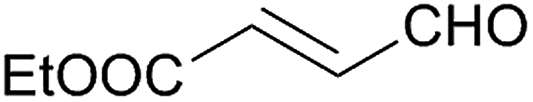 |
24 | 71d | 13 |
Most green chemistry articles solely focus on improving the synthetic step without considering the purification and isolation of the product. In order to address the inherent environmental burden of downstream processing, the isolation of the 2-imidazoline products was aimed to be performed in a single crystallisation step employing a green solvent, which eliminated the use of tedious chromatography employed in previous procedures (see Table 1). Furthermore, the considerable increase in molecular size during the reaction allowed the use of solvent-resistant nanofiltration for product purification. Four membranes, namely GMT-oNF-1, GMT-oNF-2, NF030306, and Duramem 300, were screened at 10–40 bar pressure to obtain solvent flux and solute rejection for product 1, and the corresponding benzaldehyde substrate impurity (Fig. 6a). Flux is defined as volume of solvent that permeates the membrane per unit area in a given time, while rejection is the ratio of solute concentration in the permeate and the retentate. Permeance is defined as the flux normalised by the applied pressure. The permeances (expressed in L m−2 h−1 bar−1) obtained for GMT-ONF-1, GMT-ONF-2, NF030306, and Duramem 300 were 4.39 ± 0.6, 5.2 ± 0.6, 0.35 ± 0.04, and 0.26 ± 0.01, respectively. Despite the relatively low permeance obtained for Duramem 300, it showed excellent product rejection above 99%.
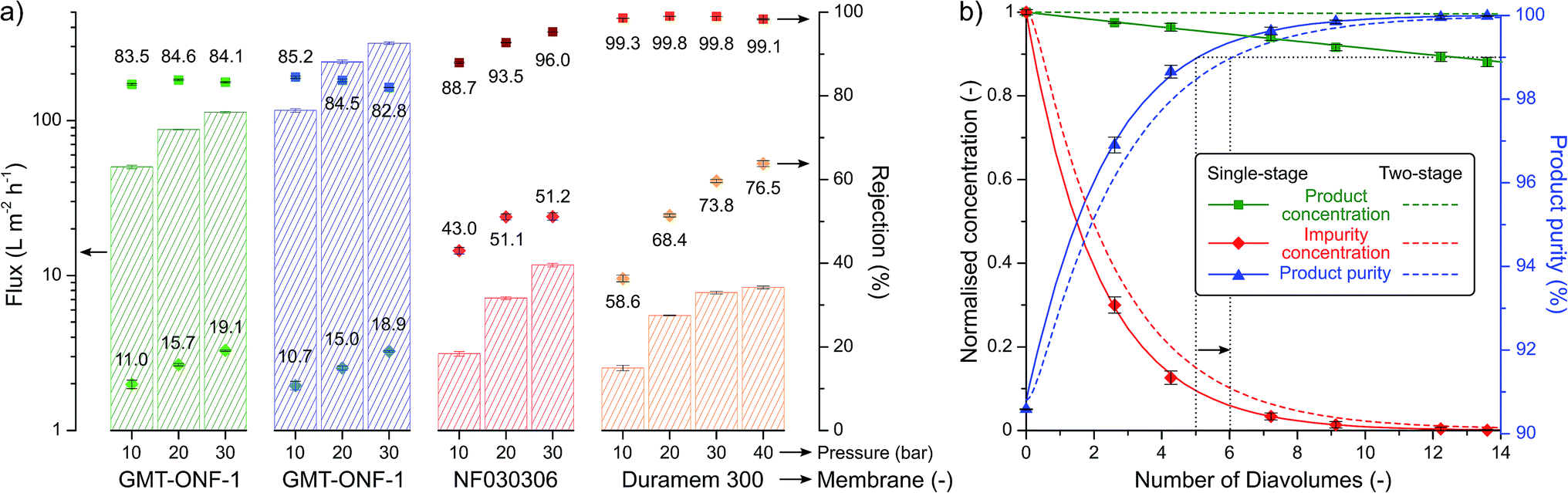 | ||
| Fig. 6 Separation performance of the solvent-resistant nanofiltration membranes for 2-imidazoline product purification: ■ and ♦ indicates rejections for 1, and the corresponding benzaldehyde substrate impurity, respectively (a). Purification performance for single-stage and two-stage diafiltration processes. Solid and dashed lines represent the simulated performance based on rejections, while symbols signify experimental points to validate the model used for simulation (b). | ||
The concentration and purity profiles for single-stage and two-stage diafiltration are shown in Fig. 6b. The detailed description of the mathematical framework can be found in the ESI.† The number of diavolumes is a time-like parameter indicating the progress of the filtration and it is defined as the ratio of permeate and retentate volume. The number of diavolumes required to obtain 99% product purity with a single-stage and two-stage diafiltration is 5 and 6, respectively (see dotted lines). In line with expectations,36 the two-stage cascade configuration markedly improved the product yield from 95% to 99.8%. The purification of a post-reaction mixture validated the calculated model. During the process, Duramem 300 at 20 bar demonstrated stable permeance and rejection performance over 8 days of continuous operation in 2-MeTHF (Fig. S118, ESI†). The solvent consumption of the filtration could be minimised by implementing an additional filtration stage for in situ solvent recovery.28,37
Conclusions
The diastereoselective synthesis of 2,4,5-trisubstituted-2-imidazolines was successfully realised from aldehydes and ammonia sources in a one-pot procedure at mild temperature employing green solvents with moderate to high polarity. The effect of the chemical nature of the aldehyde, in particular the position and polarity of aromatic substituents, was systematically investigated. It was found that strongly EWGs with high Hammett sigma constant (e.g. –NO2, –CF3) provide the 2-imidazoline product in moderate to good yield. In contrast, weakly EWGs (–Br) and electron-donating groups (–OH) poorly or did not afford the desired product, respectively. Besides the aromatic aldehydes, the synthetic methodology is applicable to sterically unhindered, conjugated analogues. The DFT results refuted two speculations and confirmed that (i) the ionic intermediate gets protonated prior to cyclisation, (ii) the most thermodynamically stable carbanion conformer is U-shaped. DFT modelling, NMR and single-crystal XRD confirmed that thermal initiation solely yields the cis isomer. It was demonstrated that the diastereoselectivity of the reaction can be changed by photoinitiation, resulting in excess trans isomer, which is a consequence of the electrocyclic nature of the reaction. The feasibility of membrane-based product purification was demonstrated, which provides a green alternative for the conventional chromatographic purification of 2,4,5-trisubstituted-2-imidazolines.Experimental section
Materials
Ammonium acetate (>98%), ammonium bicarbonate (>99.5%), ammonium trifluoroacetate (98%), benzaldehyde (>99%), 4-bromobenzaldehyde (99%), 4-formylbenzoic acid (97%), 4-hydroxybenzaldehyde (98%), 4-nitrobenzaldehyde (98%), 4-pyridinecarboxaldehyde (97%), 3-(trifluoromethyl)benzaldehyde (97%), 1-butanol (99%), butyl acetate (>99%), 1-butyl-3-methylimidazolium acetate ([bmim][OAc]; >95%), cyclopentyl methyl ether (CPME; 99.9%), dibutyl phthalate (99%), dimethyl carbonate (99%), 2-methyltetrahydrofuran (2-MeTHF; contains 250 ppm BHT stabiliser, >99%), sulfolane (99%) and 2-phenylbenzimidazole (97%) were purchased from Sigma-Aldrich. 4-Cyanobenzaldehyde (99%), 4-(dimethylamino)benzaldehyde (95%), 4-(methylsulfonyl)benzaldehyde (97%), 4-(pentafluorosulfanyl)benzaldehyde (99%), 2-(trifluoromethyl)benzaldehyde (98%), 4-(trifluoromethyl)benzaldehyde (98%) and ethyl 4-oxobut-2-enoate (95%) were purchased from Fluorochem. Ammonium carbonate (99%), thionyl chloride (99.5%) and trans-cinnamaldehyde (99%) were purchased from Acros Organics. Chloroform (HPLC gradient), dimethyl sulfoxide (DMSO; analytical gradient), methanol (MeOH; HPLC gradient) and trifluoroacetic acid (TFA; >99%) were purchased from Fisher Scientific. Cyrene™ (dihydrolevoglucosenone; >99.5%) was purchased from Apollo Scientific. Ammonium formate (97%) was purchased from Alfa Aesar. Methyl tert-butyl ether (MTBE; 99%) was purchased from Merck. Acetonitrile (HPLC gradient) was purchased from VWR. All reagents and solvents were used as received. The syntheses of methyl 4-formylbenzoate and 4-formyl-N,N,N-trimethylanilinium iodide substrates are described in the ESI.†NF010206 membrane was purchased from Solsep BV (Apeldoorn, Netherlands); GMT-oNF-1 and GMT-oNF-2 were purchased from Borsig Membrane Technology GmbH (Gladbeck, Germany); and Duramem 300 was obtained from Evonik-MET (Essen, Germany).
Analytical methods
All reactions were monitored with thin layer chromatography (TLC) using silica gel 60 F254 (Merck) plates; and with high performance liquid chromatography (HPLC). The optimum reaction time, i.e., maximum achievable conversion was determined by HPLC. HPLC analyses were measured on a VWR Hitachi Chromaster instrument with 5160 pump, 5260 autosampler, 5310 column oven, and 5430 diode array detector (DAD). See ESI† for the HPLC methods. Melting points (values without correction) were measured on a Stuart melting point apparatus SMP3 using 5 °C min−1 slope.The products were characterised by IR, 1H NMR, 13C NMR, 19F NMR, 1H COSY, 1H–13C HMBC and 1H–13C HSQC, mass spectrometry (MS) and, where possible, single-crystal XRD measurements. IR spectra were recorded from dry samples using a Thermo Fisher Nicolet iS5 iD5 ATR-FTIR spectrometer. NMR analyses were performed on a B500 Bruker Avance II+ 500 MHz instrument. Spectra were processed using the MestReNova software. Low resolution mass spectra were recorded on a Waters SQD2 Single l Quadrupole Mass spectrometer used with a Waters Acquity UHPLC using 100% methanol as mobile phase. Electrospray ionisation technique was used in negative or positive mode. High resolution mass measurements were performed on a Thermo Exactive plus EMR Orbitrap mass spectrometer, used with a Thermo Ultimate 3000 UHPLC using 100% methanol as mobile phase. Single-crystals were grown by slow evaporation at low-temperature (2 °C) from acetonitrile for X-ray analysis. CCDC 1576088 (1), CCDC 1576089 (2), CCDC 1576090 (5) and CCDC 1582071 (9) contain the supplementary crystallographic data for this paper.†
General synthetic procedure
In a typical procedure, a benzaldehyde substrate (3 mmol) was dissolved in the solvent (6 mL), followed by the addition of the ammonia source (6 mmol). The reaction mixture was stirred vigorously at the desired temperature. The reaction was followed by TLC (MeOH–CHCl3 1:10 v/v) and HPLC. The reaction was terminated when maximum conversion was reached by filtering the ammonia source. The solvent was evaporated and the crude product was recrystallised.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 1473 (aromatic CC), 1323 (C–F), 1159 (C–F), 1066 (C–F) cm−1; MS (ESI–/SQD2) m/z: 501.2 [M − H]−; MS (ESI+/SQD2) m/z: 503.3 [M + H]+; HRMS (ESI+/Orbitrap) m/z: calcd for [M + H]+ C24H16F9N2: 503.1164; found: 503.1154; D = −2.05 ppm; mp: 261–262 °C. Yield: 67% (335 mg) following recrystallisation from MTBE (2 mL). Product 1 was obtained as white crystals.
N), 1473 (aromatic CC), 1323 (C–F), 1159 (C–F), 1066 (C–F) cm−1; MS (ESI–/SQD2) m/z: 501.2 [M − H]−; MS (ESI+/SQD2) m/z: 503.3 [M + H]+; HRMS (ESI+/Orbitrap) m/z: calcd for [M + H]+ C24H16F9N2: 503.1164; found: 503.1154; D = −2.05 ppm; mp: 261–262 °C. Yield: 67% (335 mg) following recrystallisation from MTBE (2 mL). Product 1 was obtained as white crystals.1H NMR (500 MHz, DMSO-d6) δ 8.26 (d, J = 8.8 Hz, 2H), 8.19 (s, 1H) 8.18 (d, J = 9.0 Hz, 2H), 7.65 (d, J = 9.2 Hz, 2H), 7.62 (d, J = 9.3 Hz, 2H), 7.29–6.92 (m, 4H), 5.78 (d, J = 10.9 Hz, 1H), 5.54 (d, J = 10.9 Hz, 1H), 3.69 (s, 9H), 3.47 (s, 18H); 13C NMR (126 MHz, DMSO-d6) δ 162.5, 148.8, 145.8, 145.5, 141.7, 141.7, 131.1, 129.1, 128.9, 128.3, 121.0, 119.4, 119.2, 73.2, 64.0, 56.5, 56.5; IR (ATR-FTIR) νmax 3011 (CH), 1617 (CN), 1468 (C(N)–N), 955, 938 (C–N), 845 (C–N+Me3) cm−1; HRMS (ESI+/Orbitrap) m/z: calcd for [M]3+ C30H42N5: 157.4473; found: 157.4475; D = −1.01 ppm; mp: 168.7–169.4 °C decomp. Yield: 83% (590 mg) following recrystallisation from acetonitrile (5 mL). Product 2 was obtained as white crystals.
N), 1523 (NO2), 1448 (aromatic CC), 1344 (NO2), 860 (aromatic CH) cm−1; MS (ESI–/SQD2) m/z: 432.2 [M − H]−; MS (ESI+/SQD2) m/z: 434.2 [M + H]+; HRMS (ESI–/Orbitrap) m/z: calcd for [M − H]− C21H14N5O6: 432.0950; found: 432.0937; D = −2.91 ppm; mp: 195–200 °C decomp. Yield: 85% (367 mg) following recrystallisation from 2-MeTHF (2 mL). Product 3 was obtained as pale yellow crystals.N), 1292 (OSO) cm−1; MS (ESI–/SQD2) m/z: 531.3 [M − H]−; MS (ESI+/SQD2) m/z: 555.3 [M + Na]+; HRMS (HESI+/Orbitrap) m/z: calcd for [M + Na]+ C24H24N2O6S3Na: 555.0689; found: 555.0676; D = −2.29 ppm; mp: 275–277 °C. Yield: 69% (369 mg) following crystallisation from 2-MeTHF. Product 4 was obtained as white crystals.N), 1471 (aromatic CC), 816 (S–F) cm−1; MS (ESI–/SQD2) m/z: 675.1 [M − H]−, 711.1 [M + Cl]−; MS (ESI+/SQD2) m/z: 677.1 [M + H]+; HRMS (ESI+/Orbitrap) m/z: calcd for [M + H]+ C21H16F15N2S3: 677.0231; found: 677.0223, D = −1.12 ppm; mp: 264–265 °C. Yield: 87% (587 mg) following recrystallisation from heptane (2 mL). Product 5 was obtained as white crystals.![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N), 1604 (CN), 1445 (aromatic CC), 851 (aromatic CH) cm−1; MS (ESI–/SQD2) m/z: 372.3 [M − H]−; MS (ESI+/SQD2) m/z: 374.3 [M + H]+, 396.2 [M + Na]+; HRMS (ESI+/Orbitrap) m/z: calcd for [M + H]+ C24H16N5: 374.1400; found: 374.1395; D = −1.40 ppm; mp: 251–252 °C (Literature data:16 222–224 °C) yield: 68% (253 mg) following recrystallisation from butyl acetate (3 mL). Product 6 was obtained as white crystals.O), 1708 (CO), 1608 (CN), 1435 (aromatic CC), 1275 (C–C(O)–O), 1107 (C–O–C) cm−1; MS (ESI–/SQD2) m/z: 471.4 [M − H]−; MS (ESI+/SQD2) m/z: 473.3 [M + H]+; HRMS (ESI+/Orbitrap) m/z: calcd for [M + H]+ C27H25N2O6: 473.1707; found: 473.1697; D = −2.14 ppm; mp: 186–188 °C decomp. Yield: 65% (310 mg) following recrystallisation from MTBE (2 mL). Product 7 was obtained as white crystals.
N), 1604 (CN), 1445 (aromatic CC), 851 (aromatic CH) cm−1; MS (ESI–/SQD2) m/z: 372.3 [M − H]−; MS (ESI+/SQD2) m/z: 374.3 [M + H]+, 396.2 [M + Na]+; HRMS (ESI+/Orbitrap) m/z: calcd for [M + H]+ C24H16N5: 374.1400; found: 374.1395; D = −1.40 ppm; mp: 251–252 °C (Literature data:16 222–224 °C) yield: 68% (253 mg) following recrystallisation from butyl acetate (3 mL). Product 6 was obtained as white crystals.O), 1708 (CO), 1608 (CN), 1435 (aromatic CC), 1275 (C–C(O)–O), 1107 (C–O–C) cm−1; MS (ESI–/SQD2) m/z: 471.4 [M − H]−; MS (ESI+/SQD2) m/z: 473.3 [M + H]+; HRMS (ESI+/Orbitrap) m/z: calcd for [M + H]+ C27H25N2O6: 473.1707; found: 473.1697; D = −2.14 ppm; mp: 186–188 °C decomp. Yield: 65% (310 mg) following recrystallisation from MTBE (2 mL). Product 7 was obtained as white crystals.1H NMR (500 MHz, DMSO-d6) δ 11.63 (s, 2H), 8.22 (d, J = 8.7 Hz, 2H), 8.00 (d, J = 8.7 Hz, 2H), 7.37 (d, J = 8.5 Hz, 4H), 7.09 (d, J = 8.5 Hz, 4H), 5.98 (s, 2H); 13C NMR (126 MHz, DMSO-d6) δ 165.1, 134.7, 132.9, 131.6, 131.4, 130.4, 129.6, 121.8, 121.7, 64.1; IR (ATR-FTIR) νmax 3315 (NH), 2955 (CH), 1717 (CO), 1708 (CO), 1608 (CN), 1435 (aromatic CC), cm−1; MS (ESI–/SQD2) m/z: 531.1 [M − H]−; MS (ESI+/SQD2) m/z: 473.3 [M + H]+; HRMS (ESI-/Orbitrap) m/z: calcd for [M + Cl]− C21H15N2Br3Cl: 566.8471; found: 566.8479; D = −1.48 ppm; mp: 325–326 °C. Yield: 8% (45 mg). Product 8 was obtained as white crystals.
1H NMR (500 MHz, THF-d8) δ 8.22 (d, J = 8.1 Hz, 2H), 7.81 (d, J = 8.3 Hz, 2H), 7.70 (d, J = 8.2 Hz, 2H), 7.65 (d, J = 8.2 Hz, 2H), 7.58 (d, J = 8.1 Hz, 2H), 7.48 (brs, 1H), 7.47 (d, J = 8.3 Hz, 2H), 5.14 (d, J = 8.8 Hz, 1H), 4.81 (dd, J = 8.8 and 2.3 Hz, 1H); 13C NMR (126 MHz, THF-d8) δ 163.2, 149.4 (q, J = 1.3 Hz), 149.2 (q, J = 1.3 Hz), 135.1 (q, J = 1.3 Hz), 133.0 (q, J = 32 Hz), 130.6 (q, J = 32 Hz), 130.1 (q, J = 32 Hz), 129.1, 128.4, 128.1, 126.6 (q, J = 3.9 Hz), 126.2 (q, J = 3.9 Hz), 126.2 (q, J = 3.9 Hz), 125.6 (q, J = 272 Hz), 125.4 (q, J = 272 Hz), 125.2 (q, J = 272 Hz), 81.0, 71.0; 19F NMR (471 MHz, THF-d8) δ −63.1 (s, 3F), −63.2 (s, 3F), −63.6 (s, 3F); IR (ATR-FTIR) νmax 3082 (NH), 1601 (CN), 1485 (aromatic CC), 1324 (C–F), 1162 (C–F), 1067 (C–F) cm−1; MS (ESI–/SQD2) m/z: 501.2 [M−H]−; MS (ESI+/SQD2) m/z: 503.2 [M + H]+; HRMS (ASAP+/Orbitrap) m/z: calcd for [M + H]+ C24H16F9N2: 503.1164; found: 503.1148; D = −3.24 ppm; mp: 283–284 °C. Yield: 72% (108 mg) following recrystallisation from toluene (5 mL). Product 9 was obtained as white crystals.
1H NMR (500 MHz, THF-d8) δ 8.39 (s, 1H), 8.38 (d, J = 7.7 Hz, 1H), 7.86 (d, J = 7.8 Hz, 1H), 7.72 (t, J = 8.0 Hz, 1H), 7.54 (brs, 1H), 7.32–7.22 (m, 6H), 7.22–7.14 (m, 2H), 5.80 (br, 1H), 5.52 (br, 1H); 13C NMR (126 MHz, THF-d8) δ 164.2, 142.2, 132.4, 132.3 (br), 132.2 (q, J = 1.0 Hz), 131.6 (br), 131.5 (q, J = 32 Hz), 130.3, 129.3 (br), 129.0 (br), 128.2 (q, J = 3.8 Hz), 125.6 (br), 125.3 (q, J = 272 Hz), 125.1 (q, J = 4.0 Hz), 124.9 (br), 124.5 (br), 123.9 (br), 75.7, 66.6; 19F NMR (471 MHz, THF-d8) δ −63.4 (s, 3F), −63.6 (s, 3F), −63.7 (s, 3F); IR (ATR-FTIR) νmax 3137 (NH), 1615 (CN), 1114 (aromatic CC), 1326 (C–F), 1160 (C–F), cm−1; HRMS (ESI+/Orbitrap) m/z: calcd for [M + H]+ C24H16F9N2: 503.1164; found: 503.1161; D = −0.65 ppm. Yield: 61% (306 mg). Product 10 was obtained as pale yellow resin.
1H NMR (500 MHz, CD3OD) δ 8.78 (d, J = 6.1 Hz, 2H), 8.24 (d, J = 6.1 Hz, 4H), 7.99 (d, J = 6.2 Hz, 2H), 7.14 (d, J = 6.0 Hz, 4H), 5.80 (br, 1H), 5.64 (br, 1H); 13C NMR (126 MHz, CD3OD) δ 166.2, 151.1, 150.1, 149.7, 138.7, 124.8, 124.2, 123.3, 74.5, 65.7; IR (ATR-FTIR) νmax 3156 (NH), 3033 (CH), 1596 (aromatic CN), 1473 (aromatic CC) cm−1; HRMS (ESI–/Orbitrap) m/z: 300.1248 [M − H]−; HRMS (ESI+/Orbitrap) m/z: calcd for [M + Na]+ C18H15N5Na: 324.1220; found: 324.1210; D = −2.29 ppm; mp: 94.0–95.2 °C decomp. Yield: 72% (218 mg). Product 11 was obtained as white crystals.
C), 1577 (CN), 966 (aromatic C–H) cm−1; MS (ESI-/SQD2) m/z: 375.3 [M − H]−; MS (ESI+/SQD2) m/z: 377.7 [M + H]+; HRMS (ESI+/Orbitrap) m/z: calcd for [M + H]− C27H25N2: 377.2012; found: 377.2009; D = −0.86 ppm; mp: 108–109 °C. Yield: 84% (317 mg) following recrystallisation from MTBE (3 mL). Product 12 was obtained as pale yellow crystals.1H NMR (500 MHz, DMSO-d6) δ 7.54 (d, J = 15.9 Hz, 1H), 7.40 (d, J = 16.2 Hz, 1H), 7.16 (d, J = 16.2 Hz, 1H), 6.75 (d, J = 15.9 Hz, 1H), 4.23 (q, J = 7.1 Hz, 2H), 4.20 (q, J = 7.1 Hz, 2H), 4.03 (q, J = 7.1 Hz, 2H), 3.03 (t, J = 7.2 Hz, 2H), 2.75 (t, J = 7.2 Hz, 2H), 1.27 (t, J = 7.1 Hz, 3H), 1.26 (t, J = 7.1 Hz, 3H), 1.15 (t, J = 7.1 Hz, 3H); 13C NMR (126 MHz, DMSO-d6) δ 171.4, 165.8, 164.9, 141.8, 138.5, 129.4, 127.9, 126.6, 125.1, 118.3, 60.9, 60.3, 60.2, 32.7, 19.6, 14.2, 14.1, 14.0. IR (ATR-FTIR) νmax 1718 (CO), 1707 (CO), 1655 (imidazolium CC), 1185 (C–C(O)–O) cm−1; MS (ESI-/SQD2) m/z: 363.3 [M − H]−; MS (ESI+/SQD2) m/z: 387.4 [M + Na]+; HRMS (ESI+/Orbitrap) m/z: calcd for [M + Na]− C18H24N2O6Na: 387.1527; found: 387.1515; D = −2.99 ppm; mp: 96.3–97.1 °C decomp. Yield: 71% (285 mg). Product 13 was obtained as white crystals.
Reaction kinetics
The p-substituted aldehyde (3 mmol) was dissolved in 2-MeTHF (6 mL), followed by the addition of an internal standard (2-phenylbenzimidazole, 78 mg, 0.4 mmol for 4-nitrobenzaldehyde, methyl 4-formylbenzoate; and dibutyl phthalate, 278 mg, 1 mmol for 4-(trifluoromethyl) benzaldehyde and 4-(pentafluorosulfanyl)benzaldehyde). A sample of 10 μL was collected (t = 0), followed by the addition of (NH4)2CO3 (577 mg, 6 mmol) to initiate the reaction. Further samples were periodically collected, and diluted with a mixture of acetic acid, water and DMF (1:1:8 v/v). Samples were analysed by HPLC. The mathematical framework can be found in the ESI.†
Membrane separation
Membranes with an area of 52.8 cm2 were screened using a mixture of 4-(trifluoromethyl)benzaldehyde and product 1, at 0.5 g L−1 concentration each. The membrane screening was performed in a typical nanofiltration rig operated in cross-flow configuration (Fig. 7). A recirculation pump set at 1 L min−1 ensured homogeneous solute concentration in the retentate loop. The membranes were conditioned at each pressure for 24 hours prior to measurements to ensure steady-state operation. The solvent flux was obtained by measuring the volume of permeate passing through the membrane (Vp) in a definite time (t) and membrane area (A) as shown in eqn (4), while the solute rejection of solute was calculated from the ratio of solute concentration in the permeate (CP) and the solute concentration in the retentate (CR) as defined in eqn (5). The filtration of post-reaction mixture was performed at 20 bar using Duramem 300 membrane with an area of 52.8 cm2. Fresh solvent was continuously added to compensate the permeate volume, keeping the system volume constant at 60 mL. 100 μL samples of permeate and retentate were periodically taken for analysis.
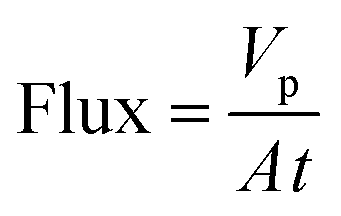 | (4) |
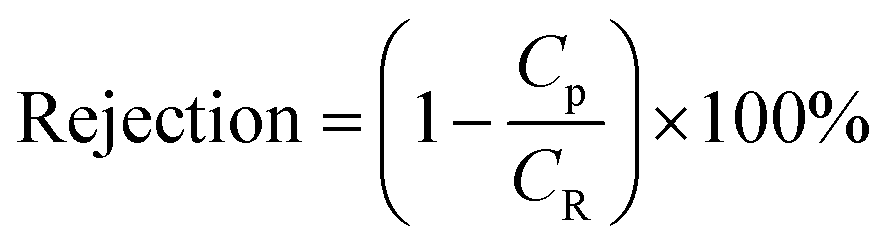 | (5) |
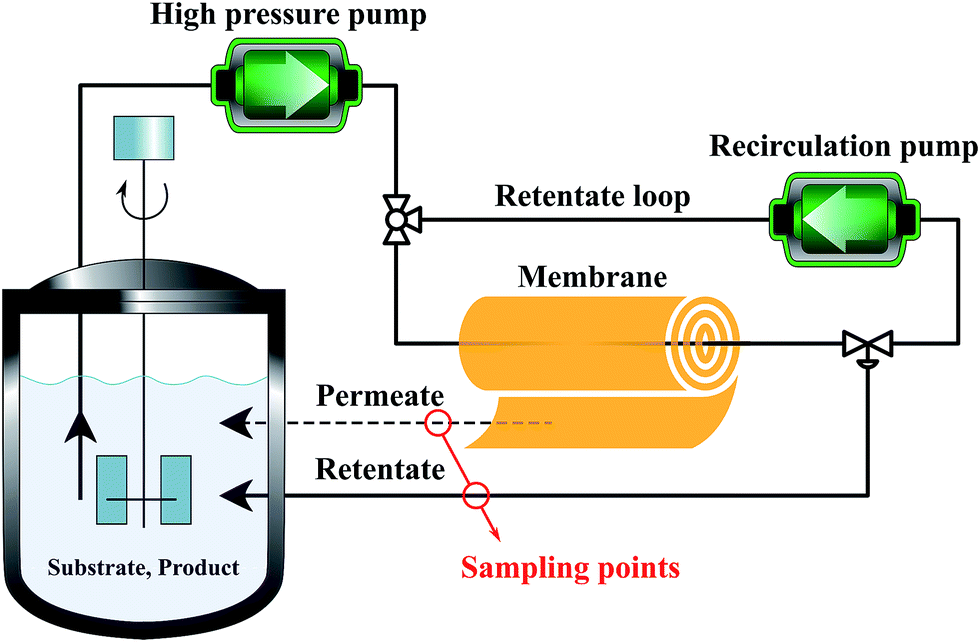 | ||
| Fig. 7 Schematic process configuration for membrane screening, i.e. determination of solute rejection and flux at 10–40 bar pressure using different membranes. | ||
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to express their gratitude to Mr Gareth David Smith and the MS laboratory (The University of Manchester) for MS measurements, to Dr Jozsef Kupai (Budapest University of Technology and Economics) for melting point measurements, to Dr Zoltan Mucsi (Femtonics Ltd., Budapest) for his guidance with DFT calculations, and Dr Peter Pogany (GlaxoSmithKline Ltd) and Prof. Krishna Persaud (The University of Manchester) for their valuable comments during the preparation of the manuscript. This work was supported by the Engineering and Physical Sciences Research Council [BioProNET BIV Nov15 Szekely]; the Biotechnology and Biological Sciences Research Council [BioProNET BIV Nov15 Szekely, BB/L013770/1].Notes and references
- X. Guinchard, Y. Vallee and J. N. Denis, J. Org. Chem., 2007, 72, 3972–3975 CrossRef CAS PubMed.
- M. Krasavin, Eur. J. Med. Chem., 2015, 97, 525–537 CrossRef CAS PubMed.
- R. Tyagi, V. K. Tyagi and S. K. Pandey, J. Oleo Sci., 2007, 56, 211–222 CrossRef CAS PubMed.
- K. Zhuo, Q. Du, G. Bai, C. Wang, Y. Chen and J. Wang, Carbohydr. Polym., 2015, 115, 49–53 CrossRef CAS PubMed.
- J. Li, D. Jia, Z. Guo, Y. Liu, Y. Lyu, Y. Zhoua and J. Wanga, Green Chem., 2017, 19, 2675–2686 RSC.
- H. Liu and D. M. Du, Adv. Synth. Catal., 2009, 351, 489–519 CrossRef CAS.
- H. Liu and D. M. Du, Adv. Synth. Catal., 2010, 352, 1113–1118 CrossRef CAS.
- X. Du, H. Liu and D. M. Du, Eur. J. Org. Chem., 2011, 2011, 786–793 CrossRef.
- X. B. Bu, Y. Yu, B. Li, L. Zhang, J. J. Chen and Y. L. Zhao, Adv. Synth. Catal., 2017, 359, 351–356 CrossRef CAS.
- A. Wang, M. Bernasconi and A. Pfaltz, Adv. Synth. Catal., 2017, 359, 2523–2529 CrossRef CAS.
- E. J. Corey and F. N. M. Kuhnle, Tetrahedron Lett., 1997, 38, 8631–8634 CrossRef CAS.
- O. F. Williams and J. C. Bailar, J. Am. Chem. Soc., 1959, 81, 4464–4469 CrossRef CAS.
- D. C. Braddock, J. M. Redmond, S. A. Hermitage and A. J. P. White, Adv. Synth. Catal., 2006, 348, 911–916 CrossRef CAS.
- J. Tsuji, K. Sakai, H. Nemoto and H. Nagashima, J. Mol. Catal., 1983, 18, 169–176 CrossRef CAS.
- R. Oi and K. B. Sharpless, Tetrahedron Lett., 1991, 32, 999–1002 CrossRef CAS.
- D. H. Hunter and S. K. Sim, Can. J. Chem., 1972, 50, 669–677 CrossRef CAS.
- M. L. Larter, M. Phillips, F. Ortega, G. Aguirre, R. Somanathan and P. J. Walsh, Tetrahedron Lett., 1998, 39, 4785–4788 CrossRef CAS.
- B. Lecea, A. Arrieta and F. P. Cossio, J. Org. Chem., 2005, 70, 1035–1041 CrossRef CAS PubMed.
- H. Uchida, T. Shimizu, P. Y. Reddy, S. Nakamura and T. Toru, Synthesis, 2003, 8, 1236–1240 Search PubMed.
- F. Wang, Q. Liao and C. Xi, Synth. Commun., 2012, 42, 905–913 CrossRef CAS.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- R. K. Henderson, C. Jimenez-Gonzalez, D. J. C. Constable, S. R. Alston, G. G. A. Inglis, G. Fisher, J. Sherwood, S. T. Binks and A. D. Curzons, Green Chem., 2011, 13, 854–862 RSC.
- C. Jimenez-Gonzalez, A. D. Curzons, D. J. C. Constable and V. L. Cunningham, Clean Technol. Environ. Policy, 2005, 7, 42–50 CrossRef CAS.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC.
- C. M. Alder, J. D. Hayler, R. K. Henderson, A. M. Redman, L. Shukla, L. E. Shuster and H. F. Sneddon, Green Chem., 2016, 18, 3879–3890 RSC.
- G. Szekely, M. F. Jimenez-Solomon, P. Marchetti, J. F. Kim and A. G. Livingston, Green Chem., 2014, 16, 4440–4473 RSC.
- G. Szekely, M. Gil, B. Sellergen, W. Heggie and F. C. Ferreira, Green Chem., 2013, 15, 210–225 RSC.
- T. Fodi, C. Didaskalou, J. Kupai, G. T. Balogh, P. Huszthy and G. Szekely, ChemSusChem, 2017, 10, 3435–3444 CrossRef CAS PubMed.
- J. Sherwood, M. De Bruyn, A. Constantinou, L. Moity, C. R. McElroy, T. J. Farmer, T. Duncan, W. Raverty, A. J. Hunt and J. H. Clark, Chem. Commun., 2014, 50, 9650–9652 RSC.
- K. L. Wilson, A. R. Kennedy, J. Murray, B. Greatex, C. Jamieson and A. J. B. Watson, Beilstein J. Org. Chem., 2016, 12, 2005–2011 CrossRef CAS PubMed.
- M. B. Smith and J. March, in March's Advanced Organic Chemistry: Reactions, Mechanism and Structure, John Wiley & Sons Ltd, Hoboken, New Jersey, 6th edn, 2007, ch. 8, pp. 356–394 Search PubMed.
- J. H. Xu, Q. Jiang and C. C. Guo, J. Org. Chem., 2013, 78, 11881–11886 CrossRef CAS PubMed.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. von Rague Schleyer, Chem. Rev., 2005, 105, 3842–3888 CrossRef CAS PubMed.
- R. B. Woodward and R. J. Hoffmann, J. Am. Chem. Soc., 1965, 87, 395–397 CrossRef CAS.
- J. F. Kim, G. Szekely, I. B. Valtcheva and A. G. Livingston, Green Chem., 2014, 16, 133–145 RSC.
- M. Schaepertoens, C. Didaskalou, J. F. Kim, A. G. Livingston and G. Szekely, J. Membr. Sci., 2016, 514, 646–658 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures; 1H NMR, 13C NMR, MS, HRMS, FTIR spectra; HPLC methods, chromatograms, kinetic calculations; DFT computational methods & raw data; single crystal XRD data, membrane separation. CCDC 1576088–1576090, 1582071. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra11827a |
| ‡ These authors equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2017 |