
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In vivo protein corona patterns of lipid nanoparticles†
A. Amici *a,
G. Caracciolo*bc,
L. Digiacomoa,
V. Gambinia,
C. Marchinia,
M. Tilioa,
A. L. Capriottid,
V. Colapicchionide,
R. Matassaf,
G. Familiarif,
S. Palchettibc,
D. Pozzibc,
M. Mahmoudi*gh and
A. Laganàd
*a,
G. Caracciolo*bc,
L. Digiacomoa,
V. Gambinia,
C. Marchinia,
M. Tilioa,
A. L. Capriottid,
V. Colapicchionide,
R. Matassaf,
G. Familiarif,
S. Palchettibc,
D. Pozzibc,
M. Mahmoudi*gh and
A. Laganàd
aSchool of Biosciences and Veterinary Medicine, University of Camerino, Via Gentile III da Varano, 62032 Camerino (MC), Italy. E-mail: augusto.amici@unicam.it
bDepartment of Molecular Medicine, Sapienza University of Rome, Viale Regina Elena 291, 00161 Rome, Italy. E-mail: giulio.caracciolo@uniroma1.it
cIstituti Fisioterapici Ospitalieri, Istituto Regina Elena, Via Elio Chianesi 53, 00144 Rome, Italy
dDepartment of Chemistry, Sapienza University of Rome, P.le A. Moro 5, 00185 Rome, Italy
eIstituto Italiano di Tecnologia, Center for Life Nano Science@Sapienza, Viale Regina Elena 291, 00161 Rome, Italy
fDipartimento di Scienze Anatomiche, Istologiche, Medico Legali e dell'Apparato Locomotore, Sapienza University of Rome, Via Alfonso Borelli 50, 00161, Rome, Italy
gDepartment of Nanotechnology and Nanotechnology Research Center, Faculty of Pharmacy, Tehran University of Medical Sciences, Tehran 13169-43551, Iran
hCenter for Nanomedicine and Department of Anesthesiology, Brigham and Women's Hospital, Harvard Medical School, Boston, Massachusetts, USA. E-mail: mmahmoudi@bwh.harvard.edu
First published on 4th January 2017
Abstract
In physiological environments (e.g. the blood), nanoparticles (NPs) are surrounded by a layer of biomolecules referred to as a ‘protein corona’ (PC). The most tightly NP-bound proteins form the so-called hard corona (HC), the key bio-entity that determines the NP's biological identity and physiological response. To date, NP-HC has been almost exclusively characterized in vitro, while NP–protein interactions under realistic in vivo conditions remain largely unexplored. In this study, we thoroughly characterized the in vivo HC of a NP formulation that forms around lipid nanoparticles with a lipid composition equal to that of clinically used liposomal amphotericin B (AmBisome®) after the recovery of the NPs from the blood circulation of FVB/N mice 10 minutes post intravenous administration. In vitro HC formed by 10 minutes incubation of NPs in FVB/N mouse plasma was used for comparison. Here we show that the biological identity (i.e. size, zeta-potential and aggregation state) of NPs in vivo is significantly different from that acquired in vitro. Furthermore, the variety of protein species in the in vivo HC was considerably larger. The present work has demonstrated that characterization of the in vivo HC is essential to provide an accurate molecular description of the biological identity of NPs in physiological environments.
Introduction
Upon entering physiological environments (e.g., blood and interstitial fluids), nanoparticles (NPs) are surrounded by a layer of biomolecules referred to as a ‘protein corona’ (PC).1 Quick binders are typically, but not necessarily, the more abundant proteins and are substituted over time by those proteins with higher affinity for the NP surface.2 At equilibrium, the long-standing PC of tightly bound proteins form the ‘hard’ corona (HC), which controls the NP fate in vivo and is the interface “seen” and processed by cells.3 Factors shaping the HC are classified in three categories, specifically NP physicochemical properties (e.g. size, zeta-potential,4 and morphology),5–8 biological media (e.g. protein source,9,10 protein concentration,11 and (local)-temperature)10–13 and exposure time.14–16 To investigate the effect of these factors on the HC, NPs are typically exposed to static plasma or serum. On the other side, the NP–protein interactions under realistic in vivo conditions have been poorly understood.17 Amongst underestimated factors, shear stress and variation of plasma proteins (in both protein content and conformation18) could be extremely relevant. Indeed, bodily fluids are dynamic in nature and exhibit macroscopic velocities that are largely dependent on body zones. In the human body, flow velocities range from a few micrometers per s (in the capillaries) up to 60 cm s−1 (in the ascending aorta). In recent investigations, we compared size, surface charge, and composition of the HCs formed under static vs. dynamic incubation.19,20 Notably, the HC formed in a dynamic flow was found to be largely different from its ‘static’ counterpart thus making a correlation between in vitro data and in vivo data challenging. Recent studies by Hadjidemetriou et al.21,22 have reported on the HC of NPs after in vivo administration. The HC of PEGylated liposomal doxorubicin (Doxil®), one of the leading approved NP product used in cancer therapy, was fully characterized after the recovery of NPs from the blood circulation of CD-1 mice. Notably, in vivo HC of Doxil® was different in morphology with respect to that formed in vitro and, most importantly, exhibited a much larger variety of molecular species. These results have demonstrated that comparing in vitro–in vivo HCs has deep implications for in vitro–in vivo extrapolations and will be of great importance to exploit the NP-HC for therapeutics and diagnostics (Fig. 1). Among other applications, this will allow understanding the drug specificity of existing, clinically approved NP formulations, as well as their therapeutic failures.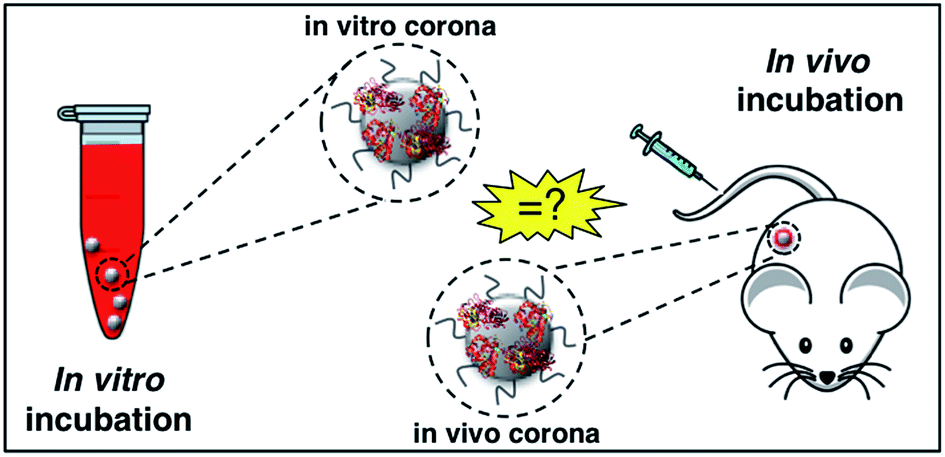 | ||
| Fig. 1 In biological media nanoparticles adsorb biomolecules to form a biomolecular ‘corona’, referred to as “protein corona”, which confers a new biological identity to the particles. The nanoparticle–protein interactions under realistic in vivo conditions have been poorly explored so far. A key question is whether the spontaneous coating of nanoparticles in vivo differs from that formed following in vitro incubation. | ||
Between NP-based drug delivery systems, liposomes are considered as a versatile delivery tool that has been investigated for formulating a vast variety of drugs and imaging agents. Recently, the liposome–HC is an emerging as an opportunity for targeted delivery on nanomedicines23 and early cancer detection.24 In this study, we thoroughly characterized the in vitro and in vivo HCs of a lipid NP made of hydrogenated soy phosphatidylcholine (HSPC), 1,2-distearoyl-sn-glycero-3-[phospho-rac-(1-glycerol)] (DSPG), and cholesterol (Chol). Lipid composition and molar ratios of the lipid species were chosen to match the exact liposome composition of the clinically-used liposomal AmBisome®.25 AmBisome® is the first amphotericin B agent and is indicated for the treatment of severe systemic and/or deep mycoses, empirical treatment of presumed fungal infections, treatment of cryptococcal meningitis in HIV-infected patients and treatment of visceral leishmaniasis. Antifungal drugs are also prophylactically administered to patients who have an increased chance of getting fungal infections such as cancer patients. Our aim was to compare the in vitro and in vivo HC of clinically used liposomal AmBisome®. This is a key issue and is addressed here for the first time.
Results and discussion
In a couple of recent papers,21,22 Hadjidemetriou et al. compared the in vitro and in vivo protein coronas formed around PEGylated liposomal formulation that constitutes the anticancer agent Doxil®. Of note, the variety of protein species in the in vivo corona was much wider than in its in vitro counterpart. In addition, different protein patterns for the most abundant common proteins were found. For instance, the in vivo formed coronas showed a drastic reduction in fibrinogen content. In summary, two main conclusions were given. First, Hadjidemetriou et al. clarified that in vivo administration could not be mimicked by in vitro incubation protocols in plasma. Second, it appeared clear that in vitro incubation with plasma proteins could be an oversimplification as a predictor of the complex in vivo protein corona formation. Anyway, some of us have recently shown that composition of the liposome–HC strongly depends on the functional chemical groups that constitute the lipid surface.26 Here we wondered whether conclusions about in vivo corona could be generalizable to all kind of liposomes. To provide new insight, we chose AmBisome® because it is reference drug for treatment of fungal infections or visceral leishmaniasis and is included in the World Health Organization (WHO) essential medicines list. The chemical composition and the physicochemical characteristics of liposomes that were fabricated for this study are summarized in Fig. S1 in the ESI.† The lipid composition and molar ratios of the individual lipid species matched the liposome composition of the clinically approved liposomal AmBisome®. Size and zeta-potential of liposomes were characterized before i.v. administration (Table S1 in the ESI†). Synthesized liposomes were small in size with a mean hydrodynamic diameter, DH, of about 190 nm. Liposomes had a negative zeta-potential of −61.2 mV. As mentioned above, the exposure time is a critical factor that shapes the composition of HC. Time-evolution of in vitro HC was recently reported for liposomes.15 We demonstrated that the HC is rapidly established (t < 1 min) and evolves with time. Exposure time was therefore considered for the experimental design of this study. To identify the appropriate circulation time in the blood, AmBisome® liposomes were i.v. administered via tail vein injection into FVB/N mice and recovered by cardiac puncture 5 min, 10 min and 20 min post-injection. Plasma was then prepared from recovered blood by centrifugation and HC was isolated as explained in the Experimental section. SDS-PAGE was used to compare the total amount of protein adsorbed at different exposure times. Temporal changes in the protein pattern let us conclude that in vivo HC of AmBisome® liposomes changes with time. As Fig. S2 in the ESI† clearly shows, the amount of proteins associated with liposomal AmBisome® was maximum at t = 10 min. Thus this exposure time was used in the following. Following 10 minutes in vitro incubation the hydrodynamic diameter of AmBisome® liposomes increased (DH ≈ 330 nm), their size distribution (Fig. 2, panel A) broadened as demonstrated by larger polydispersity index (Table S1 in the ESI†), while their surface charge was less negative than that of bare liposomes (Fig. 2, panel B; Table S1†). DLS results are consistent with particle aggregation and are in good agreement with TEM images reported in Fig. 2, panel C. The observed decrease in zeta-potential is a well know effect due protein adsorption that produces a normalization of particles' surface charge.4 On the other side, Fig. 2, panel D, shows that in vivo incubation induced a smaller increase in the mean diameter of liposomes (DH ≈ 260 nm). Zeta-potential of liposomal AmBisome® in vivo was higher than in vitro (Fig. 2, panel E; Table S1†). DLS results were supported by TEM experiments showing single particles with a layer of adsorbed proteins (Fig. 2, panel F) and, in general, a minor occurrence of particle clusters (Fig. S3 in the ESI†). The biological identity of liposomes in vivo is controlled by the structure and composition of the HC,4 which can be exploited for active targeting of NPs.27,28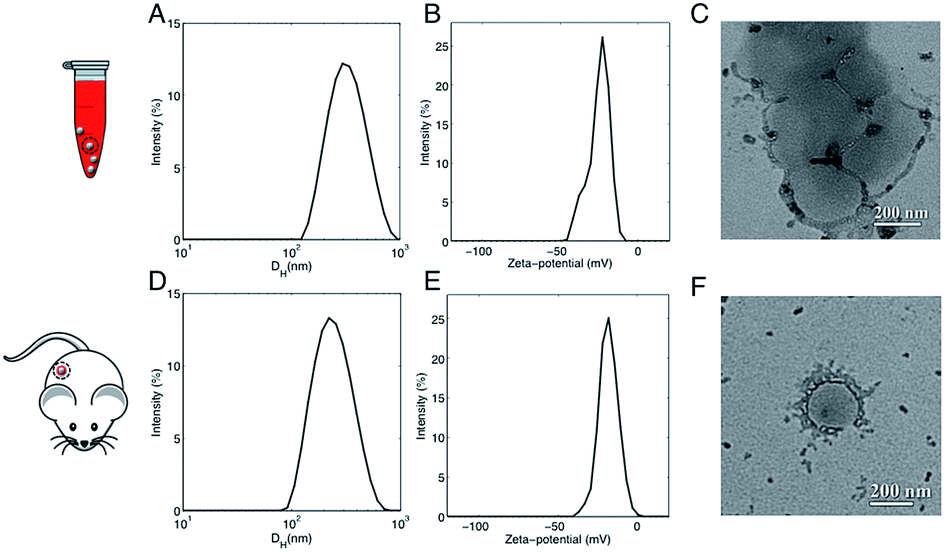 | ||
| Fig. 2 (A) Representative dynamic light scattering (DLS) intensity distribution of liposomal AmBisome® following 10 minutes incubation in FVB/N mouse plasma. (B) Representative zeta-potential distribution of liposomal AmBisome® after 10 minutes incubation in FVB/N mouse plasma. (C) Representative transmission electron microscopy (TEM) images of AmBisome®–protein complexes after 10 minutes in vitro incubation. (D) Representative DLS intensity distribution of liposomal AmBisome® after the recovery of the NPs from the blood circulation of FVB/N mice 10 minutes post intra venous (i.v.) administration. (E) Representative zeta-potential distribution of liposomal AmBisome® after the recovery from the blood circulation of FVB/N mice 10 minutes post i.v. administration. (F) Representative TEM images of AmBisome®–protein complexes after 10 minutes in vitro incubation. The plasma proteins establish a highly dynamic aura surrounding each NP, hence, the phenomenon was nicknamed protein “corona” in analogy to the aura surrounding the Sun. | ||
It could hinder the direct contact between the bare liposome surface and cell receptors or it may trigger an immune response, in which case liposomes could be eliminated from the bloodstream before reaching target cells.
A key question is also whether it is the HC itself, by proteins engaged from biological milieu, which determines the biodistribution of liposomes in vivo. Identification of proteins bound to liposome surface is a necessary step to answer this question and was made by liquid chromatography (LC) tandem mass spectrometry (MS/MS). The Venn diagram in Fig. 3, panel A, illustrates the number of common and unique proteins between the in vitro and in vivo coronas. Globally, more than five hundred proteins were identified. This result is in very good agreement with previous studies, which showed that the liposome–protein corona usually consists of some hundreds of proteins.29,30 Of note, the in vivo HC was much more enriched than its in vitro counterpart. In detail, 267 and 500 proteins were identified in the in vitro and in vivo HCs respectively, with only 40% of protein species (223 proteins) being in common between the two coronas. To predict the NP's behavior in vivo, accurate quantification of the HC is a mandatory step. To this end, the relative protein abundance (RPA) of each identified protein was determined as explained in the Experimental section. First, we classified proteins according to their molecular weight (MW). As Fig. 3, panel B, clearly shows, the in vitro HC is mainly enriched of proteins with MW between 50 and 60 kDa (37%), while the in vivo HC is preferentially composed of low MW proteins (i.e. <20 kDa, 31%). Further bioinformatic analysis (Fig. 3, panel C) showed that 88% and 73% of the in vitro and in vivo HC proteins have isoelectric point (pI) < 7.
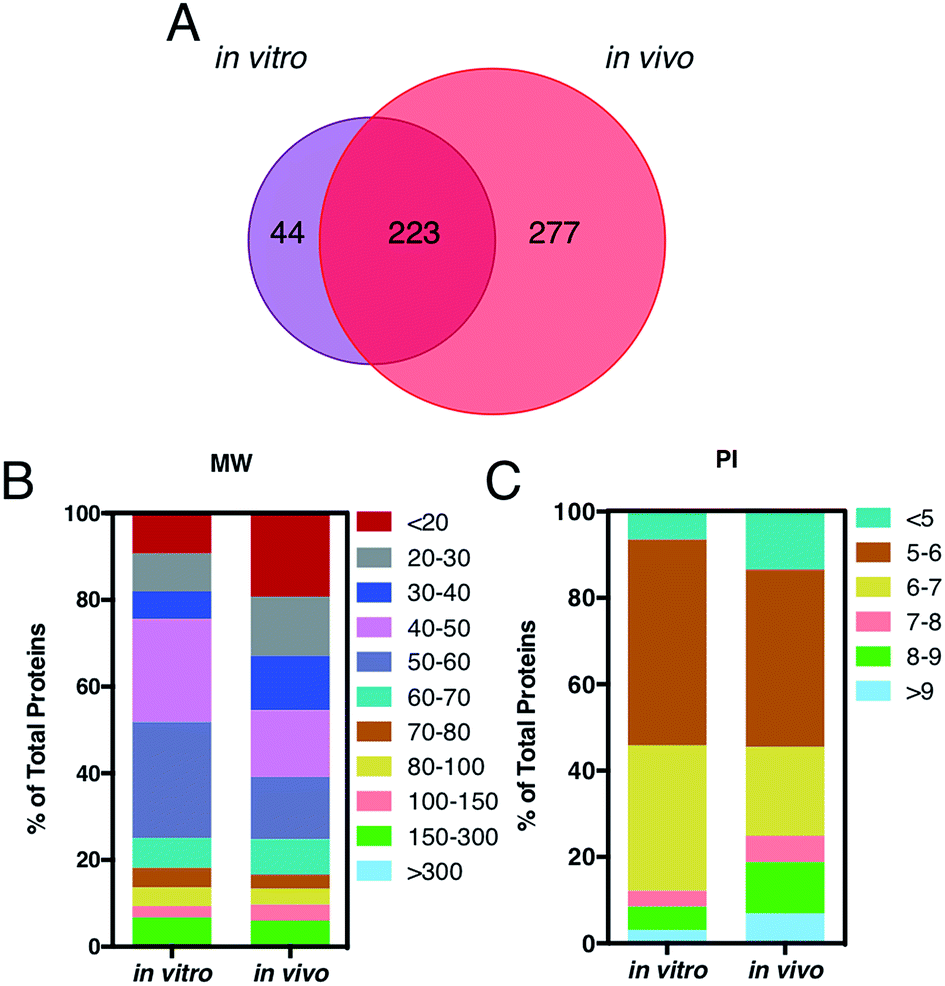 | ||
| Fig. 3 (A) Venn diagram displays the number of proteins identified onto the surface of AmBisome® following in vitro (violet) and in vivo (red) incubation. 223 proteins were found in common, while 44 and 277 plasma proteins were unique for the single coronas. (B) Percentage of total protein of corona proteins classified according to their calculated molecular mass and (C) isoelectric point. | ||
This is in good agreement with the zeta-potential results of Fig. 2, panel C, showing that, at physiological pH, the liposome–PC exhibited an overall negative charge. Tables 1 and 2 list the 25 most-abundant proteins found on the in vitro and in vivo HCs of AmBisome® liposomes. While the 25 hits constitute roughly 60% of the in vitro HC, they represent a minor fraction of the in vivo HC (≈30%). According to the results of Hadjidemetriou et al.,21 our findings confirm the higher heterogeneity of the in vivo HC on its in vitro counterpart. Among the 25 most abundant proteins, it is noteworthy to observe that only 14 proteins were in common (Fig. 4, panel A). Considering the 25 hits of both coronas, Fig. 4, panel B, shows that the largest fraction of the in vitro HC proteins have MW between 50 and 60 kDa (21%), while the in vivo HC is mainly composed of proteins with MW between 40 and 50 kDa (9%). We also observe that proteins with 5 < pI < 6 are the main components of both coronas (34% and 19% for in vitro and in vivo HCs respectively, Fig. 4, panel C). Afterward, all the HC proteins were categorized according to their physiological function (Fig. 5). First, percentages of identified proteins were appreciably altered between the two coronas. Notably, the in vivo HC was less enriched of complement proteins.
| In vitro | |||||
|---|---|---|---|---|---|
| # | Top 25 proteins | MW (kDa) | RPA (%) | SD (%) | |
| 1 | Fibrinogen beta chain | FIBB | 55 | 16.69 | 0.53 |
| 2 | Fibrinogen gamma chain | FIBG | 49 | 14.48 | 0.59 |
| 3 | Serum albumin | ALBU | 69 | 4.80 | 0.03 |
| 4 | C4b-binding protein | C4BPA | 52 | 2.37 | 0.08 |
| 5 | Coagulation factor XIII B chain | F13B | 76 | 1.51 | 0.05 |
| 6 | Coagulation factor XIII A chain | F13A | 83 | 1.47 | 0.05 |
| 7 | Actin, cytoplasmic 2 | ACTG | 42 | 1.44 | 0.11 |
| 8 | Fibronectin | FINC | 273 | 1.42 | 0.04 |
| 9 | Alpha-1-antitrypsin 1–3 | A1AT3 | 46 | 1.31 | 0.04 |
| 10 | Ig kappa chain C region | IGKC | 12 | 1.23 | 0.13 |
| 11 | Serine protease inhibitor A3K | SPA3K | 47 | 1.22 | 0.05 |
| 12 | Serotransferrin | TRFE | 77 | 1.14 | 0.04 |
| 13 | Peroxiredoxin-2 | PRDX2 | 22 | 1.11 | 0.07 |
| 14 | Alpha-1-antitrypsin 1–2 | A1AT2 | 46 | 1.11 | 0.04 |
| 15 | Plasminogen | PLMN | 91 | 1.05 | 0.01 |
| 16 | Apolipoprotein A-I | APOA1 | 31 | 1.00 | 0.13 |
| 17 | Thymosin beta-4 | TYB4 | 6 | 0.91 | 0.09 |
| 18 | Antithrombin-III | ANT3 | 52 | 0.83 | 0.02 |
| 19 | Carbonic anhydrase 2 | CAH2 | 29 | 0.83 | 0.09 |
| 20 | Beta-2-glycoprotein 1 | APOH | 39 | 0.82 | 0.03 |
| 21 | Vitronectin | VTNC | 55 | 0.78 | 0.03 |
| 22 | Apolipoprotein E | APOE | 36 | 0.77 | 0.04 |
| 23 | Alpha-1-antitrypsin 1–4 | A1AT4 | 46 | 0.68 | 0.48 |
| 24 | Band 3 anion transport protein | B3AT | 103 | 0.68 | 0.02 |
| 25 | Ig mu chain C region | IGHM | 50 | 0.67 | 0.04 |
| In vivo | |||||
|---|---|---|---|---|---|
| # | Top 25 proteins | MW (kDa) | RPA (%) | SD (%) | |
| 1 | Serum albumin | ALBU | 69 | 4.07 | 0.11 |
| 2 | Fibrinogen beta chain | FIBB | 55 | 2.28 | 0.06 |
| 3 | Apolipoprotein C-III | APOC3 | 11 | 2.14 | 0.15 |
| 4 | Actin, cytoplasmic 2 | ACTG | 42 | 2.11 | 0.22 |
| 5 | Fibrinogen gamma chain | FIBG | 49 | 1.81 | 0.08 |
| 6 | Alpha-1-antitrypsin 1–3 | A1AT3 | 46 | 1.45 | 0.14 |
| 7 | Ig kappa chain C region | IGKC | 12 | 1.41 | 0.24 |
| 8 | Alpha-1-antitrypsin 1–2 | A1AT2 | 46 | 1.34 | 0.07 |
| 9 | Actin, alpha cardiac muscle 1 | ACTC | 42 | 1.24 | 0.06 |
| 10 | Apolipoprotein A-I | APOA1 | 31 | 1.12 | 0.05 |
| 11 | Serine protease inhibitor A3K | SPA3K | 47 | 1.12 | 0.06 |
| 12 | Serotransferrin | TRFE | 77 | 1.12 | 0.02 |
| 13 | Ig mu chain C region | IGHM | 50 | 0.97 | 0.02 |
| 14 | C4b-binding protein | C4BPA | 52 | 0.96 | 0.01 |
| 15 | Apolipoprotein E | APOE | 36 | 0.96 | 0.05 |
| 16 | Apolipoprotein C-II | APOC2 | 11 | 0.78 | 0.04 |
| 17 | Vitronectin | VTNC | 55 | 0.74 | 0.08 |
| 18 | Myosin light polypeptide 6 | MYL6 | 17 | 0.70 | 0.04 |
| 19 | 14-3-3 protein zeta/delta | 1433Z | 28 | 0.70 | 0.08 |
| 20 | Ras-related protein Rap-1b | RAP1B | 21 | 0.69 | 0.05 |
| 21 | Complement C3 | CO3 | 186 | 0.66 | 0.02 |
| 22 | Peroxiredoxin-2 | PRDX2 | 22 | 0.66 | 0.09 |
| 23 | Tropomyosin alpha-4 chain | TPM4 | 28 | 0.64 | 0.13 |
| 24 | ATP synthase subunit beta, mitochondrial | ATPB | 56 | 0.64 | 0.03 |
| 25 | Alpha-2-macroglobulin | A2M | 166 | 0.63 | 0.03 |
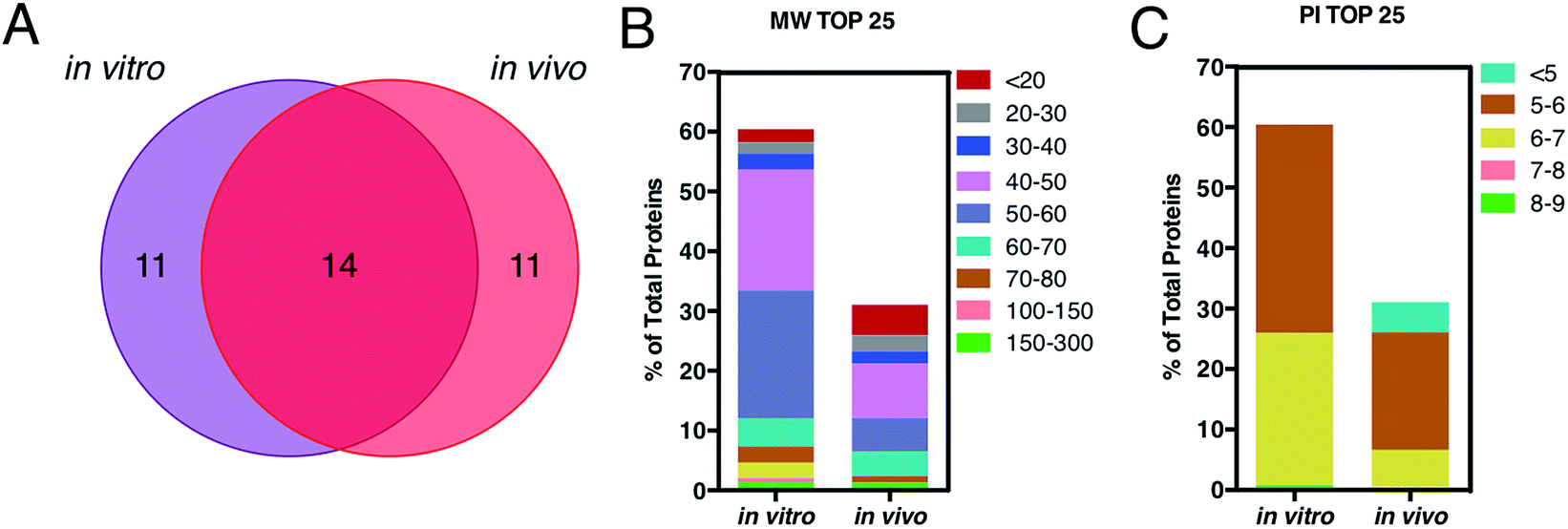 | ||
| Fig. 4 (A) Venn diagram displays the number of the top 25 proteins identified onto the surface of AmBisome® following in vitro (violet) and in vivo (red) incubation. 14 proteins were found in common, while 11 plasma proteins were unique for the single coronas. (B) Percentage of total protein of the top 25 corona proteins classified according to their calculated molecular mass. (C) Percentage of total protein of the top 25 corona proteins categorized according to their isoelectric point. | ||
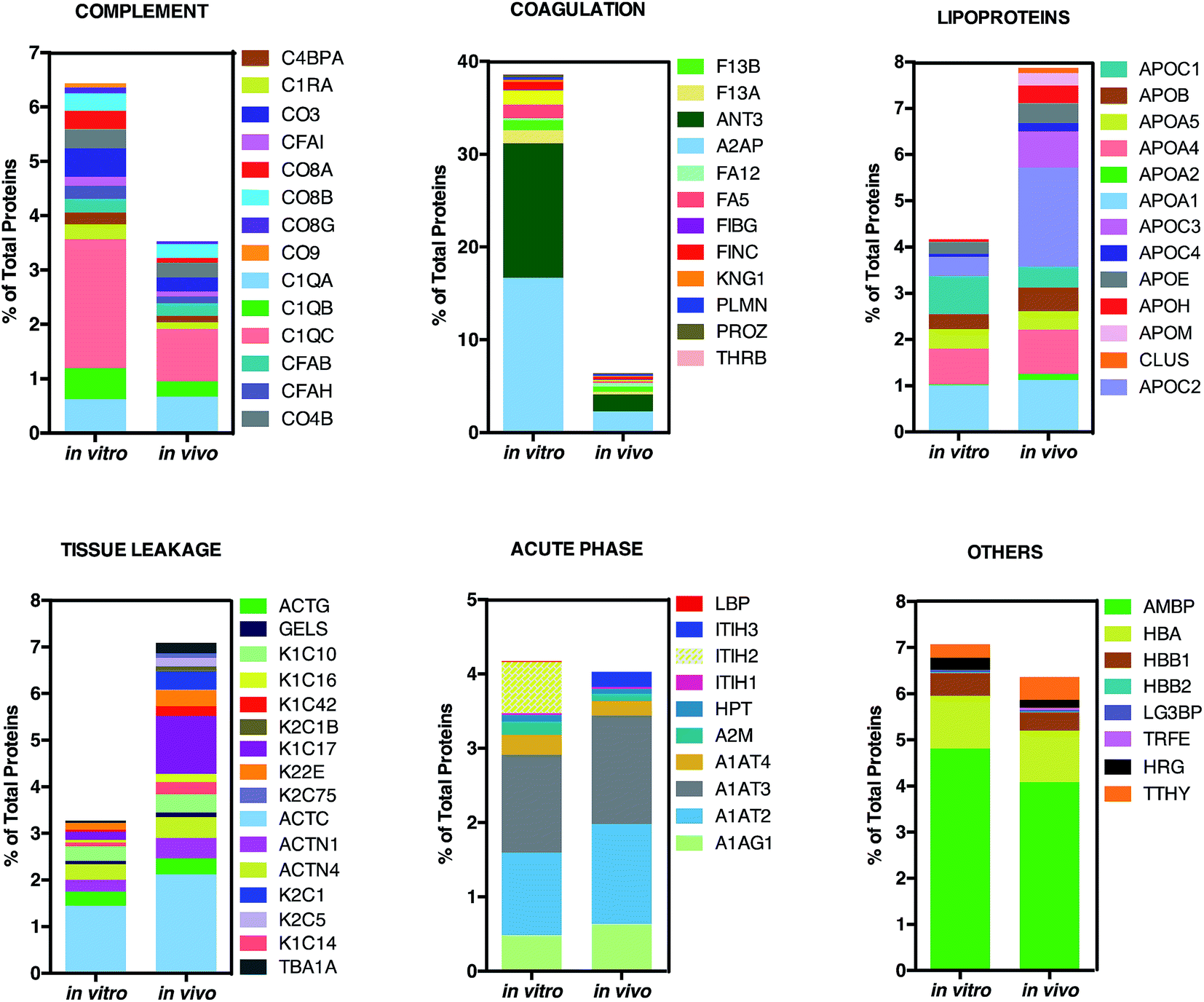 | ||
| Fig. 5 Bioinformatic classification of corona proteins. Percentage of total proteins identified in the in vitro and in vivo hard coronas of AmBisome®. | ||
This result is in very good agreement with the conclusions of a recent study19 showing that the PC that forms around NPs in circulating biological media is less enriched of complement factors with respect to its counterpart formed in vitro.17
Although being a great influential factor for PC–protein interactions, dynamical nature of bodily fluids has been largely ignored in the PC literature. Nonetheless, this result might be relevant for in vivo applications, since lower levels of complement proteins are associated with prolonged NP circulation in the blood. The main dissimilarity was found for C1QC, a protein of the classical complement pathway with a known opsonin activity. C1QC associates with the proenzymes C1r and C1s to yield C1, the first component of the serum complement system. The in vivo HC was also less enriched of coagulation proteins, a relevant class of plasma proteins that activate cells of the immune system by toll-like receptors (TLRs).31 For instance, fibrinogen induces macrophage chemokine secretion through TLR 4. A PC enriched with complement and coagulation proteins would promote the particle uptake by immune cells and, in turn, its removal from the bloodstream. Fig. 5 also shows that the in vivo HC of AmBisome® liposomes was much more enriched of lipoproteins. There are several receptors for apolipoproteins on the cell plasma membrane with potential implication in targeted drug delivery.23,26,31 Among identified lipoproteins, the main difference was observed for APOC2 that binds to scavenger receptor class B type 1 (SRBI) thus mediating the lipid transfer between low and high-density lipoproteins and cells of several tissues. Aside from relevance for targeted drug delivery, apolipoproteins have been reported to behave as dysopsonins, reducing adsorption of typical opsonins (i.e. complement proteins and immunoglobulins) and extending the circulation time of liposomes in vivo. The incubation procedure had a minor impact, if any, on the binding of acute phase proteins and ‘other proteins’ (i.e. relevant proteins not included in any of the previous classes) to the surface of AmBisome® liposomes. Lastly, we observe that neither the in vitro nor the in vivo HC were enriched of immunoglobulins (Igs). We have recently demonstrated that plasma proteins bind specifically to functional chemical groups onto the lipid surface and that the affinity of each lipid species for plasma proteins is affected by the co-presence of other species in the lipid bilayer.26 Thus, we are prompted to conclude that the deficiency of Igs is likely to be related to its specific lipid composition of liposomal AmBisome®. According to findings of Hadjidemetriou et al.21 We confirmed that the variety of proteins forming in vivo corona of AmBisome® was much wider. Equally, in vivo corona comprised considerably much lower amounts of the three fibrinogen chains compared to in vitro formed corona. On the other side, here we also show that in vivo corona was significantly less enriched of complement proteins, while it comprised higher amounts of lipoproteins with respect to its in vitro counterpart. This has not been observed before. Our findings imply that changes in protein pattern profiles can highly dependent on the lipid composition of NPs. It should be underlined that HC varies across animal species. Some of us have shown that the HC that forms around NPs in mice and humans can be markedly different from each other.10 Recently, Sahneh et al.32 have demonstrated that biocorona evolves differently in rodents and humans and that such different time evolution may have a deep impact on NPs' biodistribution across species.
Experimental
Preparation of liposomes
Hydrogenated soy phosphatidylcholine (HSPC), 1,2-distearoyl-sn-glycero-3-[phospho-rac-(1-glycerol)] (sodium salt) (DSPG), cholesterol (Chol) were purchased from Avanti Polar Lipids (Alabaster, AL). AmBisome-like liposomes were synthesized according to these molar ratios HSPC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DSPG:cholesterol (2.5:1:1.2).33 Liposomes were dissolved in chloroform and the solvent was evaporated under vacuum for 2 hours. Lipid films were then hydrated with phosphate saline buffer (PBS) to obtain a final lipid concentration of 10 mg mL−1. The obtained liposome solutions were extruded 20 times through a 0.05 μm polycarbonate filter with the Avanti Mini-Extruder (Avanti Polar Lipids, Alabaster, AL). The liposomes used in this study have a diameter of approximately 180 nm. This value is larger than that of AmBisome® (100 nm). Anyway, their lipid composition that has recently been identified as a main factor shaping the liposome–protein corona,26 is equal to that of AmBisome®.
:DSPG:cholesterol (2.5:1:1.2).33 Liposomes were dissolved in chloroform and the solvent was evaporated under vacuum for 2 hours. Lipid films were then hydrated with phosphate saline buffer (PBS) to obtain a final lipid concentration of 10 mg mL−1. The obtained liposome solutions were extruded 20 times through a 0.05 μm polycarbonate filter with the Avanti Mini-Extruder (Avanti Polar Lipids, Alabaster, AL). The liposomes used in this study have a diameter of approximately 180 nm. This value is larger than that of AmBisome® (100 nm). Anyway, their lipid composition that has recently been identified as a main factor shaping the liposome–protein corona,26 is equal to that of AmBisome®.
In vitro incubation
For in vitro incubation the following steps were carried out: (i) blood was collected from three mice by tail venipuncture using TM BD P100 Blood Collection System tubes containing K2EDTA and protease inhibitor (Franklin Lakes, NJ, USA 7). Approximately 800 L of blood was recovered from each mouse. Blood samples were not pooled; (ii) three aliquots of liposomal AmBisome® (volume = 25 μL; lipid concentration = 10 mg mL−1; total lipid/sample = 0.25 mg) were mixed with three blood samples (volume = 500 μL). Thus, this procedure resulted in the preparation of three independent samples (biological replicates) at lipid/blood ratio (w/v) = 0.5 mg mL−1; (iii) mixed solutions were incubated for 10 minutes at 37 °C. They were not pooled, but analyzed separately.In vivo incubation
Eight to ten week old female FVB/N mice were purchased from Charles River (UK). Mice were housed in groups of eight with free access to water and kept at temperature of 19–22 °C and relative humidity of 45–65%. Before performing the procedures, animals where acclimatized to the environment for at least 7 days. All animal experiments were carried out in accordance with the U.K. Animals (Scientific Procedures) Act, 1986 and associated guidelines, EU Directive 2010/63/EU for animal experiments. In vivo incubation was made of the following steps: (i) three FVB/N mice were anesthetized by inhalation of isoflurane; (ii) 100 μL liposomal AmBisome® were administered to each mouse. Since total blood volume for FVB/N mice is around 2 mL, this procedure resulted in a lipid/blood ratio = 0.5 mg mL−1 (i.e. the same lipid/blood ratio used for in vitro experiments); (iii) ten minutes after liposome injection, blood was recovered by cardiac puncture using TM BD P100 Blood Collection System tubes containing K2EDTA and protease inhibitor (Franklin Lakes, NJ, USA). Approximately 800 L of blood was recovered from each mouse. Recovered samples were not pooled. They were treated and analyzed separately.Protein assay
Immediately after in vitro an in vivo incubation, samples were centrifuged at 1800 rpm for 10 min at 4 °C. Supernatant was stored in LoBind tubes (Eppendorf) at −80 °C. When used, aliquots were thawed at 4 °C and then left to warm at room temperature. According to literature, the HC of AmBisome® was separated form excess plasma proteins by size exclusion chromatography and membrane ultrafiltration. Following both in vitro and in vivo incubation, 500 L of plasma samples was loaded onto a Sepharose CL-2B (Ge Healthcare Life Sciences) and equilibrated with PBS. Among ten collected chromatographic fractions, 3, 4 and 5 were pooled together and concentrated to 250 μL by centrifugation using Vivaspin 6 column (MWCO 10000 Da, Sigma Aldrich, USA) at 3000 rpm.
To concentrate the samples to 100 μL Vivaspin 500 centrifugal concentrator (MWCO 10000 Da, Sigma Aldrich, USA) was used at 3000 rpm. This step also allowed separating protein-coated liposomes from large unbound proteins. Liposome–protein complexes were finally washed 3 times with cold PBS to remove weekly bound proteins (the soft corona). Separation of protein-decorated liposomes from excess plasma proteins was validated using pure plasma (i.e. without liposomes) as a control. Ten microliters of each sample re-suspension was used to quantify the amount of adsorbed proteins using the Protein Assay reagent (Pierce, Thermo Scientific, Waltham, MA, USA), according to manufacturer's protocol. Briefly, 10 microliters were placed into a 96-multiwell plate and then was added 150 μL of Protein Assay reagent. The measures were performed in triplicate. The multiwell was covered, mixed on a plate shaker, and incubated at room temperature for 5 min. The absorbance of each sample, standard and blank was measured with the Glomax Discover System (Promega, Madison, WI, USA) at 660 nm. The protein concentration was calculated using the standard curve.
In solution digesting and desalting
The protein pellets obtained from precipitation were re-suspended in 50 μL of a denaturant buffer composed of 8 mol L−1 urea in 50 mmol L−1 NH4HCO3, then 2.5 μL of DTT 200 mmol L−1 in 50 mmol L−1 NH4HCO3 were added and finally incubated for 1 hour at 37 °C. Following protocols already described,27 the protein extract was enzymatically digested before nanoLC/MS/MS analysis. After overnight digestion at 37 °C, the reaction was quenched by the addition of HCOOH. Digested samples were desalted using an SPE C18 column (Bond Elut 1CC LRCC18, Varian, Palo Alto, CA, USA). Peptides were eluted from the SPE column with 0.5 mL ACN–H2O (50:50, v/v) solution containing 0.05% TFA and then vacuum dried. Each sample was reconstituted with 0.1% HCOOH solution. Digested samples were stored at −80 °C until nanoLC-MS/MS analysis.
Nanoliquid chromatography tandem mass spectrometry
Tryptic peptides were analyzed using a Dionex Ultimate 3000 (Sunnyvale, CA, USA) nanoLC system connected to a hybrid mass spectrometer LTQ Orbitrap XL (Thermo Fisher Scientific Bremen, Germany), equipped with a nanoelectrospray ion source. Peptide mixtures were enriched by injecting 10 mL (ca. 3.3 mg) of sample on line into a 300 mm i.d. 5 mm Acclaim PepMap100 C18 (5 mm particle size, 100 Å pore size) μ-precolumn (Dionex), using a premixed mobile phase H2O–ACN 95:5 (v/v) containing 0.1% HCOOH at 10 mL min−1 flow-rate. Peptide mixtures were separated by reversed-phase chromatography on in-house manufactured 25 cm fritless silica microcolumns with 75 mm i.d. The column was packed with ReproSil-Pur C18-AQ 2.2 mm resin (Dr Maisch GmbH, Ammerbuch, Germany). The mobile phase was H2O (A) and ACN (B), both with 0.1% (v/v) HCOOH. After a 5 min isocratic step at 5%, B was linearly increased from 5% to 30% within 125 min and then to 45% in 10 min. After that B was increased to 80% within 10 min and kept constant for 10 min. Then, B was decreased to 5% within 1 min and kept constant for the following 20 min to rinse the column. Separation was performed at a flow rate of 300 nL min−1. MS spectra were collected over an m/z range of 400–1800 at 60000 resolution, operating in the data dependent mode to switch automatically between Orbitrap-MS and LTQ-MS/MS acquisition. Following the “TOP5 strategy”, MS/MS spectra were collected for the twenty five most intense ions with a charge state greater than 1, using a dynamic exclusion of 60 s. CID was performed with normalized collision energy set at 35 V. All samples were analyzed in triplicate in order to assess the additional variation introduced in the measurements by the experimental procedure and to increase the number of identified proteins.
Data analysis and protein validation
Raw data files, obtained from Xcalibur software, were subjected to Proteome Discover (1.2 version, Thermo Scientific) for database search using Mascot (version 2.3.2 Matrix Science). Data were searched against the SwissProt database (57.15 version, 20266 sequences). The built-in decoy search option of Mascot was used. Enzymatic digestion with trypsin was selected, along with maximum 2 missed cleavages, peptide charges +2 and +3, a 10 ppm precursor mass tolerance and 0.8 Da fragment mass tolerance; acetylation (N-term), oxidation (M) and deamidation (N, Q) were used as dynamic modifications; carbamidomethylation (C) was used as static modification. The Scaffold software (version 3.1.2, Proteome Software Inc.) was used to validate MS/MS-based peptide and protein identifications and for label-free relative quantitation based on spectral counting. The peptide and protein probabilities were set to minimum 95% and 99%, respectively, with at least one identified peptide. For protein quantitative analysis, Scaffold software allows the normalization of the spectral countings (normalized spectral countings, NSCs) and offers various statistical tests to identify significant abundance differences in two or more categories. The mean value of NSCs obtained in the three experimental replicates for each protein was further normalized to the protein molecular weight (MWNSC) and expressed as the relative protein quantity by applying the following equation:34
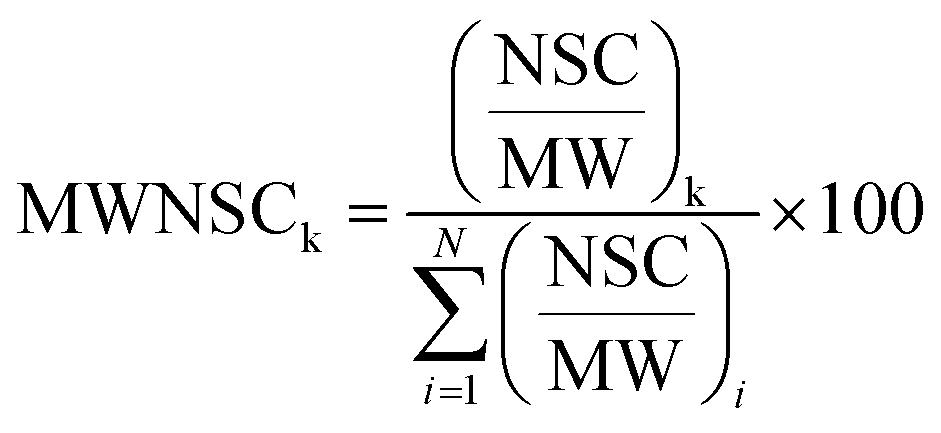 | (1) |
Size and zeta-potential experiments
All size and zeta-potential measurements were made on a Zetasizer Nano ZS90 (Malvern, U.K.) equipped with a 5 mW HeNe laser (wavelength λ = 632.8 nm) and a digital logarithmic correlator. Bare liposomes were diluted 1:100 with distilled water. Size and zeta-potential results are given as mean ± standard deviation of five replicates. To perform size and zeta-potential experiments 20 μL of liposome–HC complexes were diluted with 980 μL of distilled water (final volume = 1 mL). This experimental procedure was maintained for proteomics experiments, with the exception of using 3-fold increase volume of complexes reaching the minimum sample volume required for accurate protein identification and quantification with nanoLC-MS/MS.35 Size and zeta-potential of liposome–HC complexes were given as average ± S.D. from nine samples (i.e. three technical replicates for each of the three biological replicates).
Transmission electron microscopy
Transmission electron microscopy (TEM) images were captured fast using a ZEISS EM10 operating@60 keV. These conditions prevented any radiation damage originating from electron beam. Liposomes were visualized before and after their in vitro and in vivo interaction with plasma proteins. A drop from each liposome suspension was placed onto a Carbon Film Mesh Copper Grid (CF400-Cu, Electron Microscopy Science) and the excess suspension was removed with a filter paper. To avoid the occurrence of any fake morphological information, we did not use any staining agent (e.g. osmium tetroxide, uranyl acetate, lead citrate) of those traditionally employed for enhancing the image intensity contrast by interaction of the heavy-metal salts with lipids and proteins.All the experimental procedures carried out in this study were in compliance with the UK Animals (Scientific Procedures) Act 1986 and associated guidelines, EU Directive 2010/63/EU, and were approved by the Ethic Committee on Animal Use of the University of Camerino.
Conclusion
With a few exceptions, static incubation in biological media is the only experimental procedure followed so far to investigate the bio–nano-interactions between NPs and biological systems. This is the first study to investigate the HC of liposomal Amphotericin B (AmBisome®) under realistic in vivo conditions. Our results revealed the HC that forms around AmBisome® in vivo is markedly different from its in vitro counterpart. Specifically, the in vivo HC was found to be more heterogeneous than that formed in vitro. Notably, both the number and the relative abundance of identified proteins changed in vivo. Our results indicate that the blood flow dynamics has a tremendous impact on the HC. Thoroughly characterization of the HC protein coronas under realistic in vivo conditions for liposomal chemotherapeutics is therefore compulsory to advance our understanding of their overall clinical performance. Future exploitation of HC will need an accurate mapping of corona proteins36 as well as evaluating whether binding epitopes are presented suitably on the NP surface. This is a largely unexplored factor in the field of bio–nano-interactions that is currently under investigation in our laboratory.Acknowledgements
G. Caracciolo, S. Palchetti and D. Pozzi acknowledge support by the Italian Minister of Health (“Progetto Giovani Ricercatori 2011–2012”, Grant No. GR-2011-02350094). The authors are also pleased to thank Ezio Battaglione of the Electron Microscopy Laboratory “Pietro M. Motta”, located at Sapienza University of Rome, for fruitful discussions and experimental help in TEM measurements.Notes and references
- M. P. Monopoli, C. Åberg, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2012, 7, 779–786 CrossRef CAS PubMed.
- T. Cedervall, I. Lynch, S. Lindman, T. Berggård, E. Thulin, H. Nilsson, K. A. Dawson and S. Linse, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2050–2055 CrossRef CAS PubMed.
- I. Lynch, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2009, 4, 546–547 CrossRef CAS PubMed.
- C. D. Walkey and W. C. W. Chan, Chem. Soc. Rev., 2012, 41, 2780–2799 RSC.
- M. Lundqvist, J. Stigler, G. Elia, I. Lynch, T. Cedervall and K. A. Dawson, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14265–14270 CrossRef CAS PubMed.
- M. P. Monopoli, D. Walczyk, A. Campbell, G. Elia, I. Lynch, F. Baldelli Bombelli and K. A. Dawson, J. Am. Chem. Soc., 2011, 133, 2525–2534 CrossRef CAS PubMed.
- S. Tenzer, D. Docter, S. Rosfa, A. Wlodarski, J. R. Kuharev, A. Rekik, S. K. Knauer, C. Bantz, T. Nawroth and C. Bier, ACS Nano, 2011, 5, 7155–7167 CrossRef CAS PubMed.
- G. Caracciolo, D. Pozzi, S. Candeloro De Sanctis, A. Laura Capriotti, G. Caruso, R. Samperi and A. Laganà, Appl. Phys. Lett., 2011, 99, 033702 CrossRef.
- G. Caracciolo, G. Amiconi, L. Bencivenni, G. Boumis, R. Caminiti, E. Finocchiaro, B. Maras, C. Paolinelli and A. Congiu Castellano, Eur. Biophys. J., 2001, 30, 163–170 CrossRef CAS PubMed.
- G. Caracciolo, D. Pozzi, A. L. Capriotti, C. Cavaliere, S. Piovesana, G. La Barbera, A. Amici and A. Laganà, J. Mater. Chem. B, 2014, 2, 7419–7428 RSC.
- G. Caracciolo, D. Pozzi, A. L. Capriotti, C. Cavaliere, P. Foglia, H. Amenitsch and A. Laganà, Langmuir, 2011, 27, 15048–15053 CrossRef CAS PubMed.
- M. Mahmoudi, A. M. Abdelmonem, S. Behzadi, J. H. Clement, S. Dutz, M. R. Ejtehadi, R. Hartmann, K. Kantner, U. Linne and P. Maffre, ACS Nano, 2013, 7, 6555–6562 CrossRef CAS PubMed.
- V. Mirshafiee, R. Kim, M. Mahmoudi and M. L. Kraft, Int. J. Biochem. Cell Biol., 2016, 5, 188–195 CrossRef PubMed.
- M. Lundqvist, J. Stigler, T. Cedervall, T. Berggård, M. B. Flanagan, I. Lynch, G. Elia and K. Dawson, ACS Nano, 2011, 5, 7503–7509 CrossRef CAS PubMed.
- A. L. Barrán-Berdón, D. Pozzi, G. Caracciolo, A. L. Capriotti, G. Caruso, C. Cavaliere, A. Riccioli, S. Palchetti and A. Laganà, Langmuir, 2013, 29, 6485–6494 CrossRef PubMed.
- S. Tenzer, D. Docter, J. Kuharev, A. Musyanovych, V. Fetz, R. Hecht, F. Schlenk, D. Fischer, K. Kiouptsi and C. Reinhardt, Nat. Nanotechnol., 2013, 8, 772–781 CrossRef CAS PubMed.
- U. Sakulkhu, L. Maurizi, M. Mahmoudi, M. Motazacker, M. Vries, A. Gramoun, M.-G. Ollivier Beuzelin, J.-P. Vallee, F. Rezaee and H. Hofmann, Nanoscale, 2014, 6, 11439–11450 RSC.
- M. J. Hajipour, S. Laurent, A. Aghaie, F. Rezaee and M. Mahmoudi, Biomater. Sci., 2014, 2, 1210–1221 RSC.
- D. Pozzi, G. Caracciolo, L. Digiacomo, V. Colapicchioni, S. Palchetti, A. L. Capriotti, C. Cavaliere, R. Z. Chiozzi, A. Puglisi and A. Laganà, Nanoscale, 2015, 7, 13958–13966 RSC.
- S. Palchetti, V. Colapicchioni, L. Digiacomo, G. Caracciolo, D. Pozzi, A. L. Capriotti, G. La Barbera and A. Laganà, Biochim. Biophys. Acta, Biomembr., 2016, 1858, 189–196 CrossRef CAS PubMed.
- M. Hadjidemetriou, Z. Al-Ahmady, M. Mazza, R. F. Collins, K. Dawson and K. Kostarelos, ACS Nano, 2015, 9, 8142–8156 CrossRef CAS PubMed.
- M. Hadjidemetriou, Z. Al-Ahmady and K. Kostarelos, Nanoscale, 2016, 8, 6948–6957 RSC.
- G. Caracciolo, Nanomedicine, 2015, 11, 543–557 CAS.
- V. Colapicchioni, M. Tilio, L. Digiacomo, V. Gambini, S. Palchetti, C. Marchini, D. Pozzi, S. Occhipinti, A. Amici and G. Caracciolo, Int. J. Biochem. Cell Biol., 2016, 75, 180–187 CrossRef CAS PubMed.
- J. Adler-Moore and R. T. Proffitt, J. Antimicrob. Chemother., 2002, 49, 21–30 CrossRef CAS PubMed.
- G. Caracciolo, D. Pozzi, A. L. Capriotti, C. Cavaliere, S. Piovesana, H. Amenitsch and A. Laganà, RSC Adv., 2015, 5, 5967–5975 RSC.
- G. Caracciolo, F. Cardarelli, D. Pozzi, F. Salomone, G. Maccari, G. Bardi, A. L. Capriotti, C. Cavaliere, M. Papi and A. Laganà, ACS Appl. Mater. Interfaces, 2013, 5, 13171–13179 CAS.
- V. Mirshafiee, R. Kim, S. Park, M. Mahmoudi and M. L. Kraft, Biomaterials, 2016, 75, 295–304 CrossRef CAS PubMed.
- A. L. Capriotti, G. Caracciolo, G. Caruso, P. Foglia, D. Pozzi, R. Samperi and A. Lagan, Anal. Biochem., 2011, 419, 180–189 CrossRef CAS PubMed.
- A. L. Capriotti, G. Caracciolo, C. Cavaliere, P. Foglia, D. Pozzi, R. Samperi and A. Laganà, J. Proteomics, 2012, 75, 1924–1932 CrossRef CAS PubMed.
- S. T. Smiley, J. A. King and W. W. Hancock, J. Immunol., 2001, 167, 2887–2894 CrossRef CAS.
- F. D. Sahneh, C. M. Scoglio, N. A. Monteiro-Riviere and J. E. Riviere, Nanomedicine, 2015, 10, 25–33 CrossRef CAS PubMed.
- J. Adler-Moore and R. T. Proffitt, J. Antimicrob. Chemother., 2002, 49(suppl. 1), 21–30 CrossRef CAS PubMed.
- M. P. Monopoli, D. Walczyk, A. Campbell, G. Elia, I. Lynch, F. B. Bombelli and K. A. Dawson, J. Am. Chem. Soc., 2011, 133, 2525–2534 CrossRef CAS PubMed.
- D. Pozzi, C. Marchini, F. Cardarelli, F. Salomone, S. Coppola, M. Montani, M. E. Zabaleta, M. A. Digman, E. Gratton, V. Colapicchioni and G. Caracciolo, Biochim. Biophys. Acta, 2014, 1838, 957–967 CrossRef CAS PubMed.
- P. M. Kelly, C. Åberg, E. Polo, A. O'Connell, J. Cookman, J. Fallon, Ž. Krpetić and K. A. Dawson, Nat. Nanotechnol., 2015, 10, 472–479 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1. Size and zeta-potential distributions of bare liposomes. Table S1. The average size of liposomal AmBisome®. Fig. S2. Proteins associated with AmBisome® liposomes after in vitro and in vivo incubation. Fig. S3. Morphology and morphometric features of AmBisome® liposomes following in vitro and in vivo incubation. See DOI: 10.1039/c6ra25493d |
| This journal is © The Royal Society of Chemistry 2017 |