
Exploiting ortho-substitution effect on formation of oxygen-containing [10]paracyclophane through ring-closing metathesis†
Li
Yang‡
,
Liqiang
Song‡
,
Chong
Huang
,
Mingzheng
Huang
and
Bo
Liu
*
Key Laboratory of Green Chemistry & Technology of Ministry of Education, College of Chemistry, Sichuan University, Chengdu 610064, P. R. China. E-mail: chembliu@mail.scu.edu.cn; Fax: +86-28-85412291
First published on 22nd December 2015
Abstract
The synthesis of strained paracyclophanes is challenging and generally delivers low yields. A novel strategy of installing an ortho-substituent group to the ethereal oxygen is developed to facilitate the formation of oxygen-containing [10]paracyclophane via RCM macrocyclization. By rationally modulating the electronic and steric effects in the single and double ortho-substitution modes, efficient RCM macrocyclization was achieved for different substrates to form the corresponding [10]paracyclophane products, in which all newly generated double bonds were in the E-form. The development of this methodology will enrich the chemistry of ring-closing metathesis related to the formation of strained macrocycles.
 Bo Liu | Bo Liu: Southwest Normal University (B. S. 1998), Chengdu Institute of Organic Chemistry (M.S. 2001, Prof. Xiaoming Feng and Prof. Yaozhong Jiang), Shanghai Institute of Organic Chemistry (PhD 2004, Prof. Wei-Shan Zhou), UT Southwestern Medical Center at Dallas (Postdoc Research Fellow 2004–2007, Prof. Jef De Brabander), Sichuan University (PI, 2007–present). [Field of research] Natural Product Synthesis and Synthesis-oriented Methodology Development. |
Paracyclophanes are popular molecules among material chemists and supramolecular chemists for research, which is ascribed to their intriguing ability to interact with π-orbital-containing chemical entities, cations, anions and neutral molecules.1 They have been hot target molecules as well for synthetic chemists2 since the first naturally occurring cyclophane, cylindrocyclophane A, was reported in 1990.3 Accordingly, versatile synthetic strategies have been developed for the synthesis of paracyclophanes,2,4 which include the application of olefination as the key ring-closing step.5 Although ring-closing metathesis (RCM) is one of the favored strategies in light of its mild reaction conditions and high functional group-tolerance,6 its application in the synthesis of strained paracyclophanes with short chain tethers, such as [10]paracyclophane, is still limited thus far.
Intrigued by the highly strained paracyclophane motif and the impressive antimycobacterial activity of the hirsutellones,7 our group initiated synthetic studies toward this natural family several years ago. To date, two impressive total syntheses of hirsutellone B have been completed by the Nicolaou's group in 2009 and by the Uchiro's group in 2011.8 While elegant synthetic studies have been reported by several other groups,9 the study in our group features intramolecular ketene-capture by alcohols to construct the [11]paracyclophane intermediate,10 and the ring-closing metathesis (RCM) strategy to install the [10]paracyclophane intermediate.11 Moreover, the hirsutellone [10]paracyclophane core is embodied in the architecture of many natural products and pharmaceutically useful compounds such as piperazinomycin,12 SE205,13 mauritine A,14 and RP-66453.15
In fact, macrocyclization via the RCM strategy to construct paracyclophanes with short ring sizes, such as the [10]paracyclophane scaffolds presented in Fig. 1, is challenging if the possibility of ring-opening metathesis (ROM) and ring-opening metathesis polymerization (ROMP), which is driven by ring strain, is considered. The Suzuki's group invented an ingenious hydrogen-bonded control mode to constrain the free rotation of the two side arms in substrate 1 (Scheme 1A).16 This conformational restriction makes the two alkenes on the side arms more approachable to each other during the RCM process, and thus facilitates the formation of the strained [10]paracyclophane 2. Actually, our former success in the RCM formation of [10]paracyclophane was enlightened by the supposed arene–arene interaction during RCM macrocyclization by introducing an electro-deficient phenyl ester on the ortho-position of the ethereal oxygen (Scheme 1B).11 This synthetic strategy was exploited by the Collins’ group and applied in the versatile syntheses of natural and unnatural paracyclophanes with bigger ring sizes than that of [10]paracyclophanes.17 However, in our subsequent study, we found that the ortho-substituent on the phenyl ring displayed a substantial impact on RCM reactivity toward [10]paracyclophane, even without the arene–arene interaction proposed by the Collins’ group. Herein, we report the related research results for this ortho-substitution effect on the formation of oxygen-containing [10]paracyclophane through RCM macrocyclization (Scheme 1C).
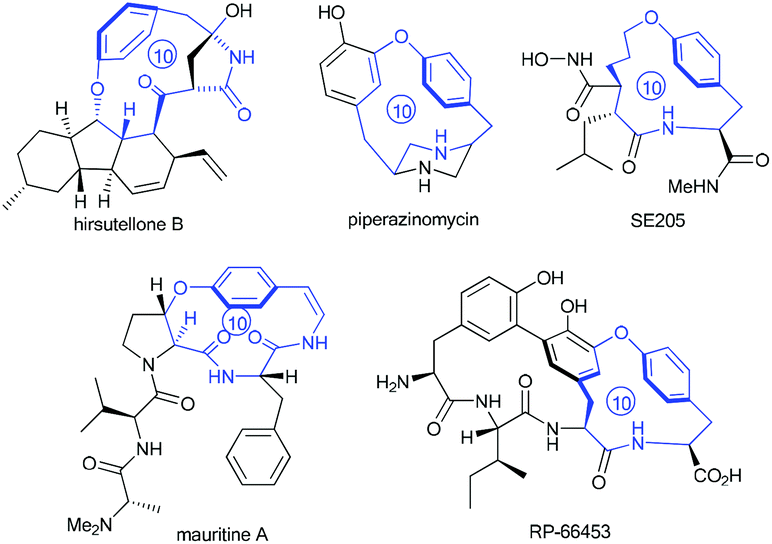 | ||
| Fig. 1 Molecular structures of natural products or pharmaceutically useful compounds with the [10]paracyclophane core. | ||
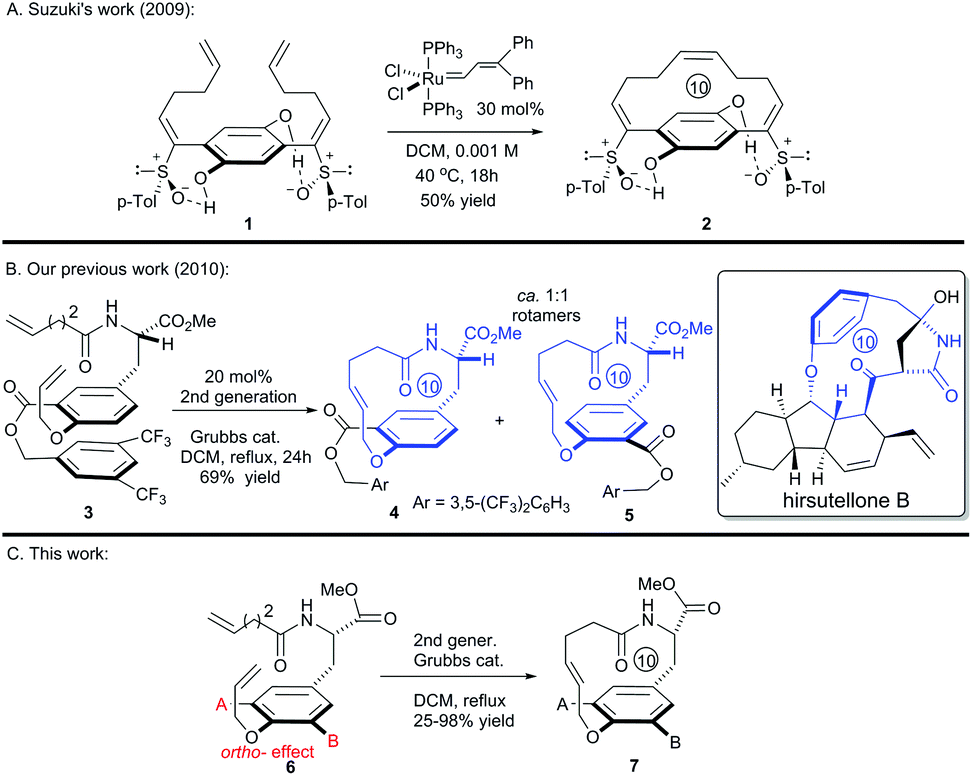 | ||
| Scheme 1 RCM strategies for the synthesis of [10]paracyclophanes. | ||
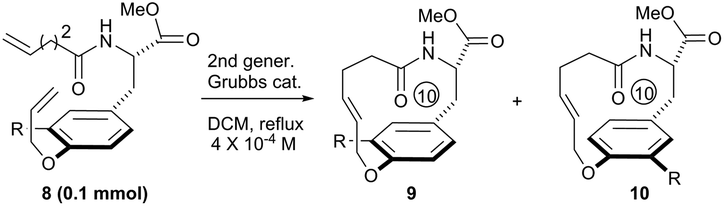
To systematically inspect the electronic and steric effect of the ortho-substituent groups, a series of substrates were prepared and examined. As illustrated in Table 1, all reactions were set up in a 0.0004 mol L−1 solution of compound 8 in dichloromethane under reflux. For each reaction, 15 mol% of second generation Grubbs catalyst was initially used.18 In some cases, an additional 10 mol% of the catalyst was supplemented after 18 hours for complete conversion of the reactants. Unexpectedly, when compound 8a, without any ortho-substituent group, was tested, the corresponding [10]paracyclophane product could be obtained in 26% isolated yield (entry 1, Table 1). This indicates the strong driving force to form a strained macrocycle by the Grubbs catalyst itself.
|
|
||||
|---|---|---|---|---|
| Entry | Substrate | Cat. loading & time | Yielda (%) | drb (9/10) |
| a Isolated yield combining 9 and 10. b Determined by 1H NMR; the molecular structures of both 9 and 10 are tentatively proposed and thus might be exchanged. c 9a = 10a. | ||||
| 1c | R = H, 8a | 15 mol%, 18 h; then 10 mol%, 13 h | 26 | — |
| 2 | R = Me, 8b | 15 mol%, 18 h; then 10 mol%, 13 h | 25 | 1/1.4 |
| 3 | R = CH2OTBS, 8c | 15 mol%, 18 h; then 10 mol%, 13 h | 33 | 1/1.2 |
| 4 | R = CO2Me, 8d | 15 mol%, 18 h | 48 | 1.2/1 |
| 5 | R = CO2CH2Ph(CF3)2, 3 | 15 mol%, 18 h | 45 | 1/1.1 |
| 6 | R = NO2, 8e | 15 mol%, 18 h | 56 | 1/1.2 |
| 7 | R = F, 8f | 15 mol%, 18 h; then 10 mol%, 5 h | 61 | 1/1 |
| 8 | R = Cl, 8g | 15 mol%, 18 h; then 10 mol%, 11 h | 41 | 1/1.3 |
| 9 | R = Br, 8h | 15 mol%, 18 h; then 10 mol%, 13 h | 39 | 1/1.2 |
| 10 | R = I, 8i | 15 mol%, 18 h; then 10 mol%, 13 h | 38 | 1/1.4 |
Theoretically, the p–π orbital conjugation between the ethereal oxygen and phenyl ring in 8a limits the free rotation of the CPh–O bond (Scheme 2A). This causes the vinyl group to swing out of the phenyl plane and be positioned faraway from the terminal alkene on the amide chain. Then, it should significantly decrease the approaching probability of these two double bonds during the RCM process, which accounts for the yield from 8a to be as low as 26%. Based on this conformational analysis, we then envisioned that the installation of an ortho-substituent should show two possible effects on the RCM macrocyclization: (1) electronic repulsion between the lone electron pair of the sp2-hybrid oxygen and the electronegative substitution group (R) in the intermediate I; and (2) steric repulsion between the R group (R ≠ H) and the allylic group in the intermediate II (Scheme 2B). Hopefully, these electronic and steric repulsions would compel rotation of the CPh–O bond to position the whole allylic group out of the phenyl plane and thus cause the two reactive alkenes to become closer for the RCM macrocyclization (intermediates III and IV, Scheme 2B).
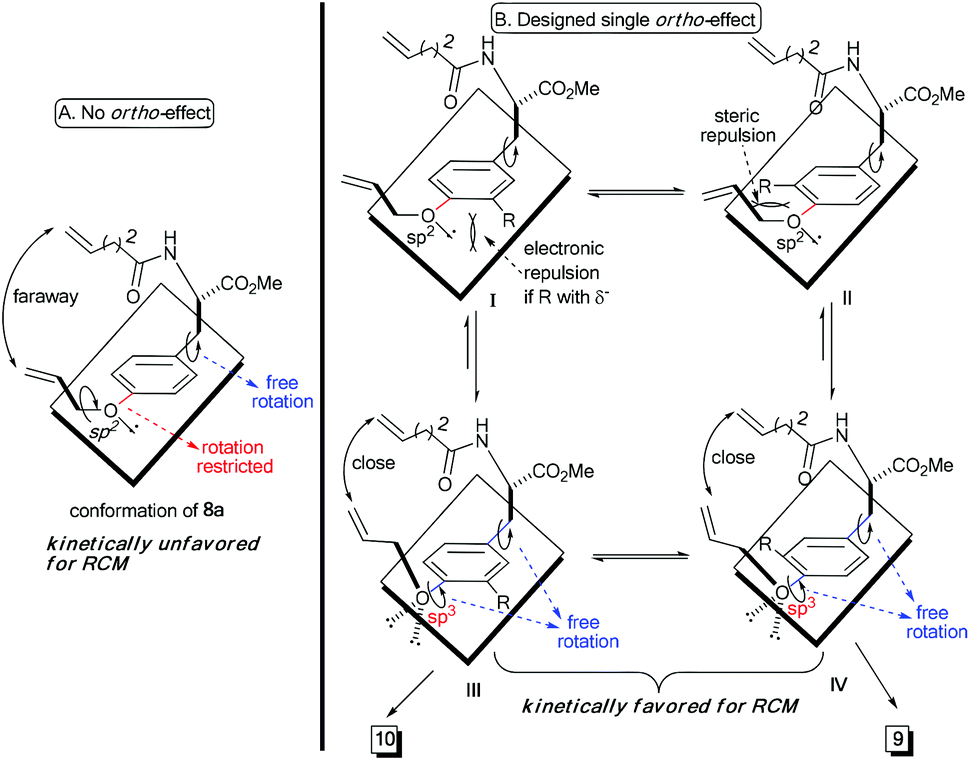 | ||
| Scheme 2 Illustration of our designed single ortho-effect to promote RCM macrocyclization. | ||
Experimentally, the substrates 8b and 8c with a methyl or substituted methyl did not afford better yields than that from 8a (entries 2 and 3, Table 1). This is reasonable because electro-neutral alkyl substitution can only show a steric effect, so that compounds 8b and 8c would take the same conformations as that in intermediate I, which is similar to that of 8a (Scheme 2). Satisfactorily, the RCM macrocyclization yields could be promoted to more than 45% when esters were installed at the ortho-position of the ether (entries 4 and 5, Table 1). It is worth noting that the presence of the electro-deficient benzyl group in our model system did not show any beneficial arene–arene interaction to form [10]paracyclophane (entry 4 vs. entry 5, Table 1), unlike the experimental findings from different model systems in the Collins group.12
We ascribed the improved yields of 8d and 3 to the π–π conjugation between the ester and phenyl ring. This provokes both steric and polar repulsions between the ester and ethereal oxygen and drives the allylic group out of the phenyl plane. Thus, the repulsion facilitates the approach of the two double bonds to each other, as we anticipated (Scheme 2B). Employing compound 8e (R = NO2) as the substrate afforded the impressive yield of 56% (entry 6, Table 1). This indicates herein that the electronic factor plays an important role in RCM macrocyclization, because the nitryl in 8e is not as bulky as the esters in 8d and 3. This conclusion can be further proven on the basis of the experimental results in entries 7–10 (Table 1). The yields decreased gradually from 61%, 41% and 39% to 38%, when the ortho-substituent was varied from the most electronegative but smallest fluorine, to the moderately electronegative but bigger chlorine and bromine to the least electronegative but biggest iodine. Considering that the size of fluorine is comparable to that of hydrogen, the yields of 8f–8i imply an extra contribution from the electron-withdrawing group, and this might be attributed to the resulting lower electronic density on the phenyl ring, which lowers the energy of the aromatic distortion in the RCM transition state and products. While both rotamers studied herein, 9 and 10, were separable through silica column chromatography, their ratios mentioned in Table 1 were determined via proton NMR. One may argue that these two rotamers are interchangeable because of their as low as, almost, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr. Actually, compound 10f or 10g could not be detected even when pure compounds 9f and 9g were heated in a solution of toluene, respectively, at 180 °C for 12 hours. This indicates that the poor values of dr in Table 1 originate from the free rotation of the aromatic ring in compound 8 before RCM macrocyclization.
:1 dr. Actually, compound 10f or 10g could not be detected even when pure compounds 9f and 9g were heated in a solution of toluene, respectively, at 180 °C for 12 hours. This indicates that the poor values of dr in Table 1 originate from the free rotation of the aromatic ring in compound 8 before RCM macrocyclization.
Accordingly, the electronic effect of the single ortho-substitution can significantly impact the yield of RCM macrocyclization towards [10]paracyclophane. However, the efficiency of the abovementioned RCM macrocyclization was not satisfactory enough. We believed that a better yield is achievable if both steric and electronic factors can be combined in a double ortho-substitution mode (Scheme 3). Rotation of the CPh–O bond would be restricted further, and the allylic group thus would be positioned almost perpendicular to the phenyl plane, so that the two alkenes on the side arms would be more approachable in the RCM process.
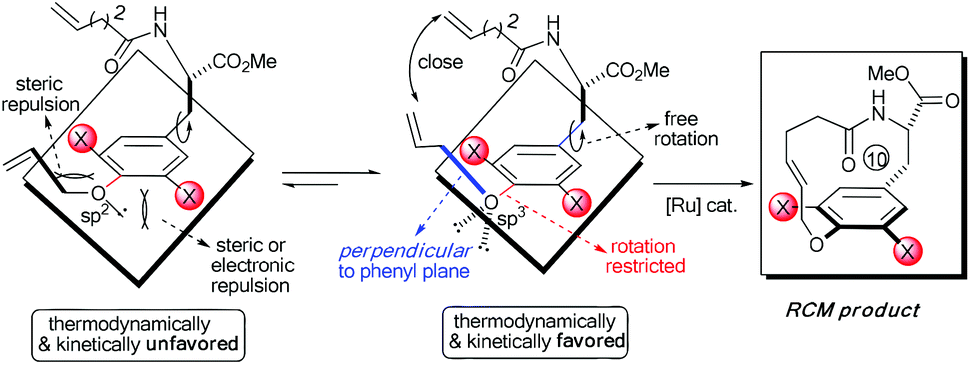 | ||
| Scheme 3 Illustration of our designed double ortho-effect to promote RCM macrocyclization. | ||
Subsequently, a series of substrates with double ortho-substitution to the ethereal oxygen were prepared and subjected to RCM conditions (Table 2). First, double ortho-halogen substitution was examined. Compared to the corresponding single ortho-substitution case, herein excellent yields were obtained in all cases (entries 1–4, Table 2). Among the four substrates, compound 11d, which is the least electronegative but has the biggest iodine, actually afforded the best results. This indicates that the steric hindrance of double ortho-iodine atoms makes the allylic group perpendicular to the phenyl plane and thus benefits the RCM macrocyclization. Thus, it is easy to understand why a much better yield was obtained from compound 11e with double ortho-methyl substitution than that from 8b (entry 5, Table 2vs. entry 2, Table 1). Interestingly, even 11f with double ortho-methoxyl substitution afforded 92% yield. Introduction of an allylic alcohol or allylic acetate on the amide arms (11g and 11h) showed a deleterious impact on the RCM process, because the yields dropped from 91% without allylic functionality (entry 3, Table 2) to the current 51% and 39% (entries 7 and 8, Table 2). Replacing one of the terminal alkenes on the side arms with a trisubstituted alkene impaired the RCM efficiency by affording 14% and 69% yields (entries 9 and 10, Table 2). These results are understandable because the increased steric hindrance at double bond hampers the [2 + 2] step in the RCM process and thus results in competitive side-reactions. Moreover, the better yield obtained with 11j (entries 10 vs. 9, Table 2) indicates that the Ru catalyst might generally react first with the double bond in the ethereal allyl group during all the RCM macrocyclization reactions mentioned in Tables 1 and 2.
|
|
|||
|---|---|---|---|
| Entry | Substrate | Cat. loading & time | Yielda (%) |
| a Isolated yield. b This compound was prepared in the racemic form. c The product is 12c, as in entry 3. | |||
| 1b | X = F, R = H; 11a | 15 mol%, 18 h | 92 |
| 2 | X = Cl, R = H; 11b | 15 mol%, 18 h | 93 |
| 3 | X = Br, R = H; 11c | 15 mol%, 18 h | 91 |
| 4 | X = I, R = H; 11d | 15 mol%, 18 h | 98 |
| 5 | X = Me, R = H; 11e | 15 mol%, 18 h | 69 |
| 6b | X = OMe, R = H; 11f | 15 mol%, 18 h | 92 |
| 7 | X = Br, R = OH; 11g | 15 mol%, 18 h | 51 |
| 8 | X = Br, R = OAc; 11h | 15 mol%, 18 h; then 10 mol%, 14 h | 39 |
| 9c |
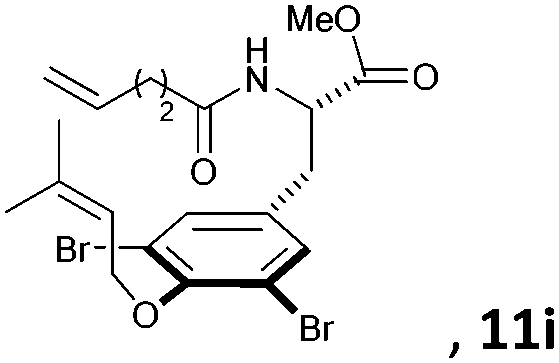
|
15 mol%, 18 h; then 10 mol%, 13 h | 14 |
| 10c |
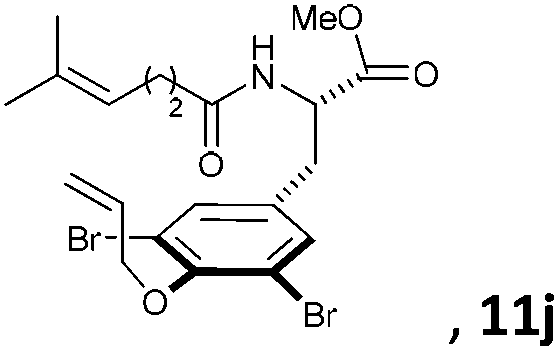
|
15 mol%, 18 h | 69 |
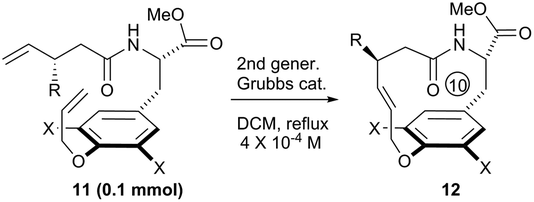
The bent phenyl rings that resulted from the macrocyclic strain and molecular structures for this type of [10]paracyclophane, mentioned in Table 2, can be further confirmed via X-ray crystallography of 12b and 12c (Fig. 2).19 Based on these ORTEP structures, we can observe that the trans configuration of the amide and E-form double bond in the tethered chain make these [10]paracyclophanes less strained than those with the Z-form double bond after RCM macrocyclization. This might account for the E-selectivity of the RCM in Tables 1 and 2. It can be noted that the halogen atoms in these [10]paracyclophanes (Table 2) are not only beneficial to the RCM macrocyclization, but also useful for further functionalization of the phenyl rings, because they can be removed or transferred to other functional groups through cross-coupling or other reactions in the presence of transition metal catalysts.
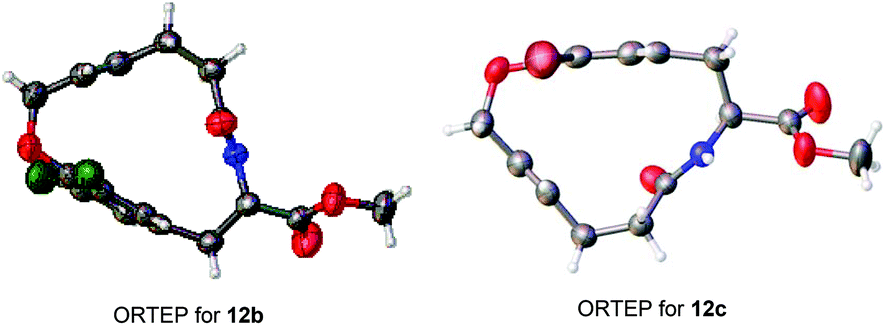 | ||
| Fig. 2 X-ray crystal structures of 12b and 12c. | ||
To further exhibit the applicability of this RCM strategy to construct [10]paracyclophane, we prepared compounds 13 and 15, without an amide on the side arm of the phenyl ring, and tested their RCM reactivity. Delightfully, both compounds afforded the corresponding macrocyclization products in respectable yields (Scheme 4).
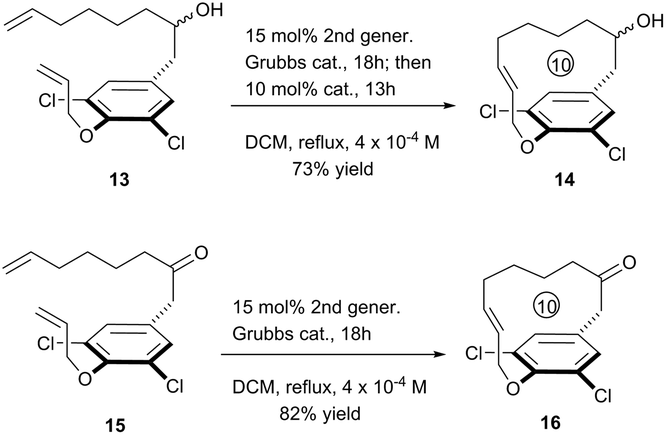 | ||
| Scheme 4 Extension of the RCM macrocyclization toward compounds 14 and 16. | ||
In conclusion, we have disclosed an interesting ortho-substitution effect on RCM macrocyclization to form oxygen-containing [10]paracyclophane. In the single ortho-substitution mode, electro-withdrawing groups show a beneficial effect, while generally good yields of the desired RCM products can be achieved in the double ortho-substitution mode. It can be noted that only the E- double bond can be detected in all the RCM products obtained. This strategy can potentially be extended to the synthesis of other strained paracyclophanes, and thus it will positively contribute to the synthesis of related functional materials and natural products. Further study on this topic is in progress in our laboratory and will be reported in the future.
Acknowledgements
We acknowledge the financial support from the NSFC (21290180, 21322205, and 21321061). We also thank the comprehensive training platform of the Specialized Laboratory in the College of Chemistry at Sichuan University for compound testing.Notes and references
- P. G. Ghasemabadi, T. Yao and G. J. Bodwell, Chem. Soc. Rev., 2015, 44, 6494 RSC.
- T. Gulder and P. S. Baran, Nat. Prod. Rep., 2012, 29, 899 RSC.
- B. S. Moore, J. L. Chen, G. M. L. Patterson, R. E. Moore, L. S. Brinen, Y. Kato and J. Clardy, J. Am. Chem. Soc., 1990, 112, 4061 CrossRef CAS.
- S. Kotha, M. E. Shirbhate and G. T. Waghule, Beilstein J. Org. Chem., 2015, 11, 1274 CrossRef PubMed.
- G. J. Bodwell and P. R. Nandaluru, Isr. J. Chem., 2012, 52, 105 CrossRef.
- (a) J. Tae and Y.-K. Yang, Org. Lett., 2003, 5, 741 CrossRef PubMed; (b) V. Martinez, J.-C. Blais and D. Astruc, Angew. Chem., Int. Ed., 2003, 42, 4366 CrossRef CAS PubMed.
- (a) M. Isaka, N. Rugseree, P. Maithip, P. Kongsaeree, S. Prabpai and Y. Thebtaranonth, Tetrahedron, 2005, 61, 5577 CrossRef CAS; (b) M. Isaka, W. Prathumpai, P. Wongsa and M. Tanticharoen, Org. Lett., 2006, 8, 2815 CrossRef CAS PubMed.
- (a) K. C. Nicolaou, D. Sarlah, T. R. Wu and W. Zhan, Angew. Chem., Int. Ed., 2009, 48, 6870 CrossRef CAS PubMed; (b) K. C. Nicolaou, Y.-P. Sun, D. Sarlah, W.-Q. Zhan and T. R. Wu, Org. Lett., 2011, 13, 5708 CrossRef CAS PubMed; (c) H. Uchiro, R. Kato, Y. Arai, M. Hasegawa and Y. Kobayakawa, Org. Lett., 2011, 13, 6268 CrossRef CAS PubMed.
- (a) S. D. Tilley, K. P. Reber and E. J. Sorensen, Org. Lett., 2009, 11, 701 CrossRef CAS PubMed; (b) K. P. Reber, S. D. Tilley and E. J. Sorensen, Chem. Soc. Rev., 2009, 38, 3022 RSC; (c) G. T. Halvorsen and W. R. Roush, Tetrahedron Lett., 2011, 52, 2072 CrossRef CAS PubMed; (d) N. Riache, I. Ndoye, X. W. Li and B. Nay, Synlett, 2011, 2686 Search PubMed; (e) X. W. Li, A. Ear and B. Nay, Nat. Prod. Rep., 2013, 30, 765 RSC; (f) K. P. Reber, S. D. Tilley, C. A. Carson and E. J. Sorensen, J. Org. Chem., 2013, 78, 9584 CrossRef CAS PubMed; (g) X. W. Li, A. Ear, L. Roger, N. Riache, A. Deville and B. Nay, Chem. – Eur. J., 2013, 19, 16389 CrossRef CAS PubMed.
- (a) M. Huang, C. Huang and B. Liu, Tetrahedron Lett., 2009, 50, 2797 CrossRef CAS; (b) L. Song, C. Huang, M. Huang and B. Liu, Tetrahedron, 2015, 71, 3603 CrossRef CAS.
- M. Huang, L. Song and B. Liu, Org. Lett., 2010, 12, 2504 CrossRef CAS PubMed.
- (a) M. Kaneda, S. Tamai, S. Nakamura, T. Hirata, Y. Kushi and T. Suga, J. Antibiot., 1982, 35, 1137 CrossRef CAS PubMed; (b) S. Tamai, M. Kaneda and S. Nakamura, J. Antibiot., 1982, 35, 1130 CrossRef CAS PubMed.
- C.-B. Xue, X. He, J. Roderick, W. F. DeGrado, R. J. Cherney, K. D. Hardman, D. J. Nelson, R. A. Copeland, B. D. Jaffee and C. P. Decicco, J. Med. Chem., 1998, 41, 1745 CrossRef CAS PubMed.
- (a) A. Kirfel and G. Will, Z. Kristallogr., 1976, 142, 368 CAS; (b) A. Kirfel, G. Will, R. Tschesche and H. Wilhelm, Z. Naturforsch., B: Anorg. Chem. Org. Chem., 1976, 31, 279 Search PubMed.
- G. Helynek, C. Dubertret, D. Frechet and J. Leboul, J. Antibiot., 1998, 51, 512 CrossRef.
- K. Mori, K. Ohmori and K. Suzuki, Angew. Chem., Int. Ed., 2009, 48, 5638 CrossRef CAS PubMed.
- (a) Y. El-Azizi, A. Schmitzer and S. K. Collins, Angew. Chem., Int. Ed., 2006, 45, 968 CrossRef CAS PubMed; (b) S. K. Collins, Y. El-Azizi and A. R. Schmitzer, J. Org. Chem., 2007, 72, 6397 CrossRef CAS PubMed; (c) Y. El-Azizi, J. E. Zakarian, L. Bouillerand, A. R. Schmitzer and S. K. Collins, Adv. Synth. Catal., 2008, 350, 2219 CrossRef CAS; (d) J. E. Zakarian, Y. El-Azizi and S. K. Collins, Org. Lett., 2008, 10, 2927 CrossRef CAS PubMed; (e) P. Bolduc, A. Jacques and S. K. Collins, J. Am. Chem. Soc., 2010, 132, 12790 CrossRef CAS PubMed.
- For seminal review articles on application of Grubbs catalyst in RCM reactions, see: (a) S. Monfette and D. E. Fogg, Chem. Rev., 2009, 109, 3783 CrossRef CAS PubMed; (b) R. H. Grubbs, Tetrahedron, 2004, 60, 7117 CrossRef CAS.
- See ESI† for details.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data of new compounds. CCDC 1436892 (12b) and 1436896 (12c). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00373c |
| ‡ These authors contributed equally to this article. |
| This journal is © the Partner Organisations 2016 |