
Iodine(III)-mediated oxidative intramolecular arene–alkene coupling exemplified in the synthesis of phenanthrenes†
Christian
Depken
,
Felix
Krätzschmar
and
Alexander
Breder
*
Institut für Organische und Biomolekulare Chemie, Georg-August-Universität, Tammannstrasse 2, 37077 Göttingen, Germany. E-mail: abreder@gwdg.de
First published on 11th January 2016
Abstract
An iodine(III)-mediated synthesis of substituted phenanthrenes from ortho-vinylated biaryl derivatives through highly 6-endo-trig selective oxidative intramolecular arene–alkene coupling is reported. The title method allows for the construction of diversely functionalized target compounds under very mild conditions and in yields ranging up to 79%. Preliminary mechanistic investigations indicate that the reaction presumably proceeds via activation of the alkene moiety followed by a Friedel–Crafts-type electrophilic aromatic substitution at the adjacent arene ring.
 Alexander Breder | Alexander Breder is a research group leader at the Georg-August-University of Göttingen, Germany. He received his diploma degree from Bielefeld University, Germany, and pursued his doctoral studies at the Swiss Federal Institute of Technology Zurich (ETH), Switzerland. Subsequently, he began his postdoctorate at Stanford University, United States, and started his independent career in late 2011. Since then his research activities are focusing on the investigation of novel concepts in metal-free catalysis and reaction design within the broader context of method development and synthesis of biologically relevant molecules. |
Introduction
The formation of phenanthrene motifs via oxidative intramolecular carbon–carbon bond coupling is a privileged strategy in contemporary synthetic methodology. Given the fact that this class of aromatic hydrocarbons i.a. plays an imperative role in the context of biologically active natural products,1 such as the phenanthroindolizidine alkaloids,2,3 many efforts have been devoted to the development of new procedures for the facile assembly of this oligocyclic core structure.4 Among the portfolio of corresponding methods, intramolecular Scholl-type oxidations5 have received considerable attention throughout recent years.6 Despite the fact that the direct merger of the arene units within a cis-stilbene precursor generally allows for a rapid and structurally diverse construction of phenanthrenes, it is important to emphasize that substitution patterns at the 4- and 5-position are typically inaccessible via this route (Scheme 1, top). This observation can be rationalized on the grounds of steric interaction between the converging arene rings. A strategically complementary approach, which obviates this shortcoming, relies on the oxidative intramolecular arene–alkene coupling of ortho-vinylated biaryl substrates. As a prime example, Georg and coworkers have recently reported a very elegant and pathbreaking vanadium(V)-mediated protocol for the oxidative cyclization of vinylated, electron-rich biphenyl derivatives (Scheme 1, middle).7 In general, this procedure requires superstoichiometric amounts of VOF3 (up to 4.5 equiv.) and reaction temperatures as low as −78 °C in order to achieve good yields and excellent 6-endo-trig selectivities. Owing to the circumstance that this particular study was designed to find a new route toward the phenanthroindolizidine alkaloid framework, the authors limited their investigations to the use of a 6-dehydroindolizidin-6-yl moiety as the olefinic component in the arene–alkene coupling. Incited by this seminal report, we became interested in the question as to whether such a cyclization mode can be generalized to a broader spectrum of alkenes in order to expand the scope of bespoken phenanthrene architectures. A decisive factor for the success of our endeavors was the establishment of reaction conditions that would entail high endo/exo selectivities in favor of the endo cyclization mode (Scheme 1, bottom). Ample precedent from the literature indicates that the competing 5-exo-trig cyclization occurs preferentially in the case of cognate Brønsted-acid or aminium radical cation promoted cyclizations involving alkyl-substituted vinyl groups as reaction partners.8 As a result of our efforts we present herein an efficient and highly selective iodine(III)-mediated protocol for the synthesis of diversely substituted phenanthrenes via an intramolecular arene–alkene coupling reaction.9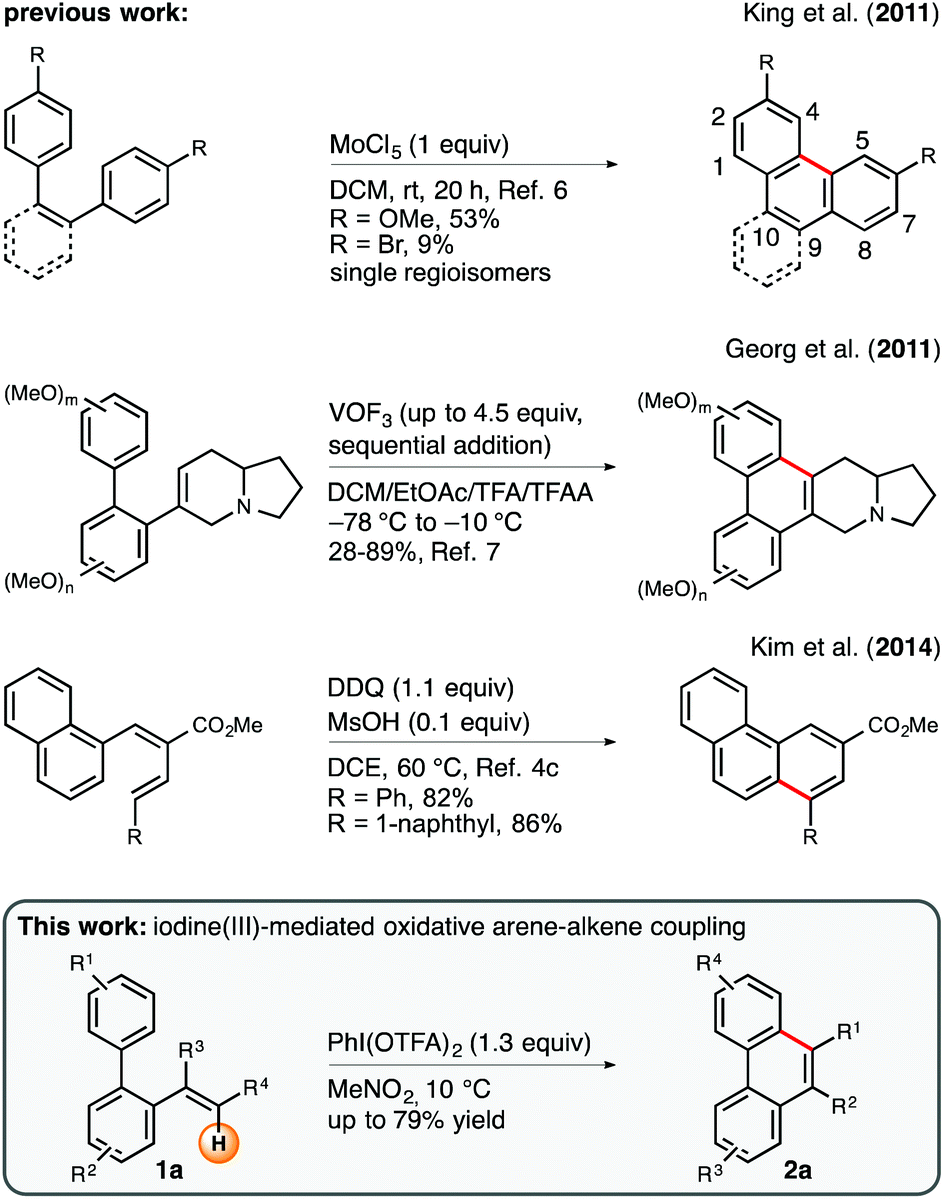 | ||
| Scheme 1 Exemplary methods for the construction of phenanthrenes via oxidative arene–arene and arene–alkene coupling. DDQ = 2,3-dichloro-5,6-dicyano-1,4-benzoquinone, TFA = trifluoroacetyl. | ||
Results and discussion
Our investigations began with the identification of an appropriate oxidant for the conversion of 2,3,4,5-tetrahydroterphenyl derivative 1a to phenanthrene 2a. In the course of orienting experiments we found that the reaction of substrate 1a with 1 equiv. of N-bromoacetamide and N-bromosuccinimide, respectively, in CH2Cl2 at ambient temperature and in the presence of 10 mol% of (PhSe)2 as a catalyst predominantly led to allylic bromination products. Changing to the less reactive N-chlorosuccinimide (NCS, 1 equiv.) and performing the reaction in toluene did not result in any reactivity at all. Therefore, we initially focused our efforts on the use of NCS in combination with methanesulfonic acid (1 equiv.) as an activating additive10 under otherwise unaltered conditions (Table 1). When toluene was used as the reaction medium still no product formation was recorded (entry 1). However, when ethereal solvents, such as diethyl ether (entry 2), THF (entry 3), and 1,4-dioxane (entry 4), were used we detected phenanthrene 2a with yields ranging from 15–24%. Slightly improved results were recorded when more polar solvents were applied. More precisely, acetonitrile (24%, entry 5), EtOAc (29%, entry 6), CH2Cl2 (32%, entry 7), and MeNO2 (35%, entry 8) generally resulted in moderate yields, however, accompanied by unspecific substrate degradation. Lowering the reaction temperature to 0 and −80 °C did not have any beneficial effects on the reaction outcome (entries 9 and 10). Furthermore, we screened several Brønsted-acid additives, such as acetic-, trifluoroacetic-, and formic acid as well as MeSO2NH2 and F3CSO2NH2 (entries 11–15), but none of the compounds tested led to a significant improvement of the yield. Next, we studied alternative chlorine-, oxygen-, and iodine-based oxidants.11 No product formation was detected when 1,3-dichloro-5,5-dimethylhydantoin (entry 16), trichloroisocyanuric acid (entry 17), and ammonium persulfate (entry 18) were employed as oxidants, respectively. Subsequently, we tested (diacetoxyiodo)benzene (PIDA, entry 19) and [bis(trifluoroacetoxy)iodo]benzene (PIFA, entry 20). In contrast to PIDA, which did not furnish phenanthrene 2a at all, use of 1 equiv. of PIFA at ambient temperature resulted in the oxidative cyclization of substrate 1a in 53%. To our surprise, when the title reaction was conducted in the absence of any catalyst a comparable yield of 51% was obtained (entry 21). In both experiments the conversion of substrate 1a was severely plagued by unspecific byproduct formation. It turned out that the reaction proceeded more selectively at 0 °C, however, to the expense of incomplete conversion (49%, entry 22). Based on these findings we conjectured that undesired reaction pathways might also be suppressible by constantly keeping the concentration of the oxidant low while conducting the reaction at moderate cooling. Thus, addition of 1 equiv. of PIFA at 10 °C to a solution of substrate 1a in MeNO2 in the course of 8 h furnished target structure 2a in 80% (entry 23).|
|
|||||
|---|---|---|---|---|---|
| Entry | Oxidant | Solvent | Temp. (°C) | Additive | Yieldb (%) |
| a Reactions were carried out on a 0.10–0.15 mmol scale in the respective solvent (0.1 M) with 1.00 equiv. of the respective oxidant, 10 mol% of (PhSe)2, and 4 Å molecular sieves for 16 h. b Yields were determined by 1H NMR using phthalide as an internal standard. c Reaction was performed without catalyst. DCDMH = 1,3-dichloro-5,5-dimethylhydantoin. TCICA = trichloroisocyanuric acid. PIDA = (diacetoxyiodo)benzene. PIFA = [bis(trifluoroacetoxy)iodo]benzene. | |||||
| 1 | NCS | Toluene | 23 | MsOH | 0 |
| 2 | NCS | Et2O | 23 | MsOH | 24 |
| 3 | NCS | THF | 23 | MsOH | 15 |
| 4 | NCS | 1,4-Dioxane | 23 | MsOH | 15 |
| 5 | NCS | MeCN | 23 | MsOH | 24 |
| 6 | NCS | EtOAc | 23 | MsOH | 29 |
| 7 | NCS | CH2Cl2 | 23 | MsOH | 32 |
| 8 | NCS | MeNO2 | 23 | MsOH | 35 |
| 9 | NCS | MeNO2 | 0 | MsOH | 34 |
| 10 | NCS | EtNO2 | −80 | MsOH | 24 |
| 11 | NCS | MeNO2 | 23 | AcOH | 25 |
| 12 | NCS | MeNO2 | 23 | TFA | 25 |
| 13 | NCS | MeNO2 | 23 | HCO2H | 20 |
| 14 | NCS | MeNO2 | 23 | MeSO2NH2 | 24 |
| 15 | NCS | MeNO2 | 23 | TfNH2 | 25 |
| 16 | DCDMH | MeNO2 | 23 | — | 0 |
| 17 | TCICA | MeNO2 | 23 | — | 0 |
| 18 | NH4S2O8 | MeNO2 | 23 | — | 0 |
| 19 | PIDA | MeNO2 | 23 | — | 0 |
| 20 | PIFA | MeNO2 | 23 | — | 53 |
| 21c | PIFA | MeNO2 | 23 | — | 51 |
| 22c | PIFA | MeNO2 | 0 | — | 49 |
| 23 | PIFA | MeNO 2 | 10 | — | 80 |

Once optimal reaction conditions were identified, investigation continued with the determination of the scope and limitations of the iodine(III)-mediated oxidative intramolecular arene–alkene coupling (Schemes 2 and 3). With the primary goal in mind to broaden the olefin-diversity of the title transformation, we initially studied the influence of varying substitution patterns at the alkene moiety (R1 and R2, Scheme 2). In addition to the cyclohexene-substituted biaryl substrate 1a, other cycloalkenes were also successfully employed. More specifically, ring sizes from five to eight carbon atoms (compounds 1a–d) were all well tolerated and resulted in isolated yields ranging from 73–78%. During this part of investigations it turned out that the use of 1.3 equiv. of PIFA generally led to more reliable conversions and isolated yields.
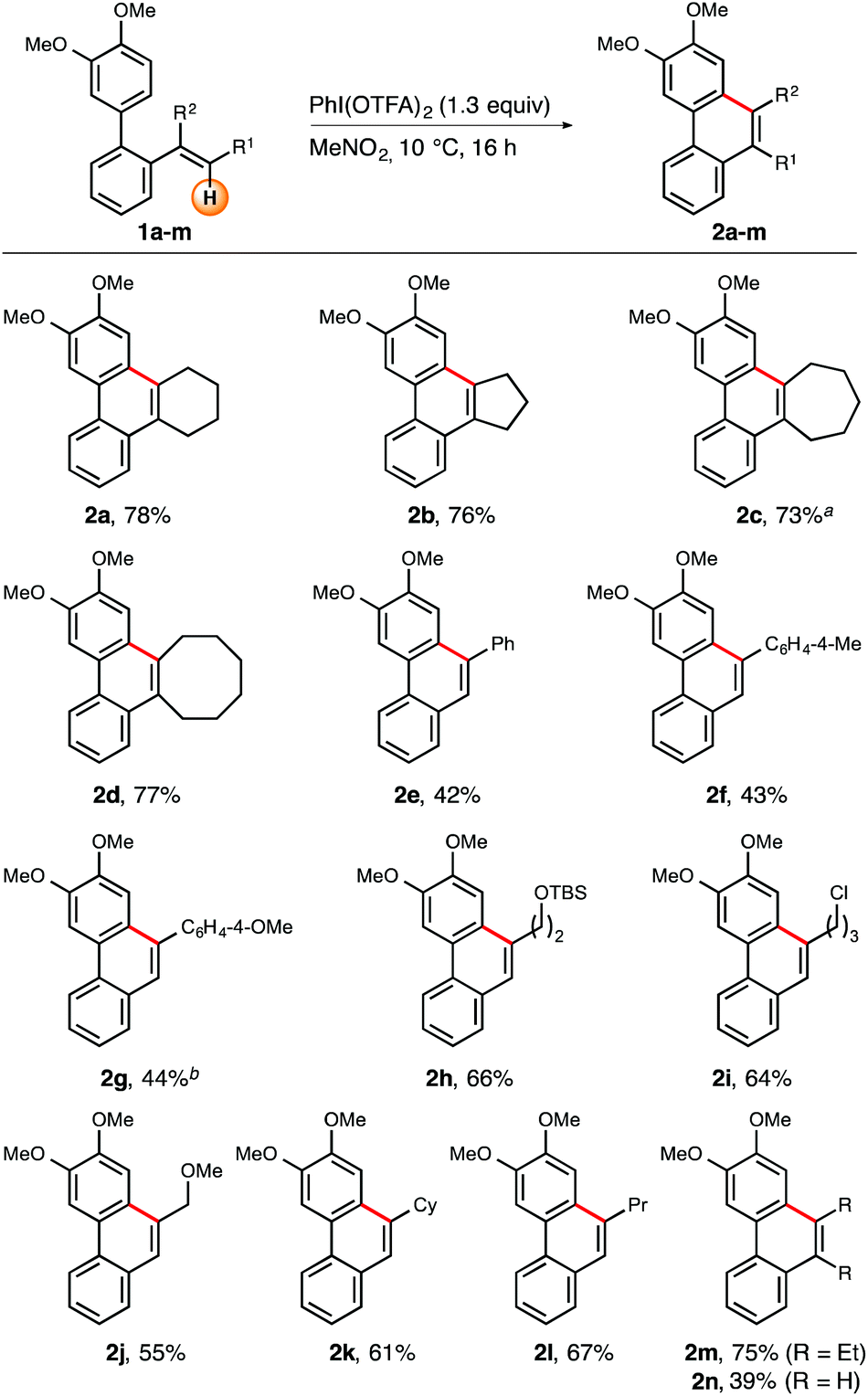 | ||
Scheme 2 Scope of the iodine(III)-mediated arene–alkene coupling with varying substituents R1 and R2. Reactions were carried out on a 0.12–0.37 mmol scale in MeNO2 (0.1 M) with 1.30 equiv. of PIFA at 10 °C. Yields refer to isolated compounds. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1.10 equiv. of PIFA were used. bReaction was performed in CH2Cl2 (0.1 M). 1.10 equiv. of PIFA were used. bReaction was performed in CH2Cl2 (0.1 M). | ||
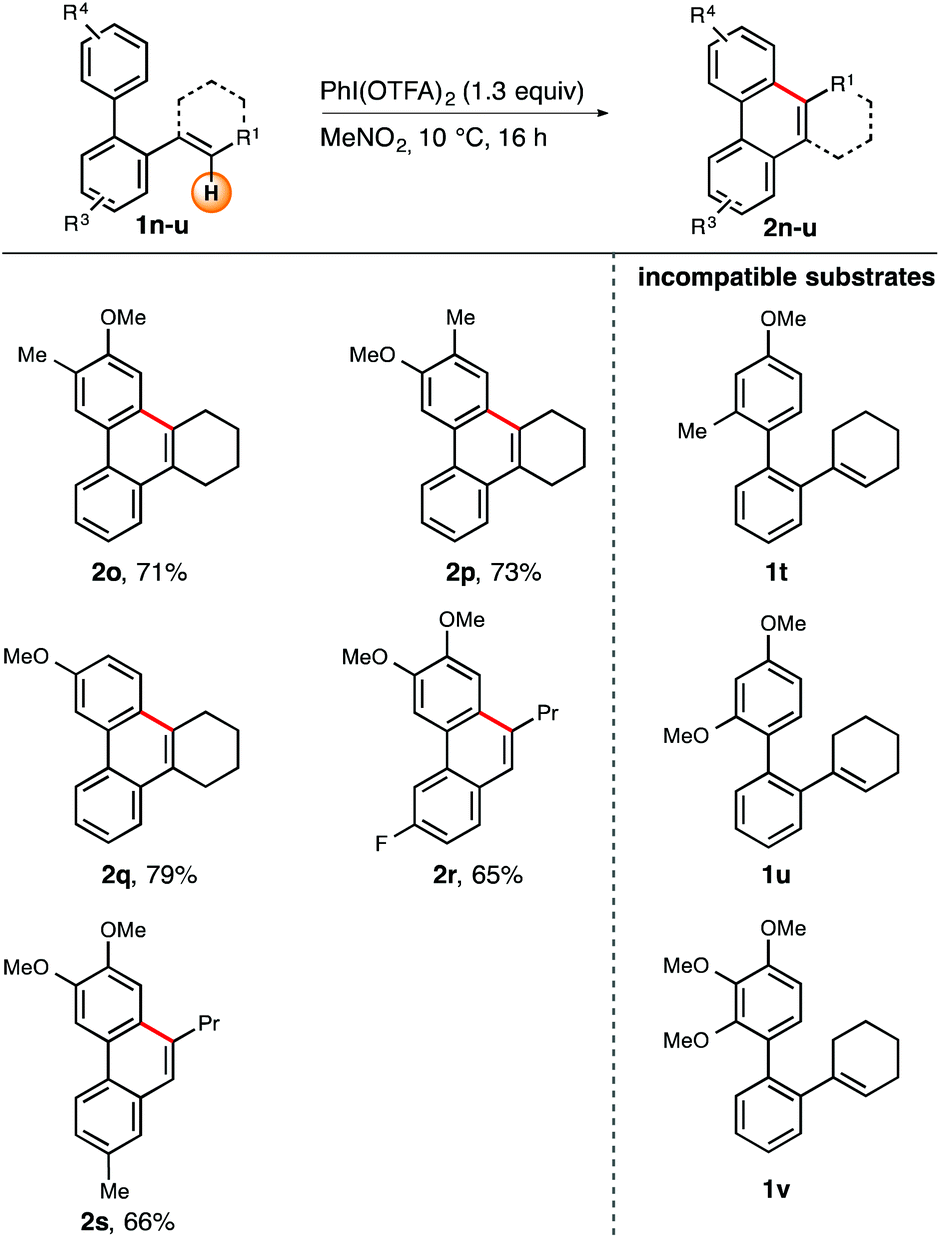 | ||
| Scheme 3 Scope of the iodine(III)-mediated arene–alkene coupling with varying substituents R1, R3, and R4. Reactions were carried out on a 0.33–0.37 mmol scale in MeNO2 (0.1 M) with 1.30 equiv. of PIFA at 10 °C. Yields refer to isolated compounds. | ||
Next, a series of 1,2-disubstituted alkenes was tested. In general, aromatic, alicyclic, and aliphatic residues at the distal position of the olefin moiety resulted in reasonable to good yields. In more detail, distally aryl- and alkyl-substituted compounds 1e–l were successively investigated. Application of arylated substrates 1e–g furnished the respective cyclization products 2e–g in moderate yields (43–48%).12 Constantly good yields were recorded for a series of β-alkyl-substituted alkene substrates (1h–l). Within this set of reactants both substituted and unsubstituted (cyclo)alkyl groups furnished the corresponding cyclization products 2h–l in reasonable yields (55–67%). Notably, even the slightly electron-deficient alkene group embedded in substrate 1j turned out to be suitable for the title reaction, giving access to phenanthrene 2j in 55% yield. A similar success was observed in the case of trisubsituted olefin 1m which resulted in an isolated yield of 75%. Even a plain vinyl group proved suitable for the oxidative cyclization reaction (substrate 1n), although the isolated yield was rather moderate (39%).
Furthermore, we wanted to briefly explore the scope and limitations with regard to the periphery of the biaryl unit (Scheme 3). Consequently, substrates 1o–v were tested in our cyclization protocol. Biaryls containing sterically undemanding, electron-rich head groups were all successfully cyclized under the title conditions. Compounds 1o–q, which vary in the substitution pattern at the top arene ring, uniformly resulted in good yields (71–79%).13 Incorporation of a fluorine residue (1r) or a methyl group (1s) into the bottom arene ring was also tolerated. The corresponding phenanthrenes 2r and 2s were obtained in 66 and 65% yield, respectively.
Unfortunately, sterically demanding arene rings, irrespective of their electron-richness, invariably led to substrate decomposition (Scheme 3, compounds 1t–v). On the basis of these observations we speculate that the above-mentioned substrates do not adopt an appropriate conformation out of which the cyclization step could occur readily. Consequently, any reactive intermediates formed during this process will then undergo undesired reaction pathways.
Finally, we wanted to determine whether the iodine(III) reagent initially activates the arene ring (e.g. via a single-electron-transfer) or the alkene unit (e.g. via an iodoiranium intermdiate) prior to the cyclization event. Kita et al. recently published a report on an iodine(III)-mediated oxidative arene–arene coupling in which it was hypothesized that the coupling occurs through the formation of a charge-transfer complex between PIFA and the corresponding arene.14,15 On the contrary, Muñiz et al. proposed the formation of an iodoiranium species in the case of iodine(III)-mediated oxidative aminations of styrene derivatives.16 Thus, we independently subjected both 4-methoxybiphenyl (3) and 1-phenylcyclohex-1-en (4) to the standard reaction conditions and subsequently analyzed the reaction mixture by 1H NMR spectroscopy (Scheme 4). While biaryl 3 displayed only marginal signs of material consumption throughout the course of 16 h in the presence of PIFA in nitromethane, we observed complete decomposition of cyclohexene derivative 4 under identical conditions. From these findings we conclude that the arene–alkene coupling most likely proceeds via initial electrophilic activation of the olefin unit followed by a SEAr vinylation of the proximal arene ring.
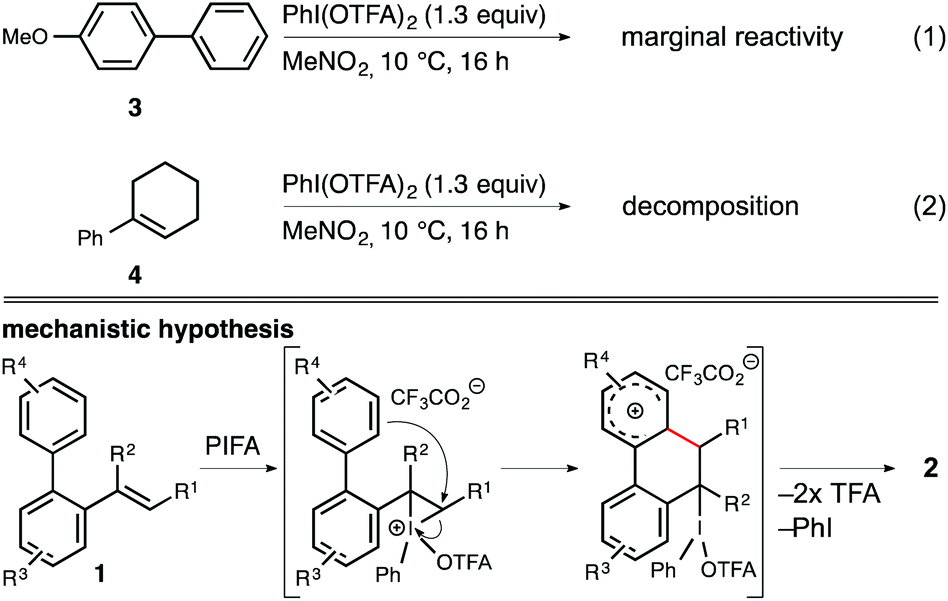 | ||
| Scheme 4 Control experiments and mechanistic hypothesis for the iodine(III)-mediated oxidative arene–alkene coupling. TFA = trifluoroacetyl (top) or trifluoroacetic acid (bottom). | ||
Conclusion
In summary, we have disclosed a novel iodine(III)-mediated oxidative arene–alkene coupling for the facile synthesis of diversely substituted phenanthrene derivatives. The title method is characterized by mild reaction conditions and outstanding 6-endo-trig selectivity. The latter observation is in stark contrast to cognate, metal-free transformations facilitated by Brønsted-acids or aminium radical cations,8a,c thus, highlighting the disparate reactivity profile exerted by PIFA. We have shown that mono-, di-, and trisubstituted alkenes are readily cyclized under the operative conditions, a feature that allows for a particularly high degree of freedom with regard to the variability of the periphery of the phenanthrene core. Consequently, we anticipate that our protocol will find ample application in the context of complex natural product synthesis. Corresponding efforts towards this goal as well as further mechanistic studies are currently ongoing in our group.Acknowledgements
This work was financially supported by the Deutsche Forschungsgemeinschaft (DFG, Emmy Noether Fellowship to A. B.) and the Fonds der Chemischen Industrie (FCI, PhD.-Fellowship to C. D. and a grant for expenditures on consumables to A. B.). We thank Prof. Dr Lutz Ackermann (University of Göttingen, Germany) for his generous infrastructural support of our work. Dr Michael John (University of Göttingen, Germany) is acknowledged for his support in the regiochemical elucidation of some of the phenanthrene products.Notes and references
- A. Kovács, A. Vasas and J. Hohmann, Phytochemistry, 2008, 69, 1084 CrossRef PubMed.
- For recent representative examples of total syntheses, see: (a) Y. Yamaoka, M. Taniguchi, K.-I. Yamada and K. Takasu, Synthesis, 2015, 47, 2819 CrossRef CAS; (b) M. Deng, B. Su, H. Zhang, Y. Liu and Q. Wang, RSC Adv., 2014, 4, 14979 RSC; (c) B. Su, H. Zhang, M. Deng and Q. Wang, Org. Biomol. Chem., 2014, 12, 3616 RSC; (d) G. Han, Y. Liu and Q. Wang, Org. Lett., 2013, 15, 5334 CrossRef CAS PubMed; (e) X. Xu, Y. Liu and C.-M. Park, Angew. Chem., Int. Ed., 2012, 51, 9372 CrossRef CAS PubMed; (f) G. Lahm, A. Stoye and T. Opatz, J. Org. Chem., 2012, 77, 6620 CrossRef CAS PubMed; (g) A. Stoye and T. Opatz, Org. Lett., 2010, 12, 2140 CrossRef CAS PubMed.
- For representative articles on the biology and biosynthesis of phenanthroindolizidine alkaloids, see: (a) A. C. B. Burtoloso, A. F. Bertonha and I. G. Rosset, Curr. Top. Med. Chem., 2014, 14, 191 CrossRef CAS PubMed; (b) A. Stoye, T. E. Peez and T. Opatz, J. Nat. Prod., 2013, 76, 275 CrossRef CAS PubMed; (c) S. R. Chemler, Curr. Bioact. Compd., 2009, 5, 2 CrossRef CAS PubMed.
- For recent representative examples, see: (a) K. Pati, C. Michas, D. Allenger, I. Piskun, P. S. Coutros, G. dos Passos Gomes and I. V. Alabugin, J. Org. Chem., 2015, 80, 11706 CrossRef CAS PubMed; (b) K. Wehming, M. Schubert, G. Schnakenburg and S. R. Waldvogel, Chem. – Eur. J., 2014, 20, 12463 CrossRef CAS PubMed; (c) K. H. Kim, C. H. Lim, J. W. Lim and J. N. Kim, Adv. Synth. Catal., 2014, 356, 697 CrossRef CAS; (d) Q. Lefebvre, M. Jentsch and M. Rueping, Beilstein J. Org. Chem., 2013, 9, 1883 CrossRef PubMed; (e) D. Ji, L. Su, K. Zhao, B. Wang, P. Hu, C. Feng, S. Xiang, H. Yang and C. Zhang, Chin. J. Chem., 2013, 31, 1045 CrossRef CAS; (f) H. Li, K.-H. He, J. Liu, B.-Q. Wang, K.-Q. Zhao, P. Hu and Z.-J. Shi, Chem. Commun., 2012, 48, 7028 RSC; (g) X. Yang, Q. Shi, K. F. Bastow and K. H. Lee, Org. Lett., 2010, 12, 1416 CrossRef CAS PubMed; (h) D. Dumoulin, S. Lebrun, A. Couture, E. Deniau and P. Grandclaudon, Eur. J. Org. Chem., 2010, 1943 CrossRef CAS; (i) A. Li, D. J. DeSchepper and D. A. Klumpp, Tetrahedron Lett., 2009, 50, 1924 CrossRef CAS.
- R. Scholl and C. Seer, Justus Liebigs Ann. Chem., 1912, 394, 111 CrossRef.
- B. T. King, J. Kroulík, C. R. Robertson, P. Rempala, C. L. Hilton, J. D. Korinek and L. M. Gortari, J. Org. Chem., 2007, 72, 2279 CrossRef PubMed.
- M. J. Niphakis and G. I. Georg, Org. Lett., 2011, 13, 196 CrossRef PubMed.
- (a) B.-H. Tan, J. Dong and N. Yoshikai, Angew. Chem., Int. Ed., 2012, 51, 9610 CrossRef CAS PubMed; (b) D. J. Gorin, I. D. G. Watson and F. D. Toste, J. Am. Chem. Soc., 2008, 130, 3736 CrossRef CAS PubMed; (c) R. Lapouyade, P. Villeneuve, A. Nourmamode and J.-P. Morand, J. Chem. Soc., Chem. Commun., 1987, 776 RSC.
- For a recent review on metal-free coupling reactions, see: C.-L. Sun and Z.-J. Shi, Chem. Rev., 2014, 114, 9219 CrossRef CAS.
- (a) S. E. Denmark, D. J. P. Kornfilt and T. Vogler, J. Am. Chem. Soc., 2011, 133, 15308 CrossRef CAS PubMed; (b) S. E. Denmark, D. Kalyani and W. R. Collins, J. Am. Chem. Soc., 2010, 132, 15752 CrossRef CAS PubMed.
- For recent review articles on iodine(III)-mediated oxidation reactions, see: (a) R. Narayana, S. Manna and A. P. Antonchick, Synlett, 2015, 26, 1785 CrossRef; (b) P. Finkbeiner and B. J. Nachtsheim, Synthesis, 2013, 45, 979 CrossRef CAS.
- Compound 2f was obtained as a 92:8 mixture of the 3-methoxy and the 1-methoxy isomer.
- Compound 2o was obtained as a 85:15 mixture of the 7-methyl and the 1-methyl isomer.
- T. Dohi, M. Ito, K. Morimoto, M. Iwata and Y. Kita, Angew. Chem., Int. Ed., 2008, 47, 1301 CrossRef CAS PubMed.
- R. Narayan, K. Matcha and A. P. Antonchick, Chem. – Eur. J., 2015, 21, 14678 CrossRef CAS.
- J. A. Souto, D. Zian and K. Muñiz, J. Am. Chem. Soc., 2012, 134, 7242 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5qo00361j |
| This journal is © the Partner Organisations 2016 |