DOI:
10.1039/C2RA20955A
(Communication)
RSC Adv., 2012,
2, 6144-6149
A facile synthesis of cysteine–ferrite magnetic nanoparticles for application in multicomponent reactions—a sustainable protocol†
Received
16th May 2012
, Accepted 16th May 2012
First published on 13th June 2012
Abstract
A facile, simple and environmentally friendly Fe3O4–cysteine MNP was synthesized without any additive or additional source of linkers. Fe3O4–cysteine MNPs were successfully used for the synthesis of β-amino carbonyl and hydroquinoline compounds, which were obtained in excellent yields via multicomponent reactions. Magnetic organocatalysts can be easily recovered by simple magnetic decantation and their catalytic activity remains unaltered after 9 consecutive cycles, making them environmentally friendly and widely applicable due to their efficiency, ease of handling, and cost effectiveness.
Introduction
Magnetic nanomaterials are envisaged to have a major impact in many areas, including biotechnology/biomedicine, environmental remediation, and especially catalysis.1 Due to their nanometre size and magnetic properties, they are useful as quasi homogeneous systems that combine the advantages of high dispersion, high reactivity, and easy separation. They are accessible from inexpensive materials via a simple synthesis and can be easily tuned by structural surface modifications.2 During the past decade, organocatalysis, a metal-free approach to the synthesis of organic molecules, has become a very significant area of research attracting worldwide attention.3 This relatively green approach has been rendered even greener by immobilization of the organocatalysts on solid supports4 and by the use of non-conventional energy sources.5 In this reaction system the catalyst is separated by filtration or centrifugation and further used for the next cycle. Functionalized magnetic nanoparticles (MNPs) are a heterogeneous catalyst support that have emerged as viable alternatives to conventional materials, being robust, readily available and having a high surface-area, which allows the design of many catalytic sites.6 They offer an added advantage of being magnetically separable, thereby eliminating the requirement for catalyst filtration after completion of the reaction. Owing to the potential features and advantages depicted above, the supporting of organocatalysts onto well-defined nano sized MNPs appears to be an interesting alternative for the synthesis of easily recoverable and recyclable catalysts for application in organic synthesis. Notably, MNPs that endorse efficient one-pot synthesis have become an important field of sustainable chemistry and catalysis science. Examples are the functionalization of ferrite MNPs by the naturally abundant glutathione, allowing the synthesis of a recyclable organocatalyst showing excellent activity for the Paal–Knorr reaction,7 surface functionalization of γ-Fe2O3 through the design of a versatile ligand for copper(I) catalyzed azide–alkyne cycloaddition,8 ferrite MNP supported (S)-diphenylprolinol trimethylsilyl ether for the organocatalytic Michael reaction,9 immobilization of a BINAP type ligand onto ferrite and its application in hydrogenation reactions,6c carbon coated cobalt nanoparticles tagged with azabis(oxazoline)-copper(II) complexes and its applications for the monobenzoylation reaction,10 immobilization of carboxyl-functionalised chiral amino alcohols onto cobalt nanoparticles for the hydrogenation reaction,11 functionalization of a siliceous mesocellular foam for Ru-mediated transfer hydrogenation,12 and a magnetic nanoparticle supported primary amine catalyst for the asymmetric aldol reaction.13 Also, functionalized MNPs can be useful in such diverse fields as, for example, for the rapid removal of metals from contaminated water by the attachment of EDTA chelators to carbon coated metal nanomagnets.14 But the main drawbacks of some of these methods are the resources needed for an expensive multistep synthesis, and the use of linkers, ligands and spacers. Hence, there is an urgent need to develop viable protocols for the coating of MNPs. In continuation of our research on the development of greener protocols,15 new methodologies and nanocatalysis,16 we have designed a new, simple, and efficient method for the functionalization of ferrite MNPs with cysteine (Cys) 1 and studied their catalytic application in multicomponent reactions.
Results and discussion
Fe3O4–Cys MNPs characterization
The organocatalyst-cysteine MNPs (Fe3O4–Cys) 2, were prepared from Fe3O4 and cysteine 1 by simple mechanical stirring (Scheme 1).17 Notably, no external additives, linkers or expensive ligands were used. Interestingly, the interaction between the ferrites and cysteine could be due to weak forces (hydrogen bonding and van der Waals interaction). The synthesized catalyst 2 was characterized by X-ray diffraction (XRD), Fourier transform infrared spectroscopy (FT-IR), secondary ion mass spectrometry (SIMS), transmission electron microscopy (TEM), field-emission gun scanning electron microscopy and electron dispersive spectrometry (FEG-SEM-EDS). The XRD spectrum of Fe3O4–Cys 2 is depicted in Fig. 1 and compared to the cysteine and Fe3O4-MNPs spectra. The XRD spectrum of Fe3O4–Cys clearly reveals most of the cysteine peaks, with the notable exception of the 2θ = 24.60° peak. The crystallite size of Fe3O4–cysteine was determined by the Debye Scherrer equation and was found to be 18 nm. To confirm the presence of cysteine on the surface of the ferrites, we have characterized the catalyst by SIMS. The spectra were collected in positive and negative modes, without any pre-sputtering in the DC mode, and are shown in Fig. 2. In both spectra the presence of cysteine was confirmed from the protonated [M+H]+ (121+1 = 122) cysteine molecule in the positive spectrum and the deprotonated [M+H]− molecule in both modes. From analysis of FEG-SEM images using a 25 kV acceleration voltage, uniform-sized magnetic nanoparticles with somewhat spherical morphology and a marked tendency to form large clusters were observed (Fig. 3). This tendency is not surprising, given the MNPs’ small size and magnetic characteristics.
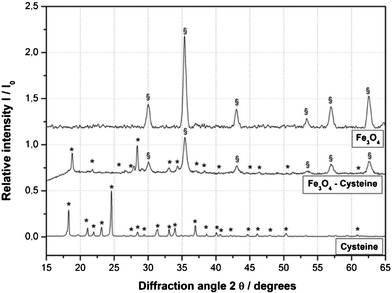 |
| | Fig. 1 XRD spectrum of Fe3O4–Cys 2. | |
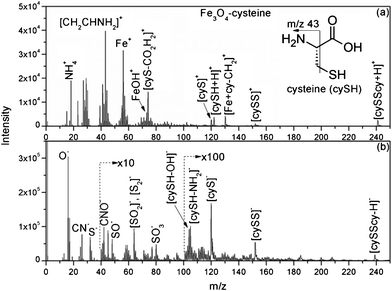 |
| | Fig. 2 (a) Positive and (b) negative TOF-SIMS mass spectra of 2. | |
 |
| | Fig. 3 SEM images of Fe3O4–Cys 2 at 100 nm. | |
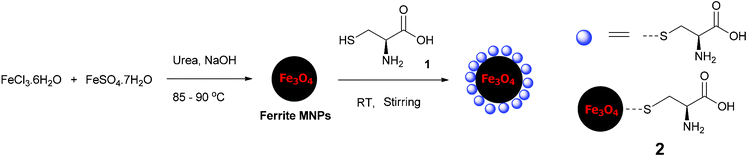 |
| | Scheme 1 Synthesis of Fe3O4–Cys MNPs 2. | |
From EDS analysis, the sulfur peak clearly confirms the presence of cysteine (Fig. 4). The TEM images of ferrite and ferrite–cysteine are shown in (Fig. 5a,b). The nanoparticles are observed in the range 10–30 nm. Notwithstanding the cluster tendency formerly observed (Fig. 3), small groups of equiaxial particles, almost entirely detached from each other, are discernible. Clustering tendencies could be due to weak forces (hydrogen bonding) between the Fe3O4–Cys molecules. A histogram based on TEM measurements was established, showing a size distribution in agreement with the XRD indications (ESI†).
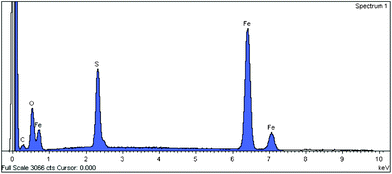 |
| | Fig. 4 EDS spectrum of Fe3O4–Cys 2. | |
 |
| | Fig. 5 (a) TEM image of Fe3O4 at 200 nm. (b) TEM image of Fe3O4–Cys at 100 nm. | |
Multicomponent reaction catalyzed by Fe3O4–Cys MNPs
The synthesized and well characterized Fe3O4–Cys MNPs were explored for the synthesis of β-amino carbonyl compounds 3via the Mannich reaction (Scheme 2). The catalytic activity of 2 was initially tested for the model reaction between aniline 4a, benzaldehyde 5a and cyclohexanone 6 under various solvents and solvent-free conditions (Table 1). Moderate to high selectivity was observed in ethanol and water although with opposite results. In water, the anti-isomer is formed preferentially (15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :85 in 75% yield, Table 1 entry (b)) and in ethanol the syn selectivity prevails (70:30 in 55% yield, Table 1 entry (f)). Although the syn intermediate is less favourable due to steric interactions, the possibility of chelation with the catalyst can lower its energy (Scheme 3). Water as a solvent can provide polar and other interactions18 which can disrupt the ferrite syn-intermediate, favouring the other and more stable anti-intermediate. The anti and syn isomers were identified by the coupling constants (J) of the vicinal protons adjacent to C
:85 in 75% yield, Table 1 entry (b)) and in ethanol the syn selectivity prevails (70:30 in 55% yield, Table 1 entry (f)). Although the syn intermediate is less favourable due to steric interactions, the possibility of chelation with the catalyst can lower its energy (Scheme 3). Water as a solvent can provide polar and other interactions18 which can disrupt the ferrite syn-intermediate, favouring the other and more stable anti-intermediate. The anti and syn isomers were identified by the coupling constants (J) of the vicinal protons adjacent to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and NH in their 1H NMR spectra.19 The coupling constant (J) for the anti-isomer is higher than that of the syn. The anti/syn ratio was determined by 1H NMR, judged by the intensity of the H1′ for the syn isomer. The reaction was extended to a series of aldehydes and arylamines, yielding the corresponding β-amino ketones 3 (Table 2) in excellent yields (89–93%) at room temperature and under solvent-free conditions, which render the chemical process more sustainable.
O and NH in their 1H NMR spectra.19 The coupling constant (J) for the anti-isomer is higher than that of the syn. The anti/syn ratio was determined by 1H NMR, judged by the intensity of the H1′ for the syn isomer. The reaction was extended to a series of aldehydes and arylamines, yielding the corresponding β-amino ketones 3 (Table 2) in excellent yields (89–93%) at room temperature and under solvent-free conditions, which render the chemical process more sustainable.
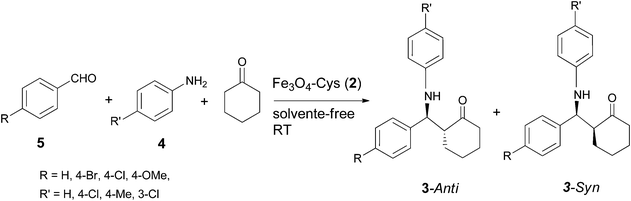 |
| | Scheme 2 Mannich reaction and synthesis of β-amino carbonyl compounds 3 catalysed by Fe3O4–Cys 2. | |
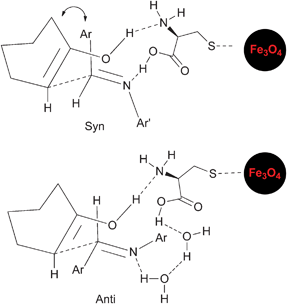 |
| | Scheme 3 Proposed syn- and anti-intermediates. | |
Table 1 Mannich reaction: optimization of reaction conditionsa
| Entry |
Catalyst |
Solvent |
Yield |
syn:anti |
|
Reaction conditions: benzaldehyde 5a (5 mmol), aniline 4a (5 mmol), cyclohexanone 6 (10 mmol), Fe3O4–Cys 2, 5 wt% with respect to aldehyde, time 2 h, room temperature (22 °C).
20% yield of imine as a side product.
10 wt% of catalyst used.
No reusability of cysteine.
|
| a |
No catalyst |
No solvent |
Trace |
— |
| b |
2
|
H2O |
75 |
15:85 |
| c |
2
|
CH3CN |
58 |
40:60 |
| d |
2
|
MeCO2Et |
62 |
54:46 |
| e |
2
|
MeOH |
60 |
43:57 |
| f |
2
|
EtOH |
55 |
70:30 |
| g |
2
|
No solvent |
93 |
45:55 |
| h |
2
|
No solvent |
95 |
45:55 |
| i |
Cysteined |
No solvent |
40 |
— |
| j |
Fe3O4 |
No solvent |
Trace |
— |
Table 2 Yields and selectivity for the β-amino ketones 3 synthesizeda
| Entry |
Aldehyde 5R |
Aniline 4R |
Time (h)/yield (%) |
syn:antic |
|
Reaction conditions: aldehyde 5 (5 mmol), arylamine 4 (5 mmol), cyclohexanone 6 (10 mmol), Fe3O4–Cys 2, 5 wt% respectively to aldehyde, room temperature.
Yield after 9 cycles.
Diastereomeric ratio determined by 1H NMR spectroscopy.
|
| a |
H |
H |
2/93 |
45:55 |
|
|
|
|
2/90b |
45:55 |
| b |
4-Br |
H |
2.5/90 |
49:51 |
| c |
4-Cl |
H |
2.5/90 |
45:55 |
| d |
H |
4-Cl |
3/92 |
49:51 |
| e |
H |
4-Me |
2.5/92 |
42:58 |
| f |
4-OMe |
H |
2/89 |
38:62 |
| g |
H |
3-Cl |
2.5/93 |
46:54 |
The stability of Fe3O4–Cys was tested for the synthesis of 3a by recycling and reuse of the catalyst nine times without any significant loss of catalytic activity or selectivity (ESI†). After each cycle, Fe3O4–Cys was separated using a simple magnet and used for the next cycle. Also the morphology of Fe3O4–Cys after nine consecutive reaction cycles was verified by TEM. Slight changes in the morphology of the particles were observed, and a significant increase in particle clustering is clearly discernible (Fig. 6).
 |
| | Fig. 6 (a) TEM image of Fe3O4–Cys at 200 nm. (b) TEM image of Fe3O4–Cys at 200 nm after 9 cycles of reuse. | |
Interestingly, the present protocol is highly chemoselective, as no aldol product was observed from the reaction of aldehyde with cyclohexanone.
The higher activity and stability of Fe3O4–Cys inspired us to explore its catalytic activity for the Hantzsch synthesis of hydroquinolines, a scaffold that has significant interest in medicinal chemistry due to the biological and pharmacological properties it can present.20 Therefore, the multicomponent synthesis between 3,4-diphenoxy benzaldehyde, ammonium acetate, ethyl acetoacetate and 5,5-dimethylcyclohexane-1,3-dione catalysed by Fe3O4–Cys under mild conditions provided the hydroquinoline 7 in 88% yield (Scheme 4).
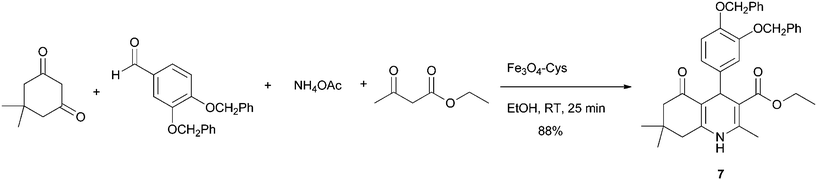 |
| | Scheme 4 Multicomponent synthesis between 3,4-bis(benzyloxy)benzaldehyde, ammonium acetate, ethyl acetoacetate and 5,5-dimethylcyclohexane-1,3-dione catalyzed by Fe3O4–Cys 2. | |
Conclusions
In summary, the cysteine homogeneous moiety was easily immobilized on the ferrite surface by simple mechanical stirring without any additive and additional source of linkers. The synthesized Fe3O4–Cys MNPs were successfully used for the synthesis of β-amino carbonyl compound derivatives and hydroquinolines. The magnetic organocatalyst is easily prepared, stable to air and magnetically recoverable by simple magnetic decantation. The catalytic activity of the catalyst remains unaltered after 9 consecutive cycles, making it environmentally friendly and widely applicable due to its efficiency, ease of handling, and cost effectiveness. We believe that this approach towards sustainable chemistry has great scope for future catalytic processes.
Experimental
General methods and experimental procedures
All commercial reagents were used as received unless otherwise mentioned. For analytical and preparative thin-layer chromatography, Merck, 0.2 mm and 0.5 mm Kieselgel GF 254 percoated were used, respectively. The spots were visualized using UV light. The X-ray powder diffraction pattern was obtained using a conventional powder diffractometer RIGAKU, model: MiniFlex™ II benchtop X-ray diffractometer; X-ray tube: Cu-Kα (30 kV/15 mA) radiation operating in Bragg–Brentano (θ/2θ) geometry. (Sample preparation: grinding when needed and compression in the sample holder with a flat glass. The sample area in the sample holder is about 2 cm2.) Transmission electron microscopy (TEM) experiments were performed on a Hitachi H8100 microscope, with a ThermoNoran light elements EDS detector and a CCD camera for image acquisition. The Fe3O4–Cys fine powder was placed on a carbon stub and the images were recorded at 5–15 kV using a LFD detector under low vacuum.
Elemental analysis was done by using an ICP-AES (inductively coupled plasma-atomic emission spectrometer) using a Horiba Jobin-Yvon, France, Ultima model equipped with a 40.68 MHz RF generator, Czerny–Turner monochromator with 1.00 m (sequential) autosampler AS500 and CMA (concomitant metals analyzer).
Scanning electron microscopy images were acquired using a JEOL JSM7001F FEG-SEM. Elemental analysis was performed using a light elements EDS detector from Oxford. The ferrite–cysteine powder was spread on double-sided carbon tape and analyzed using a 25 kV acceleration voltage.
For SIMS, (secondary ion mass spectrometry) positive and negative secondary ion spectra were collected in the mass range 0.5–500 m/z (T = 10 min) with an upgraded VG Ionex IX23LS TOF-SIMS set-up based on the Poschenrieder design. A focused liquid Ga+ gun in pulsed mode (6 kHz) was used as the source of analytical ions. A beam current in dc mode at 14 keV was ca. 15 nA with a raster size of 300 × 300 μm2. The sample potential was 5 kV. Vacuum during the experiments was maintained in the range 2–3 × 10−9 mbar in the analytical chamber.
Infrared spectra were recorded on a Perkin Elmer spectrum 1000. 1H NMR spectra were recorded on a Bruker ARX400 spectrometer at 400 MHz. 1H shifts are reported relative to internal TMS. Mass spectra were recorded at the Mass Spectrometry Unit at the University of Santiago de Compostela, Spain using a magnetic Micromass Autospec apparatus.
Preparation of ferrites/Fe3O4
FeCl3·6H2O (5.4 g) and urea (3.6 g) were dissolved in water (200 mL) at 85 to 90 °C for 2 h. The solution turned brown in color. The resultant reaction mixture was cooled to room temperature, to which was added FeSO4·7H2O (2.8 g) and then 0.1 M NaOH until the pH was 10. The molar ratio of Fe(III):Fe(II) in the above system was nearly 2.00. The obtained hydroxides were treated with ultrasound in the sealed flask at 30 to 35 °C for 30 min. After ageing for 5 h the obtained black powder of Fe3O4 was washed, and dried under vacuum.
Preparation of Fe3O4–cysteine
Fe3O4 MNPs (1 gm) were dispersed in 20 mL distilled water and L-cysteine (1 g) dissolved in 40–50 mL methanol–water (1:1) was added to the dispersed ferrite MNPs and the reaction mixture stirred at room temperature for 24 h (1200 RPM) using a magnetic stirrer device. The Fe3O4–Cys MNPs were then isolated by simple magnetic decantation and successively washed with water (2 × 10 mL) and methanol (2 × 10 mL) and dried under vacuum at 60 °C for 2 h.
Synthesis of β-amino ketones (3)
A mixture of benzaldehyde 5 (5.0 mmol), aniline 4 (5.0 mmol), cyclohexanone 6 (10 mmol), and Fe3O4–Cys 2 (5 wt % respectively to aldehyde) was stirred under solvent-free conditions at room temperature for an appropriate time. After completion of the reactions monitored by TLC (n-hexane and ethyl acetate; 70:30), 10 mL ethyl acetate was added to the reaction mixture and the was catalyst separated magnetically. The organic layer was washed with saturated NaHCO3(aq) and brine, dried with anhydrous Na2SO4 and concentrated to dryness. The crude mixture was purified by crystallization in ethanol or, if necessary, subjected to flash column chromatography.
Synthesis of hydroquinoline 7
A solution of 3,4-diphenoxy benzaldehyde (5.0 mmol), ammonium acetate (10 mmol), ethyl acetoacetate (5 mmol), 5,5-dimethylcyclohexane-1,3-dione aldehyde (5 mmol) and Fe3O4–Cys (5 wt % respectively to aldehyde) in ethanol (3 mL) was stirred at room temperature for 25 min. After terminus of the reaction monitored by TLC (n-hexane and ethyl acetate; 70:30), the catalyst was separated magnetically. To the solution was added ethyl acetate (10 mL). The organic layer was washed with a saturated aqueous NaHCO3 solution and brine, dried with anhydrous Na2SO4, and concentrated to dryness.
The crude mixture was purified by crystallization in ethanol and the ethyl 4-(3,4-bis(benzyloxy)phenyl)-2,7,7-trimethyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate 7 was obtained in 88% yield: IR(KBr) υmax: 3284, 3206, 1694, 1640, 1605 cm−1; 1H NMR (400 MHz, CDCl3) δ: 7.42-7.28 (m, 10H, PhH), 6.90 (s, 1H, ArH2), 6.80 (d, 1H, ArH5/6), 6.77 (d, 1H, ArH5/6), 5.87 (bs, 1H, NH), 5.08 (d, 4H, OCH2Ph), 4.96 (s, 1H, H4), 4.01 (q, 2H, OCH2CH3), 2.31 (s, 3H, CH3), 2.28-2.05 (m, 4H, CH2, CH2), 1.17 (t, 3H, OCH2CH3), 1.05 (s, 3H, CH3), 0.90 (s, 3H, CH3); 13C NMR δ: 14.23 (CH3), 19.4 (CH3), 27.2 (CH3), 29.3 (CH3), 31.8 (C7), 35.6 (C4), 41.0 (C6/8), 50.45 (C6/8), 59.8 (OCH2), 71.36 (OCH2), 106.3 (C10), 112.05 (C3), 114.5 (ArC5), 115.36 (ArC2), 120.9 (ArC6), 127.3 (Ph), 127.5 (Ph), 127.6 (Ph), 128.4 (Ph), 137.6 (Ph), 137.8 (Ph), 140.5 (C2), 142.9 (C9), 147.3 (ArC4), 148.4 (ArC3), 167.3 (CO), 195.4 (CO); MSEI(+) m/z: 551 [M]+, 262 [M-PhCH2O)2C6H3]+, 91 [C7H7]+. HRMSEI(+) calcd for C35H37NO5 [M]+ 551.2672 found 551.2668.
Acknowledgements
This work has been supported by Fundação para a Ciência e a Tecnologia through grant no. PEst-C/EQB/LA0006/2011. The NMR spectrometers are part of the National NMR Network (RNRMN) and are funded by Fundação para a Ciência e a Tecnologia (FCT). Manoj B. Gawande also thank the PRAXIS program for the award of research fellowship SFRH/BPD/64934/2009.
References
-
(a) A. H. Lu, E. L. Salabas and F. Schuth, Angew. Chem., Int. Ed., 2007, 46, 1222–1244 CrossRef CAS;
(b) J. M. Perez, Nat. Nanotechnol., 2007, 2, 535–536 CrossRef CAS.
- A. H. Latham and M. E. Williams, Acc. Chem. Res., 2008, 41, 411–420 CrossRef CAS.
- D. W. C. MacMillan, Nature, 2008, 455, 304–308 CrossRef CAS.
- M. Gruttadauria, F. Giacalone and R. Noto, Chem. Soc. Rev., 2008, 37, 1666–1688 RSC.
- A. Bruckmann, A. Krebs and C. Bolm, Green Chem., 2008, 10, 1131–1141 RSC.
-
(a) A. H. Lu, W. Schmidt, N. Matoussevitch, H. Bonnemann, B. Spliethoff, B. Tesche, E. Bill, W. Kiefer and F. Schuth, Angew. Chem., Int. Ed., 2004, 43, 4303–4306 CrossRef CAS;
(b) H. M. R. Gardimalla, D. Mandal, P. D. Stevens, M. Yen and Y. Gao, Chem. Commun., 2005, 4432–4434 RSC;
(c) A. G. Hu, G. T. Yee and W. B. Lin, J. Am. Chem. Soc., 2005, 127, 12486–12487 CrossRef CAS;
(d) D. Lee, J. Lee, H. Lee, S. Jin, T. Hyeon and B. M. Kim, Adv. Synth. Catal., 2006, 348, 41–46 CrossRef CAS;
(e) R. Abu-Reziq, H. Alper, D. S. Wang and M. L. Post, J. Am. Chem. Soc., 2006, 128, 5279–5282 CrossRef CAS;
(f) M. Kawamura and K. Sato, Chem. Commun., 2006, 4718–4719 RSC;
(g) N. T. S. Phan, C. S. Gill, J. V. Nguyen, Z. J. Zhang and C. W. Jones, Angew. Chem., Int. Ed., 2006, 45, 2209–2212 CrossRef CAS;
(h) M. Shokouhimehr, Y. Z. Piao, J. Kim, Y. J. Jang and T. Hyeon, Angew. Chem., Int. Ed., 2007, 46, 7039–7043 CrossRef CAS;
(i) F. Shi, M. K. Tse, M. M. Pohl, A. Bruckner, S. M. Zhang and M. Beller, Angew. Chem., Int. Ed., 2007, 46, 8866–8868 CrossRef CAS;
(j) S. Z. Luo, X. X. Zheng, H. Xu, X. L. Mi, L. Zhang and J. P. Cheng, Adv. Synth. Catal., 2007, 349, 2431–2434 CrossRef CAS;
(k) G. Chouhan, D. S. Wang and H. Alper, Chem. Commun., 2007, 4809–4811 RSC;
(l) T. Arai, T. Sato, H. Kanoh, K. Kaneko, K. Oguma and A. Yanagisawa, Chem.–Eur. J., 2008, 14, 882–885 CrossRef CAS;
(m) J. M. Liu, X. G. Peng, W. Sun, Y. W. Zhao and C. G. Xia, Org. Lett., 2008, 10, 3933–3936 CrossRef CAS.
- V. Polshettiwar, B. Baruwati and R. S. Varma, Chem. Commun., 2009, 1837–1839 RSC.
- M. A. White, J. A. Johnson, J. T. Koberstein and N. J. Turro, J. Am. Chem. Soc., 2006, 128, 11356–11357 CrossRef CAS.
-
(a) P. Riente, C. Mendoza and M. A. Pericas, J. Mater. Chem., 2011, 21, 7350–7355 RSC;
(b) B. G. Wang, B. C. Ma, Q. O. Wang and W. Wang, Adv. Synth. Catal., 2010, 352, 2923–2928 CrossRef CAS.
- A. Schatz, R. N. Grass, Q. Kainz, W. J. Stark and O. Reiser, Chem. Mater., 2010, 22, 305–310 CrossRef.
- F. Michalek, A. Lagunas, C. Jimeno and M. A. Pericas, J. Mater. Chem., 2008, 18, 4692–4697 RSC.
- J. Li, Y. M. Zhang, D. F. Han, Q. Gao and C. Li, J. Mol. Catal. A: Chem., 2009, 298, 31–35 CrossRef CAS.
- Y. Jiang, C. Guo, H. Xia, I. Mahmood and H. Liu, Ind. Eng. Chem. Res, 2008, 47, 9648–9635 Search PubMed.
- F. M. Koehler, M. Rossier, M. Waelle, E. K. Athanassiou, L. K. Limbach, R. N. Grass, D. Gunther and W. J. Stark, Chem. Commun., 2009, 4862–4864 RSC.
-
(a) M. B. Gawande, S. S. Deshpande, S. U. Sonavane and R. V. Jayaram, J. Mol. Catal. A: Chem., 2005, 241, 151–155 CrossRef CAS;
(b) M. B. Gawande and R. V. Jayaram, Catal. Commun., 2006, 7, 931–935 CrossRef CAS;
(c) M. B. Gawande, V. Polshettiwar, S. V. B. Rajender and R. V. Jayaram, Tetrahedron Lett., 2007, 48, 8170–8173 CrossRef CAS;
(d) P. S. Branco, V. P. Raje, J. Dourado and J. Gordo, Org. Biomol. Chem., 2010, 8, 2968–2974 RSC;
(e) M. B. Gawande and P. S. Branco, Green Chem., 2011, 13, 3355–3359 RSC.
-
(a) S. U. Sonavane, M. B. Gawande, S. S. Deshpande, A. Venkataraman and R. V. JayaraM, Catal. Commun., 2007, 8, 1803–1806 CrossRef CAS;
(b) M. B. Gawande, P. S. Branco, K. Parghi, J. J. Shrikhande, R. K. Pandey, C. A. A. Ghumman, N. Bundaleski, O. Teodoro and R. V. Jayaram, Catal. Sci. Technol., 2011, 1, 1653–1664 RSC.
- O. Gleeson, R. Tekoriute, Y. K. Gun'ko and S. J. Connon, Chem.–Eur. J., 2009, 15, 5669–5673 CrossRef CAS.
- X. Y. Zhang and K. N. Houk, J. Org. Chem., 2005, 70, 9712–9716 CrossRef CAS.
- H. Wu, Y. Shen, L. Y. Fan, Y. Wan, P. Zhang, C. F. Chen and W. X. Wang, Tetrahedron, 2007, 63, 2404–2408 CrossRef CAS.
-
(a) R. D. Shan, C. Velazquez and E. E. Knaus, J. Med. Chem., 2004, 47, 254–261 CrossRef CAS;
(b) M. Kawase, A. Shah, H. Gaveriya, N. Motohashi, H. Sakagami, A. Varga and J. Molnar, Bioorg. Med. Chem., 2002, 10, 1051–1055 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.