
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIdentification of new inhibitors of NS5 from dengue virus using saturation transfer difference (STD-NMR) and molecular docking studies†
Asmat Ullah a,
Atia-tul-Wahab*a,
Peng Gongc,
Abdul Mateen Khanb and
M. Iqbal Choudhary*abd
a,
Atia-tul-Wahab*a,
Peng Gongc,
Abdul Mateen Khanb and
M. Iqbal Choudhary*abd
aDr Panjwani Center for Molecular Medicine and Drug Research, International Center of Chemical and Biological Sciences, University of Karachi, Karachi, 75270, Pakistan. E-mail: atia.tulwahab@iccs.edu; iqbal.choudhary@iccs.edu
bH.E.J. Research Institute of Chemistry, International Center for Chemical and Biological Sciences, University of Karachi, Karachi, 75270, Pakistan. E-mail: iqbal.choudhary@iccs.edu
cWuhan Institute of Virology, Chinese Academy of Sciences, Wuhan, Hubei 430071, China
dDepartment of Biochemistry, Faculty of Science, King Abdulaziz University, Jeddah-21589, Saudi Arabia
First published on 21st December 2022
Abstract
The rapid spread of dengue virus has now emerged as a major health problem worldwide, particularly in tropical and sub-tropical regions. Nearly half of the human population is at risk of getting infection. Among the proteomes of dengue virus, nonstructural protein NS5 is conserved across the genus Flavivirus. NS5 comprises methyltransferase enzyme (MTase) domain, which helps in viral RNA capping, and RNA-dependent RNA polymerase (RdRp) domain, which is important for the virus replication. Negative modulation of NS5 decreases its activity and associated functions. Despite recent advances, there is still an immense need for effective approaches toward drug discovery against dengue virus. Drug repurposing is an approach to identify the new therapeutic indications of already approved drugs, for the treatment of both common and rare diseases, and can potentially lower the cost, and time required for drug discovery and development. In this study, we evaluated 75 compounds (grouped into 15 mixtures), including 13 natural compounds and 62 drugs, by using biophysical methods, for their ability to interact with NS5 protein, which were further validated by molecular docking and simulation studies. Our current study led to the identification of 12 ligands, including both 9 US-FDA approved drugs and 3 natural products that need to be further studied as potential antiviral agents against dengue virus.
1 Introduction
Dengue virus belongs to the positive strand RNA family of viruses Flaviviridae. This family includes dengue virus (DENV), West Nile virus (WNV), yellow fever virus (YFV), Japanese encephalitis virus (JEV), and tick-borne encephalitis virus. According to a WHO report, over 390 million people are infected annually from dengue, 96 million of whom show severe clinical symptoms, while 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 people die annually.1 DENV affects the whole world, although 67% of the actual clinical burden is reported in Asian countries (https://www.who.int/emergencies/disease-outbreak-news/item/dengue-fever-pakistan, 2021). According to a recent report (2021), a total of 48906 cases of dengue, including 183 deaths, have been reported in Pakistan (https://www.who.int/, 2021). DENV is transferred to humans through the bite of infected Aedes aegypti and Aedes albopictus mosquitos,2 as well as via the maternal–fetal mode of transmission.3,4 There are 4 serotypes of DENV (DENV-1, DENV-2, DENV-3, and DENV-4), sharing 65% of their genome similarity, while all are antigenically distinct.5 All the serotypes have the capacity to cause mild symptomatic dengue fever (DF) to severe dengue hemorrhage fever (DHF), coagulopathy, increased vascular fragility and permeability, followed by a condition called dengue shock syndrome DSS (hypovolemic shock).
000 people die annually.1 DENV affects the whole world, although 67% of the actual clinical burden is reported in Asian countries (https://www.who.int/emergencies/disease-outbreak-news/item/dengue-fever-pakistan, 2021). According to a recent report (2021), a total of 48906 cases of dengue, including 183 deaths, have been reported in Pakistan (https://www.who.int/, 2021). DENV is transferred to humans through the bite of infected Aedes aegypti and Aedes albopictus mosquitos,2 as well as via the maternal–fetal mode of transmission.3,4 There are 4 serotypes of DENV (DENV-1, DENV-2, DENV-3, and DENV-4), sharing 65% of their genome similarity, while all are antigenically distinct.5 All the serotypes have the capacity to cause mild symptomatic dengue fever (DF) to severe dengue hemorrhage fever (DHF), coagulopathy, increased vascular fragility and permeability, followed by a condition called dengue shock syndrome DSS (hypovolemic shock).
Dengue virus harbors 10–11 kb long RNA genome, flanked by untranslated regions at both the 5′ and 3′ ends. It is encoded as a single polyprotein, and is processed into three structural and seven nonstructural proteins via host and viral proteases. Structural proteins, such as capsid (C), precursor membrane (prM), and envelope (E), while the nonstructural proteins are NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5 proteins.6 Among these nonstructural proteins, NS5 is the most conserved protein, sharing 67% amino acid sequence similarity across all the four DENV serotypes. The crystal structures have shown a high degree of similarity for NS5 across the genera of family Flaviviridae, from the hepatitis C virus (HCV) to bovine viral diarrhea virus (BVDV).7–10 NS5 is a monomeric 104.387 kDa protein, having a methyl transferase (MTase) domain at the N-terminal, while the C-terminal harbors the RNA-dependent RNA polymerase (RdRP) region. RdRp is further divided into three subdomains, such as finger palm and thumb, while MTase consists of the GTP binding domain, and catalytic domain (SAM binding domain). The catalytic domain of MTase contains seven β-sheets surrounded by four-helices folds in such a way that the K–D–K–E catalytic tetrad is positioned in the center of MTase catalytic cleft with a GTP binding site on the opposite site. This interaction assists the RdRp in the unstable initiation phase, however, MTase is not involved in the RdRp catalysis, and both the domains work independently from each other.11 MTase is necessary for the methylation of the RNA cap at guanosine N-7 to form N-7-methylguanosine and at ribose to form 2′-O-methyl-adenosine by using S-adenosyl-L-methionine as a methyl donor. Mutational studies have shown that Phe-133, Ser-56, Gly81, Gly85, Trp87, Thr104, His110, Glu111, Asp131, and Glu149 are the key residues that are vital for both the methyl transfer activities.12 This process of methylation occurs subsequently as N7 methylation before 2′-O-methylation, which shows the enzyme has higher catalytic efficiency for N7 methylation. Site-directed mutagenesis studies showed that N7 methylation is vital for virus replication, while 2′-O-methylation facilitates the host immune clearance. RdRp is important for de novo RNA synthesis.13 It triggers the nuclear localization signal (NLS), which helps the virus to interact with its host and other viral proteins.14,15 Reverse genetic, biochemical, and structural studies of NS5 have revealed that RdRp and MTase interact via a linker of 10 residues domain among several flaviviruses.11,16–19 Along with that, NS5 exhibits interactions with other viral replication proteins, including the nonstructural protein 3 (NS3) protease/helicase, as well as the host proteins of the ubiquitin proteasome pathway, such as NF90, and eEF1A.20 NS5 helps the dengue virus to evade the innate immune response by decreasing the STAT2 level of expression through promoting its degradation via ubiquitination and proteasomal activity of the cell.21 Thus NS5 is important in the context of antiviral research because of its key role in replication, capping, and immune invading.
Currently, despite global efforts toward the development of a vaccine, there is neither any cross-protective vaccine nor clinically approved antiviral therapy available against DENV infections.22 In such a context, the development of potent antiviral drugs to treat DENV infection is urgently needed. Drug repurposing is an important approach for the identification of new therapeutic indications for already known drugs. Recently, several repurposed drugs were evaluated against dengue virus, including sinefungin, ivermectin, lovastatin, prednisolone, modipafant, ketotifen, balapiravir, ribavirin, celgosivir, UV-4B, and chloroquine, but no significant success has been reported yet.23
Saturation transfer difference (STD-NMR) is a robust technique for the analysis of protein–ligand interactions, as well as for the identification of binding epitopes of a ligand. In this study, 75 compounds were evaluated against NS5 from our in-house library, including US-FDA approved drugs and natural products, using saturation transfer difference (STD)-NMR spectroscopy. Compounds 1–12 showing the STD effect were then further validated via computational studies of the ligand with the protein, as mentioned in Table 1.
| Name | Structures |
|---|---|
| Atenolol (1) | 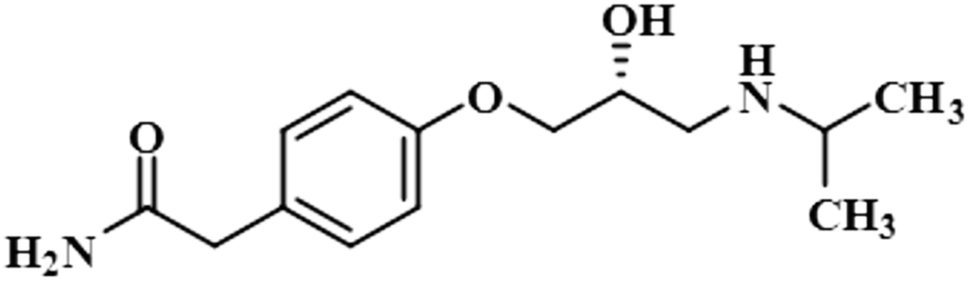 |
| Itopride hydrochloride (2) | 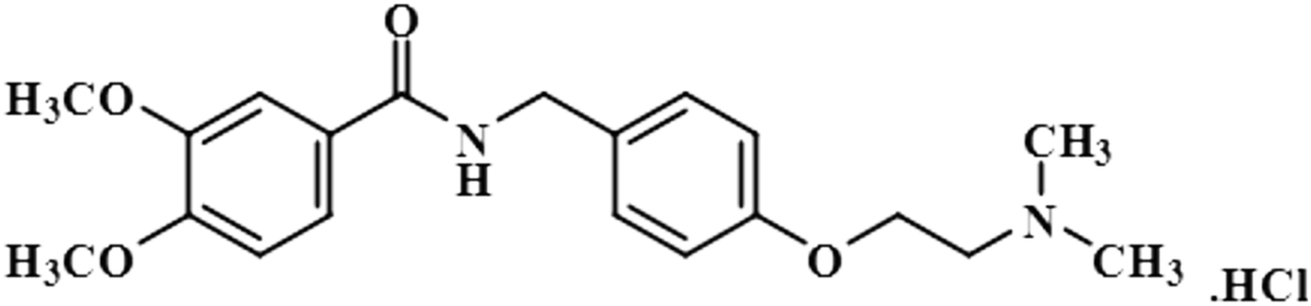 |
| Scopolamine hydrobromide trihydrate (3) | 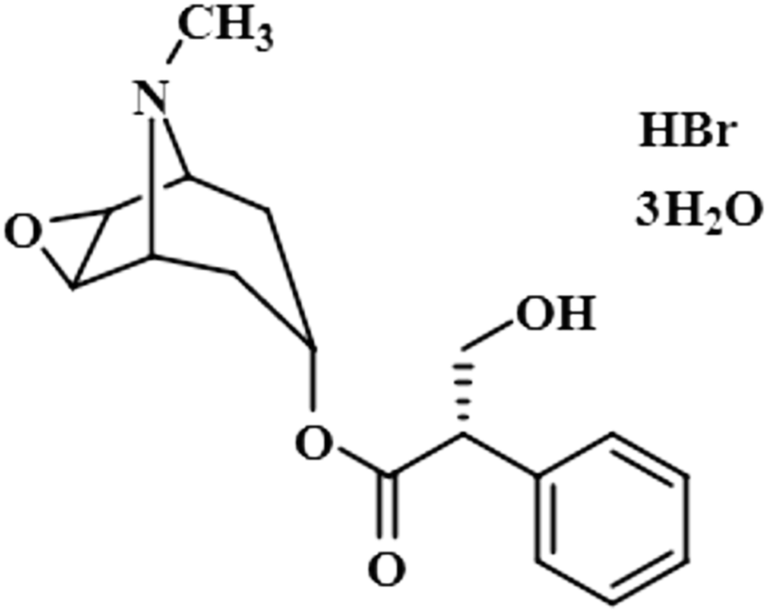 |
| Phloridzin (4) | 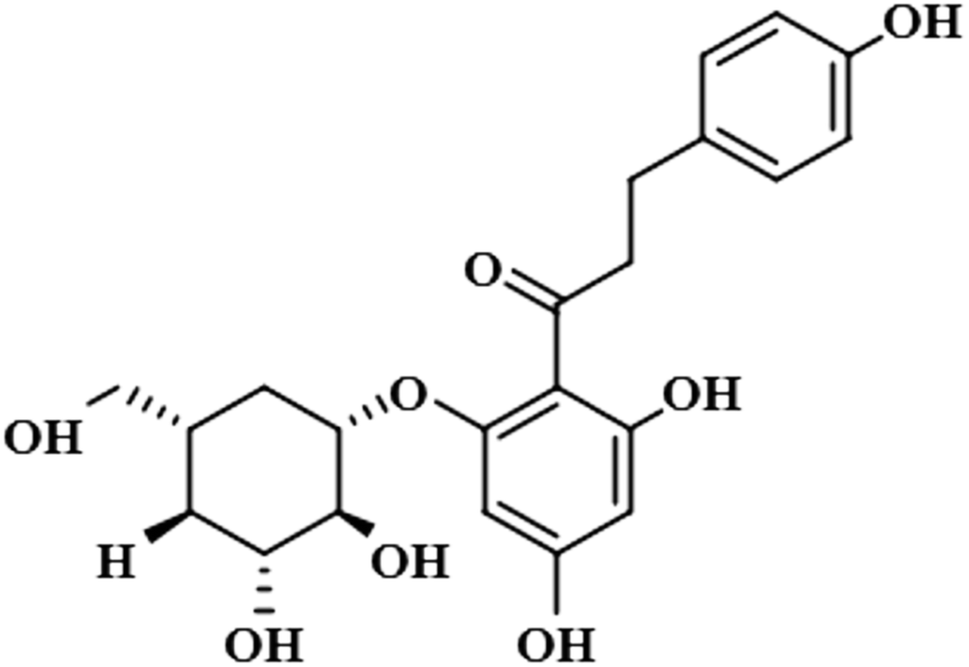 |
| Cefadroxil monohydrate (5) | 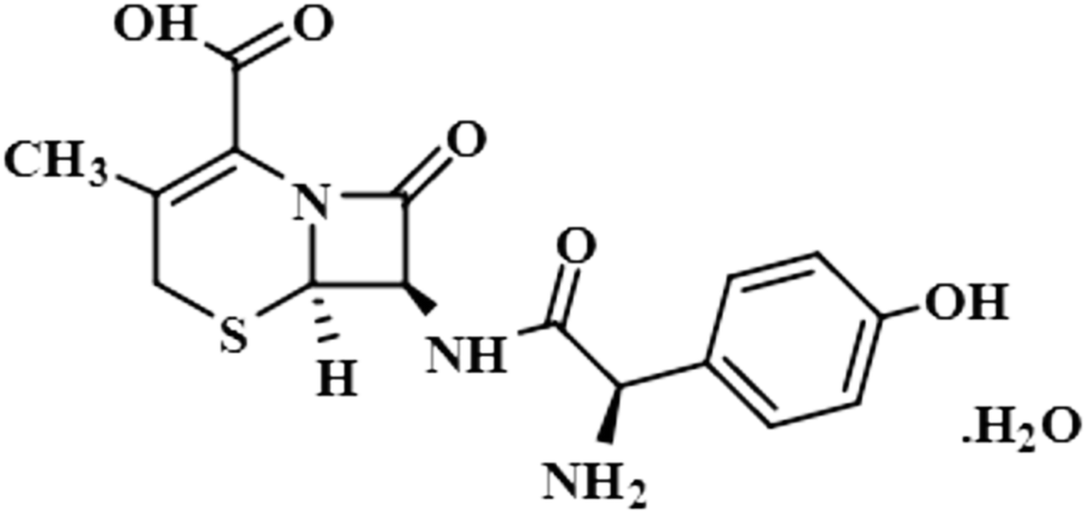 |
| Diclofenac sodium (6) | 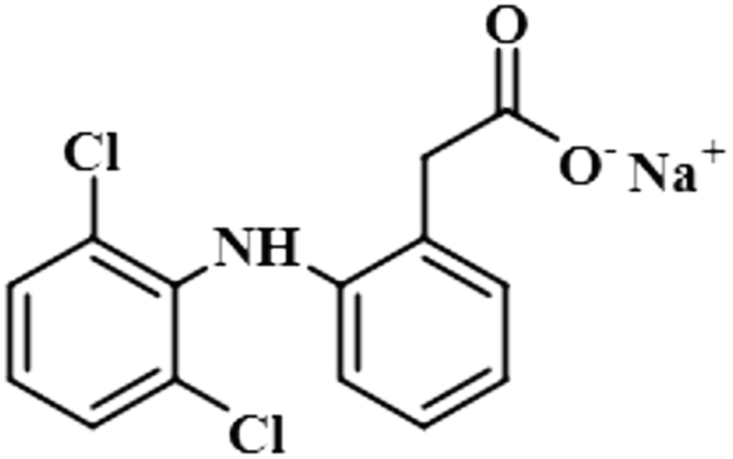 |
| Epinephrine bitartrate/adrenaline bitartrate (7) |  |
| Cloxacillin (8) | 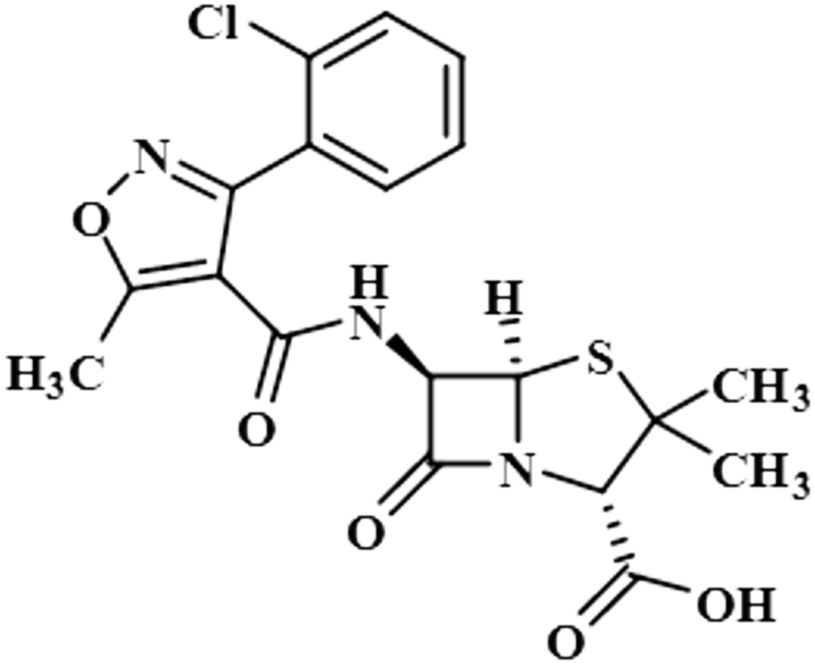 |
| Levosulpiride (9) | 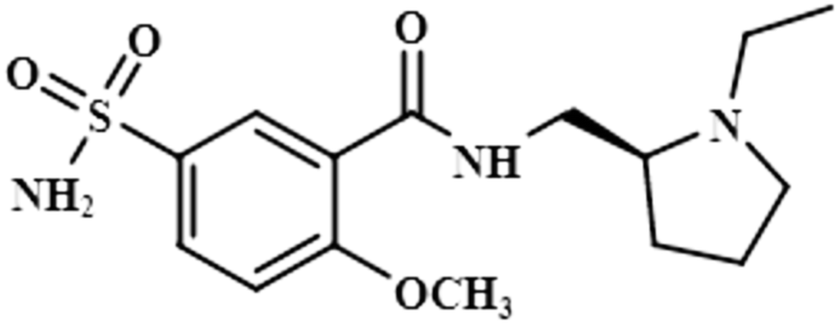 |
| Cinitapride (10) | 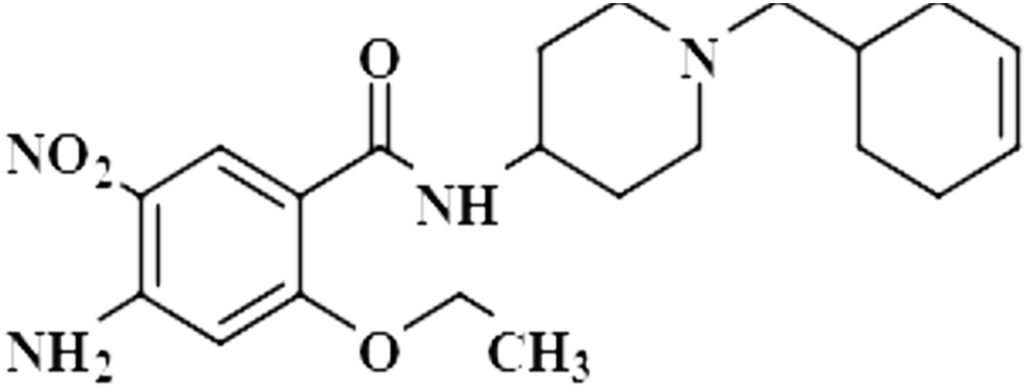 |
| Boldine (11) | 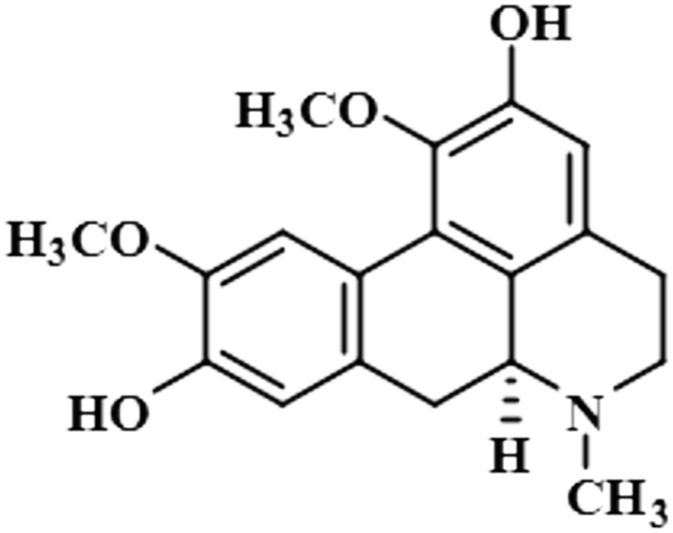 |
| Neohesperidin dihydrochalcone (12) | 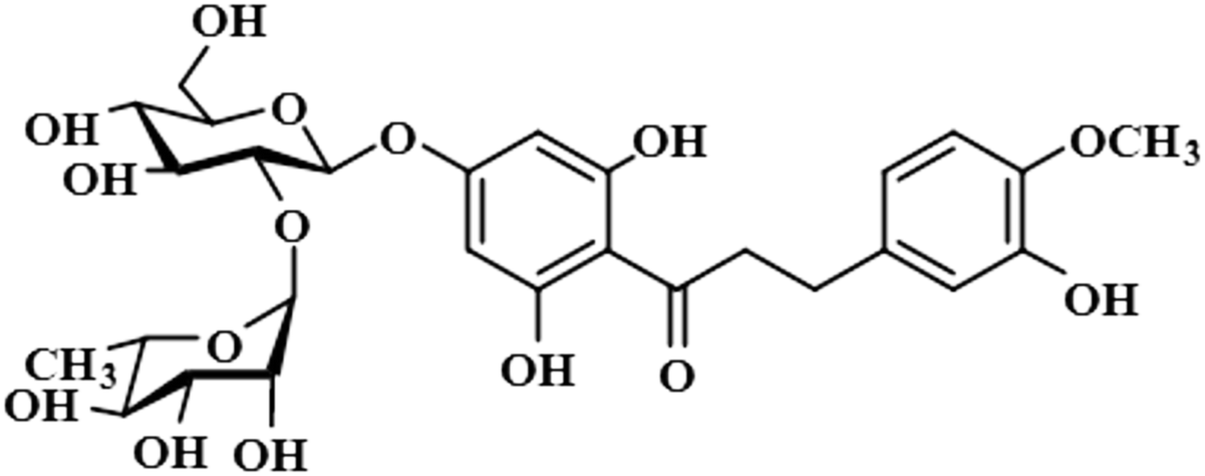 |
2 Material and methods
2.1 Expression and purification of DENV NS5
The full-length sequence of a gene WT DENV2 NS5 cloned vector was transformed and expressed into Escherichia coli strain BL21-CodonPlus (DE3)-RIL carrying the hexa-histidine tag at the C-terminus (Genescript USA) (Fig. S1†). Cells were grown in NZCYM medium, containing 25 μg mL−1 kanamycin and 20 μg mL−1 chloramphenicol, at 37 °C in a shaking incubator till the optical density at 600 nm (OD600) reached 1.0. Target protein expression was then induced with isopropyl-β-D-thiogalactopyranoside (IPTG) with a final concentration of 0.5 mM, and the cells were further incubated for an additional 18 h at 18 °C before harvesting. The cells were lysed via ultra-sonication in lysis buffer of 50 mM Tris HCl pH 8.0, 300 mM NaCl, 10 mM imidazole, 20% (v/v) glycerol, 1 mM PMSF, and 0.02% (w/v) NaN3. The lysate was pelleted down for 40 min at 20000 rpm in an SS-630 rotor (Thermo Scientific, USA). The soluble lysate was loaded into an affinity chromatography column (HisTrap™ HP GE Healthcare, USA). The protein was eluted by a linear gradient of 50 mM Tris HCl, pH 8.0, 300 mM NaCl, 300 mM imidazole, 20% (v/v) glycerol, and 0.02% (w/v) NaN3.
The pooled fractions were analyzed via sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and concentrated up to 5 mL. Then, the sample was loaded on a HiTrap 16/600 Superdex 200 pg gel filtration column (GE Healthcare, Germany), equilibrated with a buffer 20 mM Tris HCl pH 8.0, 20 mM NaCl, and 5 mM imidazole. Fractions containing protein with an extinction coefficient of 220990 M−1 cm−1 were then concentrated to 2 mL which yielded 6–8 mg mL−1 of pure protein from 1 liter of bacterial culture.
2.2 Screening procedure
A total of 75 compounds, comprising 62 drugs and 13 natural compounds, were obtained from our in-house PCMD Molecular Bank. These compounds were grouped into 15 mixtures, each with 5 compounds based on their non-reactivity toward each other, solubility in buffer, and non-overlapping of their 1H-NMR signals (details are mentioned in the ESI†). All the compounds were solubilized in deionized water with a stock concentration of 1 M. Each mixture was further diluted to a final concentration of 100 mM in NMR buffer (20 mM Tris HCl, pH 8.0, 20 mM NaCl; 5 mM imidazole). NMR buffer was prepared in deuterium oxide (D2O) to ensure the maximum magnetization transfer from the protein (receptor) to ligand. The purpose of grouping was to achieve a maximum screening efficiency using the shortest NMR measurement time.2.3 Saturation transfer difference (STD) NMR experiments
All the STD-NMR spectra were acquired on a Bruker Avance spectrometer, operating at a proton frequency of 600 MHz (Bruker, Biospin, Switzerland). The NMR spectrometer was equipped with a triple channel (TCI) cryogenically cooled probe, an automated SampleCase™, and Topspin 4.5 NMR (Bruker, Biospin, Switzerland).The STD-NMR experiments were performed at 298 K using the pulse program (stddiffesgp.3), selected from the Bruker library with excitation sculpting for water suppression. For both the mixtures and individual compounds, 32 and, 1024 scans were recorded, respectively. The spin lock pulse was set to 25 ms, and the saturation time to 3 s with an interpulse delay of 10 s. A narrow specific pulse of 50 ms, Gaussian in nature with a width of 200 Hz, was used to drive the excitation of the protein. The saturation point was set to −24.67 Hz, corresponding to the protein signals. First, in order to subtract any false positive signal, all the STD-NMR experiments were recorded without the protein (control), followed by with the protein STD-NMR experiments. The difference spectrum was obtained by subtraction of the second spectrum from the former, which indicated the interactions of the protein with the ligand as the signal intensities of interacting protons of ligands were enhanced. The STD amplification factor of each proton was calculated by using the following formula:
| STD amplification factor = (I0 − Isat/I0) × ligand excess |
2.4 Molecular docking studies
Molecular docking studies were performed to examine the interactions of each ligand with the NS5 protein via the Glide 6.9 module in the Schrödinger suite of programs.24,25 The crystal structure of the NS5 protein (PDB ID: 6kr2) was used for the ligand docking studies.26 The protein structure was prepared and minimized through Protein Preparation Wizard in Maestro Schrödinger 10.4. The OPLS4e force field was used to assign the missing partial charges, as well as missing protons.27,28 The prime module was used to fill the missing loops, and to produce zero order bonds to metal ions. PROPKA was used to predict the pKa of the proteins. Using the OPLS4e force field, the complex was subjected to restricted minimization for optimizing the heavy atoms and hydrogens to ease steric hinderance.29 The LigPrep tool (Schrödinger) was used to prepare the ligands by altering their torsions, and assigning them suitable protonation states. The tautomeric and ionization states were generated for each ligand through Epik (Schrödinger (2015c) Prime. The Glide_XP dock tool was used for docking, and the results were analyzed for the best docked pose.3 Results and discussion
Among all the nonstructural proteins, NS5 is the most important as it helps the dengue virus to replicate, and protect against the host immune reaction. Due to its highest homology among all the serotypes, NS5 is considered a promising target for anti-DENV therapy.Our current study was therefore focused on analyzing how well different drugs and natural products could interact and destabilize NS5. A total of 75 compounds from an in-house molecular bank were selected, out of which 9 US-FDA approved drugs and 3 natural products had shown interactions with NS5 (PDB ID 6kr2) in STD NMR spectra. All these compounds were never reported earlier for their binding to NS5 of dengue virus. Molecular docking was then performed to predict the features of the ligand–protein complex.30
3.1 Identification of the protein (NS5)–ligand interactions using (STD)-NMR spectroscopy
STD-NMR is a robust technique to identify weak to moderate ligand–receptor interactions. Through spin–spin relaxation, saturation applied on protein resonance diffuses to different residues and might transfer to the protons of ligands that are interacting with these residues. The STD-NMR estimates the ligand protons proximity on the protein surface through group epitope mapping (GEM). The intensity of the ligand protons indicates its closeness with the surface of the protein. The closer to the protein surface, the higher the intensity of those ligand protons in STD-NMR spectra. The percentages of saturation transfer of various ligand protons were determined through GEM analysis. The proton with the highest relative STD integral value was set as the 100% value.Compound 1 (Atenolol), a US-FDA approved drug, is a beta-1 adrenergic blocker used for the treatment of hypertension. It decreases the blood pressure and stops the relapse of supraventricular arrhythmias once controlled.31 Recent studies have also shown the significant therapeutic effect of atenolol in the treatment of infantile hemangiomas.32 In this compound, aliphatic protons of CH2 at C-9 have shown maximum (100%) saturation from protein. Whereas, H-10 received a relative saturation of 99.5%, followed by 98.8% for CH3 at C-13 and C-14, respectively. Aromatic H-2/H4 and H3/H5 received 91.5% and 80.6% saturation successively. The rest of the aliphatic protons H-7/H-11 exhibited a relative saturation of 80.9% and 79.1%, respectively. The degree of saturation received by H-12 was calculated as 75.2% relative to CH2-9. Thus the GEM analysis showed CH2 at C-9 lay in very close proximity to the protein (Fig. 1).
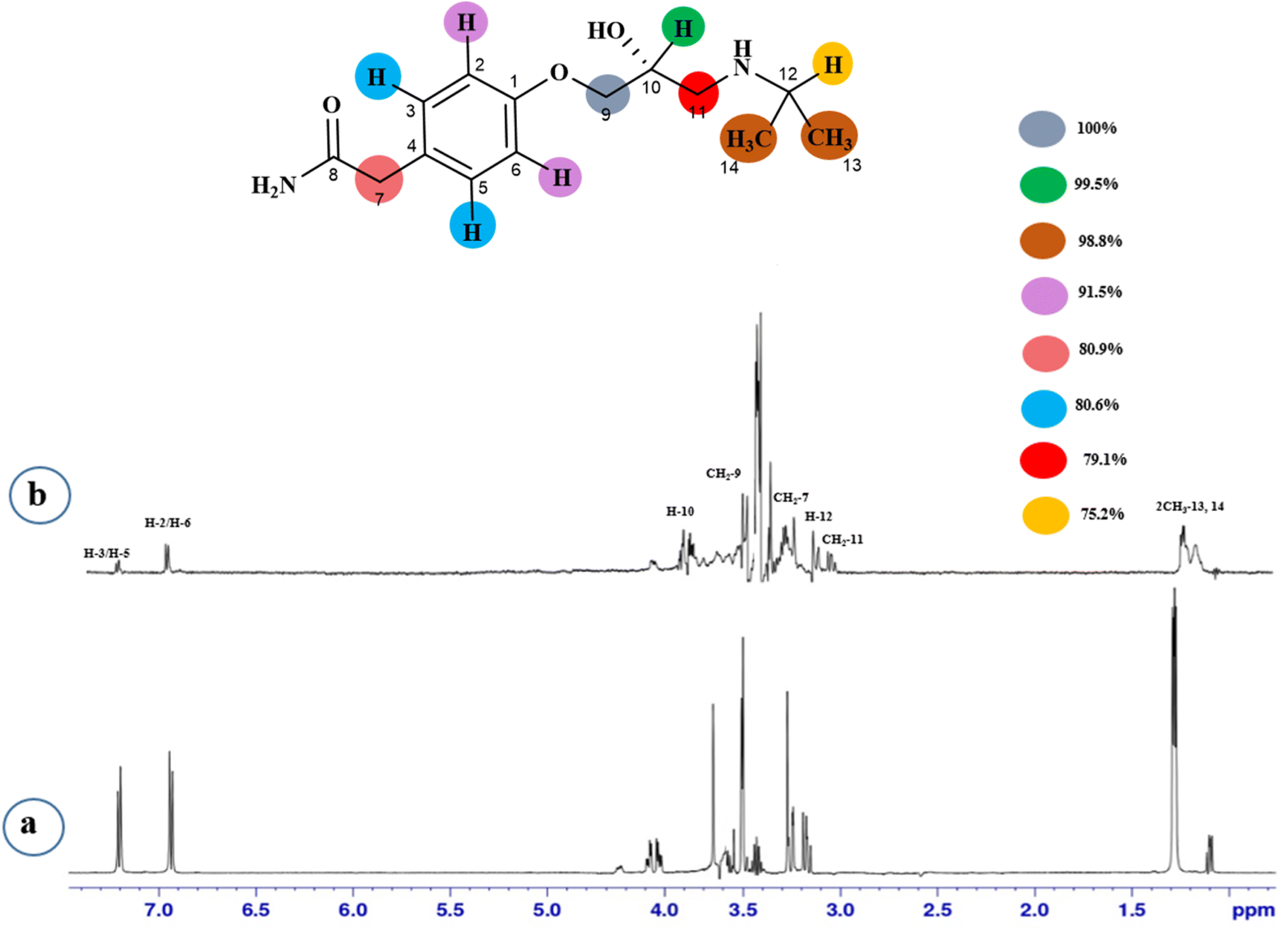 | ||
| Fig. 1 STD-NMR of compound 1. (a) 1H NMR reference spectrum of compound 1. (b) STD difference spectrum recorded with NS5 protein with 2 μM concentration. H-9 had the largest STD integral value, which was set to 100%. All other interacting protons are normalized against H-9 and indicated with a color code. | ||
Compound 2 (itopride hydrochloride) is a US-FDA approved drug for gastrointestinal motility disorders. It is involved in the stimulation of gastric motility and enhances the contraction of the gastrointestinal tract, inhibiting the acetylcholine (AChE)-induced pathway.33 The aromatic protons H-10/H-14 with the integral value of 100% achieved maximum interactions. All the other interactions were normalized, such that H-10/H-14. H-11/H-13 displayed 89% saturation, while H-5 and H-6 received 87% and 81% saturation, respectively. The terminal methyl protons achieved 83.2% saturation relative to the maximum integral. H-2 received 79% saturation. Thus the GEM analysis indicated the proximity of the aromatic H-10/H-14 protons with the NS5 protein (Fig. 2)
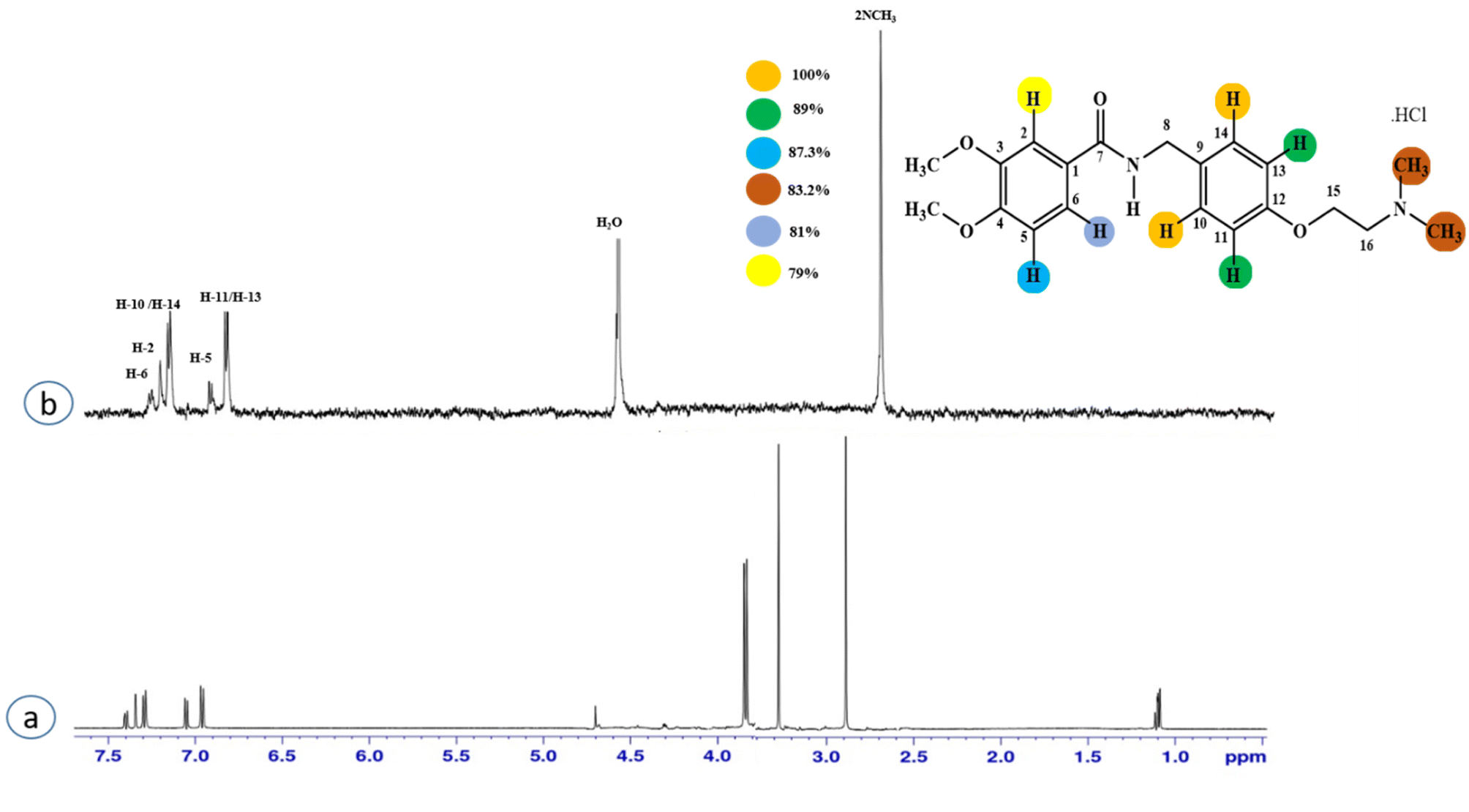 | ||
| Fig. 2 STD-NMR of compound 2. (a) 1H NMR reference spectrum of compound 2. (b) STD difference spectrum recorded with the NS5 protein with 2 μM concentration. H-14 had the largest STD integral value, and was set to 100%. All the other interacting protons were normalized against H-14, and indicated here with the shown color code. | ||
Compound 3 (scopolamine hydrobromide trihydrate) is a potent anticholinergic drug that works on CNS to produce a calming effect on the muscles in the gut and stomach. It inhibits the muscarinic acetylcholine receptors (M-AChR), specifically M1 and M2, to give relief to patients with depression in Parkinson's disease. Scopolamine hydrobromide trihydrate has also shown an inhibitory affect against the NS5 protein of Japanese encephalitis virus by regulating the TLR and INF signaling pathways34,35 The aliphatic protons H-2′/H-3′ received a maximum saturation of the relative STD integral value, set to be 100%. Ha-8/Ha-6 achieved a relative saturation of 94%. All the aromatic protons had a saturation of 91% each. Hb-8/Hb-6 experienced 87.8 and 86.9%, respectively. The methyl group at C-10 showed the least saturation of 84%. Hence the GEM analysis predicted that the aliphatic protons H-2′/H-3′ were in very close proximity (Fig. 3).
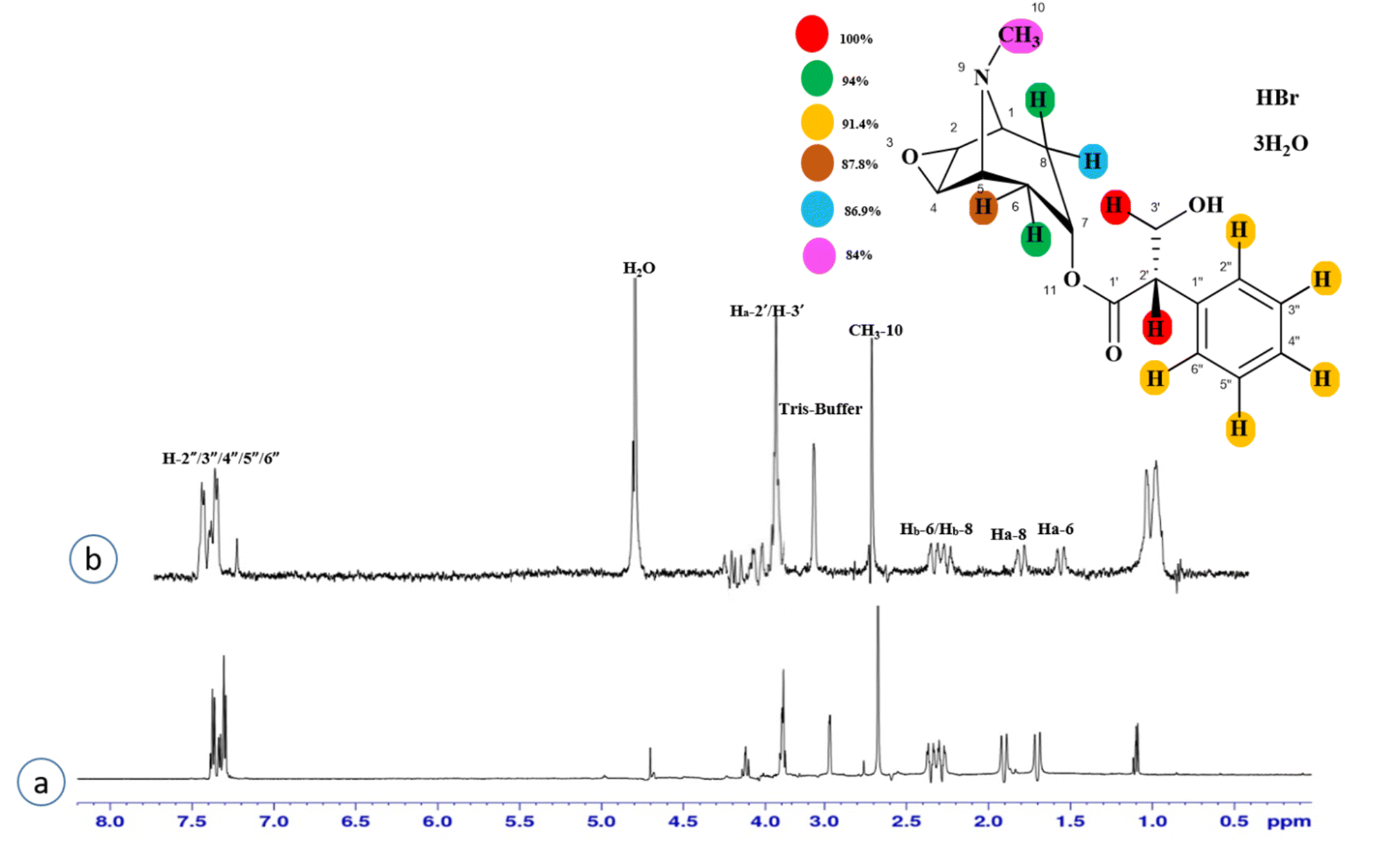 | ||
| Fig. 3 STD-NMR of compound 3. (a) 1H NMR reference spectrum of compound 3. (b) STD difference spectrum recorded with the NS5 protein with 2 μM concentration. H-2′/H-3′ exhibited the largest STD integral value, and this was set to 100%. All the other interacting protons were normalized against H-2′/H-3′ and indicated with the color code shown. | ||
Compound 4 (phloridzin) is a flavonoid, extracted from the bark of Cinchona and apple trees. It is mainly used in the treatment of diabetes mellitus and functions by restricting the intestinal glucose absorption by inhibiting sodium–glucose symporters in the proximal renal tubule and small intestine mucosa. In some studies, phloridzin has also shown an antipyritic effect, particularly for the treatment of malaria. It has also exhibited an antiviral effect against SARS-CoV-2 proteases.36,37 H-12 received the highest STD integral value, thus referred to as 100% saturation. This was followed by H-3/H-5, which received 84.46% saturation, while H-2/H-6 received 83.8% relative saturation. Thus the group epitope mapping results analysis predicted that H-12 is in close proximity (Fig. 4).
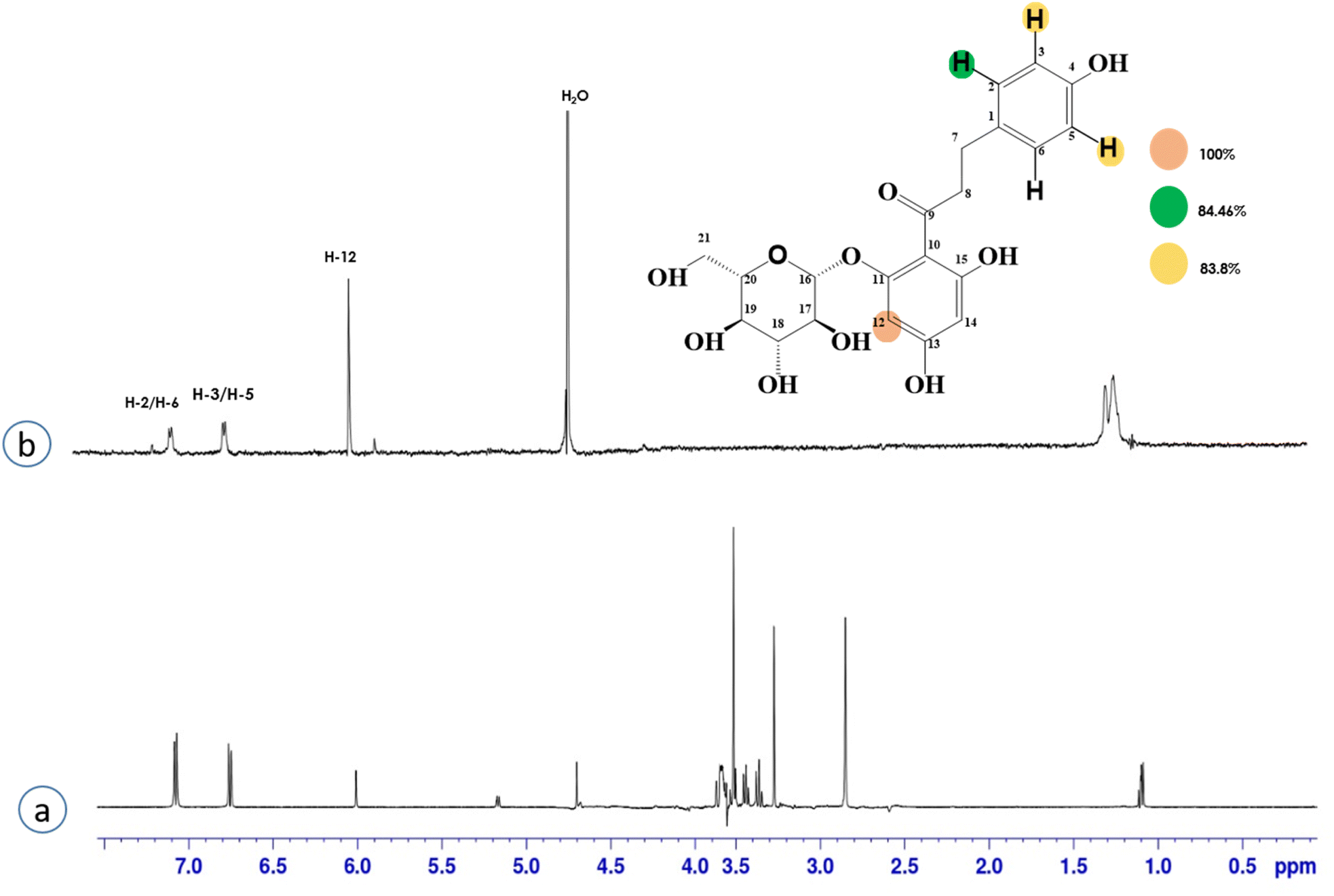 | ||
| Fig. 4 STD-NMR of compound 4. (a) 1H NMR reference spectrum of compound 4. (b) STD difference spectrum recorded with the NS5 protein with 2 μM concentration. H-12 had the largest STD integral value, and this was set to 100%. All the other interacting protons were normalized against H-12, indicated with the color code shown. | ||
Compound 5 (cefadroxil monohydrate) is a US-FDA approved drug that is primarily administered as an antidote to organophosphorus poisoning, pharyngitis, and for respiratory tract infections, skin infections, and tonsillitis. Due to its bactericidal activity, cefadroxil monohydrate is used as an antibacterial agent to treat various infections, such as urinary tract infections.38 In this study, only the aromatic protons H-10/H-14 of compound 5 showed interactions with the protein, and received 100% saturation. The GEM results revealed the close proximity of H-10/H-14 with the protein residues (Fig. S2†).
Compound 6 (diclofenac sodium) is a US-FDA approved non-steroidal anti-inflammatory agent (NSAID) with antipyretic and analgesic effects. It prevents the synthesis of prostaglandin through the inhibition of cyclo-oxygenase, which is important for inflammatory activity.39 The ligand-based STD-NMR experiment showed that H-12 of compound 6 received the maximum saturation with the STD integral value, which was set to 100% because of its closest proximity with the protein. The degree of saturation received by H-4 was 57%, followed by H-10/H-11 with a relative saturation of 36%, both because of their same chemical environment. Compared to H-12, the degree of saturation received by H-3/H-5 was 19.7%, while H-9 received the lowest saturation of 7.8%. GEM analysis indicated that H-12 lay in close proximity to the NS5 protein (Fig. S3†).
Compound 7 (epinephrine bitartrate/adrenaline bitartrate) is an active sympathomimetic hormone that stimulates both α-and β-adrenergic systems and causes systemic vasoconstriction and gastrointestinal relaxation. It is used to reduce the production of aqueous humor in glaucoma via the alpha-adrenergic receptor. It also slows the absorption of local anesthetic during asthma and cardiac arrest (National Center for Biotechnology Information, 2022). The maximum integral value of 100% was shown by the aromatic H-2 and H-6, while the rest of the ring protons had a relative saturation of 50.3%. The minimum saturation of 35.4% was displayed by the terminal methyl group. Group epitope mapping revealed that the aromatic H-2/H-6 were in close proximity to the protein (Fig. S4†).
Compound 8 (cloxacillin) is an antibiotic used for the treatment of several bacterial infections, such as S. aureus, N. gonorrhea, and N. meningitides. Computational studies revealed the potential interaction of this drug with the SARS-CoV-2 protease.40 In this compound, the methyl at C-2′ showed the maximum saturation of STD with an integral value of 100%. H-3′/CH3-7 received 77% and 7.3% saturation, respectively. H-4′ displayed the lowest saturation of 4.8%. GEM analysis indicated that the methyl group is in close proximity of the NS5 protein (Fig. S5†).
Compound 9 (levosulpiride), a levo enantiomer of sulpiride, is used for the treatment of depression, anxiety, emesis, psychosis, somatoform diseases, and dyspepsia. It prevents the binding of dopamine by blocking the dopaminergic receptors, which is vital for the treatment of psychotic illness.41 In this compound, the C-14 methoxy of the aromatic ring displayed the largest integral value of 100%. The relative saturations for the other protons H-9 and H-10 were 80.7% and 71.3%, respectively. The methoxy present was near the protein surface as inferred from the GEM analysis (Fig. S6†).
Compound 10 (cinitapride), a US-FDA approved drug, is used for the treatment of nausea, and vomiting and is effective against dyspepsia. It prevents gastroesophageal refluxes via blocking the 5-HT2 receptors, which generate excitatory potential through interaction with a neurotransmitter, called serotonin.42 In this compound, only the aromatic H-3 had interacted with the protein, and was assigned as 100% saturation. Group epitope mapping revealed the presence of the aromatic H-3 in close proximity to the NS5 protein (Fig. S7†).
Compound 11 (boldine) is an aporphine alkaloid, extracted from the plant Peumus boldus, that exhibits anti-inflammatory, bacterial pyrogen-induced hyperthermia activities. Boldine is widely known for its hepatoprotective effect by lowering the production of lactate-dehydrogenase and malondialdehydes.43 Boldin has also shown an antiviral effect against both HCV and HIV by halting the process of replication.44 In this compound, aromatic H-3 acquired 100% saturation, followed by H-11 and H-8, which received saturations of 86% and 56%, respectively. Group epitope mapping showed the aromatic H-3 was in the vicinity of the protein (Fig. S8†).
Compound 12 (neohesperidin dihydrochalcone) is a natural product that is used as a sweetener in food item (Borrego, 2001). In this compound, the anomeric H-1′′ of the attached sugar moiety acquired 100% saturation, followed by H-3′/H-5′, which received a saturation of 62%. The aromatic protons H-6/H-2 displayed 49% saturation. GEM analysis predicted the anomeric protein was within close proximity of the protein (Fig. S9†).
In this study, we predicted through GEM analysis that the binding pocket of the protein would be folded in such a way that the aromatic rings would fit into it, as the maximum interactions were shown by the aromatic protons. Atenolol (1) and scopolamine hydrobromide trihydrate (3) are the two compounds with a single aromatic ring. They displayed maximum interactions. The protons of aliphatic regions also contributed significantly, but not to the same level as the aromatic regions. Among all the drugs, cefadroxil monohydrate (5) exhibited the least interaction with the protein, in which only two aromatic protons interacted slightly with the binding cleft of the protein.
3.2 Molecular docking on NS5 (PDB ID 6kr2)
The active site/substrate binding site of NS5 is divided into two subdomains, i.e., the GTP binding domain and SAM binding domain. Among those, the SAM-dependent methyl transferase subdomains were folded into a seven-stranded β-sheet, surrounded by four α-helices.20 The ligand S-Adenosyl-L-Homocysteine (SAH) bound crystal structure of the NS5 protein (PDB ID: 6KR2) was selected for docking. The ligands were docked against the SAH binding site. All the compounds showed significant interactions with the binding pocket residues.NS5 exists in the monomeric form in which both the RdRp and MTase domains work independently from each other. The MTase domain contains a catalytic site, which contains four alpha helices and seven beta strands.,15 which helps in the methylation reaction in which SAH is used as a methyl donor, as depicted in Fig. 5. This ligand binding domain is conserved among all the flavivirus.
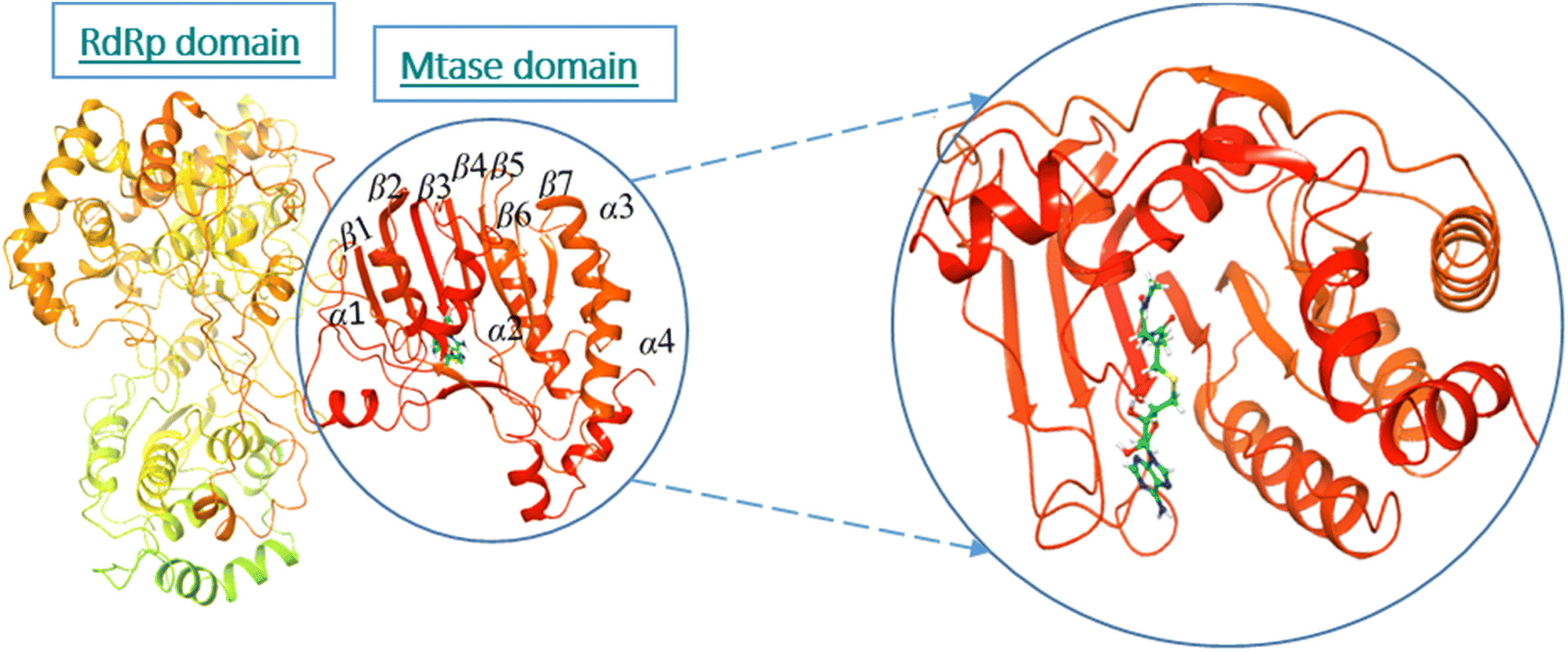 | ||
| Fig. 5 Ribbon presentation of NS5 (PDB: 6kr2): red color indicates alpha helices, while orange shows beta strands. | ||
This sequence alignment (Fig. 6) showed a wide range of residues that are 100% conserved among the MTase domain of the above-mentioned flaviviruses. Here, α1 contained eight conserved residues across all the strains while α2 possessed only two conserved residues, and α3 and α4 contained one and five conserved residues respectively, as presented in Fig. 6. The beta sheets also contained a large amount of conserved residues as β1 was fully conserved, except for two amino acids where all the residues were present across all the flaviviruses. β2 and β3 had a low number of conserved regions, while β4 and β5 were well conserved in all the strains. Beta 6 contained three residues that were conserved, while β7 possessed four conserved amino acids. Overall this similarity profile showed that there is a wide region conserved across all the flavivirus, which makes it a very attractive target for therapeutic implications.
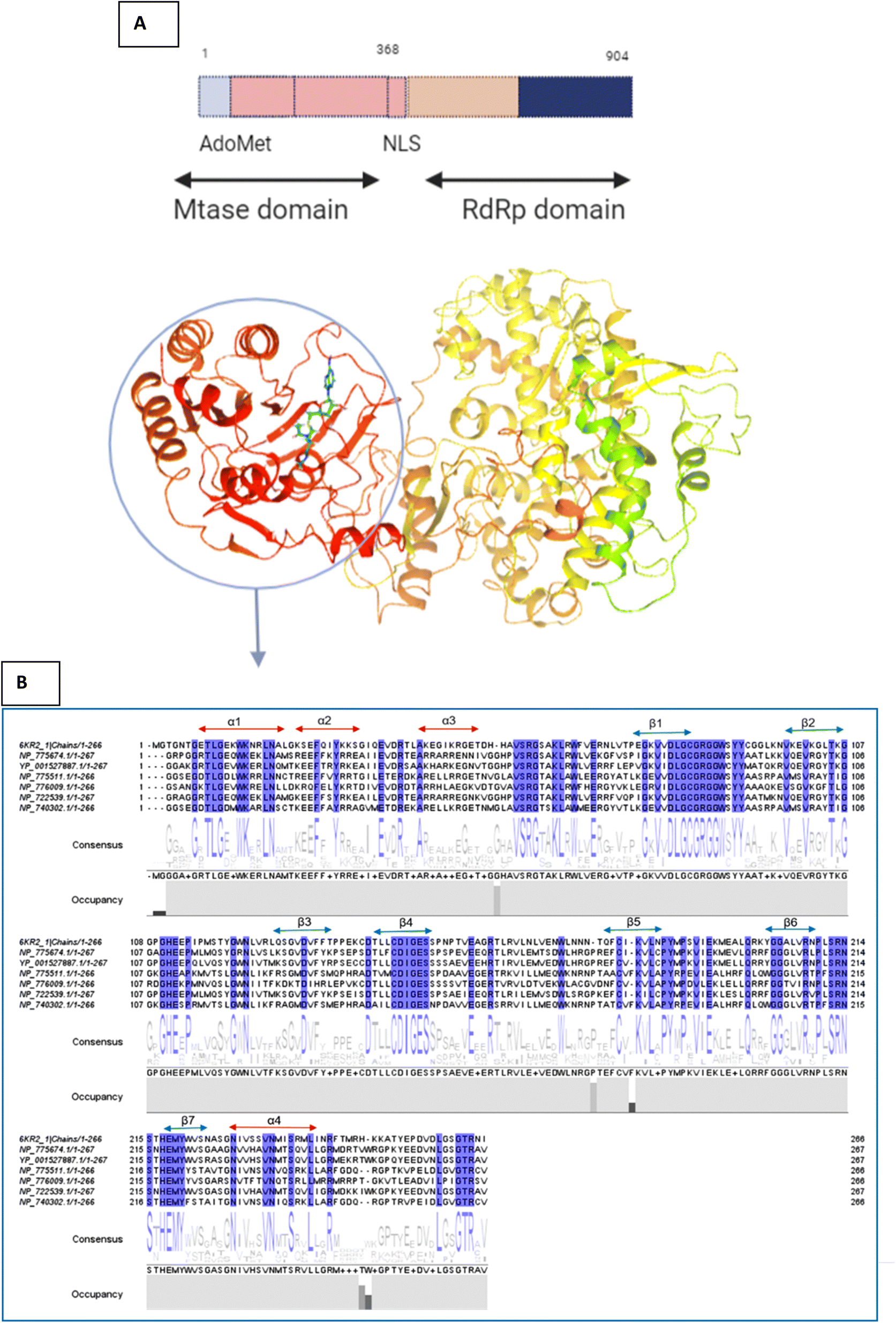 | ||
| Fig. 6 (A) Depiction of the two subunits of NS5, i.e., the methyl transferase (MTase) domain at the N-terminal and the RNA-dependent RNA polymerase (RdRP) domain at the C-terminal (B) Amino acid alignment of NS5MTase: The sequences are NS5MTase domain from dengue virus 6KR2, Japanese encephalitis virus NP_775674.1, West Nile virus YP_001527887.1, tick-borne encephalitis virus NP_775511.1, yellow fever virus NP_776009.1, Murray Valley encephalitis virus NP_722539.1, and Langat virus NP_740302.1. | ||
Compound 4 showed a significant interaction with the protein, Val 132, Gly81, Cys82, Trp87, and Ser56 amino acids via H-bond interaction, while Glu149 interacted both through hydrogen bonding and aromatic hydrogen interactions, showing the highest docking score of −11.103, as shown in Fig. 7 and Table 2.
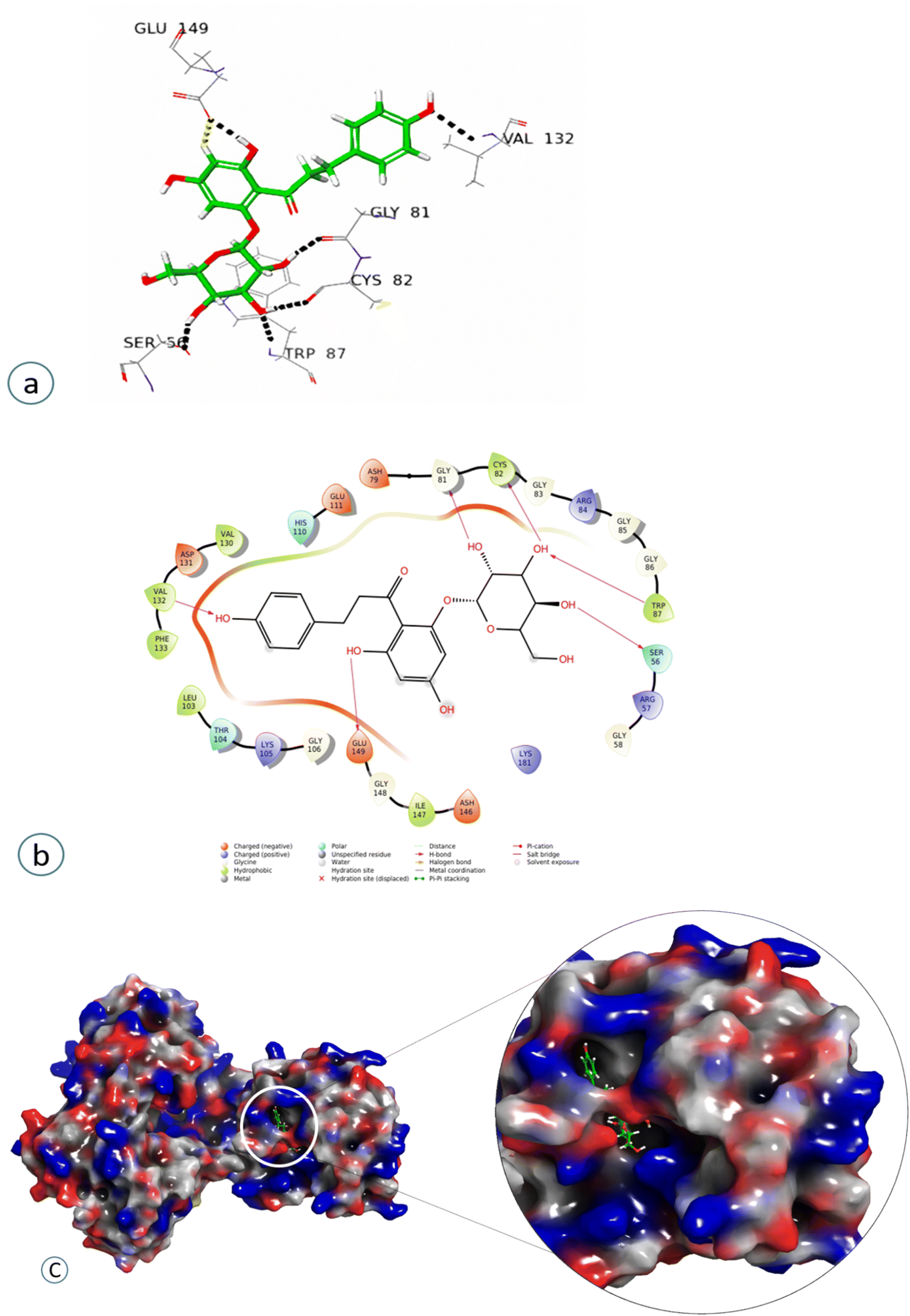 | ||
| Fig. 7 Docked pose of compound 4 (Phloridzin): (a) 3D Representation of the ligand–protein interaction in dotted lines indicating hydrogen bonds (black), and aromatic hydrogen bonds (yellow). (b) 2D Representation of the ligand–protein profile. (c) Solid surface representation of the ligand–protein profile, depicting the electrostatic potential distribution over the surface (red, negative regions; blue, positive regions). | ||
| Compounds | Interacting amino acids | Type of interactions |
|---|---|---|
| Atenolol (1) | His110, Glu149, Asp146 | Aromatic hydrogen, |
| Phe133 | Aromatic hydrogen π–π interaction | |
| Itopride hydrochloride (2) | Glu149 | Hydrogen bonding, aromatic hydrogen interactions |
| Scopolamine hydrobromide trihydrate (3) | Glu111, Glu149 | H-bond |
| His110 | π-cation | |
| Phloridzin (4) | Val 132, Gly81, Cys82, Trp87, Ser56 | Aliphatic H-bond |
| Glu149 | Aliphatic H-bond, aromatic hydrogen interactions | |
| Cefadroxil monohydrate (5) | His110 | H-bond, π–π stacking |
| Glu111, Glu149 | Aromatic hydrogen interaction | |
| Diclofenac sodium (6) | Glu111, Gly106, His110 | H-bond, |
| Asp131 | Aromatic H-bond | |
| Epinephrine bitartrate/adrenaline bitartrate (7) | Glu149, Thr104, Asp79, Gly 86 | H-bond |
| Trp87 | Aromatic hydrogen interaction | |
| Cloxacillin (8) | Glu149 | H-bond |
| Levosulpiride (9) | His110 | H-bond and π cation |
| Cinitapride (10) | Phe133 | π–π stacking |
| His110, Asp148, Glu149 | Aromatic hydrogen interaction | |
| Boldine (11) | Asp131, Glu149 | H-bond linkages and aromatic hydrogen |
| Neohesperidin dihydrochalcone (12) | Phe133 | π–π stacking |
| Gly81, Trp87, Lys105, Arg163, Glu149 | H-bond interaction |
Compound 1 interacted via aromatic hydrogen interactions with His110, Glu149, Asp146, and with the Phe133 through both aromatic hydrogen bonds and π–π interactions, showing a docking score of −4.3 (Fig. S10†) (Table 2), while compound 2 interacted only through Glu149 via hydrogen bonding and aromatic hydrogen interactions, with a docking score of −3.8 (Fig. S11†) (Table 2).
Compond 3 interacted with Glu111 and Glu149 through H-bond and via π-cation with His110, showing a docking score of −4.2 (Fig. S12†) (Table 2), while compound 5 interacted with His110 via H-bond along with π–π stacking, while it interacted with Glu111 and Glu149 through aromatic hydrogen interactions, showing a higher docking score of −6.31 (Fig. S13†) (Table 2). The ligand showed H-bond interaction with Asp146. Compound 6 interacted with Glu111, Gly106, and His110 via H-bonds, and with Asp131 through aromatic H-bond interaction, having a docking score of −4.7 (Fig. S14†) (Table 2).
Compound 7, epinephrine bitartrate, interacted with Glu149 and Thr104 via only H-bond interactions showing a slightly higher docking score of −6.4 (Fig. S15†) (Table 2), while adrenaline bitartrate interacted with Asp79 and Gly 86 through hydrogen-bond interactions and had aromatic H-bond linkages with Trp87 (Fig. S16†) (Table 2). Compound 8 interacted with Glu149 via H-bonds, showing a docking score of −3.3 (Fig. S17†) and compound 9 interacted with His110, showing both H-bond and π cation interaction and a docking score of −0.69 (Fig. S18†) (Table 2). Compound 10 interacted with Phe133 through π–π stacking, while His110, Asp148, and Glu149 showed aromatic interactions and a docking score of −3.06 (Fig. S19†) (Table 2). Compound 11 showed H-bond linkages and aromatic hydrogen bonds with Asp131 and Glu149 and a docking score of −4.44 (Fig. S20†) (Table 2). Compound 12 interacted with Phe 133 via π–π stacking, while Gly81, Trp87, Lys105, Arg163, and Glu149 showed H-bond interactions and a higher docking score of −10.4 (Fig. 8) (Table 2).
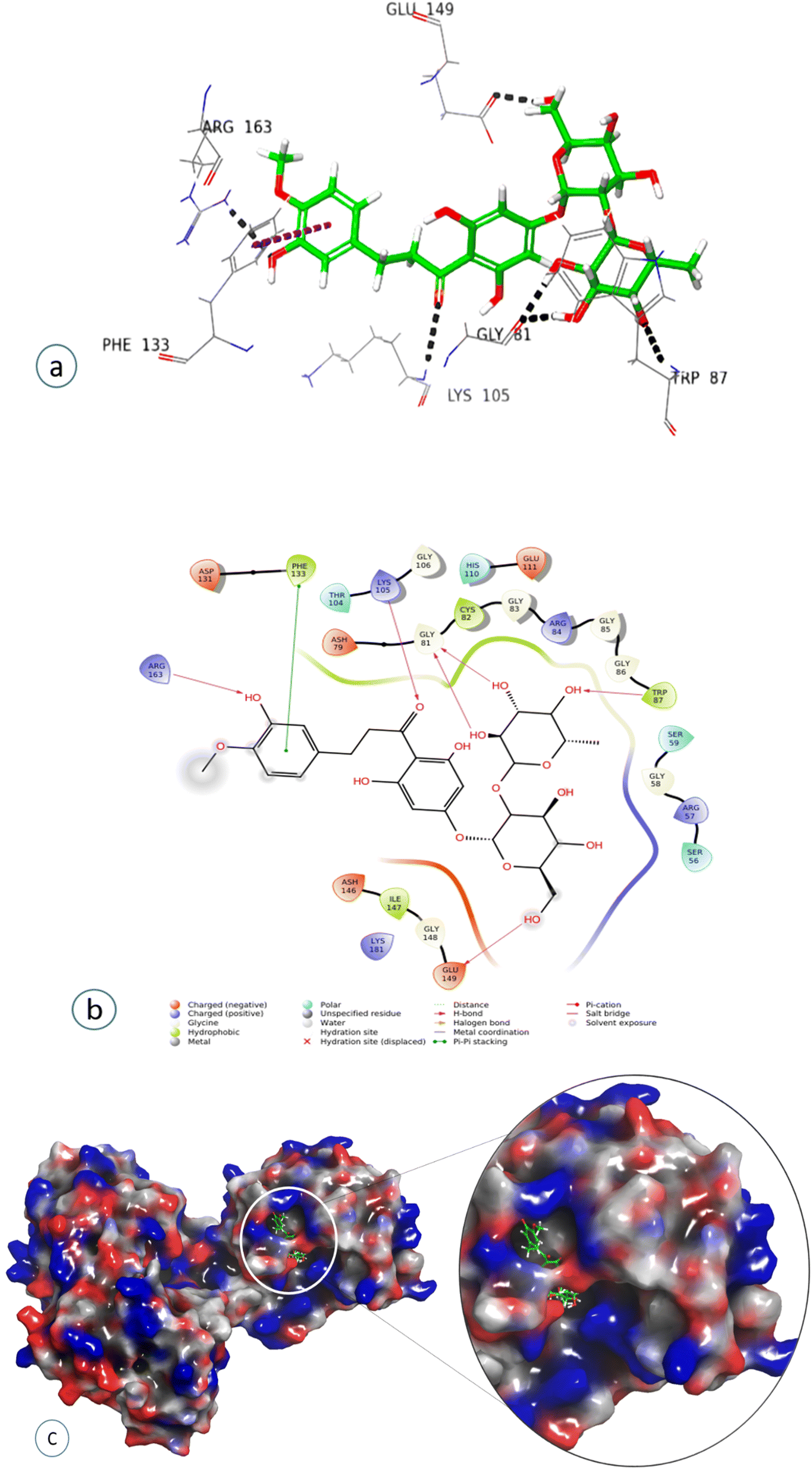 | ||
| Fig. 8 Docked pose of compound 12 (neohesperidin dihydrochalcone): (a) 3D representation of ligand–protein interaction in dotted lines indicating hydrogen bonds (black) and π–π stacking (magenta). (b) 2D representation of ligand–protein profile. (c) Solid surface representation of the ligand–protein profile, depicting the electrostatic potential distribution over the surface (red, negative regions; blue, positive regions). | ||
All the selected ligands shown interactions with MTase residues involved in the methylation of RNA, except atenolol, which also bound with the Asp146 of the catalytic tetrad. Furthermore, we found that along with all the above residues, Glu149 and Glu111 interactions were very frequent among all the interacting compounds.
In our study, we found that all the compounds interacted through various types of non-covalent interactions, including π–π stacking, aromatic hydrogen bonds, and hydrogen bonding with important active site residues, such as Glu 149, Glu 111, His 110, and Trp 187. This further validated that these compounds might have a role in destabilizing the NS5, and thus can serve as leads against DENV.
4 Conclusions
NS5 protein has a naturally unique fusion of MTase and RdRP enzymes, which are responsible for maintaining various functions in dengue virus. As NS5 is conserved across all the dengue serotypes, it could be a promising target for anti-DENV therapy. In this study, 75 compounds were evaluated by STD-NMR spectroscopy followed by molecular docking studies, which showed various interactions of NS5 protein with all the compounds 1–12. All the ligands interacted with the key amino acid residues of NS5, such as Glu149, His110, Glu111, and Phe133. Atenolol (1), itopride hydrochloride (2), scopolamine hydrobromide trihydrate (3), and phloridzin (4) were found to possess substantial interactions, while the rest of the compounds showed relatively weak interactions with NS5. Therefore, we suggest that some of these compounds may be optimized as a potential drug candidates against DENV.Author contributions
ATW and MIC were involved in conceptualization, investigation, supervision, analysis, reviewing, and editing of manuscript. AU was involved in experimentation, analysis and manuscript writing. AM had helped in acquiring and processing the NMR experiments. PG has provided the construct of NS5 and involved in reviewing, and editing of manuscript.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
Authors acknowledge the enabling role of the Searle Company through research funding and also financial support of Higher Education Commission, Pakistan, through research project no. 12589. We thank to Dr Pei-Yong Shi and Dr Bo Zhang for providing the cloning material for DENV2-NS5 gene.References
- E. Ma and G. Cheng, J. Infect. Dis., 2022, 1, 50–58, DOI:10.1016/j.imj.2021.12.003.
- S. J. Gan, Y. Q. Leong, M. F. H. bin Barhanuddin, S. T. Wong, S. F. Wong, J. W. Mak and R. B. Ahmad, J. Parasitol. Vector, 2021, 14, 1–19, DOI:10.1186/s13071-021-04785-4.
- B. Cao, M. S. Diamond and I. U. J. Mysorekar, J. Interferon Cytokine Res., 2017, 37, 287–294, DOI:10.1089/jir.2017.0011.
- N. M. Ferguson, Nature, 2018, 559, 490–497, DOI:10.1038/s41586-018-0318-5.
- N. T. Darwish, S. D. Sekaran and S. M. Khor, Sens. Actuators, B, 2018, 255, 3316–3331, DOI:10.1016/j.snb.2017.09.159.
- R. Perera and R. Kuhn, Curr. Opin. Microbiol., 2008, 11, 369–377, DOI:10.1016/j.mib.2008.06.004.
- C. A. Lesburg, M. B. Cable, E. Ferrari, Z. Hong, A. F. Mannarino and P. C. Weber, Nat. Struct. Mol. Biol., 1999, 6, 937–943, DOI:10.1038/13305.
- K. H. Choi, J. M. Groarke, D. C. Young, R. J. Kuhn, J. L. Smith, D. C. Pevear and M. G. Rossmann, Proc. Natl. Acad. Sci., 2004, 101, 4425–4430 CrossRef CAS PubMed.
- G. Lu and P. Gong, PLoS Pathog., 2013, 9, e1003549, DOI:10.1371/journal.ppat.1003549.
- W. Liu, X. Shi and P. Gong, Nucleic Acids Res., 2018, 46, 10840–10854, DOI:10.1093/nar/gky848.
- J. Wu, H. Q. Ye, Q. Y. Zhang, G. Lu, B. Zhang and P. Gong, PLoS Pathog., 2020, 16, e1008484, DOI:10.1371/journal.ppat.1008484.
- M. Bollati, K. Alvarez, R. Assenberg, C. Baronti, B. Canard, S. Cook, B. Coutard, E. Decroly, X. de Lamballerie and E. A. Gould, Antiviral Res., 2010, 87, 125–148, DOI:10.1016/j.antiviral.2009.11.009.
- A. Parihar, Z. F. Sonia, F. Akter, M. A. Ali, F. T. Hakim and M. S. Hossain, Comput. Biol. Med., 2022, 145, 105468, DOI:10.1016/j.compbiomed.2022.105468.
- D. Ray, A. Shah, M. Tilgner, Y. Guo, Y. Zhao, H. Dong, T. S. Deas, Y. Zhou, H. Li and P. Y. J. Shi, J. Virol., 2006, 80, 8362–8370, DOI:10.1128/JVI.00814-06.
- M. P. Egloff, E. Decroly, H. Malet, B. Selisko, D. Benarroch, F. Ferron and B. Canard, J. Mol. Biol., 2007, 372, 723–736, DOI:10.1016/j.jmb.2007.07.005.
- C. S. Tan, J. M. Hobson-Peters, M. J. Stoermer, D. P. Fairlie, A. A. Khromykh and R. A. Hall, J. Gen. Virol., 2013, 94, 1961–1971, DOI:10.1099/vir.0.054395-0.
- S. P. Lim, J. H. K. Koh, C. C. Seh, C. W. Liew, A. D. Davidson, L. S. Chua, R. Chandrasekaran, T. C. Cornvik, P. Y. Shi and J. Lescar, J. Biol. Chem., 2013, 288, 31105–31114, DOI:10.1074/jbc.M113.508606.
- J. Wu, G. Lu, B. Zhang and P. Gong, J. Virol., 2015, 89, 249–261, DOI:10.1128/JVI.02085-14.
- Y. Zhao, T. S. Soh, K. W. K. Chan, S. S. Y. Fung, K. Swaminathan, S. P. Lim, P. Y. Shi, T. Huber, J. Lescar and D. Luo, J. Virol., 2015, 89, 10717–10721, DOI:10.1128/JVI.01239-15.
- C. V. Filomatori, M. F. Lodeiro, D. E. Alvarez, M. M. Samsa, L. Pietrasanta and A. V. Gamarnik, Genes Dev., 2006, 20, 2238–2249, DOI:10.1101/gad.1444206.
- J. Ashour, M. Laurent-Rolle, P. Y. Shi and A. García-Sastre, J. Virol., 2009, 83, 5408–5418, DOI:10.1128/JVI.02188-08.
- M. A. Behnam, C. Nitsche, V. Boldescu and C. D. Klein, J. Med. Chem., 2016, 59, 5622–5649, DOI:10.1021/acs.jmedchem.5b01653.
- Q. Li and C. Kang, Membranes, 2022, 12(2), 231, DOI:10.3390/membranes12020231.
- R. A. Friesner, R. B. Murphy, M. P. Repasky, L. L. Frye, J. R. Greenwood, T. A. Halgren, P. C. Sanschagrin and D. T. Mainz, J. Med. Chem., 2006, 49, 6177–6196, DOI:10.1021/jm051256o.
- M. T. Khan, M. J. Islam, A. Parihar, R. Islam, T. J. Jerin, R. Dhote, M. A. Ali, F. K. Laura and M. A. Halim, Inform. Med. Unlocked., 2021, 24, 100578, DOI:10.1016/j.imu.2021.100578.
- A. Parihar, Z. F. Sonia, N. kumar Choudhary, P. Sharma, I. Mahdi, F. T. H. Hakim, M. A. Ali, R. Khan, M. S. Alqahtan and M. Abbas, Res Sq., 2022 DOI:10.21203/rs.3.rs-1517448/v1.
- E. Harder, W. Damm, J. Maple, C. Wu, M. Reboul, J. Y. Xiang, L. Wang, D. Lupyan, M. K. Dahlgren and J. L. Knight, J. Chem. Theory Comput., 2016, 12, 281–296, DOI:10.1021/acs.jctc.5b00864.
- D. Shivakumar, J. Williams, Y. Wu, W. Damm, J. Shelley and W. Sherman, J. Chem. Theory Comput., 2010, 6, 1509–1519, DOI:10.1021/ct900587b.
- A. Parihar, R. Shrivastava and A. Dube, J. Photochem. Photobiol., 2021, 5, 100013, DOI:10.1016/j.jpap.2020.100013.
- B. Zhang, H. Li, K. Yu and Z. Jin, CCF Trans. High Perform. Comput., 2022, 4, 63–74, DOI:10.1007/s42514-021-00086-5.
- A. N. Wadworth, D. Murdoch and R. N. Brogden, Drugs, 1991, 42, 468–510, DOI:10.2165/00003495-199142030-00007.
- Z. L. Zhao, C. Liu, Q. Z. Wang, H. W. Wu and J. W. Zheng, Ann. Transl. Med., 2021, 9 DOI:10.21037/atm-20-5359.
- Y. Iwanaga, T. Kimura, N. Miyashita, K. Morikawa, O. Nagata, Z. Itoh and Y. kondo, Jpn. J. Clin. Pharmacol., 1994, 66, 317–322, DOI:10.1254/jjp.66.317.
- S. Liu, D. Shi, Z. Sun, Y. He, J. Yang and G. Wang, Front. Psychiatry, 2021, 12, 601985, DOI:10.3389/fpsyt.2021.601985.
- A. Bhattacharjee, R. Chaudhuri, J. J. Dash, M. Saha, L. Choudhury and S. Roy, Appl. Biochem. Biotechnol., 2021, 193, 1654–1674, DOI:10.1007/s12010-021-03526-8.
- J. R. Ehrenkranz, N. G. Lewis, C. Ronald Kahn and J. Roth, Diabetes/Metab. Res. Rev., 2005, 21, 31–38, DOI:10.1002/dmrr.532.
- B. G. Vijayakumar, D. Ramesh, A. Joji and T. Kannan, Eur. J. Pharmacol., 2020, 886, 173448, DOI:10.1016/j.ejphar.2020.173448.
- B. Tanrisever and P. Santella, Drugs, 1986, 32, 1–16, DOI:10.2165/00003495-198600323-00003.
- R. Roškar and V. Kmetec, J. Chromatogr. B: Biomed. Sci. Appl., 2003, 788, 57–64, DOI:10.1016/S1570-0232(02)01015-2.
- J. Saraswat, P. Singh and R. J. Patel, J. Mol. Liq., 2021, 326, 115298, DOI:10.1016/j.molliq.2021.115298.
- F. Rossi and A. Forgione, J. Pharm. Res., 1995, 31, 81–94, DOI:10.1016/1043-6618(95)80052-2.
- A. G. Fernández and R. Massingham, J. Life Sci., 1985, 36, 1–14, DOI:10.1016/0024-3205(85)90280-2.
- N. Backhouse, C. Delporte, M. Givernau, B. Cassels, A. Valenzuela and H. Speisky, Agents Actions, 1994, 42, 114–117, DOI:10.1007/BF01983475.
- I. Tietjen, F. Ntie-Kang, P. Mwimanzi, P. A. Onguéné, M. A. Scull, T. O. Idowu, A. O. Ogundaini, L. M. Meva’a, B. M. Abegaz and C. M. Rice, PLoS One, 2015, 10, e0121099, DOI:10.1371/journal.pone.0121099.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ra04836a |
| This journal is © The Royal Society of Chemistry 2023 |