
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegulating the preparation of antibacterial poly(amidoxime) for efficient uranium extraction from seawater†
Xue
Zhang
and
Dadong
Shao
 *
*
School of Environmental and Biological Engineering, Nanjing University of Science and Technology, Nanjing 210094, P R China. E-mail: shaodadong@126.com
First published on 20th July 2023
Abstract
Seawater is a huge store of uranium, and the related uranium retraction technology has become a critical step in the sustainable development of nuclear power. For obtaining uranium U(VI) from seawater cheaply and environmentally, we introduced K2FeO4 during the polyamidoxime (PAO) preparation process in water to endow PAO with high antibacterial and low agglomeration properties. In this reaction, only water was used as the solvent with the goal of extracting U(VI) from seawater in a low-cost, pollution-free manner. Studies showed that the adsorption of U(VI) on K2FeO4@PAO conformed to a pseudo-second-order model, and the maximum adsorption capacity calculated by the Langmuir model was 137 mg g−1 at 298 K and pH 8.2. Moreover, K2FeO4@PAO showed high selectivity for U(VI) compared with a range of metal ions. K2FeO4@PAO also showed good recyclability, and the recovery rate only decreased by 3% after six cycles. In addition, antibacterial experiments indicated that K2FeO4@PAO could effectively inhibit the growth of Escherichia coli (E. coli) and Vibrio alginolyticus (V. alginolyticus) that are commonly found in seawater.
Introduction
As a low-carbon and efficient energy, the sound development of nuclear power is crucial for a future low-carbon society.1,2 As the most important nuclear fuel for nuclear power operations, the safe supply of uranium is key to guaranteeing the sustainable development of nuclear power. Traditional terrestrial uranium resources are limited and would be exhausted in few decades. It is thus a worldwide task to explore new uranium resources. As a typical uranium resource, seawater has attracted worldwide attention because an estimated ∼4.5 billion tons of U(VI) in seawater can solve the global shortage of uranium.3,4 There is thus an urgent need to develop appropriate methods and materials for seawater U(VI) extraction.5–7 Thus far, several technologies have been reported to recover U(VI) from seawater, such as ionic exchange, electrochemical process, co-precipitation, and adsorption method.8–11 Among them, adsorption has been widely adopted due to its low cost and practicability. However, the complexity of the marine environment, such as very low U(VI) concentration (∼3 ppb), presence of many coexisting competing ions, high salinity, and easy biofouling, makes the U(VI) extraction process extremely challenging.12–14 Other factors, such as the technical economy of the extraction process, also need to be seriously considered. Therefore, it is necessary to develop high-performance materials to improve comprehensive properties of the used adsorbents to enable the economical extraction of U(VI) from seawater.15Polyamidoxime (PAO)-based materials can adsorb U(VI) due to the O and N atoms of PAO having the same distance and lone pair of electrons, and these are thus among the hot materials in U(VI) extraction research. Resulting from polyacrylonitrile's (PAN) low cost, good chemical stability, and easy industrial production, numerous adsorbents select it to prepare PAO-based materials for U(VI) recovery.16–18 There are many methods for synthesizing PAO in the laboratory. The classic synthesis method is –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N amidoximation, which can be divided into homogeneous and heterogeneous processes according to the solvent. The homogeneous method mainly involves the reaction of PAN and NH2OH in an organic solvent with dimethyl sulfoxide and N,N′-dimethyl formamide generally chosen in the laboratory. The heterogeneous method mainly involves the reaction of PAN with NH2OH in water and methanol/water solutions. Organic solvents are often required in this method, which makes it not only harmful to the environment but also increases the cost of U(VI) recovery, dramatically reducing the competitiveness of PAO in U(VI) extraction. Thus, we considered selecting water as a solvent during the amidoximation process. PAN, as a macromolecule material, is prone to agglomerate in water, leading to intramolecular bond accumulation without the reaction of its groups.19,20 In this work, K2FeO4 was adopted to inhibit the aggregation of PAN in water with an aim to obtain a higher conversion rate.
N amidoximation, which can be divided into homogeneous and heterogeneous processes according to the solvent. The homogeneous method mainly involves the reaction of PAN and NH2OH in an organic solvent with dimethyl sulfoxide and N,N′-dimethyl formamide generally chosen in the laboratory. The heterogeneous method mainly involves the reaction of PAN with NH2OH in water and methanol/water solutions. Organic solvents are often required in this method, which makes it not only harmful to the environment but also increases the cost of U(VI) recovery, dramatically reducing the competitiveness of PAO in U(VI) extraction. Thus, we considered selecting water as a solvent during the amidoximation process. PAN, as a macromolecule material, is prone to agglomerate in water, leading to intramolecular bond accumulation without the reaction of its groups.19,20 In this work, K2FeO4 was adopted to inhibit the aggregation of PAN in water with an aim to obtain a higher conversion rate.
In the U(VI) extraction process, the interaction between U(VI) extraction materials and marine microorganisms should be considered in addition to improving the U(VI) recovery rate and cost.21,22 Seawater is a very harsh corrosive environment, which is typically characterized by high biological activity, with 106 colony-forming units (CFU) per mL level microbes (including bacteria and microalgae) found to exist in seawater. The microbes and their metabolisms affect material surface properties, which can eventually affect the U(VI) extraction performance. Thus, there is a necessity to design a novel adsorbent to achieve an antifouling goal.15,23,24 The corresponding standard electrode potentials of FeO42− both in acid (2.20 V) and alkali (0.72 V) media are higher than the well-known strong oxidant KMnO4 under the same conditions. This strong oxidation ensures the strong bactericidal ability of FeO42−. K2FeO4 can kill both Gram-positive and Gram-negative bacteria, fungi, viruses, and spores.25,26 The introduced K2FeO4 in PAN can inhibit the corrosion from marine microorganisms.
The structure of a material determines its properties, and ultimately affects its application optimizing the structure of a material is an effective way to enhance its properties and expand its application fields. Herein, we constructed the anti-biofouling nanoscale adsorbent K2FeO4@PAO for U(VI) recovery by introducing K2FeO4 during the amidoximation method with only water as a solvent (Fig. 1). The added K2FeO4 could overcome PAN agglomeration in water, so that PAN could well contact with NH2OH, which could help realize the economic and environmental recovering of uranium. Meanwhile, the high oxidation ability of K2FeO4 could exterminate bacterial. The obtained antibacterial K2FeO4@PAO particles exhibited a higher adsorption capacity and better anti-biofouling properties than PAO under the experimental conditions.
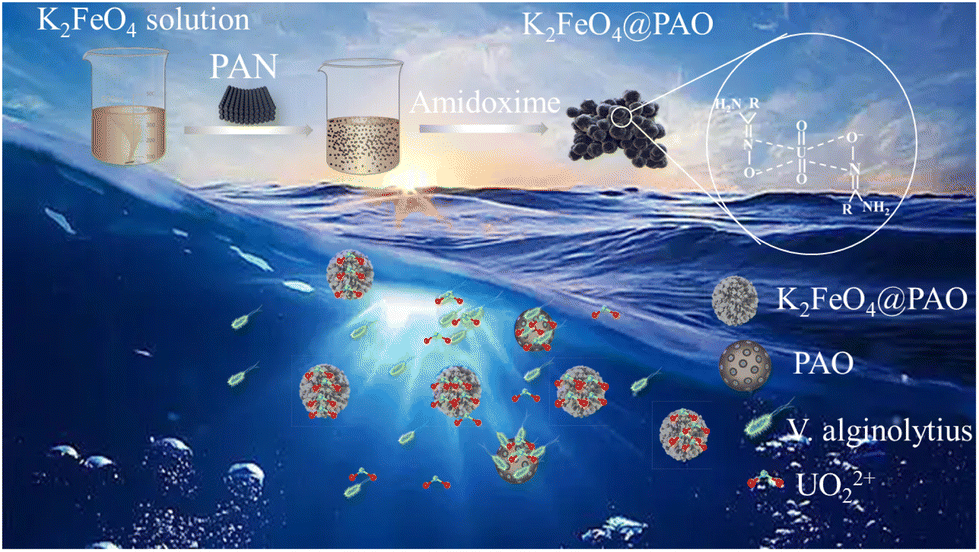 | ||
| Fig. 1 Scheme for the fabrication and application of K2FeO4@PAO in the extraction of U(VI) from seawater. | ||
Results and discussion
Characterization
As shown in Fig. S1A and B,† PAN tends to aggregate into large particles and is extremely unstable in aqueous solution, which is detrimental to the conversion of PAN into PAO, and ultimately affects the adsorption capability of PAO. Therefore, K2FeO4 was introduced to inhibit the agglomeration of PAN during the amidoximation process in this work. This is a simple way to obtain a high conversion of PAN, which would accelerate the practical application of PAO as a candidate for U(VI) extraction from seawater. As shown in Fig. S1C,† after introducing K2FeO4, the obtained PAO-based material could uniformly disperse in aqueous solution and the PAO appearance was improved overall. Given the effect of K2FeO4 on the inhibition of PAN particle agglomeration, the obtained PAO morphology may be regulated by K2FeO4.To study the effect of K2FeO4 on the PAO surface topologies, the microstructures of PAN and PAO prepared at different conditions were analyzed by SEM (Fig. 2A), with varying the addition amounts of K2FeO4 and PAN in the preparation process, as shown in Table 1. The SEM images indicated that the addition of 0.20 g K2FeO4 had no significant effect on PAO surface topologies, and the obtained K2FeO4@PAO was uniformly dispersed. As the mass of K2FeO4 increased, PAN agglomeration became more evident, and eventually, a PAN block was formed. The three reactions include the formation of –CN from unsaturated amine, the redox reaction between NH2OH and K2FeO4, and the conversion of –CN to –C(NH2)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–OH, which restrict and promote each other in water. When the amounts of PAN and NH2OH were much more than K2FeO4, the amidoximation reaction was the primary reaction in solution, and finally, uniform dispersed PAO particles were obtained. When the K2FeO4 amount was higher than PAN and NH2OH, the solution reaction was dominated by the redox reaction of K2FeO4 and NH2OH, which consumed most of the NH2OH and made it impossible to complete the conversion of –CN, instead forming –CN rings on the PAN macromolecular chains and thus creating bulk materials. Besides SEM, TEM was also applied to study the morphologies of PAO and K2FeO4@PAO. As shown in Fig. 2B, the single PAO presents a block form, but transformed into nanoscale particles (K2FeO4@PAO) after K2FeO4 was introduced. It was found that C, N, and O elements were uniformly distributed on PAO and K2FeO4@PAO surfaces by the TEM element mapping. Moreover, Fe was evenly distributed on the K2FeO4@PAO surface, proving that K2FeO4 recombined with PAO successfully. It is considered that the –C(NH2)N–OH unit in K2FeO4@PAO is the core of its application in U(VI) extraction. The adsorption capacity of K2FeO4@PAO for U(VI) gradually increased with increasing the PAN amount when the K2FeO4 amount was fixed at 0.20 g (Fig. S2A†) and decreased with increasing the K2FeO4 amount when the PAN amount was set at 2.00 g (Fig. S2B†). Therefore, the K2FeO4 amount should be well controlled during the K2FeO4@PAO preparation process. The aggregation of PAN could be significantly inhibited, and PAO particles with smaller particle sizes and better adsorption capability could be obtained at the composition of 0.20 g K2FeO4 and 2.00 g PAN. This reactant amount was selected in the following experiment.
N–OH, which restrict and promote each other in water. When the amounts of PAN and NH2OH were much more than K2FeO4, the amidoximation reaction was the primary reaction in solution, and finally, uniform dispersed PAO particles were obtained. When the K2FeO4 amount was higher than PAN and NH2OH, the solution reaction was dominated by the redox reaction of K2FeO4 and NH2OH, which consumed most of the NH2OH and made it impossible to complete the conversion of –CN, instead forming –CN rings on the PAN macromolecular chains and thus creating bulk materials. Besides SEM, TEM was also applied to study the morphologies of PAO and K2FeO4@PAO. As shown in Fig. 2B, the single PAO presents a block form, but transformed into nanoscale particles (K2FeO4@PAO) after K2FeO4 was introduced. It was found that C, N, and O elements were uniformly distributed on PAO and K2FeO4@PAO surfaces by the TEM element mapping. Moreover, Fe was evenly distributed on the K2FeO4@PAO surface, proving that K2FeO4 recombined with PAO successfully. It is considered that the –C(NH2)N–OH unit in K2FeO4@PAO is the core of its application in U(VI) extraction. The adsorption capacity of K2FeO4@PAO for U(VI) gradually increased with increasing the PAN amount when the K2FeO4 amount was fixed at 0.20 g (Fig. S2A†) and decreased with increasing the K2FeO4 amount when the PAN amount was set at 2.00 g (Fig. S2B†). Therefore, the K2FeO4 amount should be well controlled during the K2FeO4@PAO preparation process. The aggregation of PAN could be significantly inhibited, and PAO particles with smaller particle sizes and better adsorption capability could be obtained at the composition of 0.20 g K2FeO4 and 2.00 g PAN. This reactant amount was selected in the following experiment.
 | ||
| Fig. 2 SEM images of K2FeO4@PAO with different K2FeO4 and PAO contents (A) and TEM image and EDS mappings of PAO and K2FeO4@PAO (B). The preparation conditions for K2FeO4@PAO are shown in Table 1. | ||
| K2FeO4 (g) | PAN (g) | Name | |
|---|---|---|---|
| Fixed K2FeO4 series | 0.20 | 0.50 | 0.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.5 :0.5 |
| 1.00 | 0.2:1 |
||
| 1.50 | 0.2:1.5 |
||
| 2.00 | 0.2:2 |
||
| Fixed PAN series | 0 | 2.00 | 0:2 |
| 0.50 | 0.5:2 |
||
| 1.00 | 1:2 |
||
| 1.50 | 1.5:2 |
||
| 2.00 | 2:2 |
||
| 4.00 | 4:2 |
||
| 5.00 | 5:2 |
||
The effect of K2FeO4 on the PAO particle-size distribution was also studied. Dynamic light scattering (DLS) measurements showed that the PAO particle sizes were concentrated around 1600 nm (Fig. 3A). The particle diameter of K2FeO4@PAO was reduced ∼31% at 500 nm (Fig. 3B) compared with PAO. The introduced K2FeO4 could thus inhibit the agglomeration of PAN in water and promote the formation of nano-sized K2FeO4@PAO particles. The smaller K2FeO4@PAO particle size will help the adsorbent application in the target solution. The Tyndall phenomenon was used to further verify the size of the two particles. Here, if the particle is smaller than the wavelength of incident light, light scattering occurs. In this case, a light wave around the particle is observed and radiates around it, which is called scattered light and the Tyndall phenomenon is the scattering phenomenon of light. In terms of the macroscopic performance for the same incident light, if the particles are small, have a larger scattering angle, the scattered light has a weak flood angle, and its brightness is high, then the Tyndall phenomenon is obvious. If the particles are large, have a small scattering angle, the scattered light has a large flood angle, and a low centre brightness, then the Tyndall phenomenon is poor. As shown in Fig. S3,† the light-scattering angle of the particles of K2FeO4@PAO was large, the centre brightness was high, the flooding was small, and the Tyndall phenomenon was obvious. However, PAO had a poor particle size uniformity, and the light tended to be scattered by large particles. The light-scattering angle of the particles was small, the flooding area was large, the centre brightness was low, and the Tyndall path was dark.
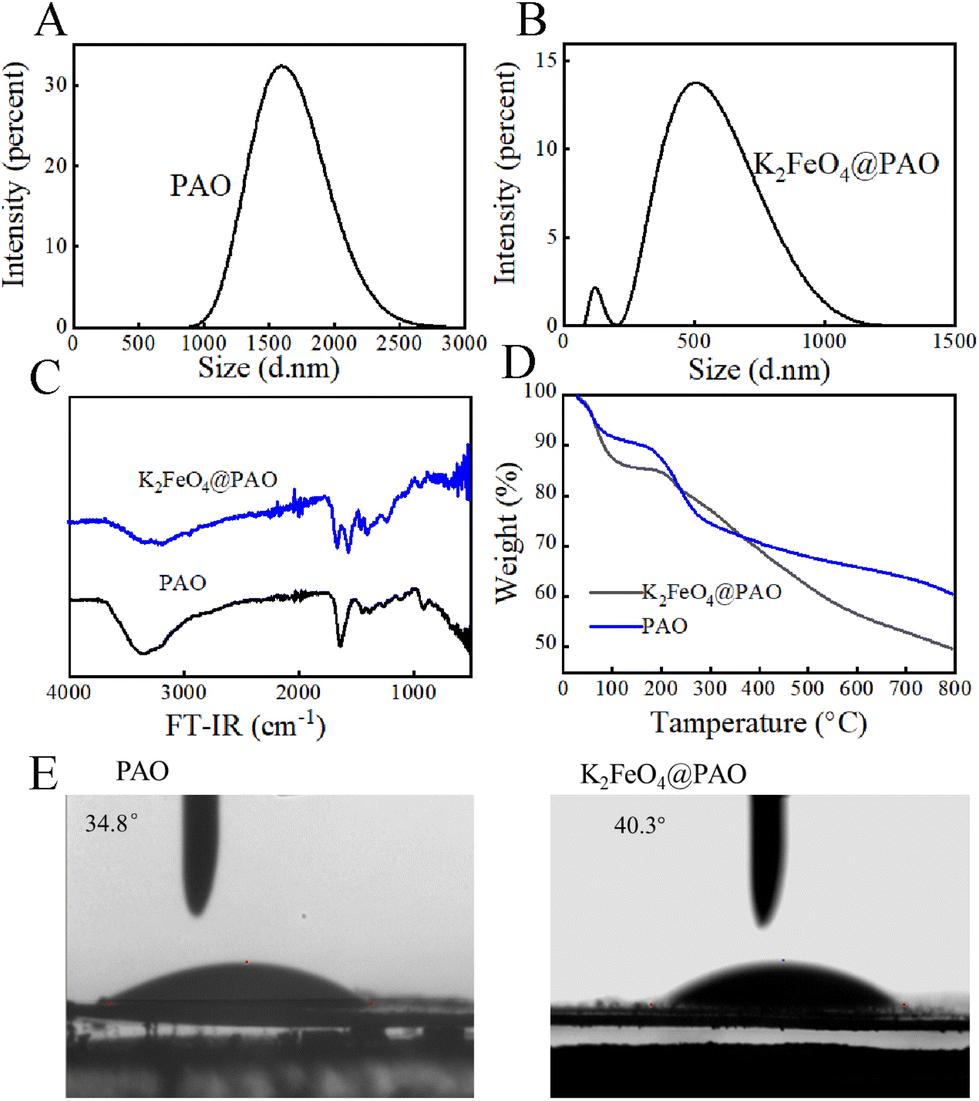 | ||
| Fig. 3 Size distributions (A and B), FT-IR spectra (C), TGA curves (D), and water contact angles (E) of PAO and K2FeO4@PAO. | ||
FT-IR and TGA were used to analyze the effects of K2FeO4 on the surface functional groups and thermal stability of PAO. As shown in Fig. 3C, The FT-IR spectra of K2FeO4@PAO and PAO were similar. In the K2FeO4@PAO spectrum, the peaks of N–H and O–H were at 3500–3000 cm−1, the peak of –CN was at ∼1668 cm−1, and the characteristic peak of N–O was at ∼932 cm−1, which were all consistent with the PAO infrared spectrum. Fig. 3D shows the TGA curves of PAO and K2FeO4@PAO. The first ∼4% weight losses of PAO and K2FeO4@PAO could be assigned to moisture loss. PAO and K2FeO4@PAO showed good thermal stability at 100–200 °C. The second weight loss at 200–500 °C was related to the cyclization dehydrogenation of PAO. PAO and K2FeO4@PAO then underwent severe oxidative cleavage at 500–700 °C.19
Static contact-angle experiments were used to quantify the surface hydrophilic property of PAO and K2FeO4@PAO. Before the test, each powder material was pressed into a tablet with a tablet press and the pressure of 20 MPa was maintained for 40 s. During the contact-angle measurement, 2 μL ultrapure water was injected for each test. Each sample was tested three times, and every test used a new sample to exclude residual effects. The results of the three tests are shown in Fig. 3E, whereby the average contact angles of PAO and K2FeO4@PAO were 34.8° and 40.3°, respectively. The lower water contact angles of PAO and K2FeO4@PAO indicated they both have good hydrophilicity, which would benefit their application in aqueous solution.
XPS was used to infer the binding states of the atoms and the electron distribution in K2FeO4@PAO and PAO by measuring the chemical shift of electrons in their layers (Fig. 4A). The high resolution of Fe 2p in the K2FeO4@PAO spectrum revealed the characteristic Fe 2p3/2 and Fe 2p1/227–29 of Fe signals located at 711.8 and 724.7 eV, respectively (Fig. S4†). A new peak appeared at ∼390 eV (Fig. S5†) related to XPS U 4f4 after U(VI) adsorption, revealing the high enrichment of U(VI) on PAO and K2FeO4@PAO surfaces. The XPS peaks of O 1s, N 1s, and C 1s were located at ∼532, ∼400, and ∼285 eV, respectively. The C 1s spectra (Table S1† and Fig. 4B) could be divided into five components at 284.6 ± 0.1, 285.3 ± 0.1, 286.4 ± 0.1, 287.4 ± 0.1, and 288.6 ± 0.1 eV corresponding to –CN, C–C, C–OH and –C(NH2)NOH, –CO, and –COO− groups of PAO and K2FeO4@PAO surfaces, respectively.30,31 As indicated in the N 1s spectra (Fig. 4C and Table S2†), the characteristic peaks with binding energies at 398.7 ± 0.1, 399.3 ± 0.1, and 400.5 ± 0.1 eV were attributed to the –CN, N–H, and –C(NH2)NOH bonds, respectively.31,32 The three fitting peaks in the O 1s spectra (Fig. 4D and Table S3†) belonged to –COO− (531.2 ± 0.1 eV), CO (532.3 ± 0.1 eV), and –OH (533.4 ± 0.1 eV), respectively.33,34 The XPS results showed that the chemical environments of PAO and K2FeO4@PAO were similar to each other, revealing that K2FeO4 did not affect PAO's chemical properties.
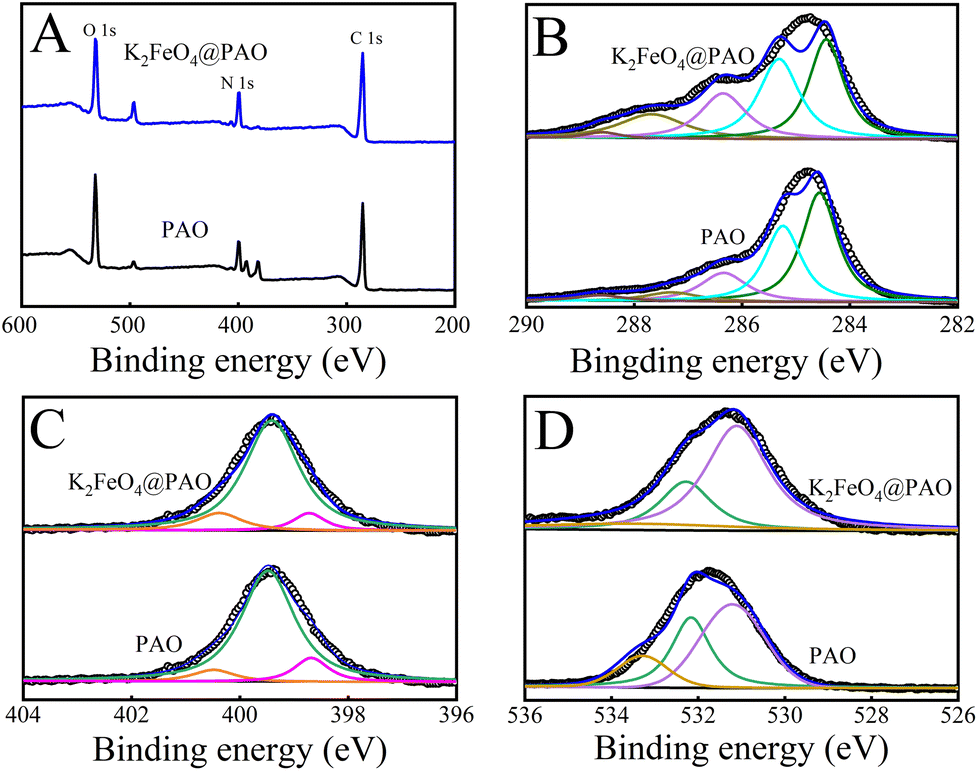 | ||
| Fig. 4 XPS survey (A), C 1s (B), N 1s (C), and O 1s (D) spectra of K2FeO4@PAO and PAO. | ||
Adsorption performance of K2FeO4@PAO
The solution pH can affect the forms of U(VI) existing in solution and on the adsorbent surface, therefore affecting the U(VI) adsorption behaviours.35Fig. 5A shows that the U(VI) extraction capacity of K2FeO4@PAO displayed an increasing trend at pH 2–5. UO22+ was the dominant form of U(VI) at pH < 4, and K2FeO4@PAO was positive due to protonation. Electrostatic repulsion would lead to a poor recovery effect for K2FeO4@PAO to U(VI). At pH 4.0–6.0, K2FeO4@PAO surface was gradually deprotonated and the electrostatic attraction of the solution-dominant positively charged U(VI) species was enhanced, resulting in increased U(VI) adsorption on K2FeO4@PAO. The U(VI) uptake decreased when the pH value increased from 6.0 to 9.0. The main reason for this phenomenon was that the negatively charged U(VI) species was the dominant substance, and so the adsorption capacity of K2FeO4@PAO for U(VI) was inhibited by electrostatic repulsion with the pH increasing. The maximum adsorption capacity of K2FeO4@PAO (17.1 mg g−1) was obtained at pH ∼ 5.0. The higher maximum adsorption capacity of K2FeO4@PAO over PAO illustrated that nano-sized K2FeO4@PAO favours U(VI) extraction.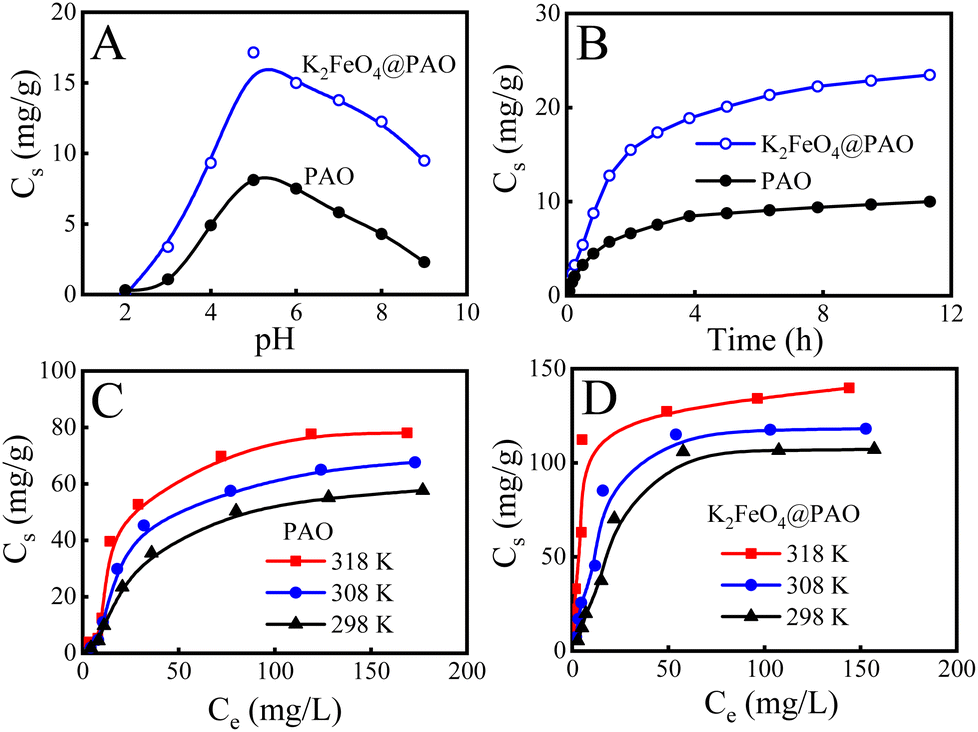 | ||
| Fig. 5 Effects of pH (A), contact time (B), adsorption isotherms, and reaction temperature on the adsorption of U(VI) on PAO (C) and K2FeO4@PAO (D) from solution. m = 0.02 g, V = 50 mL, I = 0.1 mol L−1 NaCl. (A) T = 298 ± 1 K, contact time: 24 h, [U(VI)]initial = 10.0 mg L−1. (B) T = 298 ± 1 K, pH = 8.2 ± 0.1, C[U(VI)]initial = 20.0 mg L−1. (C and D) pH = 8.2 ± 0.1, contact time: 24 h. | ||
The adsorption performances of K2FeO4@PAO at different contact times were also studied. U(VI) adsorption on PAO and K2FeO4@PAO rose sharply in the first 2 h, and then increased slowly with further increasing the reaction time (Fig. 5B), reaching equilibrium within 8 h. The adsorption kinetics were analyzed by pseudo-first-order (qt = qe × (1 − exp(−k1t)), where qe and qt (mg g−1) are the adsorbed amounts of U(VI) at equilibrium time and time t (h), respectively, and k1t (1 h−1) is the pseudo-first-order kinetic constant) and pseudo-second-order (qt = qe × t/(1/(K′ × qe) + t), where K′ (g (mg h)−1) is the pseudo-second-order kinetic constant) models.36,37 The relevant these parameters coefficients are generalized in Table S4.† The adsorption process of U(VI) by K2FeO4@PAO and PAO conformed to the pseudo-second-order model and chemisorption was the primary adsorption mode for U(VI) adsorption on K2FeO4@PAO and on PAO. According to the relevant results of the pseudo-second-order model, the maximum adsorption capacities of U(VI) on K2FeO4@PAO and PAO were 26.9 and 10.9 mg g−1 at pH 8.2 and 298 K, respectively.
Given the above excellent U(VI) recovery performance of K2FeO4@PAO, its effect for U(VI) recovery in various environmental solutions was also studied. As shown in Table 2, K2FeO4@PAO could effectively extract most U(VI) from this complex system. Therefore, the real application of K2FeO4@PAO in extraction U(VI) from seawater was performed, and seawater was prepared with sea salt. We found that K2FeO4@PAO could effective uptake ∼57% of 5 mg L−1 U(VI) from seawater. Experimental results further confirmed the high efficiency of K2FeO4@PAO in the extraction of U(VI) from seawater.
| Sample | C[U(VI)] (mg L−1) | ||
|---|---|---|---|
| Initial | Final | Adsorption (%) | |
| Diluted U(VI) sloution | 20 | 10.62 | 46.90 |
| 15 | 7.07 | 52.87 | |
| 10 | 5.04 | 49.60 | |
| 5 | 2.85 | 43.00 | |
| Running water | 20 | 13.39 | 33.05 |
| 15 | 8.65 | 42.33 | |
| 10 | 6.40 | 36.00 | |
| 5 | 2.87 | 42.60 | |
| Seawater | 20 | 14.51 | 27.45 |
| 15 | 8.78 | 41.47 | |
| 10 | 5.78 | 42.20 | |
| 5 | 2.17 | 56.60 | |
Adsorption isotherms was used to depict the adsorption capacities of K2FeO4@PAO and PAO at three different temperatures (i.e. 298, 308, and 318 K). As shown in Fig. 5C and D, with increasing the U(VI) initial concentration and temperature, the adsorption capacities of K2FeO4@PAO and PAO for U(VI) increased. Langmuir (Cs = b × Cs,max × Ceq/(1 + b × Ceq), where Ceq is the equilibrium concentration of U(VI) after adsorption, Cs,max (mg g−1) and b (L mg−1) are the maximum adsorption capability of the adsorbent and the Langmuir constant, respectively) and Freundlich (Cs = K × C1/ne, K (mg g−1) and 1/n are the constants indicative of the adsorption capability and intensity, respectively) models were used to fit the experimental data, and the mechanism of the adsorption process is discussed.36,37 According to the R2 values in Table S5,† the Langmuir isotherm model could better describe the experimental data, suggesting there was no interaction between the adsorption sites, and the U(VI) adsorption process on K2FeO4@PAO was a monolayer adsorption. The Cs,max value K2FeO4@PAO for U(VI) at pH 8.2 and 298 K was calculated to be 137 mg g−1.
According to the adsorption results, the adsorption capacity of PAO prepared in water was lower than that reported when prepared in organic reagents.38–40 From the perspective of economic cost and environmental protection, obtaining PAO in water is more conducive to the long-term development of PAN as a material for extracting U(VI) from seawater. More importantly, the PAO's preparation in water has not been reported yet. PAN is widely used in U(VI) recovery, mainly because its surface contains a large number of –CN groups, which can be converted into –C(NH2)NOH by chemical reaction with NH2OH in solution and –C(NH2)NOH can coordinate with U(VI) to recover uranium. However, one cannot ignore that PAN is easy to agglomerate and most –CN is not reacted with NH2OH in water, resulting in PAO having a low adsorption capacity for U(VI). Here, K2FeO4@PAO nanoparticles were obtained after K2FeO4 was introduced into the PAN transformation process. Compared with PAO, the adsorption capacity of K2FeO4@PAO was significantly increased, indicating that PAN reacted more fully with NH2OH and the amount of –C(NH2)NOH on its surface increased.
The relevant thermodynamic parameters,41 standard enthalpy change (ΔH°), standard Gibbs free energy change (ΔG°), and entropy change (ΔS°) of K2FeO4@PAO and PAO for U(VI) adsorption at three different temperatures (i.e. 298, 308, and 318 K) were investigated and are discussed, and the results are shown in Table S6.† The energy released or adsorbed in the chemical reaction process can be expressed by heat, called the heat of reaction, also known as the “enthalpy change” (ΔH°). ΔH° > 0 means that the adsorption of U(VI) on K2FeO4@PAO and PAO surfaces is an endothermic process, which is consistent with the improved U(VI) adsorption at higher reaction temperature. For a chemical reaction, the ΔG° value determines the reaction direction. The negative ΔG° value indicated that the adsorption of U(VI) on K2FeO4@PAO and PAO is a spontaneous process, which also confirmed the strong affinity between –C(NH2)NOH and U(VI).
To evaluate the stability of K2FeO4@PAO, the desorption–regeneration properties of K2FeO4@PAO were studied. Different concentrations and solvents were used to eluate K2FeO4@PAO. First, 0.5 mol L−1 HCl, HNO3, NaOH, and Na2CO3 were selected as eluting agents for the desorption of U(VI) from the K2FeO4@PAO surface. As shown in Fig. S6,† Na2CO3 showed the best resolution rate at 99%. Different concentrations (0.1–1 mol L−1) of Na2CO3 solution were used to eluate the K2FeO4@PAO. The elution results are shown in Fig. 6A, where it can be seen that the elution rate gradually increased with the increase in Na2CO3 concentration. When the Na2CO3 concentration reached 0.5 mol L−1, the solution elution rate could reach almost 100%, so 0.5 mol L−1 Na2CO3 solution could achieve effectively regenerate K2FeO4@PAO. The adsorption capability of the regenerated K2FeO4@PAO for U(VI) is shown in Fig. 6B. The adsorption capacity of K2FeO4@PAO only decreased by 3% after its regeneration and reuse six times.
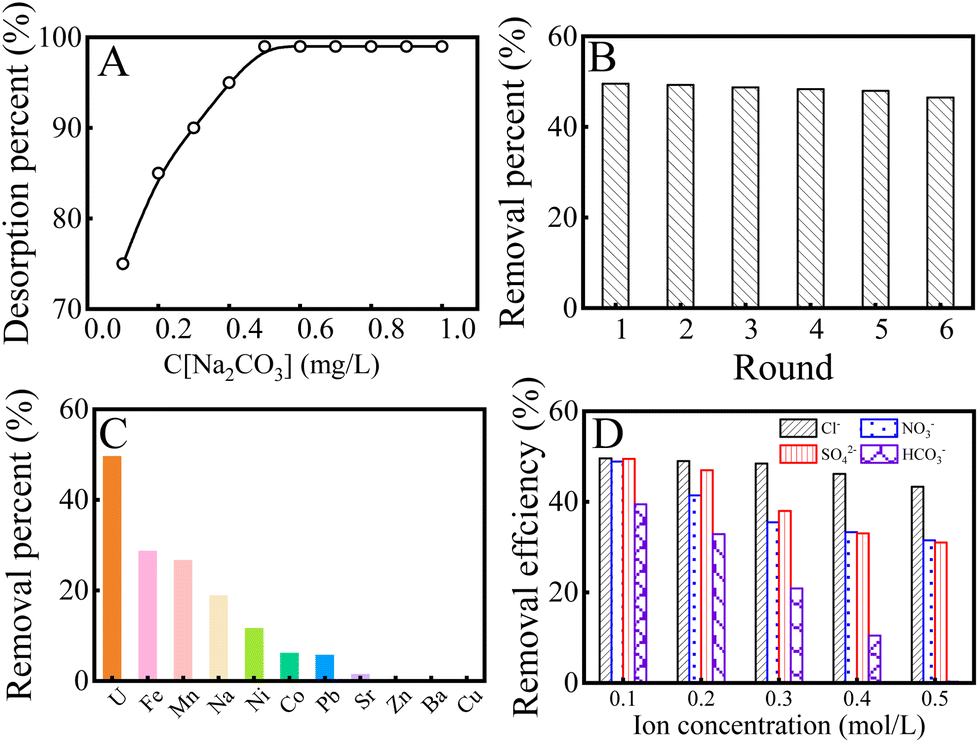 | ||
| Fig. 6 Effect of Na2CO3 concentration on the desorption of U(VI) from K2FeO4@PAO (A); recycling of K2FeO4@PAO in the extraction of U(VI) from solution (B); comparison of K2FeO4@PAO enrichment performance for 10 mg L−1 U(VI) and heavy metal ions (C); U(VI) removal efficiency for K2FeO4@PAO with the coexistence of different anions (D). I = 0.1 mol L−1 NaCl, T = 298 ± 1 K, contact time: 24 h. C[U(VI)]initial = 10.0 mg L−1, m = 0.02 g, V = 50 mL, pH = 8.2 ± 0.1. | ||
Based on the complex compositions of seawater, the coexistence of ions in seawater can affect the U(VI) adsorption. With various metal ions coexisting in seawater (such as Na(I), Ba(II), Sr(II), Ni(II), Co(II), Zn(II), Cu(II), Mn(II), Pb(II), and Fe(III)), ICP-MS was used to detect the concentration of each ion and then to evaluate the selectivity of K2FeO4@PAO for U(VI). As depicted in Fig. 6C, the adsorption capacity of K2FeO4@PAO for U(VI) was higher than that of the other ions, indicating that K2FeO4@PAO had excellent selective adsorption capacity for U(VI) in the coexistence ion system. The recovery of U(VI) from anionic solutions was also investigated for K2FeO4@PAO. As shown in Fig. 6D, with the increase in ion concentration, the influence of HCO3− on U(VI) recovery was undeniable due to the formation of new U(VI) species. In natural seawater, the concentration of various anions is far lower than 0.1 mol L−1, which means that anions have little effect on recycling U(VI).42 K2FeO4@PAO was obtained by introducing K2FeO4 into PAN to inhibit PAN agglomeration in water. The overall structure and morphology of the adsorbent were optimized. The particle size of K2FeO4@PAO can reach the nanometre scale, and it can be evenly dispersed in solution, so that the adsorbent can better contact with U(VI) and promote the occurrence of chemical reactions. The introduced K2FeO4 did not damage the stability and selectivity of PAO. As we expected, the adsorption capability of K2FeO4@PAO was much higher than PAO in various U(VI) solutions. In K2FeO4@PAO, PAO ensures the excellent selectivity of K2FeO4@PAO for U(VI), and the nanoscale particle size enables K2FeO4@PAO to fully react with U(VI) in solution, ultimately ensuring that K2FeO4@PAO has better adsorption effect on U(VI) than other ions in complex systems. Furthermore, K2FeO4@PAO also showed good cycling and selectivity for U(VI). In summary, K2FeO4@PAO can not only reduce the cost of the PAO preparation process and make the process greener, but can also further optimize the surface structure of the PAO and improve its adsorption properties in a variety of environments.
Antibacterial activity of K2FeO4@PAO
Many marine microorganisms in seawater will accumulate on the surface of materials, resulting in the biological contamination of materials, which will lead to materials corrosion and finally affect the U(VI) adsorption performance. Conventionally obtained PAO powders have no ability to resist biological fouling and often need to work synergistically with other antibacterial materials in the field of anti-biological fouling U(VI) extraction from seawater. As is well known, K2FeO4 presents excellent properties in decontamination, off-colour, and flocculation applications etc. Researchers have also found that the strong oxidation of K2FeO4 can destroy bacterial structures (such as cell walls and cell membranes) and suppress the reproduction of bacteria.24,25 Combining K2FeO4 with PAO can be expected to break through the inhibition of PAO adsorption by marine bacteria and maintain good U(VI) recovery performance in bacteria-containing solutions. Vibrio marine is a kind of bacteria commonly living in the ocean. Here, we took V. alginolyticus as an example to investigate the effects of marine microorganisms on the U(VI) recovery performance by PAO and K2FeO4@PAO. We adjusted the V. alginolyticus concentration to 1.0 × 106 CFU mL−1 (close to its concentration in offshore areas) and balanced this with PAO and K2FeO4@PAO for 24 h. As shown in Fig. 7A, V. alginolyticus resulted in severe biological contamination and significantly decreased the adsorption capacity compared with the samples without bacteria. These results indicated that biofouling has a significant effect on PAO-based materials. Unlike PAO, the recovery capability of K2FeO4@PAO for U(VI) did not significantly decrease compared with the data results for tests performed under sterile conditions, indicating that the K2FeO4 ensured K2FeO4@PAO presented excellent anti-biofouling properties (Fig. 7B). Therefore, we studied the effect of the K2FeO4 and PAO contents on the anti-biofouling properties of K2FeO4@PAO, using two Gram-negative bacteria (E. coli and V. alginolyticus). Fig. S7† shows the change in colonies of E. coli and V. alginolyticus on agar plates at different conditions. Fig. 7C exhibits the bactericidal rates calculated by the Petri dish counting method to quantitatively analyze different materials antibacterial properties. The results showed that K2FeO4 is critical for the anti-biofouling properties of K2FeO4@PAO for E. coli and V. alginolyticus, and the anti-boifouling performance increased with increasing the K2FeO4 content in K2FeO4@PAO. K2FeO4@PAO could kill almost all E. coli and V. alginolyticus when the K2FeO4 dosage was increased to 4.0 g. The introduced K2FeO4 not only promoted formatting PAO with good recovery performance for U(VI), but also directly endowed the PAO with antibacterial properties to maintain good recovery for U(VI) in bacterial-containing solutions.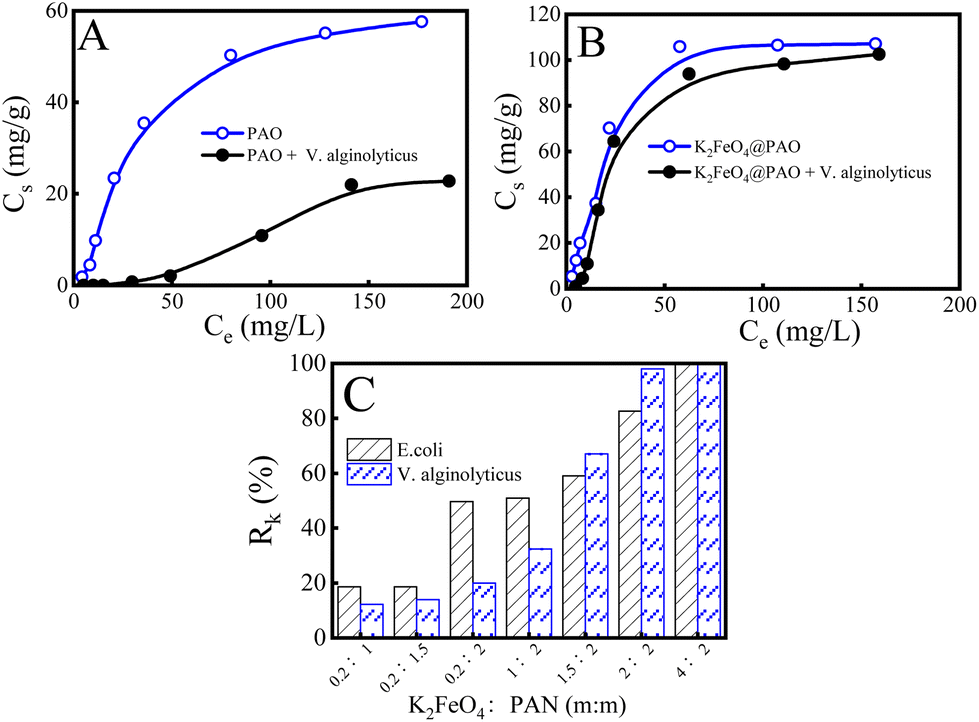 | ||
| Fig. 7 Adsorption of U(VI) by PAO (A) and K2FeO4@PAO (B) in solutions of different concentrations. m = 0.02 g, V = 50 mL, I = 0.1 mol L−1 NaCl. T = 298 ± 1 K, pH = 8.2 ± 0.1, contact time: 24 h; bactericidal rates against E. coli and V. alginolyticus of different particles (C). The K2FeO4@PAO materials were named according to the used amounts of K2FeO4 and PAN (Table 1). | ||
Conclusions
Considering the limitation of PAO as a single material for U(VI) extraction from seawater, we further optimized the structure of PAN during the conversion of PAN into PAO to further enable the application of PAO to be used for multiple relationships. The results showed that fine control of K2FeO4 dosage during the preparation process can effectively enhance the anti-biofouling property and adsorption capability of the PAO-based material (K2FeO4@PAO) for extracting U(VI) from seawater. K2FeO4@PAO displayed good adsorption and circulatory capacities under laboratory conditions. K2FeO4@PAO also showed remarkable selectivity for U(VI) against multiple coexisting metal ions. Due to the K2FeO4 effect, K2FeO4@PAO exhibited excellent anti-biofouling property, and could kill almost all E. coli and V. alginolyticus. In summary, research attempts to convert PAN into PAO in water and inhibit PAN agglomeration, improve the conversion rate of surface functional groups, improve its U(VI) adsorption capacity, and confer antibacterial properties will promote the further development of low-cost, pollution-free, and biofouling resistant U(VI) extraction materials from seawater.Experimental section
Synthesis of K2FeO4@PAO
Two series of K2FeO4@PAO materials with different mass ratios (Table 1) were prepared by regulating PAN and K2FeO4. Specific amounts of K2FeO4 and PAN powder, 3.20 g NH2OH·HCl, and 1.75 g NaOH were added into 20 mL de-gassed water. The solution was reacted at 75 °C for 6 h. After washing with de-gassed water, the K2FeO4@PAO materials were vacuum dried at 75 °C. To evaluate the effect of K2FeO4, PAO was also prepared by the same method.Synthesis of PAO in methanol/water solution
NH2OH solution was prepared by dissolving 4.0 g NH2OH·HCl in 50 mL methanol/water (4:1, V/V) solution, and the pH was adjusted to ∼7.0 with NaOH under stirring condition. Then, 2.0 g PAN powder was added to the solution. Amidoximation reactions were performed at 75 °C for 6 h in a 100 mL glass flask under nitrogen and electromagnetic stirring condition. The resulting material was repeatedly washed with methanol/water (4:1, V/V) to remove the residual NH2OH, and then vacuum dried at 50 °C.
Synthesis of PAO in H2O
After 3.2 g NH2OH·HCl was dissolved in 20 mL de-gassed water, NaOH was slowly added to pH ∼ 7.0 under stirring condition. Then, 2.0 g PAN powder was added and reacted at 75 °C for 6 h. The resulting material was washed with de-gassed water, and vacuum dried at 50 °C.Characterization
Scanning electron microscopy was adopted to analyze the morphology of the particles (SEM, FEI Quanta 250 FEG) and transmission electron microscopy (TEM, FEI Tecnai G2 F20) was used to obtain the morphology of the particles. FT-IR spectroscopy was mounted on a Bruker Tensor II system with a platinum ATR instrument at room temperature. Thermogravimetric analysis (TGA, TGA 5500) was performed from room temperature to 800 °C under N2 atmosphere at a uniform heating rate of 10 °C min−1. X-Ray photoelectron spectroscopy (XPS) measurements were obtained with an ESCALab220i-XL surface microanalysis system (VG Scientific) equipped with an Al Kα (hλ = 1486.6 eV) source at a chamber pressure of 3 × 10−9 mbar. The surface charging effects were corrected with the C 1s peak at 284.4 eV as a reference. Dynamic light scattering (DLS) measurements were used to measure the distribution of particles. DLS was operated on a Malvern Instruments Zetasizer operated in the backscatter (173°) mode. The PAO and K2FeO4@PAO were dispersed in methanol to prepare 250 mg L−1 and 500 mg L−1 sample slurries. A 10 mm sample pool was used for the measurements. The hydrophilicity of the adsorbents was determined by the dynamic contact angle of pure water droplets on a solid surface with a contact angle meter (JC200d Zhongchen Digital Technology, China).Batch adsorption experiments
The adsorption capabilities of K2FeO4@PAO for U(VI) were studied by batch adsorption technique, with UO2(NO3)2·6H2O (>99% purity) used as the U(VI) source. U(VI) stock solution was prepared from UO2(NO3)2·6H2O (>99% purity). Different concentrations of U(VI) sample solutions were prepared from the stock solution. Briefly, after K2FeO4@PAO and PAO were pre-equilibrated with NaCl for 24 h, a solution containing U(VI) diluted to a certain concentration with ultrapure water was added. The salinity of seawater is ∼3.5%, and most of it is NaCl. Because of the considerable amount of water used, the adsorbent must be competent for normal seawater values and salinity conditions. The equilibrium of the adsorbent in NaCl solution lays the foundation for K2FeO4@PAO for U(VI) extraction from seawater. The solution pH values were adjusted by adding 0.1 mol L−1 NaOH or HCl. After shaking for the desired time in a thermostatic oscillation incubator, the suspension was filtered with a 0.22 μm needle filter. The final U(VI) concentration in the supernatant was measured by inductively coupled plasma mass spectrometry (ICP-MS, Thermo Scientific X-Series II, λ = 358.8 nm). The adsorption capacity of U(VI) was calculated as follows: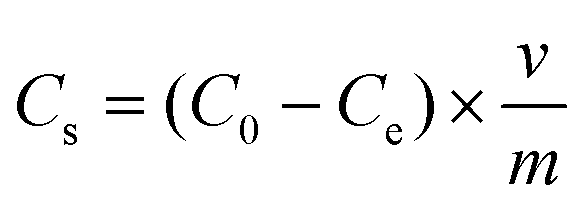 | (1) |
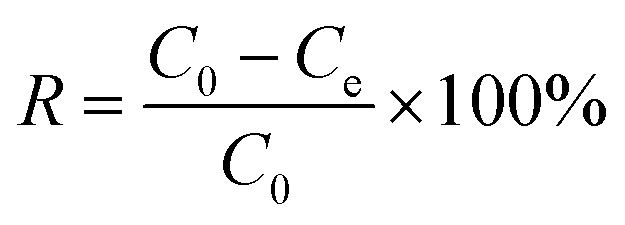 | (2) |
Desorption of U(VI) from K2FeO4@PAO
Here, 0.5 mol L−1 Na2CO3 was used as the eluting agent to study the desorption of U(VI) from the K2FeO4@PAO surface. Briefly, 0.02 g U(VI)-laden K2FeO4@PAO was immersed in 50 mL 0.5 mol L−1 Na2CO3 and shaken at 200 rpm for 24 h. The regenerated K2FeO4@PAO was washed with de-gassed water and vacuum dried at 50 °C.Antibacterial property
The two kinds of Gram-negative bacteria E. coli and V. alginolyticus were chosen to verify the antibacterial properties of K2FeO4@PAO. The bacteria were severally cultured in Luria–Bertani (LB) medium and shaken at 37 °C for 12 h. The resulting bacterial suspension was subsequently centrifuged at 10000 rpm for 10 min. The obtained bacterial precipitate was diluted by sterile physiological saline and the cell density was adjusted to ∼1.0 × 106 CFU mL−1. Next, 0.02 g K2FeO4@PAO and 50 mL diluted bacterial solution at 37 °C were incubated for 24 h to determine the antibacterial activity. The cultured bacterial solution (0.1 mL) was taken out and diluted and spread on an LB nutrient agar plate, followed by cultivation at 37 °C for 24 h.
Effect of biofouling on U(VI) adsorption
To obtain the target compositions, the adsorbents (K2FeO4@PAO) and NaCl were pre-reacted for 24 h at first; and then deionized water, U(VI), and exponential growth phase V. alginolyticus were injected, finally adjusting the suspension pH. After reacting for 24 h, the suspensions were centrifugated at 9000 rpm for 30 min at 25 °C, and then filtered. Except for specific evaluations, the reaction temperature, time, m/V, pH, and ionic strength were 298 ± 1 K, 24 h, 0.04 g L−1, 8.2 ± 0.1, and 0.10 mol L−1 NaCl, respectively. The final U(VI) concentrations were analyzed by inductively coupled plasma mass spectrometry (ICP-MS, Thermo Scientific X-Series II, λ = 358.8 nm).Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial supports from National Natural Science Foundation of China (21976089, 22176098).References
- Z. H. Liu, S. Shinde, S. Xie, N. Hao, F. Lin, C. G. Yoo, A. J. Regaukes and J. S. Yuan, Sustainable Energy Fuels, 2019, 3, 2024–2037 RSC.
- X. Luo, J. Zhang, J. Tao, X. Wang, S. Zhao, Z. Chen, S. Liu, J. Li and S. Li, Chem. Eng. J., 2021, 416, 129486 CrossRef CAS.
- R. Yu, Y. Lu, X. Zhang, W. Chen, X. Chen and L. Li, Desalination, 2022, 539, 115965 CrossRef CAS.
- N. Li, P. Gao, H. Chen, F. Li and Z. Wang, Chemosphere, 2022, 287, 116153 Search PubMed.
- Z. Mi, D. Zhang, J. Wang, S. Bi, J. Liu, X. Gao, D. Zhang, Y. Jiang, Z. Li, Y. Zhu and Z. Liu, New J. Chem., 2022, 46, 6296–6306 RSC.
- X. Chen, C. Wan, R. Yu, L. Meng, D. Wang, T. Duan and L. Li, Desalination, 2020, 486, 114447 CrossRef CAS.
- C. Liu, P. C. Hsu, J. Xie, J. Zhao, T. Wu and H. Wang, Nat. Energy, 2017, 2, 17007–17014 CrossRef CAS.
- F. Chi, S. Zhang, J. Wen, J. Xiong and S. Hu, Ind. Eng. Chem. Res., 2018, 57, 8078–8084 CrossRef CAS.
- X. Liu, H. Liu, H. Ma, C. Cao, M. Yu, Z. Wang, B. Deng, M. Wang and J. Li, Ind. Eng. Chem. Res., 2012, 51, 15089–15095 CrossRef CAS.
- X. Lu, D. Zhang, A. T. Reda, C. Liu, Z. Yang, S. Guo, S. Xiao and Y. Ouyang, Ind. Eng. Chem. Res., 2017, 56, 11936–11947 CrossRef CAS.
- F. Ma, B. Dong, Y. Gui, M. Cao, L. Han, C. Jiao, H. Lv, J. Hou and Y. Xue, Ind. Eng. Chem. Res., 2018, 57, 17384–17393 CrossRef CAS.
- F. Endrizzi and L. F. Rao, Chem. – Eur. J., 2014, 20, 14499–14506 CrossRef CAS PubMed.
- S. Shi, B. Li, Y. Qian, P. Mei and N. Wan, Chem. Eng. J., 2020, 397, 9–17 CrossRef.
- C. Ma, J. Gao, D. Wang, Y. Yuan, J. Wen, B. Yan, S. Zhao, X. Zhao, Y. Sun, X. Wang and N. Wang, Adv. Sci., 2019, 6, 1900085 CrossRef PubMed.
- F. Endrizzi, C. J. Leggett and L. Rao, Ind. Eng. Chem. Res., 2016, 55, 4249–4256 CrossRef CAS.
- J. Zhang, J. Ma, G. Jiao, K. Liu, R. Cui, S. Zhai and R. Sun, Desalination, 2023, 548, 116243 CrossRef CAS.
- G. Jiao, J. Ma, J. Zhang, S. Zhai and R. Sun, Carbohydr. Polym., 2023, 300, 120244 CrossRef CAS PubMed.
- X. Chen, Y. Lin, W. Li, G. Zhang, Y. Wang, J. Ma, Z. Meng, S. Wu, S. Wang, X. Zhang and H. Pang, Sustainable Mater. Technol., 2022, 34, e00521 CrossRef CAS.
- D. Shao, G. Hou, F. Chi, X. Lu and X. Ren, RSC Adv., 2021, 11, 1909–1915 RSC.
- G. Wang, C. Lu, T. Sun and Y. Li, J. Appl. Polym. Sci., 2022, 139, 52129 CrossRef CAS.
- C. Bi, C. Zhang, W. Xu, F. Ma, L. Zhu, R. Zhu, Q. Qi, L. Liu, J. Bai and H. Dong, Desalination, 2023, 545, 116169 CrossRef CAS.
- C. Zhang, Y. Song, Q. He, F. Wang, S. Zhan and F. Zhou, J. Environ. Chem. Eng., 2021, 9, 105973 CrossRef CAS.
- W. Sun, L. Feng, J. Zhang, K. Lin, H. Wang, B. Yan, T. Feng, M. Cao, T. Liu, Y. Yuan and N. Wang, Adv. Sci., 2022, 9, 2105008 CrossRef CAS PubMed.
- Q. Chen, X. Xue, Y. Liu, A. Guo, K. Chen, J. Yin, F. Yu, H. Zhu and X. Guo, J. Hazard. Mater., 2022, 438, 129524 CrossRef CAS PubMed.
- M. Thomas, P. Drzewicz, A. Więckol-Ryk and K. Schwochau, Environ. Sci. Pollut. Res., 2022, 29, 8514–8524 CrossRef CAS PubMed.
- V. G. Petrov, Y. D. Perfiliev, S. K. Dedushenko, T. S. Kuchinskay and S. N. Kalmykov, J. Radioanal. Nucl. Chem., 2016, 310, 347–352 CrossRef CAS.
- N. Wang, L. Tan, R. Zhang, Q. Zhao and H. Wang, Sci. Total Environ., 2020, 726, 138541 CrossRef CAS PubMed.
- Z. Viktor, L. Wang and J. Ma, J. Hazard. Mater., 2020, 384, 121264 CrossRef CAS PubMed.
- Y. Tan, Z. Xu, L. He and H. Li, J. Energy Storage, 2022, 52, 104889 CrossRef.
- D. Wang, J. Song, J. Wen, Y. Yuan, Z. Liu, S. Lin, H. Wang, H. Wang, S. Zhao, X. Zhao, M. Fang, M. Lei, B. Li, N. Wang, X. Wang and H. Wu, Adv. Energy Mater., 2018, 8, 1802607 CrossRef.
- J. Zeng, H. Zhang, Y. Sui, N. Hu, D. Ding, F. Wang, J. Xue and Y. Wang, Ind. Eng. Chem. Res., 2017, 56, 5021–5032 CrossRef CAS.
- W. Li, Q. Liu, J. Liu, H. Zhang, R. Li, Z. Li, X. Jing and J. Wang, Appl. Surf. Sci., 2017, 403, 378–388 CrossRef CAS.
- D. Shao, X. Wang, X. Ren, S. Hua, J. Wen, Z. Tan, J. Xiong, A. M. Asiric and H. M. Marwani, J. Ind. Eng. Chem., 2018, 67, 380–387 CrossRef CAS.
- D. Shao, X. Liu, T. Hayat, J. Li and X. Ren, J. Radioanal. Nucl. Chem., 2019, 319, 379–386 CrossRef CAS.
- C. W. Abney, R. T. Mayes, T. Saito and S. Dai, Chem. Rev., 2017, 117, 13935–14013 CrossRef CAS PubMed.
- H. Wang, T. Xu, B. Zheng, M. Cao, F. Gao, G. Zhou, C. Ma, J. Dang, W. Yao, K. Wu, T. Liu, Y. Yuan, Q. Fu and N. Wang, J. Hazard. Mater., 2022, 433, 128789 CrossRef CAS PubMed.
- N. Li, J. Wu, R. Su, N. Zhang, J. Zhao and Z. Wang, Desalination, 2023, 545, 116153 CrossRef CAS.
- Y. Zeng, S. Liu, J. Xu, A. Zhang, Y. Song, L. Yang, A. Pu, Y. Ni and F. Chi, J. Environ. Chem. Eng., 2021, 9, 106490 CrossRef CAS.
- X. Wei, Q. Liu, H. Zhang, Z. Lu, J. Liu, R. Chen, R. Li, Z. Li, P. Liu and J. Wang, Dalton Trans., 2017, 46, 15746–15756 RSC.
- J. Yu, H. Zhang, Q. Liu, J. Zhu, J. Yu, G. Sun, R. Li and J. Wang, J. Hazard. Mater., 2022, 440, 129735 CrossRef CAS PubMed.
- M. N. Sahmoune, Environ. Chem. Lett., 2019, 17, 697–704 CrossRef CAS.
- L. Feng, H. Wang, T. Feng, B. Yan, Q. Yu, J. Zhang, Z. Guo, Y. Yuan, C. Ma, T. Liu and N. Wang, Angew. Chem., Int. Ed., 2022, 61, 82–86 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3lp00060e |
| This journal is © The Royal Society of Chemistry 2023 |