
Targeting vitamin E TPGS–cantharidin conjugate nanoparticles for colorectal cancer therapy
Shihou Shenga,
Tao Zhangb,
Shijie Lic,
Jun Weid,
Guangjun Xud,
Tianhong Sune,
Yahong Chena,
Fengqing Lua,
Yongchao Lif,
Jinghui Yangg,
Huiqiu Yuh,
Tongjun Liu*a and
Gang Han*i
aDepartment of Colorectal, Rectal and Anal Surgery, China-Japan Union Hospital of Ji Lin University, ChangChun 130000, China. E-mail: tongjunliu@163.com; Tel: +86-13943090285
bDepartment of Gastrointestinal Surgery, China-Japan Union Hospital of JiLin University, XinMin District, ChangChun 130000, China
cDepartment of Thyroid Surgery, China-Japan Union Hospital of JiLin University, ChangChun 130000, China
dDepartment of Neurosurgery, China-Japan Union Hospital of JiLin University, ChangChun 130000, China
eDepartment of Operating Room, China-Japan Union Hospital of JiLin University, ChangChun 130000, China
fDepartment of Gastrointestinal Surgery, China-Japan Union Hospital of JiLin University, ChangChun 130000, China
gDepartment of Hepatopancreatobiliary Surgery, China-Japan Union Hospital of JiLin University, ChangChun, 130000, China
hDepartment of Rehabilitation Medicine, China-Japan Union Hospital of JiLin University, ChangChun 130000, China
iDepartment of Gastrointestinal Surgery, The Second Hospital of JiLin University, ChangChun 130000, China. E-mail: gmail1973@163.com; Tel: +86-18743099891
First published on 8th June 2015
Abstract
A traditional Chinese medicine cantharidin which was previously found to be effective on colorectal cancer cells was translated into nanoparticles for drug delivery to reduce its side effects and enhance its drug efficacy. By further introducing the folate targeting ligands to the nanoparticles, targeting delivery of cantharidin to colorectal cancer is realized. Cantharidin loaded nanoparticles can increase the cytotoxicity of cantharidin on the folate over-expressed HT-29 cells; moreover, introducing folate to the nanoparticles can further increase its efficacy, while this obvious enhancement cannot be found on the MCF-7 cells with lower folate receptors.
Introduction
Nowadays, colorectal cancer has become the third most commonly diagnosed cancer in both men and women in the US excluding skin cancer. Moreover, it is the second leading cause of death in cancer patients in the US for both men and women.1,2 Though hundreds of potential anticancer drugs are available on the market, most of them are not ideal, and can cause serious adverse effects.3 A very promising candidate is cantharidin, which is a type of terpenoid, a chemical compound secreted by many species of blister beetle.4 Cantharidin has long been used as a traditional Chinese medicine for the treatment of a variety of cancers, including liver, lung, intestinal, and digestive tract tumors.4,5 Researchers around the world have found that cantharidin as well as its derivatives demonstrated strong affinity and specificity for protein phosphatase 2A (PP2A). It is reported that the level of PP2A inhibition parallels its cytotoxicity.6 Recently, cantharidin has also been found to be effective against several colorectal cancer cells through inhibition of heat shock protein and Bcl-2 associated athanogene domain 3 expressions by blocking heat shock factor 1 binding to promoters.7 Nevertheless, cantharidin has displayed serious side effects such as dysphasia, hematemesis, and dysuria.8 Therefore, a potential way of reducing the side effects but retain its activity is urgent.Nanoparticle based drug delivery system (DDS), defined as DDS, with particle diameters of approximately 100 nm or less, is attracting considerable attention worldwide as efficient cancer therapeutics for overcoming some of the limitations of conventional anticancer drug therapy.9,10 For an efficient DDS, the right choice of a drug carrier is vital. A good example is D-alpha-tocopheryl polyethylene glycol 1000 succinate monoester (TPGS), which is an amiphiphilic PEGylated vitamin E and can work as best surfactant for hydrophobic drugs both via encapsulating the drugs or conjugating with the drugs and then assembling the conjugates into nanoparticular drug delivery systems.11 What's more, the drug carrier TPGS has almost no toxic issues as it is widely applied in the food and drug industry.12
Folate is a small molecular compound, which is very important for tumor cell proliferation and survival. Several cancer cells were over-expressed folate receptors as much as 200-fold on the cell surfaces compared to the normal cells. The over-expressed receptors are vital to the intake of folate to the tumor cells.13,14 Typically, folate receptor is over-expressed in ovarian cancer, breast cancer, head and neck cancer, and some childhood cancers.15 Given these attributes of folate receptors, folic acid has been conjugated to many delivery systems for cancer therapy including liposomes, polymeric micelles and capsules, upconversion nanoparticles and carbon nanotubes, etc.16–19 Moreover, previous study revealed that the folate receptor was highly expressed in colorectal cancers,20 which makes it possible to utilize folate for target delivery of drugs for treatment of colorectal cancer.
Taking into account the unique anticancer property of cantharidin and the drug delivery technology developed till far, researchers around the world have tried tremendous ways of delivering cantharidin or its derivatives directly to the cancer cells for cancer therapy. Zhu, et al. reported preparation of cantharidin-loaded solid lipid nanoparticles (CA-SLNs) with oral bioavailability by a film dispersion-ultrasonication method.21 The result showed that CA-SLNs had a sustained release profile without a burst effect and a higher bioavailability than free cantharidin after oral administration. Later, Zhu, et al. reported an inclusion complex of cantharidin with β-cyclodextrin for drug delivery.22 However, the in vitro and in vivo drug efficacy was not studied. More recently, norcantharidin, a demethyl derivative of cantharidin with lower toxicity was conjugated to polyethylenimine (PEI) and polylysine (PLL) for acid liable drug release by Shen, et al.23 Though this system showed reasonable acid responsiveness, the drug itself was not the more efficient cantharidin and the polymers used are not FDA approved, making further clinical use harder. Taken together, we here show the first example of rational design of cantharidin loaded TPGS nanoparticles for targeting delivery of cantharidin and effective treatment of colorectal cancer. As cantharidin is an anhydride which can undergo readily reaction with hydroxyl groups. Via a simple one step ring opening reaction of cantharidin with the end hydroxyl group of TPGS, a cantharidin–TPGS conjugate (Can–TPGS, Scheme 1a) was obtained. Due to the amphiphilic nature of TPGS, this Can–TPGS conjugate can self-assemble in aqueous solution into nanoparticles as effective cantharidin delivery systems (Scheme 1b). To further increase the efficacy of this system, folate was introduced to the nanoparticles via assembly of FA–TPGS and Can–TPGS together (Scheme 1c). The novel cantharidin loaded nanoparticles were systematically characterized and studied in vitro on two cancer cell lines, HT-29 (human colorectal cancer cell, folate receptor over-expressed)24 and MCF7 (human breast cancer, folate receptor low-expressed).25 Results showed that cantharidin loaded nanoparticles can increase the cytotoxicity of cantharidin on the folate over-expressed HT-29 cells; moreover, introducing folate to the nanoparticles can further increase its efficacy, while this obvious enhancement cannot be found on the MCF-7 cells.
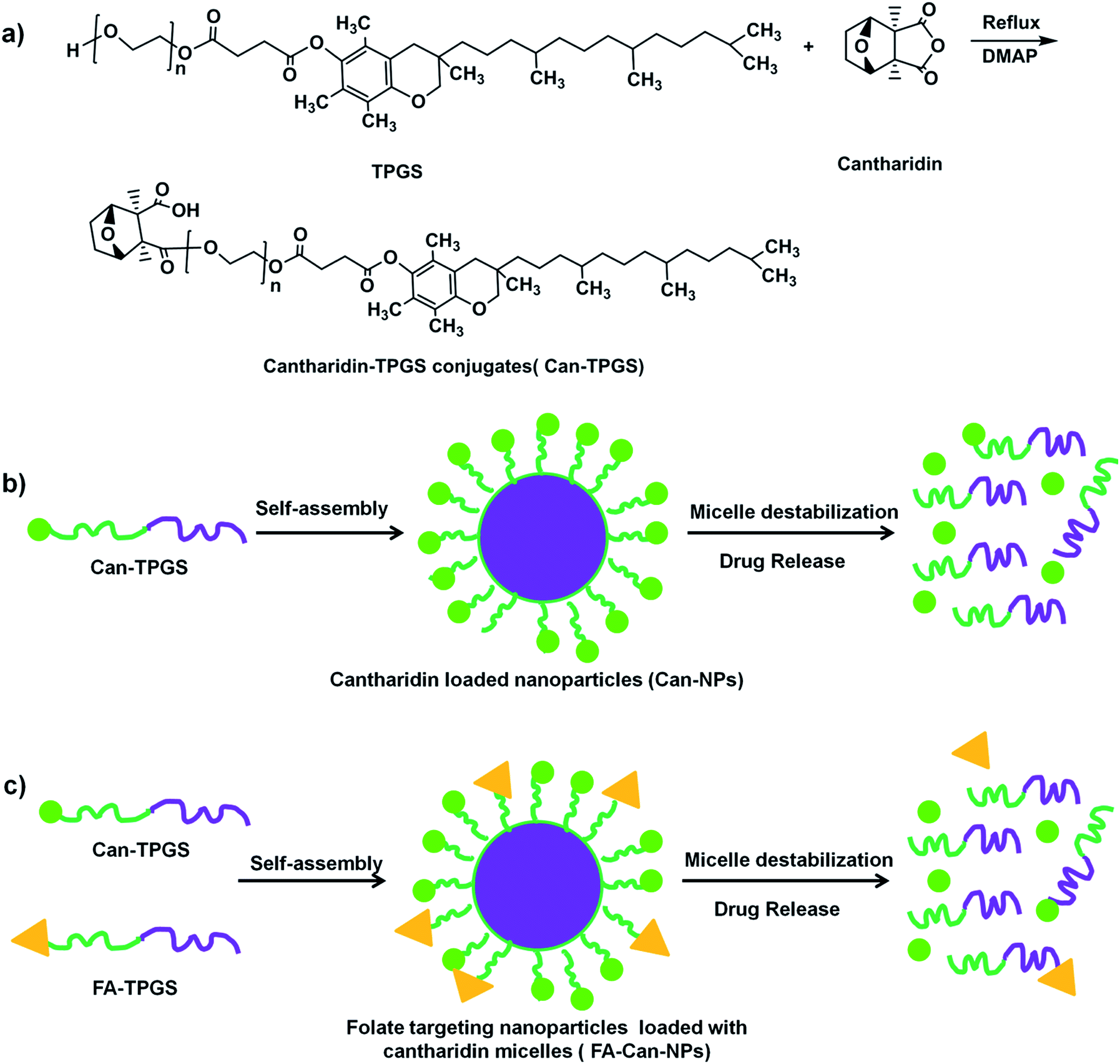 | ||
| Scheme 1 Preparation of the folate targeted nanoparticles loaded with cantharidin. (a) Synthesis of the cantharidin TPGS conjugates (Can–TPGS); (b) self-assembly of the Can–TPGS conjugates to cantharidin loaded nanoparticles (Can-NPs); (c) self-assembly of the Can–TPGS conjugates with folate–TPGS (FA–TPGS) to folate targeting nanoparticles (FA–Can-NPs). | ||
Materials
4-Dimethylaminopyridine (DMAP), folic acid, D-alpha-tocopheryl polyethylene glycol 1000 succinate monoester (TPGS) was purchased from Sigma-Aldrich. Cantharidin was purchased from Santa Cruz Biotech. All other chemicals and reagents were used as reagent grade without further purification in all of the experiments.Instrumentation
1H NMR spectra were recorded at ambient temperature on a JEOL JNM LA400 NMR spectrometer. The nanoparticles were analyzed using Transmission electron microscopy (TEM) (TEM; JEOL JEM-1011). The size and size distribution of the nanoparticles was then determined by dynamic light scattering (DLS) with a vertically polarized He–Ne laser (DAWN EOS, Wyatt Technology, USA). The scattering angle was fixed at 90° and the measurement was carried out at a constant temperature of 25 °C. The zeta potential of the nanoparticles prepared here was conducted on a Malvern Zetasizer Nano ZS which was calibrated using a 60 nm polystyrene standard. Nanoparticle samples are prepared by dissolving into de-ionized water and the particle size and zeta potential were measured simultaneously in triplicate.Synthesis of Can–TPGS conjugates
TPGS (1.5 g, 1 mmol) was dissolved in 50 mL anhydrous CH2Cl2 in a round flask, to which 1 N cantharidin anhydride (0.196 g, 1 mmol) and DMAP (0.122 g, 1 mmol) were added. The reaction mixture was heated to reflux under stirring overnight in an oil bath. After that, the reaction mixture was left cooled down to room temperature and subjected to rotation evaporation. Then, the obtained product was dissolved in 5 mL dimethylformamide (DMF) and subjected to 3 days of dialysis against water in a dialysis bag with a molecular cut-off at 1000 Da. Then the purified product was collected and lyophilized as white powder for further use.Synthesis of FA–TPGS conjugates
TPGS (0.15 g, 0.1 mmol) was dissolved in 10 mL anhydrous dimethyl sulfoxide (DMSO) in a round flask, to which 1 N folic acid (0.044 g, 0.1 mmol), DCC (0.041 g, 0.2 mmol) and DMAP (0.024 g, 0.2 mmol) were added. The reaction mixture was kept in ice bath under stirring overnight. After that, the reaction mixture was filtered to remove dicyclohexyl urea. Then the DMSO solution of the FA–TPGS conjugate was put into a dialysis bag with a molecular cut-off at 1000 Da against DMSO for 3 days to remove excess DCC and DMAP. In the meanwhile, DMSO outside the dialysis bag was changed several times. After that, the product was dialyzed against water for 3 days. Then, it was collected and lyophilized to get FA–TPGS powder.Preparation of TPGS NPs and FA–TPGS NPs
For preparation of the blank carrier nanoparticles (TPGS NPs) and FA targeting blank nanoparticles (FA–TPGS NPs), a nano-precipitation method was used.26 Taking TPGS NPs as an example, briefly, 20 mg of TPGS was dissolved in 2 mL of acetone under vigorously stirring, after that, 15 mL of de-ionized water was added. Then this solution was left stirring at room temperature for volatizing the acetone. Thereafter, the nanoparticles were lyophilized for further use. For making the targeting nanoparticles, the staring materials become TPGS and FA–TPGS at a ratio of 0.95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.05. All the other steps were the same.
:0.05. All the other steps were the same.
Preparation of Can-NPs and FA–Can-NPs
For preparation of the cantharidin loaded nanoparticles (Can-NPs) and folate targeting nanoparticles (FA–Can-NPs), a nano-precipitation method was used.26 Taking Can-NPs as an example, briefly, 20 mg of Can–TPGS conjugates was dissolved in 2 mL of acetone under vigorously stirring, after that, 15 mL of de-ionized water was added. Then this solution was left stirring at room temperature for volatizing the acetone. Thereafter, the nanoparticles were lyophilized for further use. For making the targeting nanoparticles, the staring materials become Can–TPGS and FA–TPGS at a ratio of 0.95:0.05. All the other steps were the same.
Preparation rhodamine B loaded nanoparticles of Can/RhB-NPs and FA–Can/RhB-NPs
To prepare fluorescent molecule labeled nanoparticles for further in vitro study, rhodamine B (RhB) labeled Can-NPs and FA–Can-NPs called Can/RhB-NPs and FA–Can/RhB-NPs were prepared respectively using the method described above.26 First, RhB–TPGS conjugate was prepared as previously described for FA–TPGS conjugates. Then the RhB–TPGS conjugate was used together with Can–TPGS or Can–TPGS as well as FA–TPGS to prepare Can/RhB-NPs and FA–Can/RhB-NPs respectively.Cantharidin drug release and zeta potential monitoring during drug release
The hydrolysis of cantharidin from Can-NPs was monitored by 1H NMR spectroscopy as previously described.23 Briefly, Can-NPs (200 mg) were dissolved in 20 mL PBS (pH = 7.4, 10 mM) or acetate buffered solution (pH = 5.0, 10 mM). The samples were put into the dialysis bag at a molecular cutoff at 1000 Da with shaking. At desirable time intervals, 2 mL of samples was removed from the dialysis bag. The collected polymer was subjected to ultra-centrifugation and washed 3 times by water to remove salt and then lyophilized. Their 1H NMR spectra were recorded. The degree of hydrolysis was calculated by comparing the peak at 4.7 ppm (peak c in cantharidin in Fig. 1a) with the peak at 3.65 ppm of PEG (–CH2–CH2–O–) in TPGS. | ||
| Fig. 1 1H NMR spectra of cantharidin (a), TPGS (b) and Can–TPGS conjugate (c). | ||
To monitor the zeta potential change in drug release process, 0.5 mL of samples at pH5.0 and pH7.4 respectively were collected at desirable time points and diluted to 3 mL for zeta potential monitoring. The initial samples prepared at t = 0 were set as controls.
Cell culture conditions and cell lines
The cancer cell lines HT-29 (human colon cancer, folate receptor over-expression) and MCF-7 (human breast cancer, folate receptor negative expression) were maintained in McCoy's 5a media and Eagle's minimum essential medium respectively. L-929 cells (mouse fibroblast) were cultured in DMEM. All the culture media were supplemented with 10% FBS (fetal bovine serum), 0.03% L-glutamine and 1% penicillin/streptomycin in 5% CO2 at 37 °C in a 95% humidified atmosphere.In vitro cytotoxic evaluation via MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay
HT-29, MCF-7 and L-929 cells were harvested in a logarithmic growth phase and seeded on a 96-well plate in 100 μL media at a density of 5000 cells per well and incubated overnight. All the drugs were dissolved in culture media and subjected for use. The cells were treated in three ways separately: (1) for testing the compatibility of the blank micelles, the cells were treated with TPGS NPs and FA–TPGS NPs from 15.6 μg mL−1 to 500 μg mL−1; (2) for testing drug loaded micelles, Can-NPs and FA–Can-NPs with a cantharidin concentration from 0.097 μM to 50 μM were used to treat the cells; (3) for folate competing experiments, the cells were pre-treated with 2 mM sodium folate for 4 h and then the culture media was removed, washed by PBS and new culture media was added. After that, FA–Can-NPs with a final cantharidin concentration from 0.097 μM to 50 μM were added to the plates. After treatment of drugs, the cells were transferred to an incubator at 37 °C for further incubation. At the end of the incubation for 48 h at 37 °C, 10 μL of MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) stock solution (5 mg mL−1) is added to each culture well and further incubated for 4 hours. 100 μL of acidified isopropyl alcohol-sodium dodecyl sulfate (SDS) solution was added to each well and incubated for another 12 h. After that, the plate was read by a micro-plate reader at a wavelength of 570 nm.Intracellular localization and uptake efficiency study
HT-29 cells were seed onto a glass in 6-wells plate at a density of 1 × 105 cells per well in 2 mL culture media overnight before use. Then cells were then treated with Can/RhB NPs and FA–Can/RhB NPs at an equal RhB concentration of 2 μg mL−1 for 1 h. Then the cells were washed twice with cold PBS, and fixed with 4% formaldehyde. Cell nucleus was stained by DAPI (2-(4-amidinophenyl)-6-indolecarbamidine dihydrochloride). Cells were then thoroughly washed by PBS for three times and observed using confocal laser scanning microscope (CLSM, Olympus FV1000).For flow cytometry study, HT-29 cells were seed onto a glass in 6-wells plate at a density of 2 × 105 cells per well in 2 mL culture media overnight before use. Then cells were treated with Can/RhB-NPs, FA–Can/RhB-NPs at an equal RhB concentration of 5 μg mL−1. For FA blocking study, the cells were pretreated with 2 mM FA and washed 3 times by cold PBS and then treated with FA–Can/RhB-NPs. For lower temperature controls, cells were treated with the same procedures but the plates were put at 4 °C instead. For NaN3 mediated inhibition of endocytosis, before treatment of the drugs, cells were pretreated with 20 mM NaN3 and washed for 3 times by cold PBS prior to use. Each sample was analyzed on a BD FACSCalibur™ flow cytometer (BD Biosciences).
PP2A inhibition assay
A million of HT-29 cells were seeded in a 6-well plate and incubated with cantharidin, Can-NPs, FA–Can-NPs at an equal cantharidin concentration of 10 μM for 6 h. Then the cells were washed for several times, the cytosolic contents were extracted for PP2A activity assay. The PP2A activity is the percentage of residual PP2A activity compared with untreated control groups. PP2A inhibition adds PP2A activity equals to 1.Results and discussion
Synthesis of Can–TPGS conjugates
Secreted by blister beetle, most notably by the “Spanish fly”,4 cantharidin works by inhibiting protein phosphatases 1 and 2A (PP1, PP2A).6 It has been a long time for people to use blister beetle to treat Molluscum contagiosum virus (MCV) infections and associated warts. Nowadays, cantharidin has also shown potent anticancer activities on many types of human cancer cells.4,5Although cantharidin possesses potent anti-tumor properties, the clinical application of cantharidin is limited till far due to severe side-effects and its highly toxic nature.8 Therefore, to find a way to reduce its severe side effects becomes urgent. Recently, drug delivery systems (DDS) using liposomes, biodegradable polymers, inorganic nano-materials such as nanotubes, nanocrystals, etc., have drawn particular audience in the scientific world because they have the ability to reduce systemic toxicity and improve tumor-targeting efficiency of therapeutic agents such as drugs, antibodies, proteins, siRNA and imaging agents.27–31 For this specific reason, we show here to utilize the most widely accepted TPGS as a drug carrier to prepare a Can–TPGS conjugate. To prove the successful synthesis of the Can–TPGS conjugate, 1H NMR spectra of cantharidin, TPGS and Can–TPGS were collected in Fig. 1a–c. As shown in Fig. 1a, the typical chemical proton shifts of –CH3 at 1.24 ppm, –O–CH–CH2–CH2–CH–O– at 1.79 ppm and –CH2–CH–O– at 4.73 ppm were assigned. Fig. 1b collects the 1H NMR of TPGS. The peak at 3.65 ppm could be assigned to the –CH2– protons of PEO in TPGS. The lower peaks in the aliphatic region belong to various moieties of vitamin E tails. Fig. 1c collects the 1H NMR of Can–TPGS. There appears a peak in 4.7 ppm which is typical chemical shift of –CH2–CH–O– in cantharidin. By integrating the peak compared to the PEO in TPGS, we can calculate that there is about 0.9 cantharidin per TPGS, which means ca. 90% of the TPGS is end capped with cantharidin and a drug content of 11.5% weight by weight.
Preparation and characterization of TPGS NPs and FA–TPGS NPs
TPGS is an amphiphilic molecule which can self-assemble into micelles in aqueous solution itself.11 To introduce the targeting ligand folate to the micelles, TPGS and FA–TPGS were co-assembled. The TPGS NPs and FA–TPGS NPs were characterized by DLS and zeta potential analyzer. As shown in Table 1, the mean diameter of TPGS is 65.7 ± 2.5 nm with a polydispersity index (PDI) of 0.125. TPGS NPs showed a zeta potential of −22.1 ± 1.5 mV. For the FA–TPGS NPs, the mean diameter is 72.4 ± 1.9 nm with a PDI of 0.135. Zeta potential of FA–TPGS NPs was −29.2 ± 3.1 mV, slightly lower than that of TPGS NPs.| Code | DLS (nm) | PDI | Zeta potential (mV) |
|---|---|---|---|
| TPGS NPs | 65.7 ± 2.5 | 0.125 | −22.1 ± 1.5 |
| FA–TPGS NPs | 72.4 ± 1.9 | 0.135 | −29.2 ± 3.1 |
| Can-NPs | 114.7 ± 1.2 | 0.104 | −35.6 ± 2.4 |
| FA–Can-NPs | 130.4 ± 3.2 | 0.216 | −28.4 ± 3.5 |
Preparation and characterization of Can-NPs and FA–Can-NPs
Cantharidin is conjugated to TPGS to make a Can–TPGS conjugate which can be further assembled into micelles. Due to the hydrophilic nature of the free carboxyl group in the cantharidin molecule, it will stay with PEG as the outer layer and the vitamin E can form the hydrophobic inner core. Similarly, the size and size distribution of Can-NPs and FA–Can-NPs were characterized by DLS. Data were shown in Fig. 2a and b and Table 1. Can-NPs and FA–Can-NPs had a mean diameter of 114.7 ± 1.2 nm and 130.4 ± 3.2 nm with a poly-dispersity index of 0.104 and 0.216 respectively. FA–Can-NPs had a slightly larger diameter possibly due to the introducing of folate ligands onto the surface. From the TEM images, spherical structures of both Can-NPs and FA–Can-NPs can be found with diameters of 102.5 nm and 110.7 nm respectively (Fig. 2c and d). The zeta potential of Can-NPs and FA–Can-NPs were −35.6 ± 2.4 mV and −28.4 ± 3.5 mV. The negative charge is considered beneficial for blood circulation. | ||
| Fig. 2 Characterization of cantharidin loaded nanoparticles Can-NPs (a and c) and folate targeting nanoparticles FA–Can-NPs (b and d) by DLS (a and b) and TEM (c and d). Can-NPs showed a mean diameter of 114.7 nm with a polydispersity index of 0.104 by DLS (a) and a mean diameter of 102.5 nm by TEM (c). Similarly, FA–Can-NPs showed a mean diameter of 130.4 nm with a polydispersity index of 0.216 by DLS (b) and a mean diameter of 110.7 nm by TEM (d). | ||
Drug release profiles of cantharidin and zeta potential change
In order to study the drug release profiles of cantharidin from Can-NPs and FA–Can-NPs, we simplify to study only Can-NPs rather than both of Can-NPs and FA–Can-NPs on the assumption that FA could not change the drug release behavior of cantharidin. Here, a dialysis method was utilized for the drug release study and two pH values at pH5.0 and pH7.4 for the dialysis buffer solution were chosen to simulate the drug release in blood and in the tumor cells respectively due to the extensive report that the pH in the tumor cells is around 5.0, while it is 7.4 in the blood.23 Due to release and liberation of the drug molecule cantharidin into the buffered solution, most of the released cantharidin can go through the dialysis bag. At desirable time points, we collected the polymer solution in the dialysis bag and washed the polymer via ultracentrifugation to remove the salt and residual cantharidin released, and then the nanoparticles were collected and lyophilized for 1H NMR measurement. By simply integrating the peak of cantharidin at 4.7 ppm as compared to 3.65 ppm of PEG in TPGS, we can calculate the drug release percentage. In Fig. 3a, results showed that cantharidin was released much faster at pH5.0 than at pH7.4. To be more specific, it can be found that at 48 h, 19% cantharidin was released at pH7.4, while this was 48% at pH5.0. The drug release difference between the two pH values can be explained by the possible hydrolysis of the linkage of the ester bond at two different pH values. The fact that cantharidin is released much faster at pH5.0 is would be beneficial for drug delivery to cancer cells due to the acidic environment in the cancer cells.32 | ||
| Fig. 3 Representative drug release of cantharidin from Can-NPs studied by 1H NMR at pH5.0 and pH7.4. Experimental details were shown in the text. Data were shown as mean value ± standard deviation (n = 3) (a). To give an insight into the nanoparticle change, the zeta potential of the nanoparticles were monitored during this process and the results were listed in (b). | ||
As cantharidin was linked to TPGS with a bare carboxyl group on the end, there are many carboxyl groups on the surfaces of Can–TPGS NPs. Once more and more cantharidin molecules are released, less and less carboxyl groups would be present at the surfaces of Can–TPGS NPs. Therefore, monitoring the zeta potential during the drug release process may give some additional information to the drug release process. As shown in Fig. 3b, the freshly prepared Can–TPGS NPs at pH5.0 and pH7.4 had a zeta potential of −25.3 mV and −39.6 mV respectively. The difference at the initial stage can be attributed to the pH dependence of zeta potential. At lower pH values (pH5.0), less carboxyl groups can be de-protonated, therefore higher zeta potential could be found (−25.3 mV at pH5.0 vs. −39.6 mV at pH7.4). As the incubation time was prolonged, the drugs with carboxyl group were liberated from the surface of the nanoparticles, the zeta potential became higher and higher both at pH5.0 and pH7.4. However, at pH5.0, the zeta potential increased much greater than that that at pH7.4, indicating more drugs were released at 5.0.
In vitro evaluation of TPGS NPs and FA–TPGS NPs
TPGS is widely used in pharmaceutical applications. TPGS has shown proven properties to improve bioavailability of poorly absorbed drugs. As a water soluble compound, TPGS is also used as an efficient source of natural vitamin E both for therapeutic purposes and nutrition. To prove that TPGS NPs and FA–TPGS NPs are non-toxic to the two cancer cell lines HT-29 and MCF-7, TPGS NPs and FA–TPGS NPs were treated with the cells for 48 h at a concentration ranging from 500 μg mL−1 to 16.25 μg mL−1. As shown in Fig. 4a and b, we can find that even up to 500 μg mL−1, the cell viability for the two cell lines are larger than 90%. And it can be further found that HT-29 and MCF-7 cells displayed no difference in cell viability on the two kinds of nanoparticles, TPGS NPs and FA–TPGS NPs. This can be explained by the fact that the two nanoparticles had no drugs and cells cannot be killed though there is possible targeting effect. From the above results, we can find that TPGS NPs and FA–TPGS NPs are safe enough as drug carriers. | ||
| Fig. 4 In vitro cytotoxicity of TPGS NPs and FA–TPGS NPs on HT-29 (a) and breast cancer MCF-7 (b) cells at 48 h (b). Data were shown as mean value ± standard deviation (n = 4). | ||
In vitro evaluation of Can-NPs and FA–Can-NPs
The anti-cancer therapeutic promise of cantharidin is limited because of its high mammalian toxicity.8 Recent study has shown that cantharidin displayed considerable toxicity on some colorectal cancer cell lines.7 To reduce the side effects and further possibly increase the efficacy of cantharidin, here we prepared a Can–TPGS conjugate and self-assembled this conjugate into nanoparticles to deliver cantharidin to the cancer cells. In this way, we believe that the Can-NPs can release the drug cantharidin to kill the cancer cells. Further by targeting the cells with folate ligand, the efficacy of FA–Can-NPs would be higher than that of Can-NPs. To test the targeting effect of folate ligand, here we choose a colorectal cancer cell line, HT-29 and a breast cancer cell line MCF-7 because the former one is reported to have over-expressed folate receptors while the latter one has low-expressed folate receptors.24,25 Moreover, pretreatment of the cells with 2 mM folate was utilized to block the folate receptors. The results of Can-NPs and FA–Can-NPs on two cell lines were shown as in Fig. 5a and b. In Fig. 5a, it was found that cantharidin showed a dose dependent cytotoxicity towards HT-29 cells at a concentration ranging from 0.097 μM to 50 μM to (2-fold dilution). Can-NPs were much more effective than cantharidin almost in all the concentration range. For the targeting nanoparticles, FA–Can-NPs, the cytotoxicity is increased to a greater extent. Pretreatment of the cells with 2 mM folate to block the folate receptors on the cell surfaces can reduce the efficacy of FA–Can-NPs. In this situation, the efficacy of FA–Can-NPs is somewhat the same as non-targeting nanoparticles Can-NPs. The results here demonstrated that the efficacy of the drugs are in the order of FA–Can-NPs > Can-NPs ≈ FA–Can-NPs + FA > cantharidin on HT-29 cells. This can be possibly explained by the targeting effect of the folate ligands and the delivery of drugs to the cancer cells. | ||
| Fig. 5 Representative in vitro cytotoxicity of cantharidin loaded nanoparticles Can-NPs and folate targeting nanoparticles with cantharidin (FA–Can-NPs) on breast cells (a) and HT-29 cells (b) at 48 h and representative cytotoxicity of FA–Can-NPs via blocking the folate receptor by pre-incubating the cells with 2 mM folate for 1 h. The IC50 values of cantharidin, Can-NPs, FA–Can-NPs and FA–Can-NPs blocked by free 2 mM FA on breast cancer MCF-7 (c) and HT-29 cells (d) at 48 h were collected. Data were shown as mean value ± standard deviation (n = 3). *** indicates p < 0.001, ** indicates p < 0.01. To further show the preference to kill the cancer cells, L-929 cells were treated with cantharidin and Can-NPs, MTT assay was performed (e). | ||
To further prove that, MCF-7 cells with low-expression of folate receptors was tested and the results were shown in Fig. 5b. We can find that the efficacy of the drugs was in the order of FA–Can-NPs ≈ FA–Can-NPs + FA ≈ Can-NPs > cantharidin. The results mean that folate targeting ligand has minimum targeting effect on the folate receptor low-expressed cells. However, all the nanoparticles are better than the free drug cantharidin, this is possibly due to the intracellular delivery of more drugs to the cells via the nanoparticles.
To make it clearer, the IC50 values of cantharidin, Can-NPs, FA–Can-NPs and FA–Can-NPs blocked by free FA on HT-29 and MCF-7 cells at 48 h were listed in Fig. 5c and d. As shown in Fig. 5c, the IC50 values for cantharidin, Can-NPs and FA–Can-NPs and FA–Can-NPs + FA (blocked by 2 mM folate) were 15.3, 9.2, 3.6 and 8 μM on HT-29 cells at 48 h. Delivery of cantharidin by nanoparticles (Can-NPs) can increase the efficacy of cantharidin by ca. 1.7 fold. Further targeting the nanoparticles can increase its efficacy up to approximately 4.3 fold. Therefore, the extra 2.5 fold increase in efficacy can be attributed to the targeting effect of folate. Further, we can take a look at the MCF-7 cell line as shown in Fig. 5d. The IC50 values for cantharidin, Can-NPs and FA–Can-NPs and FA–Can-NPs + FA (blocked by 2 mM folate) were 32.4, 16.5, 13.1 and 14.7 μM respectively at 48 h. One can clearly find that delivery of cantharidin by nanoparticles (Can-NPs) can increase the efficacy of cantharidin by ca. 1.9 fold. However, further targeting the nanoparticles did not show any obvious increase in its efficacy and blocking the cells with 2 mM folate also demonstrated no effect on the enhancement of drug efficacy. This is possibly due to the low-expression of folate receptors on MCF-7 cells.
Moreover, to further show the benefit of delivery cantharidin, we have chosen a mouse fibroblast cells to test the toxicity of cantharidin and Can-NPs. The results were shown in Fig. 5e. We can clearly see that both cantharidin and Can-NPs showed less toxicity on this cell lines than on MCF-7 and HT-20 at the same doses, indicating the preference to kill the cancer cells by the Can-NPs.
Intracellular uptake of Can-NPs and FA–Can-NPs
To give some insight into the intracellular uptake of Can-NPs and FA–Can-NPs, first the cells were treated with RhB loaded micelles Can/RhB-NPs and FA–Can/RhB-NPs at an equal RhB concentration of 2 μg mL−1. After treatment of them for 1 h, the cells were imaged via confocal laser scanning microscope. The results were shown in Fig. 6a. The blue fluorescence comes from DAPI in the cell nucleus. The red fluorescence, which comes from RhB and stands for the nanoparticles, diffused into the whole cells and mainly in the cytosol, indicating that Can-NPs entered the cancer cells. It should be noted that at the same 1 h, cells treated with FA–Can-NPs displayed brighter red fluorescence, suggesting more FA–Can/RhB-NPs were in the cells and existence of possible FA targeting of the cancer cells. | ||
| Fig. 6 Intracellular uptakes of Can-NPs by HT-29 cancer cells. Representative confocal laser scanning images of Can/RhB-NPs and FA–Can/RhB-NPs with an equal RhB concentration of 2 μg mL−1 at 1 h (a). After treatment of Can/RhB-NPs and FA–Can/RhB-NPs for 1 h, cells were thoroughly washed and fixed by with 4% formaldehyde. Cell nucleus was stained by DAPI (blue fluorescence) and then observed using confocal laser scanning microscope (CLSM, Olympus FV1000). The red fluorescence comes from RhB. The scale bar is 40 μm. The relative uptake study of Can-NPs, FA–Can-NPs and FA–Can-NPs blocked by 2 mM FA at 4 h via flow cytometry was shown in (b). Cells were treated with the drug cantharidin loaded micelles at 1 μM. RhB was 5 μg mL−1. Results were shown as relative to Can-NPs at 37 °C for 4 h. Data were shown as mean value ± standard deviation (n = 3). *** indicates p < 0.001, ** indicates p < 0.01. | ||
Free anticancer drugs such as doxorubicin, paclitaxel, cisplatin as well as cantharidin are widely believed to enter the cancer cells via passive diffusion.33 However, the nanoparticles are extensively reported to be internalized by the cancer cells via a different uptake pathway, via so-called endocytosis, which is an energy dependent process. To explain why the nanoparticle formulation of cantharidin is better than the free drug and FA–Can-NPs internalized more drugs than Can-NPs, here we studied intracellular uptake of the drugs via flow cytometry. To track the nanoparticles in this process, we labeled the Can-NPs and FA–Can-NPs with RhB (Can/RhB-NPs and FA–Can/RhB-NPs). Furthermore, to prove the endocytosis of Can-NPs and FA–Can-NPs, 4 °C and 20 mM NaN3 (ATP depleting agent) were used to inhibit the energy dependent process.34 As shown in Fig. 6b, taking HT-29 as a representative cancer cell line, we can found that FA–Can/RhB-NPs had more than 1.8-fold uptake of nanoparticles compared to the non-targeting nanoparticles of Can/RhB-NPs (control). Blocking the cells with 2 mM folate can greatly reduce the relative uptake of the Can/RhB-NPs to 1.1. The energy inhibition can greatly affect the uptake as is can be seen from rapid reduction of the relative uptake values. It can be further found that 4 °C had shown a more profound effect on uptake than pretreatment of cells with 20 mM NaN3. The results above clearly revealed that Can/RhB-NPs were internalized by endocytosis and folate targeting can increase the endocytosis efficiency of cantharidin loaded nanoparticles.
PP2A inhibition assay
It is generally believed cantharidin exert its anticancer efficacy via a strong affinity and specificity for protein phosphatase 2A (PP2A).6 Moreover, scientist around the world have found that he level of PP2A inhibition parallels its cytotoxicity for cantharidin.6 To find whether Can-NPs and FA–Can-NPs showed the same PP2A inhibition mechanism. A PP2A inhibition assay was studied on the HT-29 cells by treatment of cantharidin, Can-NPs and FA–Can-NPs at 10 μM for 6 h. As shown in Fig. 7, compared to the non-treated cells (control), the PP2A activity of the cells treated with cantharidin, Can-NPs, FA–Can-NPs, FA–Can-NPs + FA were 52%, 41%, 25% and 33% respectively. Therefore, the nanoparticles loaded with cantharidin have shown PP2A inhibition efficacy. In accordance with the cell viability assay, targeting cantharidin nanoparticles increase the inhibition of PP2A, while blocking the cells with 2 mM folate will reduce the inhibition rate. | ||
| Fig. 7 The PP2A activity of HT-29 cells treated with cantharidin, Can-NPs, FA–Can-NPs and FA–Can-NPs blocked by 2 mM FA. The non-treated cells were set as control. Data were shown as mean value ± standard deviation (n = 3). *** indicates p < 0.001, ** indicates p < 0.01. | ||
Conclusion
Taken together, we have shown here rational design of nanoparticles loaded with cantharidin via a Can–TPGS conjugate for drug delivery. By further introducing the folate targeting ligand, we have shown here folate targeting nanoparticles FA–Can-NPs loaded with cantharidin. The drug conjugate was thoroughly characterized and two kinds of nanoparticles were systematically studied in vitro. We have found that folate targeting nanoparticles with cantharidin to kill colorectal cancer via a PP2A dependent way. Further in vivo evaluation of this system is on-going and this cantharidin loaded nanoparticles may find the potential use in the future.Acknowledgements
The authors thank the financial support provided by Youth Foundation of Jilin Province (Project no. 201201046) and Jilin Province Natural Science Foundation of China (Project no. 201215065).References
- S. Rebecca, M. Kimberly and J. Ahmedin, Ca-Cancer J. Clin., 2015, 65, 5 CrossRef PubMed.
- S. Rebecca, D. Carol and J. Ahmedin, Ca-Cancer J. Clin., 2014, 64, 104 CrossRef PubMed.
- V. MacDonald, Can. Vet. J., 2009, 50, 665 Search PubMed.
- E. P. Cherniack, Alternative Med. Rev., 2010, 15, 124 Search PubMed.
- C. E. P. Puerto, L. Y. V. Méndez and V. V. Kouznetsov, Chem. Biol. Drug Des., 2013, 82, 477 Search PubMed.
- O. Kadioglu, N. S. Kermani, G. Kelter, U. Schumacher, H. H. Fiebig and H. J. Greten, et al., Biochem. Pharmacol., 2014, 1, 399 CrossRef PubMed.
- J. A. Kim, Y. Kim, B. M. Kwon and D. C. Han, J. Biol. Chem., 2014, 288, 28713 CrossRef PubMed.
- G. S. Wang, J. Ethnopharmacol., 1989, 26, 147 CrossRef CAS.
- G. Tiwari, R. Tiwari, B. Sriwastawa, L. Bhati, S. Pandey and P. Pandey, et al., Int. J. Pharm. Invest., 2012, 2, 2 CrossRef PubMed.
- T. M. Allen and P. R. Cullis, Science, 2004, 19, 1818 CrossRef PubMed.
- L. Mu and S. S. Feng, J. Controlled Release, 2002, 80, 129 CrossRef CAS.
- Z. Zhang, S. Tan and S. S. Feng, Biomaterials, 2012, 33, 4889 CrossRef CAS PubMed.
- L. E. Kelemen, Int. J. Cancer, 2006, 119, 243 CrossRef CAS PubMed.
- Q. Q. Zhu, C. Feng, W. W. Liao, Y. Zhang and S. Q. Tang, Cancer Cell Int., 2013, 13, 65 CrossRef CAS PubMed.
- G. L. Zwicke, G. A. Mansoori and C. J. Jeffery, Nano Rev., 2012, 3, 10 Search PubMed.
- J. Sudimack and R. J. Lee, Adv. Drug Delivery Rev., 2000, 41, 147 CrossRef CAS.
- S. Wang and P. S. Low, J. Controlled Release, 1998, 53, 39 CrossRef CAS.
- A. Gabizon, A. T. Horowitz, D. Goren, D. Tzemach, H. Shmeeda and S. Zalipsky, Clin. Cancer Res., 2003, 9, 6551 CAS.
- X. Q. Yang, S. Pill, J. J. Grailer, D. A. Steeber, S. Q. Gong and Y. H. Chen, et al., J. Mater. Chem., 2009, 19, 5812 RSC.
- M. D. Angelica, J. Ammori, M. Gonen, D. S. Klimstra, P. S. Low, L. Murphy, M. R. Weiser, P. B. Paty, Y. Fong, R. P. Dematteo, P. Allen, W. R. Jarnagin and J. Shia, Mod. Pathol., 2011, 24, 1221 CrossRef PubMed.
- Y. J. Dang and C. Y. Zhu, Chin. Med., 2013, 8, 1 CrossRef CAS PubMed.
- Y. J. Dang, L. N. An, C. H. Hu and C. Y. Zhu, J. Appl. Polym. Sci., 2012, 123, 1557 CrossRef CAS PubMed.
- K. D. Lu, M. Z. Cao, W. W. Mao, X. R. Sun, J. B. Tang, Y. Q. Shen and M. H. Sui, J. Mater. Chem., 2012, 22, 15804 RSC.
- M. D. Nonancourt-Dition, J. L. Gueant, C. Adjalla, C. Chery, R. Hatier and F. Namour, Cancer Lett., 2001, 171, 139 CrossRef.
- G. A. Mansoori and K. S. Brandenburg, Cancers, 2010, 2, 1911 CrossRef CAS PubMed.
- D. Lin, G. Li, L. Qin, Z. Wen, J. Wang and X. Sun, J. Biomed. Nanotechnol., 2013, 9, 2017 CrossRef CAS PubMed.
- K. P. Singh, P. Panwar, P. Kohli and Sanjesh, J. Biomed. Nanotechnol., 2011, 7, 60–62 CrossRef CAS PubMed.
- L. Zhang, F. X. Gu, J. M. Chan, A. Z. Wang, R. S. Langer and O. C. Farokhzad, Clin. Pharmacol. Ther., 2008, 83, 761 CrossRef CAS PubMed.
- Y. Wen and W. S. Meng, Journal of Pharmaceutical Innovation, 2014, 9, 158 CrossRef PubMed.
- Y. Wen, H. R. Kolonich, K. M. Kruszewski, N. Giannoukakis, E. S. Gawalt and W. S. Meng, Mol. Pharm., 2013, 10, 1035 CrossRef CAS PubMed.
- A. Balducci, Y. Wen, Y. Zhang, B. M. Helfer, T. K. Hitchens, W. S. Meng, A. K. Wesa and J. M. Janjic, OncoImmunology, 2013, 2, e23034 CrossRef PubMed.
- S. F. Yua, G. L. Wua, X. Gua, J. J. Wang, Y. N. Wang and H. Gao, et al., Colloids Surf., B, 2013, 103, 15 CrossRef PubMed.
- M. J. Ratain and W. K. Plunkett, Holland-Frei Cancer Medicine, 6th edn, 2003 Search PubMed.
- S. Vranic, N. Boggetto, V. Contremoulins, S. Mornet, N. Reinhardt and F. Marano, et al., Part. Fibre Toxicol., 2013, 10, 2 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |