
An exonuclease III-aided “turn-on” fluorescence assay for mercury ions based on graphene oxide and metal-mediated “molecular beacon”†
Ying Zhou,
Xiao-Jing Xing,
Dai-Wen Pang and
Hong-Wu Tang*
Key Laboratory of Analytical Chemistry for Biology and Medicine (Ministry of Education), College of Chemistry and Molecular Sciences, State Key Laboratory of Virology, Wuhan Institute of Biotechnology, Wuhan University, Wuhan, 430072, P. R. China. E-mail: hwtang@whu.edu.cn; Fax: +86 27 68754067; Tel: +86 27 68756759
First published on 5th January 2015
Abstract
A novel fluorescence “turn-on” strategy, which is based on the formation of Hg2+-mediated molecular-beacons (MBs), the preferable cleavage capacity of exonuclease III to double-stranded DNA compared with single-stranded one, and the remarkable difference in the binding ability of graphene oxide (GO) with single-stranded DNA and the mononucleotides, is designed for Hg2+ assay. The Hg2+-mediated base pairs facilitate the dye labeled MBs to fold into a hairpin structure, which is more likely to be digested by exonuclease III, and an obvious increase in the fluorescence intensity is observed after incubating with GO due to the weak affinity of the product-mononucleotides to GO. A fluorescent “turn-on” method based on graphene oxide and exonuclease III was designed for Hg2+ assay. The introduction of GO greatly increases the signal-to-background ratio, and the sensitivity is significantly improved due to the amplified capability of exonuclease III. Under the optimal conditions, Hg2+ is specifically and sensitively detected with a detection limit of 0.83 nM. Compared with the reported Hg2+ assay methods, the proposed strategy is simple, cost effective and selective, which might provide a new platform for developing a sensitive Hg2+ biosensor. Mercury level in the blood is an important indicator of mercury poisoning in clinical study. To testify the possibility of the use of this method for the assay of Hg2+ in real samples, detection of Hg2+ in 1% human serum was investigated and satisfactory results were obtained, which suggests that this method has great potential for bioanalysis.
Introduction
Mercury ions, which represent a widespread highly toxic contaminant in aquatic ecosystems, pose severe risk for human health and the environment.1 Mercury exposure can cause a number of adverse health effects such as damage in the brain, nervous system, immune system, kidney, and many other organs.2 The concern for the worldwide spread of mercury pollution and its damage to human health make the development of efficient, selective and sensitive methods for the detection of Hg2+ urgent. In the past decades, many methods, including atomic fluorescence spectroscopy,3 inductively coupled plasma mass spectrometry (ICP-MS),4 and selective cold vapor atomic fluorescence spectrometry,5 have been developed. However, most of them are rather expensive, complex, and not suitable for on-site analyses. The past few years have witnessed great progress in the development of optical and electrochemical techniques for the detection of Hg2+ based on small molecules,6,7 DNAzymes,8 oligonucleotides,9 polymers10 and functional nanoparticles.11 Despite this progress, some limitations, which include insufficient resolution in solution, relatively low selectivity among other metal ions, the need to use organic solvents or sophisticated synthesis of the probe, still exist. Therefore, a simple sensor with high sensitivity and selectivity for facile on-site and real-time mercury detection is still needed.Recently, synthetic oligodeoxyribonucleotides (ODNs) containing artificial bases have been used to form metal-mediated base pairs in which hydrogen bonds in Watson Crick (W–C) type base pairs in natural DNA are replaced by metal–base bonds.12 There is no need for time-consuming organic synthesis for the formation of metal-mediated base pairs, which makes its process simpler compared with base pair formation. Hairpin–DNA molecular-beacons (MBs), which are dually labeled single-stranded oligonucleotide with a loop-and-stem structure that become fluorescent upon binding to a complementary sequence or target as a result of the disruption of the stem–loop structure and an increased distance between the fluorophore and quencher tagged at either end,13 have been applied as promising probes because they not only provide a variety of analytical signal transduction options but also have better selectivity than linear DNA probes. It has been proven that the DNA incorporating one or more metal-mediated base pairs shows higher stability than the natural DNA.14 A previous research has reported that Hg2+ can be specifically inserted into two DNA thymine bases and facilitates these T–T mismatches to form stable base pairs.15 This special property opens a new way for utilizing T-containing oligonucleotides for the development of Hg2+-mediated MBs for Hg2+ detection. Some T–Hg2+–T based Hg2+ detection methods, including fluorescence biosensors,16 gold nanoparticle-based colorimetric method,17–19 anodic stripping voltammetry,20 amperometric biosensor based on electrically contacted enzyme and electrochemical DNA structure switching sensors,21,22 have been reported. Despite these developments, most of them might suffer relatively small signal-to-background ratio. Thus, it is still desirable to develop a detection system that is not only simple and sensitive, but also selective and reliable.
Graphene oxide (GO), a single-atom-thick two-dimensional nanosheet prepared by the acid exfoliation of graphite,23 has attracted great interests in biological applications owing to its unique characteristics, such as good water-solubility, versatile surface modification and superior fluorescence quenching ability.24,25 Furthermore, GO was reported to interact with single-stranded DNA (ssDNA) by π–π stacking interactions between nucleotide bases and GO but hardly interacts with rigid double-stranded DNA (dsDNA) or aptamer-target complexes.26 Nowadays, graphene oxide (GO)-based sensing platforms are being widely applied for the detection of various analytes attributed to its unique ability of adsorbing DNA species as well as its super quenching capacity for a wide range of fluorophores.27 In our previous report,28 we developed a novel fluorescence turn-on strategy for the biothiols assay, which is based on the resistance of metal-mediated molecular-beacons (MBs) toward nuclease digestion and the specific cleavage capacity of exonuclease I towards single-stranded DNA compared with double-stranded one. We found that GO shows remarkable difference in the binding ability toward single-stranded DNA and mononucleotides, and thus it displays a different fluorescence quenching capacity. Exonuclease III (Exo III) is an enzyme with specific exodeoxyribonuclease activity for dsDNA in the direction from 3′ to 5′.29 Inspired by our previous study, taking advantage of the specific affinity between Hg2+ and T–T mismatches, the preferable cleavage capacity of exonuclease III to double-stranded DNA compared with single-stranded one, and the superb fluorescence quenching ability of GO, we present an Exo III-aided “turn-on” fluorescence detection method for Hg2+. This strategy relies on the principle that Exo III shows different cleavage capacity for single-stranded DNA and double-stranded one in the absence and presence of Hg2+ and the preferential binding ability of GO to single-stranded DNA (ssDNA) over mononucleotides. Because Hg2+ could be released from the T–Hg2+–T hybrid by Exo III, it facilitates the formation of a new T–Hg2+–T hybrid, which may make this conformation-dependent enzymatic activity triggered by metal ions a signal-amplified strategy.
Experimental section
Chemicals and materials
Graphite powder, sulfuric acid, potassium persulfate, phosphorus pentoxide, hydrogen peroxide, mercuric nitrate, and potassium permanganate were purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). All the chemicals were of analytical grade or of the highest purity available. The oligonucleotides were purchased from Sangon Biological Engineering Technology and Services Co., Ltd. (Shanghai, China); their sequences are listed in Table S1.† GelRed Nucleic Acid Gel Stain (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×) was purchased from Biotium, Inc. (USA). Agarose was purchased from Invitrogen Corporation (USA). 6× loading buffer, 20 bp DNA ladder marker and Exo III were purchased from Takara Biotechnology Co., Ltd. (Dalian, China). The buffer solutions used in this work are as follows: 10× Exo III buffer consisted of 500 mM Tris–HCl (pH 8.0), 50 mM MgCl2, and 10 mM DTT.
000×) was purchased from Biotium, Inc. (USA). Agarose was purchased from Invitrogen Corporation (USA). 6× loading buffer, 20 bp DNA ladder marker and Exo III were purchased from Takara Biotechnology Co., Ltd. (Dalian, China). The buffer solutions used in this work are as follows: 10× Exo III buffer consisted of 500 mM Tris–HCl (pH 8.0), 50 mM MgCl2, and 10 mM DTT.
Apparatus
A Hitachi F-4600 fluorescence spectrophotometer (Hitachi Company, Tokyo, Japan) was used to record the fluorescence spectra and measure the fluorescence intensity. The sample cell was a 700 μL quartz cuvette. The fluorescence intensity was monitored by exciting the sample at 480 nm and measuring the emission at 520 nm. The slits for excitation and emission were set at 5 nm and 10 nm, respectively. The digital image of the gel was captured with the AlphaImager IS-2200 (Alpha Innotech, CA, USA).Synthesis of graphene oxide
Graphite oxide was synthesized from natural graphitic powder according to the Hummers method and our previous report (Hummers and Offeman, 1958; Xing et al., 2012). The resulting mixture was filtered and washed with 10% HCl aqueous solution and deionized water. Finally, the as-synthesized product was purified by dialysis for one week, dried in a desiccator and then dispersed in water with sonication for 5 h.Test of the general feasibility of this approach
The digestion of MB by Exo III in the absence and presence of Hg2+ were investigated, respectively. In detail, 2 μL of MB (10 μM) without and with 1 μL of Hg2+ (91.5 μM) were first mixed at 25 °C for 1 h, respectively. Then, 6 U Exo III was added and incubated with the abovementioned solution in 1× Exo III buffer at 25 °C for 15 min. Finally, all the reacted samples were heated to 80 °C for 10 min to inactivate Exo III and stop the digestion reaction. 6 μL of each reacted sample was mixed with 1.2 μL 6× loading buffer and was loaded onto a 3.5% agarose gel mixed with GelRed. Following electrophoresis in 0.5% TBE buffer at 100 volt for 60 min, the gel image was captured with the AlphaImager IS-2200.General confirmation of amplified mechanism of Exo III
To confirm the hypothesized mechanism of the release of Hg2+ and its cyclic reaction with MB, a gel electrophoresis experiment was conducted. 2 μL of MB (10 μM) with 1 μL of Hg2+ (3.66 μM) were first mixed at 25 °C for 1 h in two microcentrifuge tubes, respectively. Then, 6 U Exo III was added to incubate with the abovementioned solution in 1× Exo III buffer at 25 °C for 10 min and 70 min. 2 μL of MB (10 μM) with 1 μL of Hg2+ (91.5 μM) was incubated with 6 U Exo III for 10 min after mixing for 1 h at 25 °C. Each reacted sample was heated to 80 °C for 10 min to inactivate Exo III and stop the digestion reaction. 6 μL of each reacted sample was mixed with 1.2 μL 6× loading buffer and was loaded on a 3.5% agarose gel mixed with GelRed. Following electrophoresis in 0.5% TBE buffer at 100 volt for 60 min, the gel image was captured with the AlphaImager IS-2200.Study of Exo III activity on a DNA duplex with T–Hg2+–T base pairs
3.33 μM DNA 2 and 7.625 μM Hg2+; 1.67 μM DNA 1, 1.67 μM DNA 2 and 7.625 μM Hg2+; 1.67 μM DNA 1, 1.67 μM DNA 2 and 7.625 μM Hg2+; 1.67 μM DNA 3, 1.67 μM DNA 1 and 7.625 μM Hg2+; 1.67 μM DNA 4, 1.67 μM DNA 1 and 7.625 μM Hg2+; 1.67 μM DNA 5, 1.67 μM DNA 1 and 7.625 μM Hg2+; 1.67 μM DNA 6, 1.67 μM DNA 1 and 7.625 μM Hg2+ were first mixed at 25 °C for 1 h. Then, 6 U Exo III was added to incubate with these five samples in 1× Exo III buffer at 25 °C for 15 min. The other two samples were incubated with 1× Exo III buffer of the same volume. Finally, all the reacted samples were heated to 80 °C for 10 min to inactivate Exo III and stop the digestion reaction. 6 μL of each reacted sample was mixed with 1.2 μL 6× loading buffer and was loaded on a 3.5% agarose gel mixed with GelRed. Following electrophoresis in 0.5% TBE buffer at 100 volt for 60 min, the gel image was captured with the AlphaImager IS-2200.Detection procedure
For Hg2+ assay, a solution containing 10 μL of the probe stock solution was incubated with different concentrations of Hg2+ solution at 25 °C for 1 h; then, 0.5 U of Exo III was added into the abovementioned mixture solution. After the mixture was incubated for 15 min at 25 °C, 10 μg mL−1 of GO was added and incubated for another 30 min at room temperature. Then, the fluorescence emission spectra were immediately recorded for excitation at 480 nm. The same procedures were repeated in the presence of other metal ions instead of Hg2+ to assess the selectivity of the analytical procedure.Results and discussion
Working principle
Scheme 1 illustrates the principle of this strategy. The detection system consists of a dye labeled single-stranded oligonucleotide with a T–T mismatch in the region of stem (5′-ACA CTGTAA AAA AAA AAA AAA AAC ACT GT〈FAM〉 G-3′) (MB), GO, and Exo III. Hg2+, a widespread highly toxic contaminant in aquatic ecosystems, was chosen as a model system to demonstrate the proof-of-concept of our approach. In the absence of Hg2+, the base-pair binding energy is not sufficient to fully hybridize under room temperature, leading to the probe adopting a random coil structure, which shows resistance to the digestion of Exo III, and subsequently it is adsorbed onto the GO surface after being incubated with GO due to the strong binding strength between single-stranded DNA and GO, resulting in a significant quenching of the fluorescence. | ||
| Scheme 1 Schematic illustration of GO/Exo III based sensing strategy. | ||
In the presence of Hg2+, T–Hg2+–T base pairs are formed, and the linear probe transforms into a hairpin structure. This conformational switch triggers the occurrence of nuclease digestion of Exo III from the 3′ terminus of the nucleotide probe. No obvious fluorescence change is observed after the introduction of GO due to the weak affinity of the product mononucleotides to GO. Therefore, Hg2+ detection could be easily obtained by monitoring the fluorescence signal enhancement. Because Hg2+ could be released from the T–Hg2+–T hybrid by Exo III, it could get involved in the formation of new T–Hg2+–T hybrids, which may make this conformation-dependent enzymatic activity triggered by metal ions a signal-amplified method.
Test of the general feasibility of this approach
The fluorescence intensities of MB under different conditions were measured to testify the general feasibility of this approach, and the corresponding fluorescence spectra are shown in Fig. 1. The initial fluorescence of MB (curve a) is greatly quenched to 4.9% in the presence of 10 μg mL−1 GO (curve d), resulting from the strong binding of MB with GO and the high quenching ability of GO. When Exo III digestion is included in the assay steps, just as expected, only a slight fluorescence signal change is observed (curve c), indicating the poor cleavage capacity of Exo III to the single stranded DNA. However, when Hg2+ is added, the assay system shows dramatically higher fluorescence intensity (curve b). The enhanced fluorescence suggests the occurrence of nuclease digestion, where MB is digested into mononucleotides exhibiting a weak affinity to GO surface. These observations confirm that the stem–loop structure formed by the coordination of T–Hg2+–T is more likely to be digested by Exo III than the random coil ssDNA, and this strategy can be used for Hg2+ assay. | ||
| Fig. 1 Fluorescence intensity of MB under different conditions: (a) MB; (b) MB + Hg2+ + Exo III + GO; (c) MB + Exo III + GO; (d) MB + GO. | ||
In addition, gel electrophoresis was used to further testify the feasibility of this approach. The results are shown in Fig. 2. As can be seen in Fig. 2A, only the band in Lane 3 corresponding to MB/Hg2+/Exo III complex disappears. From Fig. 2B, which was obtained under ultraviolet light, the yellow band of FAM labelled on MB can be clearly seen. This indicates that MB was digested by Exo III in the presence of Hg2+. The gel electrophoresis results further prove that the existence of Hg2+ prompts the enzyme reaction of Exo III.
 | ||
| Fig. 2 Image of the gel electrophoresis to verify the feasibility of this method, Lane 1: 3.3 μM MB only; Lane 2: 3.3 μM MB with 15.25 μM Hg2+; Lanes 3 and 4: 3.3 μM MB treated with 6 U Exo III without Hg2+ (Lane 4) and with 15.25 μM Hg2+ (Lane 3). | ||
To generally confirm the hypothetical amplified mechanism of this method, a gel electrophoresis experiment was conducted (Fig. S1†). The result shows that Exo III performs the same cleavage effect on the system with low concentration of Hg2+ compared with the system with a relatively high concentration of Hg2+ after a considerably longer incubation time, which implies that the cyclic reaction may occur.
To investigate the influence of T–Hg2+–T base pairs on the activity of Exo III, five different DNA containing different numbers of T–T mismatches at different sites with DNA 2 were introduced. Each of these DNA was incubated with an excessive amount of Hg2+ to ensure that each T–T mismatched pair has one Hg2+ incorporated. The Exo III digestion of these DNA duplexes was analyzed by gel electrophoresis. As shown in Fig. S2,†T–Hg2+–T base pairs have no obvious inhibiting effect on exonuclease activity.
Optimization of experiment conditions
In an attempt to obtain an optimal assay condition and achieve a high sensitivity, the concentration of GO and Exo III and the enzymatic reaction time were investigated. According to the results, 10 μg mL−1 GO (Fig. S3†) and 0.5 U Exo III (Fig. S4†) were used in the following experiment. The enzymatic reaction time was set at 15 min (Fig. S5†).The reaction temperature is an important aspect involved in the detection system. A relatively low temperature may lead to a high background signal caused by the intramolecular base pairing of MB and a poor activity of Exo III, while a very high temperature may not be beneficial to the stability of Hg2+-induced hairpin strand. Thus, we need to find a suitable temperature that is conducive to the formation of hairpin structure as well as ensuring a low background signal. Fig. S6† shows the different fluorescence intensities of the detection system under different temperatures. A downward trend of fluorescence intensity in the system without Hg2+ can be seen under relatively low temperature, which may be caused by the dissociation of the intramolecular pairing base of MB, and increasing amount of MBs adopt a random coil structure, which is resistant to the digestion of Exo III. In the system with Hg2+, fluorescence intensity slowly changes at the beginning stage. During this period, two processes may be involved. First, some intramolecular pairing bases of MB dissociate slowly with the increase of temperature, which results in the resistance to the digestion of Exo III. Moreover, the formation of hairpin structure mediated by Hg2+ facilitates the cleavage reaction, which contributes to the increase of fluorescence intensities. These two complementary effects made the fluorescence intensities remain stable. However, under the temperature between 15 °C and 25 °C, an obvious increase of fluorescence signal can be observed, which shows the formation of more hairpin structure mediated by Hg2+. However, with the continuous growth of temperature, the hairpin structure of MB will be opened with the approach of its melting temperature; moreover, a high background signal was shown due to the enhanced Exo III activity. As a result, the decrease of F/F0 value can be observed (inset, Fig. S6†). We choose 25 °C as the ultimate enzyme reaction temperature after the comprehensive analysis of the abovementioned results.
Sensitivity and selectivity of the detection system
The quantitative detection of Hg2+ using this method under the optimized experimental conditions was evaluated by monitoring the fluorescence change under different concentrations of Hg2+. As shown in Fig. 3A, by increasing the concentration of Hg2+, the fluorescence intensity dramatically increases, implying that increasing amount of MB is digested by Exo III. Furthermore, the fluorescence intensity shows a clear linear dependence on Hg2+ concentration in the range of 1.83–109.8 nM (Fig. 3B). The linear regression equation is y = 7.39x + 199.33 with the correlation coefficient of 0.9776. The limit of detection (LOD) based on the 3σ/S calculation (σ is the standard deviation for the blank solution, and S is the slope of the calibration curve) is about 0.83 nM; an improved sensitivity compared with some of the reported fluorescence methods (Table 1). The satisfying sensitivity of our method may be mainly attributed to the low background signal induced by GO and amplified signal by Exo III.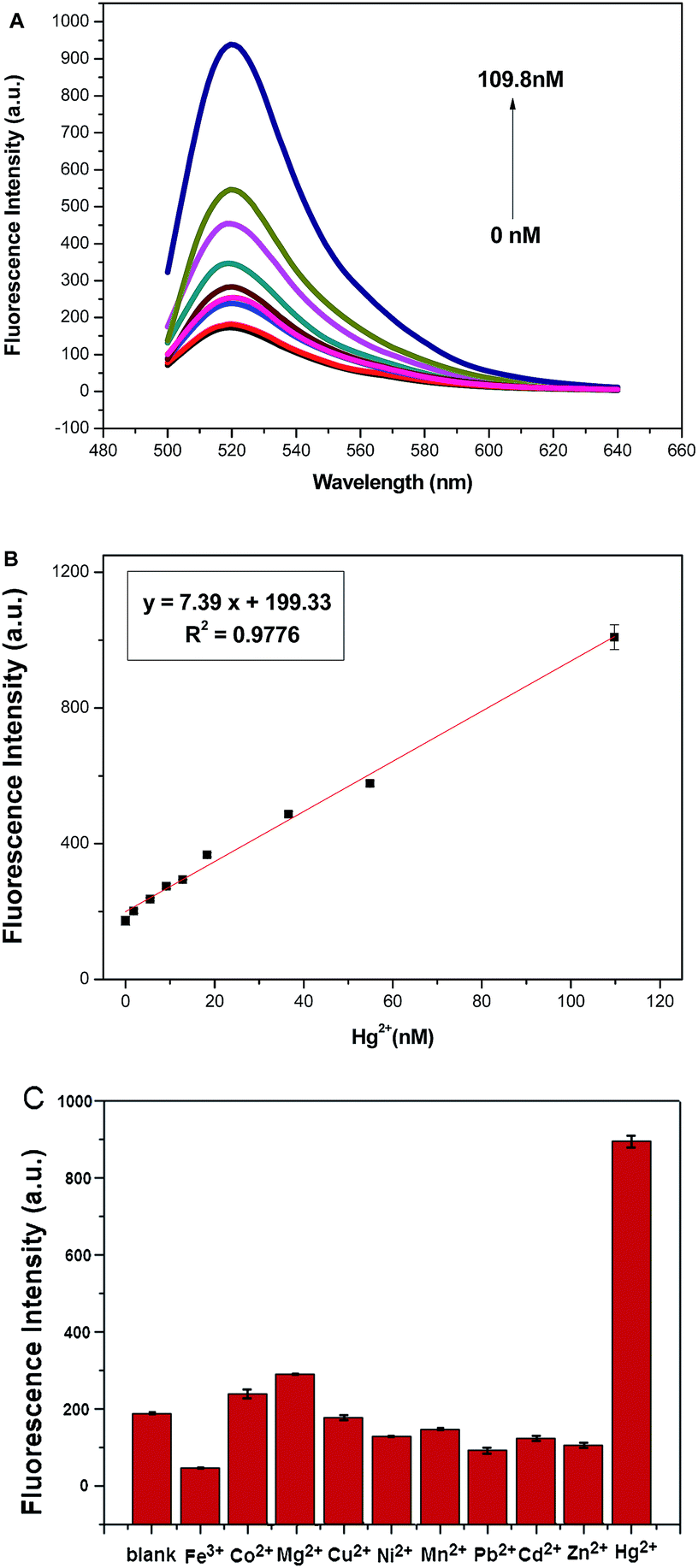 | ||
| Fig. 3 (A) Fluorescence spectra of the assay system in the presence of different concentrations of Hg2+. Bottom to top: 0, 1.83, 5.49, 9.15, 12.81, 18.3, 36.6, 54.9, and 109.8 nM. (B) Linear relationship between the fluorescence intensity and Hg2+ concentrations. (C) Fluorescence intensity of the assay system in the presence of different metal ions. | ||
| Method | Linear range | LOD | Ref. |
|---|---|---|---|
| Oligodeoxyribonucleotide (ODN)-based | 40–100 nm | 40 nM | 9 |
| DNA/single-walled carbon nanotubes based | 0.5–5.0 μM | 250 nM | 30 |
| Non-crosslinking aggregation of double-stranded | 45–720 nM | 10 nM | 31 |
| Application of a DNA-based luminescence switch-on method | 0.03–2.5 μM | 500 nM | 32 |
| Gold nanoparticle-based colorimetric and “turn-on” fluorescent probe | 96–6400 nM | 40 nM | 33 |
Considering the structural similarity of some other metal ions, the selectivity of the detection was investigated by substituting the Hg2+ in the system with various metal ions of the same concentration (3.66 μM), such as Mg2+, Pb2+, Zn2+, Ni2+, Cu2+, Fe3+, Co2+, Cd2+ and Mn2+. No significant increase in fluorescence intensity is observed in comparison with that of Hg2+ (Fig. 3C). These results indicate that the proposed strategy has high selectivity for Hg2+, which is attributed to the specific T–Hg2+–T base pairing and the preferential activity of Exo III.
The detection of Hg2+ in human serum
To evaluate the practicability of the proposed method, the detection of Hg2+ in human serum was performed, and the detection process was the same as that in the pure buffer. Fig. 4A shows the fluorescence intensities of MB in the presence of different concentrations of Hg2+ in 1% human serum. A good linear relationship with a calibration equation y = 3.64x + 313.37 (R2 = 0.9819) is still obtained in 1% human serum (Fig. 4B). These results demonstrate the potential value of the proposed method for detecting Hg2+ in real samples.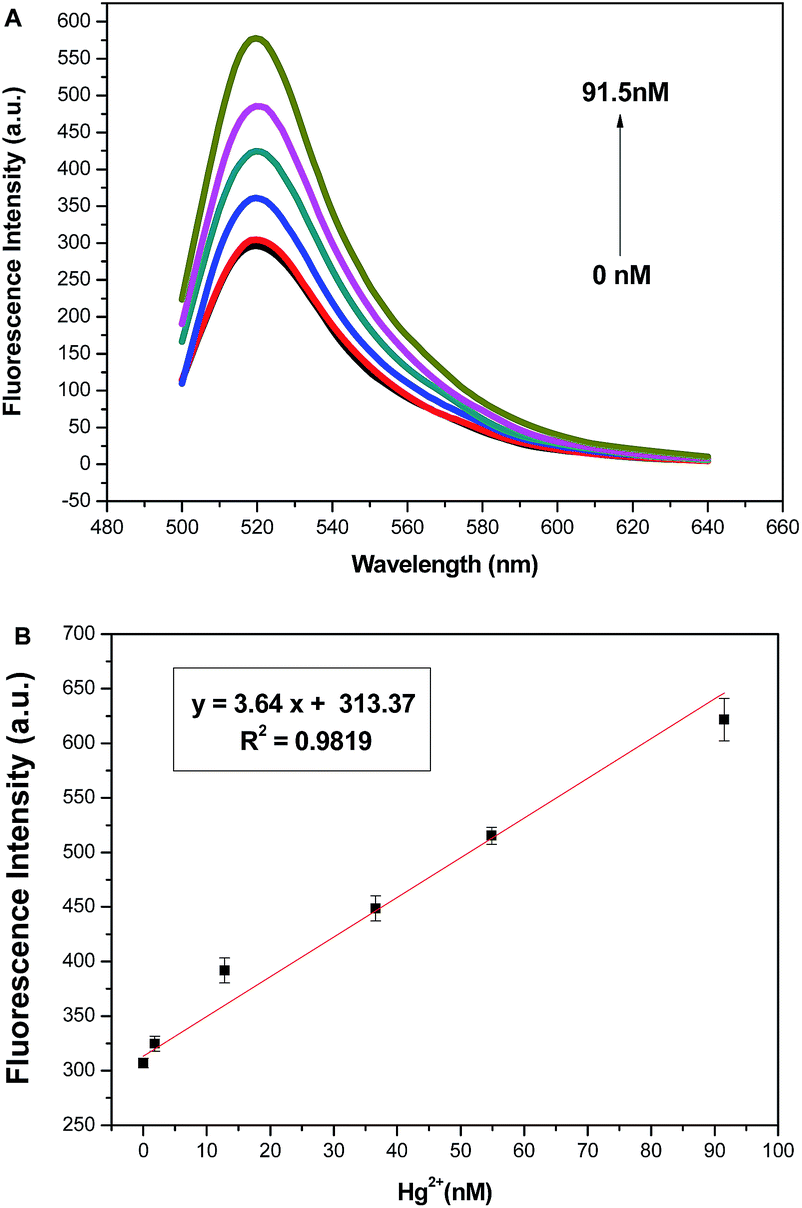 | ||
| Fig. 4 (A) Fluorescence spectra of the assay system in the presence of different concentrations of Hg2+ in 1% human serum. Bottom to top: 0, 1.83, 12.81, 36.6, 54.9, and 91.5 nM. (B) Linear relationship between fluorescence intensity and Hg2+ concentrations in 1% human serum. | ||
Conclusions
In summary, we have proposed a novel and sensitive fluorescence turn-on strategy for the assay of Hg2+ by employing metal-mediated MB, GO and Exo III. An obvious improvement on the selectivity of Hg2+ was shown in this strategy. The design mechanism is mainly based on the preferential activity of Exo III to dsDNA compared with ssDNA and the remarkable difference in the affinity of GO with ssDNA and mononucleotides. A dye labeled single-stranded oligonucleotide with a T–T mismatch was designed for the detection of Hg2+. Under the optimal conditions, Hg2+ could be specifically and sensitively detected with a limit of 0.83 nM. More importantly, the proposed method was also applied to the determination of Hg2+ in human serum with satisfactory results, which suggests that this strategy has great potential for bioanalysis. Furthermore, this new approach will greatly contribute to the application of metal-mediated base pairs-based biosensors in biochemical and biomedical studies as long as the collective problems involved in the application of GO could be properly addressed.Acknowledgements
We gratefully acknowledge the support of the National Basic Research Program of China (973 Program, nos 2011CB933600 and 2006CB933100), the National Natural Science Foundation of China (21275110), and the Fundamental Research Funds for the Central Universities (2042014kf0246).References
- K. Yoshizawa, E. B. Rimm, J. S. Morris, V. L. Spate, C. C. Hsieh, D. Spiegelman, M. J. Stampfer and W. C. Willett, N. Engl. J. Med., 2002, 347, 1755–1760 CrossRef CAS PubMed.
- H. H. Harris, I. J. Pickering and G. N. George, Science, 2003, 301, 1203 CrossRef CAS PubMed.
- J. L. Gómez-Ariza, F. Lorenzo and T. García-Barrera, Anal. Bioanal. Chem., 2005, 382, 485–492 CrossRef PubMed.
- B. M. W. Fong, T. S. Siu, J. S. K. Lee and S. Tam, J. Anal. Toxicol., 2007, 31, 281–287 CrossRef CAS PubMed.
- N. Bloom and W. F. Fitzgerald, Anal. Chim. Acta, 1988, 208, 151–162 CrossRef CAS.
- J. H. Huang and Y. F. Xu, J. Org. Chem., 2009, 74, 2167–2170 CrossRef CAS PubMed.
- Y. K. Yang, K. J. Yook and J. Tae, J. Am. Chem. Soc., 2005, 127, 16760–16761 CrossRef CAS PubMed.
- J. M. Thomas, R. Ting and D. M. Perrin, Org. Biomol. Chem., 2004, 2, 307–312 CAS.
- A. Ono and H. Togashi, Angew. Chem., Int. Ed., 2004, 43, 4300–4302 CrossRef CAS PubMed.
- C. D. Geary, I. Zudans, A. V. Goponenko, S. A. Asher and S. G. Weber, Anal. Chem., 2005, 77, 185–192 CrossRef CAS PubMed.
- C. C. Huang, Z. Yang, K. H. Lee and H. T. Chang, Angew. Chem., 2007, 119, 6948–6952 CrossRef.
- E. Meggers, P. L. Holland, W. B. Tolman, F. E. Romesberg and P. G. Schultz, J. Am. Chem. Soc., 2000, 122, 10714–10715 CrossRef CAS.
- S. Tyagi, D. P. Bratu and F. R. Kramer, Nat. Biotechnol., 1998, 14, 303–308 CrossRef PubMed.
- Y. X. Wang, J. S. Li, H. Wang, J. Y. Jin, J. H. Liu, K. M. Wang, W. H. Tan and R. H. Yang, Am. Chem., 2010, 82, 6607–6612 CAS.
- J. Anichina, Z. Dobrusin and D. K. Bohme, J. Phys. Chem., 2010, 114, 15106–15112 CrossRef CAS PubMed.
- B. C. Ye and B. C. Yin, Angew. Chem., Int. Ed., 2008, 47, 8386–8389 CrossRef CAS PubMed.
- X. J. Xue, F. Wang and X. G. Liu, J. Am. Chem. Soc., 2008, 130, 3244–3245 CrossRef CAS PubMed.
- X. W. Xu, J. Wang, K. Jiao and X. R. Yang, Biosens. Bioelectron., 2009, 24, 3153–3158 CrossRef CAS PubMed.
- J. S. Lee, M. S. Han and C. A. Mirkin, Angew. Chem., 2007, 119, 4171–4174 CrossRef.
- A. Mandil, L. Lidrissi and A. Amine, Microchim. Acta., 2010, 170, 299–305 CrossRef CAS.
- S. J. Liu, H. G. Nie, J. H. Jiang, G. L. Shen and R. Q. Yu, Anal. Chem., 2009, 81, 5724–5730 CrossRef CAS PubMed.
- Z. Q. Zhu, Y. Y. Su, J. Li, D. Li, J. Zhang, S. P. Song, Y. Zhao, G. X. Li and C. H. Fan, Anal. Chem., 2009, 81, 7660–7666 CrossRef CAS PubMed.
- K. S. Kim, Y. Zhao, H. Jang, S. Y. Lee, J. M. Kim, K. S. Kim, J. H. Ahn, P. Kim, J. Y. Choi and B. H. Hong, Nature, 2009, 457, 706–710 CrossRef CAS PubMed.
- Z. Liu, J. T. Robinson, X. M. Sun and H. J. Dai, J. Am. Chem. Soc., 2008, 130, 10876–10877 CrossRef CAS PubMed.
- N. Mohanty and V. Berry, Nano Lett., 2008, 8, 4469–4476 CrossRef CAS PubMed.
- S. J. He, K. K. Liu, S. Su, J. Yan, X. H. Mao, D. F. Wang, Y. He, L. J. Li, S. P. Song and C. H. Fan, Anal. Chem., 2012, 84, 4622–4627 CrossRef CAS PubMed.
- J. R. Zhang, W. T. Huang, W. Y. Xie, T. Wen, H. Q. Luo and N. B. Li, Analyst, 2012, 137, 3300–3305 RSC.
- X. J. Xing, Y. Zhou, X. G. Liu, D. W. Pang and H. W. Tang, Small, 2014, 10, 3412–3420 CrossRef CAS PubMed.
- H. J. Lee, Y. Li, A. W. Wark and R. M. Corn, Anal. Chem., 2005, 77, 5096–5100 CrossRef CAS PubMed.
- S. Y. Niu, Q. Y. Li, R. Ren and K. C. Hu, Anal. Lett., 2010, 43, 2432–2439 CrossRef CAS.
- N. Kanayama, T. Takarada and M. Maeda, Chem. Commun., 2011, 47, 2077–2079 RSC.
- H. Z. He, K. H. Leung, W. C. Fu, D. S. H. Chan and D. L. Ma, Environ. Res. Lett., 2012, 7, 044032 CrossRef.
- H. Wang, Y. X. Wang, J. Y. Jin and R. H. Yang, Anal. Chem., 2008, 80, 9021–9028 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra14024a |
| This journal is © The Royal Society of Chemistry 2015 |