
Hybrid nanoporous polystyrene derived from cubic octavinylsilsesquioxane and commercial polystyrene via the Friedel–Crafts reaction†‡
Abstract
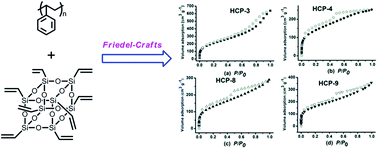
A series of hybrid nanoporous polystyrenes were synthesized from commercial polystyrene (PS) and cubic octavinylsilsesquioxane (OVS) via the Friedel–Crafts reaction. The porosity of the resulting hybrid polymers could be fine-tuned by varying the ratio of PS and OVS. The resulting polymers, HCP-1 to HCP-9 had apparent Brunauer–Emmett–Teller surface areas (SBET) in a range of 2.6 to 767 m2 g−1, with total pore volumes in the range of 0.36 to 0.90 cm3 g−1. The SBET was maximum when OVS loading was 9.1 wt%; it then decreased to almost no porosity before increasing again with the increase of OVS loading. The thermal decomposition temperatures of the hybrid polymers were close to pure PS under a N2 atmosphere, but the residues increased with the increase of OVS loading. The gas sorption applications revealed that HCP-3 possessed H2 uptake of 3.01 mmol g−1 (0.60 wt%) at 77 K and 1.01 bar and CO2 uptake of 1.12 mmol g−1 (4.93 wt%) at 298 K and 1.01 bar.
 Please wait while we load your content...
Please wait while we load your content...

