DOI:
10.1039/C4RA11484A
(Paper)
RSC Adv., 2015,
5, 12747-12754
Low density solvent based dispersive liquid–liquid microextraction followed by vortex-assisted magnetic nanoparticle based solid-phase extraction and surfactant enhanced spectrofluorimetric detection for the determination of aflatoxins in pistachio nuts†
Received
29th September 2014
, Accepted 6th January 2015
First published on 8th January 2015
Abstract
A simple and efficient two-step extraction method, namely a low density solvent based dispersive liquid–liquid microextraction (DLLME) followed by a vortex-assisted dispersive solid-phase extraction (VA-D-SPE) combined with analysis by surfactant enhanced spectrofluorimetry, was developed for the determination of the total aflatoxins in pistachio samples. The analytes were first extracted with methanol–water (80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20, v/v) from solid pistachio matrices and this solution was directly used as the dispersing solvent accompanied with 1-heptanol as the low density extracting solvent in the DLLME procedure. In the VA-D-SPE approach, hydrophobic Fe3O4 nanoparticles (i.e. oleic acid modified magnetic nanoparticles) were used to retrieve the analytes from the DLLME step. It was noticed that the target of the hydrophobic nanoparticles was 1-heptanol rather than the aflatoxins directly. The main parameters affecting the efficiency in DLLME and VA-D-SPE and the signal enhancement of the analytes were investigated and optimized. Under the optimum conditions, the calibration curve showed a good linearity in the range of 0.05–500 μg L−1 (R2 = 0.9984) with a low detection limit of 21 ng L−1. The repeatability and reproducibility of the extraction (as RSD%) were in the range of 2.3–4.6% and high recoveries ranging from 91.6 to 99.6% were obtained. Finally, the proposed method was successfully applied to the determination of the total aflatoxins in commercial pistachio samples. The obtained results revealed that the method is simple, inexpensive, accurate and remarkably free from interference effects. Furthermore, the proposed method reclaimed the versatility of DLLME because the selection of extraction solvent was not limited to high density solvents.
:20, v/v) from solid pistachio matrices and this solution was directly used as the dispersing solvent accompanied with 1-heptanol as the low density extracting solvent in the DLLME procedure. In the VA-D-SPE approach, hydrophobic Fe3O4 nanoparticles (i.e. oleic acid modified magnetic nanoparticles) were used to retrieve the analytes from the DLLME step. It was noticed that the target of the hydrophobic nanoparticles was 1-heptanol rather than the aflatoxins directly. The main parameters affecting the efficiency in DLLME and VA-D-SPE and the signal enhancement of the analytes were investigated and optimized. Under the optimum conditions, the calibration curve showed a good linearity in the range of 0.05–500 μg L−1 (R2 = 0.9984) with a low detection limit of 21 ng L−1. The repeatability and reproducibility of the extraction (as RSD%) were in the range of 2.3–4.6% and high recoveries ranging from 91.6 to 99.6% were obtained. Finally, the proposed method was successfully applied to the determination of the total aflatoxins in commercial pistachio samples. The obtained results revealed that the method is simple, inexpensive, accurate and remarkably free from interference effects. Furthermore, the proposed method reclaimed the versatility of DLLME because the selection of extraction solvent was not limited to high density solvents.
1 Introduction
Today, more than 300 mycotoxins, which are toxic metabolites of various fungi, grow on a wide range of food and animal feedstuffs.1 Among them, aflatoxins (AFs, Fig. 1), produced by some Aspergillus moulds such as Aspergillus flavus and Aspergillus parasiticus, represent the main threat worldwide owing to their occurrence and toxicity.2 AFs are potentially hazardous to humans and animals and display strong immunosuppressive, mutagenic, teratogenic and carcinogenic effects.3 Among AF compounds, aflatoxin B1 (AFB1) has been reported to be the most toxic and is classified as a group Al human carcinogen.4 Many countries and international organizations have set stringent regulations about the level of AFs permitted in food commodities. The European Commission has established the maximum levels for AFs in groundnuts, nuts, dried fruits and cereals as 2 ng g−1 for AFB1 and 4 ng g−1 for total AFs.5
 |
| | Fig. 1 The molecular structures of aflatoxin B1, B2, G1 and G2. | |
The simultaneous determination of multiple aflatoxins in a single test considerably reduces the time and cost of each analysis and is the most attractive approach practically.6 Currently, many simultaneous methods, such as high-performance liquid chromatography (HPLC), liquid chromatography-mass spectrometry (LC-MS) and immunoassay based analysis have been developed for the detection and identification of mycotoxins in food and feedstuffs.7–11 However, most of these methods are time-consuming, costly, laborious, and require expensive instruments.12 On the other hand, these methods are not sufficiently sensitive for the direct determination of these compounds in food samples. In this context, the development of methods for the preconcentration of AFs is necessary. Thus, methods normally used to analyze aflatoxins in sample matrices are based on extraction/preconcentration/clean-up with solid phase extraction (SPE) or immunoaffinity columns, followed by concentration steps.7,8,13 On the other hand, modern trends in analytical chemistry are toward the simplification and miniaturization of sample preparation procedures. Liquid-phase microextraction (LPME) has emerged in these last few years as a powerful tool for preconcentration and matrix separation prior to detection. A new mode of LPME, namely dispersive liquid–liquid microextraction (DLLME), was developed by Assadi et al. in 2006 (ref. 14) and is based on ternary component solvent systems. Here, an appropriate mixture of extraction solvent and dispersive solvent is injected rapidly into an aqueous solution, resulting in a cloudy state consisting of fine droplets of the extraction solvent dispersed in the aqueous phase, which markedly increases the contact surface between the phases and reduces the extraction time with the increasing enrichment factors.15,16 The advantages of the DLLME method are simplicity of operation, rapidity, low cost, high recovery and enrichment factors.17 But, as in conventional DLLME the density of the extraction solvent should be higher than water,18 the application of DLLME in most cases was limited to water samples and the volume of the sedimented phase in some cases was dependent on the surrounding temperature. These limitations caused some development of the DLLME method. Some modification techniques resulting in DLLME improvement are the use of organic solvents with lower density than water and applying SPE in combination with DLLME.19,20
In this study, a DLLME procedure using 1-heptanol as the extraction solvent was applied to extract AFs from pistachio samples and a vortex-assisted dispersive solid phase extraction (VA-D-SPE) using hydrophobic oleic acid modified Fe3O4 nanoparticles as the adsorbent was applied to retrieve the AF-containing extracting solvent from the DLLME step. Since 1-heptanol is a large alcohol with a non-polar hydrophobic chain, a hydrophobic interaction can occur between this solvent and the hydrophobic nanoparticles and the analytes were rapidly partitioned on the surface of the magnetic nanoparticles (MNPs). Separation was quickly carried out by the application of an external magnetic field overcoming the need for centrifugation, refrigeration to freeze, manual collection of the extractant or specialized apparatus. Then, a surfactant enhanced spectrofluorimetric determination using Triton X-100 micelle formation was applied for the determination of AFs. All the experimental parameters affecting the two-step extraction procedure were investigated in detail and the analytical characteristics of the method were evaluated. The method was successfully applied for the determination of AFs in pistachio samples.
2 Experimental
2.1 Standards and materials
Standards of AFB1, AFB2, AFG1, and AFG2 and all HPLC-grade solvents including acetone (Me2CO), acetonitrile (MeCN), dichloromethane (CH2Cl2), methanol (MeOH), ethanol (EtOH), ethyl acetate (C4H8O2), toluene (C6H5–CH3), 1-heptanol (C7H16O), 1-octanol (C8H18O), 2-ethylhexanol (C8H18O), diethyl ether ((C2H5)2O), and trichloromethane (CHCl3) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Iron(III) chloride hexahydrate (FeCl3·6H2O), iron(II) chloride tetrahydrate (FeCl2·4H2O), Triton X-100, oleic acid and the other chemicals used were supplied by Merck (Darmstadt, Germany). Deionized water was used throughout the experiments.
After the preparation of standard solutions of each aflatoxin, their concentrations were determined using an UV-Vis spectrophotometer through the AOAC Official method no. 971.22, chap. 49.2.03.21 These standards were used to prepare a tertiary stock solution of mixed standards as total AFs 1000 μg mL−1 (AFB1, AFG1 = 400 μg mL−1; AFB2, AFG2 = 100 μg mL−1), and the working standard solution was prepared by diluting the stock solution with methanol and water.
Since AFs are potential carcinogen compounds, extreme handling precautions must be warranted. Gloves and other protective clothing must be worn as a safety precaution and it is necessary to protect analytical work from sunlight because of degradation in light. All glassware should be soaked in 5% sodium hypochlorite solution to destroy AF residues before re-use.
2.2 Instrumentation
A Varian Cary Eclipse fluorescence spectrophotometer (Palo Alto, CA, USA) equipped with a xenon lamp was used for recording the fluorescence spectra of the AFs with a scan rate of 1200 nm min−1. All measurements were performed using 10 mm quartz microcells at room temperature and spectra recordings were carried out with slit widths of 5 nm. The excitation and emission wavelengths were 360 and 450 nm respectively. The modified magnetic nanoparticles were characterized by a Hitachi H-800 (Tokyo, Japan) transmission electron microscope. Chemical interactions were studied using a Perkin Elmer Spectrum one Bv5.3.0 FT-IR spectrometer (Waltham, Massachusetts, US) in the range of 400–4000 cm−1 with KBr pellets. The UV-Vis spectra of the AF standards were measured using an UV-240 Shimadzu spectrophotometer (Tokyo, Japan). A Labinco BV L46 Vortex mixer (Breda, Netherlands) was used to mix and accelerate the reactions between the reagents.
2.3 Synthesis of oleic acid modified MNPs
The Fe3O4 nanoparticles were prepared via a previously reported simple chemical co-precipitation method22 with slight modifications. Briefly, FeCl3·6H2O (5.8 g) and FeCl2·4H2O (2.1 g) were dissolved in 100 mL deionized water under a nitrogen atmosphere with vigorous stirring at 85 °C. Then, 20 mL of aqueous ammonia solution (25% w/w) was added to the solution. The color of the bulk solution changed from orange to black immediately. The magnetic precipitate was washed with deionized water (2 × 100 mL) and 0.02 mol L−1 (1 × 100 mL) sodium chloride. Then, oleic acid (1.0 g) was introduced and the reaction was kept at 80 °C for 3 h. Finally, the suspension was cooled to room temperature. The resulting nanoparticles were washed sequentially with deionized water (2 × 100 mL), methanol (2 × 100 mL) and deionized water (3 × 100 mL) and separated from the solution with the help of an external magnet. Finally, oleic acid modified magnetite nanoparticles were stored in deionized water at a concentration of 80 mg mL−1.
2.4 Real sample pretreatment
For the preparation of the pistachio samples (oily sample), 50 g of thoroughly homogenized nuts and 5 g NaCl were dissolved in 200 mL of methanol–water (80:20, v/v) and then, the mixture was added to 100 mL of n-hexane in a blender and mixed thoroughly for 3 min. The mixture was transferred to a separating funnel and the lower aqueous phase was filtered and diluted to 150 mL with methanol–water (80:20, v/v) and shaken intensively. The separated phase was then passed through a glass microfiber filter paper and an appropriate aliquot of the filtrate was used for the DLLME step.
2.5 Analytical procedure
310 μL of 1-heptanol was added to 3 mL of a sample solution (i.e. MeOH–H2O (80:20, v/v) containing analytes) and the mixture was rapidly injected into a conical-bottom vial containing 15 mL of deionized water. Then, the vial was sealed and swirled on a vortex agitator at 3500 rpm for 1 min (equilibration time). After that, 750 μL of the adsorbent (containing 60 mg of modified MNPs) was quickly added to the vial. The solution was swirled again using the vortex agitator for 2 min to facilitate the interaction of the organic solvent containing AFs with the surface of the oleic acid modified MNPs. Then, the magnetic adsorbent was collected by applying an external magnet and the supernatant was removed. The adsorbed AFs were desorbed from the adsorbent by the addition of 2 mL of MeCN for 3 min. After desorption, the eluent was separated by magnetic decantation and evaporated to dryness under a nitrogen gas flow at room temperature. The dry residue was dissolved in 2 mL of 0.5 mM Triton X-100 in 15% (v/v) acetonitrile–water and the solution was stirred for 5 min. The final solution was evaporated to 500 μL under a nitrogen flow and used for recording fluorescence spectra.
3 Results and discussion
3.1 Characterization of the adsorbent
The size and morphology of the oleic acid modified Fe3O4 nanoparticles were characterized by TEM images. As can be seen from Fig. 2, the modified nanoparticles had a uniform size distribution and most of them were quasi-spherical in shape with a mean diameter of 9 ± 1.2 nm. FT-IR spectroscopy was used to characterize the chemical interaction between the Fe3O4 nanoparticles and oleic acid. As can be seen from Fig. 3, the characteristic peak of the Fe3O4 nanoparticles can be observed as a strong absorption band at 583 cm−1, which corresponds to the Fe–O band of bulk magnetite. This band can be observed in the spectrum of the oleic acid modified MNPs too. The two sharp bands at 2923 and 2853 cm−1 are attributed to the asymmetric and symmetric CH2 stretches, respectively. It is worth noting that the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching band of the carboxyl group, which generally appears at 1700–1750 cm−1 was absent in spectrum (b) of the oleic acid modified MNPs and two new bands at 1541 and 1630 cm−1 appeared, which were characteristic of the asymmetric νas(COO–) stretch and the symmetric νs(COO–) stretch, instead.23,24 These results reveal that oleic acid was chemisorbed onto the Fe3O4 nanoparticles as a carboxylate and its hydrocarbon tail is free to interact with the analyte containing 1-heptanol solvent.
O stretching band of the carboxyl group, which generally appears at 1700–1750 cm−1 was absent in spectrum (b) of the oleic acid modified MNPs and two new bands at 1541 and 1630 cm−1 appeared, which were characteristic of the asymmetric νas(COO–) stretch and the symmetric νs(COO–) stretch, instead.23,24 These results reveal that oleic acid was chemisorbed onto the Fe3O4 nanoparticles as a carboxylate and its hydrocarbon tail is free to interact with the analyte containing 1-heptanol solvent.
 |
| | Fig. 2 TEM image of oleic acid modified MNPs. | |
 |
| | Fig. 3 FT-IR spectra of MNPs (a) and oleic acid modified MNPs (b). | |
3.2 Signal enhancement conditions
The fluorescence of mycotoxins is quenched in water and addition of some compounds like surfactants or complexing agents like β-cyclodextrin enhances the fluorescence intensity.25,26 This confirms that the microenvironment around AFs in these solutions is quite different from pure aqueous solutions. In this study, Triton X-100 was selected as the signal enhancement agent. It possesses a long tail length which forms large micelles around AF molecules, providing a better environment to encapsulate and restrict the intramolecular rotation of AFs to boost emission. The effect of surfactant addition on the AF fluorescence signal was investigated by adding different amounts of Triton X-100 (0.02–1 mM) to the desorbed AFs. As can be seen from Fig. 4, significant fluorescence enhancement occurred with increasing Triton X-100 concentration and reached a maximum at 0.5 mM, which is above the critical micellar concentration (CMC) value of 0.2 mM for this surfactant. The effect of micelle formation time on the fluorescence signal of AFs was also investigated in the range of 1–15 min. The results revealed that 5 min was enough for maximum signal enhancement and used for subsequent experiments.
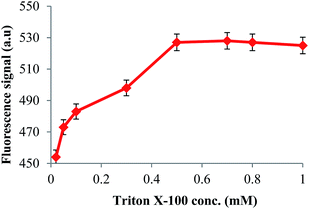 |
| | Fig. 4 The effect of Triton X-100 concentration on the fluorescence intensity of the AFs. The excitation and emission wavelengths were 360 and 450 nm respectively. | |
3.3 Optimization of the DLLME procedure
3.3.1 Selection of the dispersing solvent. Since the solvent used to extract AFs from the solid pistachio matrix is applied directly as the disperser solvent in DLLME, its selection must take into account both the properties required for AF extraction from the solid sample matrix and as the DLLME dispersant.27 Generally, an aqueous mixture of MeOH is applied for the extraction of AFs, and Me2CO, MeCN and MeOH are the commonly used disperser solvents for DLLME. Thus, the applicability of several solvents, including Me2CO, MeOH, MeCN, EtOH, MeOH–water (80:20 v/v) and MeCN–water (80:20 v/v), was investigated in the preliminary experiments. The results (Fig. 5) revealed that the extraction efficiency achieved by MeOH–water (80:20 v/v) was higher than the other solvents and, therefore, it was selected to act as both AF extractant from the pistachio samples and as the disperser solvent in DLLME for subsequent experiments.
 |
| | Fig. 5 The effect of the dispersing solvent on the recovery of total AFs. Conditions: extraction solvent, 310 μL of 1-heptanol; water volume, 15 mL; equilibration time, 60 s; adsorbent amount, 60 mg; adsorption time, 2 min; desorption time, 3 min; desorption solvent volume and type, 2 mL of MeCN. | |
Furthermore, the effect of the disperser volume on the AF recovery was investigated in the range of 1–5 mL. The obtained results (see Fig. S1, ESI†) revealed that the extraction efficiency increases with increasing the volume of MeOH–water (80:20 v/v) up to 3 mL and then decreases due to the increase in solubility of AFs in the aqueous phase and decrease in their distribution ratio. Based on the results, further studies were performed using 3 mL of MeOH–water (80:20 v/v) as the dispersing solvent.
3.3.2 Selection of the extracting solvent. An appropriate extraction solvent has a crucial role in the DLLME procedure. It should meet special conditions and have several characteristics. For example, it must have a good emulsification efficiency in the aqueous sample, high affinity for compounds of interest, low solubility in water, low density and low vapor pressure to prevent loss during agitation. Five organic solvents were evaluated as extraction solvents including ethyl acetate (density, d = 0.897 g mL−1), toluene (d = 0.865 g mL−1), 1-heptanol (d = 0.818 g mL−1), 1-octanol (d = 0.827 g mL−1), and 2-ethylhexanol (d = 0.833 g mL−1) in the preliminary experiments. As can be seen from Fig. 6, 1-heptanol gave the highest fluorescence signal for the analytes, followed by 1-octanol and toluene, and finally ethyl acetate and 2-ethylhexanol. Thus, 1-heptanol was considered as the most suitable extraction solvent for the subsequent experiments.
 |
| | Fig. 6 The effect of the extracting solvent on the recovery of total AFs. Conditions: dispersive solvent, 3 mL of MeOH–water (80:20 v/v); water volume, 15 mL; equilibration time, 60 s; adsorbent amount, 60 mg; adsorption time, 2 min; desorption time, 3 min; desorption solvent volume and type, 2 mL of MeCN. | |
The volume of the extracting solvent is another important parameter affecting the cloudy state formation and efficiency of the extraction process. So, the effect of 1-heptanol volume on the extraction of AFs was investigated in the range of 250–350 μL. The obtained results (Fig. S2, ESI†) revealed that the fluorescence intensity of AFs increases with increasing 1-heptanol volume in the range of 290 to 330 μL. The decrease in signal intensity above 330 μL is due to dilution effects and the decreased signal intensity below 290 μL corresponds to the dissolution of the organic phase in the aqueous media. Based on the results, 310 μL was selected as the optimum volume for further studies.
3.3.3 Effect of salt addition. Addition of salt to the sample may have several effects on the extraction efficiency of the analyte. Generally, salt addition can decrease the solubility of the target analyte in the aqueous phase and promote analyte transfer toward the organic phase resulting in improvement in the extraction efficiency, which is known as the salting-out effect.28 On the other hand, salt addition increases the viscosity and density of the sample solution and it can reduce the efficiency of the emulsification phenomenon because of the lower solubility of the extracting solvent in the aqueous phase. In this study, the effect of salt addition on the extraction efficiency of AFs was investigated by adding different amounts of NaCl in the range of 0–5% (w/v) to the samples. The obtained results (see Fig. S3, ESI†) showed that the extraction efficiency of AFs was not affected by the presence of NaCl. Thus, the experiments were carried out without adding salt.
3.3.4 Effect of water volume. The recovery of AFs was also affected by the water volume used in DLLME because it can influence the solubility of AFs in the aqueous phase. The effect of water volume on the extraction efficiency of the analytes was investigated using different volumes in the range of 2.5–25 mL. The results (Fig. S4, ESI†) showed that the extraction efficiency was constant in the range of 12.5–18 mL. Based on the results, 15 mL was selected for the subsequent experiments.
3.3.5 Effect of equilibration time. In this study, the equilibration time is defined as the interval time from the occurrence of the cloudy state and just before addition of the hydrophobic magnetic nanoparticles. The equilibration time was investigated in the range of 0–200 s maintaining the rotational speed at 3500 rpm to maximize mass transfer and reduce mixing time. As can be seen in Fig. S5, ESI,† the intensity of the fluorescence signal was not affected remarkably in different extraction times above 60 s, showing that the mass transfer from the sample solution to the extracting solvent is very fast. Based on the results, 60 s was selected for the subsequent experiments.
3.4 Optimization of the MNP based VA-D-SPE procedure
3.4.1 Effect of MNP amount and vortex time. The amount of hydrophobic MNPs is a key parameter to accomplish quantitative separation of the extraction solvent containing AFs in the DLLME step. Thus, different amounts of oleic acid modified Fe3O4 nanoparticles were added in the range of 10–100 mg to the sample solution. The results showed that the extraction efficiency increases with increasing amount of magnetic adsorbent up to 60 mg and then levels off (Fig. S6, ESI†). MNPs have a significantly higher surface area and short diffusion route compared to ordinary sorbents. Thus, satisfactory results with a lower adsorbent amount can be achieved with these sorbent materials. Based on the results, 60 mg was selected for the next experiments. In order to investigate the effect of adsorption time on the recovery of the analyte, the vortex time was varied in the range of 1–10 min. The experimental results (see Fig. S7, ESI†) indicate that 2 min is sufficient for achieving appropriate adsorption of AFs and it was used for the next experiments.
3.4.2 Desorption conditions. After extraction, the AFs containing extracting solvent were desorbed from the adsorbent into a suitable organic solvent. The desorption capabilities of different solvents were investigated using five commonly used organic solvents including Me2CO, EtOH, MeOH, MeCN and CHCl3. As can be seen from Fig. 7, the best result was found with MeCN as the desorbing solvent.
 |
| | Fig. 7 The effect of desorption solvent type on the recovery of total AFs. Conditions: dispersing solvent volume and type, 3 mL of MeOH–water (80:20 v/v); extraction solvent, 310 μL of 1-heptanol; water volume, 15 mL; equilibration time, 60 s; adsorbent amount, 60 mg; adsorption time, 2 min; desorption time, 3 min; desorption solvent volume, 2 mL. | |
Furthermore, the effect of the desorbing solvent volume on the recovery of AFs was investigated in the range of 0.5–5 mL and the maximum recovery was obtained with volumes higher than 2 mL (Fig. S8, ESI†). Thus, 2 mL of acetonitrile was selected as the optimum desorbing solvent for the subsequent experiments. In addition, the effect of desorption time on the recovery of the analytes was examined in the range of 1–10 min (Fig. S9, ESI†). As can be seen, the duration time of 3 min appeared to be sufficient for complete desorption. Since, magnetic nanoparticles can be easily and rapidly collected from the solution using an external magnetic field, the analysis time is greatly reduced compared to the conventional SPE methods and the combination of MNP based SPE with DLLME eliminates many time-consuming steps such as centrifugation, refrigeration to freeze and then thawing or manual collection of the extracting solvent which usually accompany the DLLME procedure.
3.4.3 Reusability of the adsorbent. In order to investigate the re-applicability of the hydrophobic adsorbent, the oleic acid modified MNPs which were used in one VA-D-SPE procedure were further desorbed and analyzed under the same conditions and the reproducibility of the recovery data was investigated. The experimental results show that the oleic acid modified MNPs are capable of being used for up to 10 extractions without sacrificing the analytical results (obtained RSD% less than 3.1% for the recovery results), which illustrates the capacity of these materials as an alternative sorbent for immunoaffinity columns.
3.5 Effect of interferences
Selectivity and competitive extraction experiments were carried out using total AFs as well as zearalenone (ZEN), ochratoxin A (OTA) and deoxynivalenol (DON), which are other mycotoxins that may exist in pistachios. Therefore, the possible interference effects of ZEN, DON and OTA were studied by the co-existing of each of them alone and in a mixture. The obtained results (Table 1) showed that the recoveries of total AFs in the presence of ZEN, OTA, DON and a mixture of these were not significantly affected by the presence of the interferences, indicating a good selectivity for the determination of total AFs in pistachios. Two possible reasons are: the difference in excitation/emission wavelengths of these mycotoxins with the corresponding ones for total AFs and unsuitability of the solvent medium (i.e. the desorbing solvent in the VA-D-SPE step) for both quantitative desorption from the adsorbent and taking the analytes fluorescence.
Table 1 The effect of mycotoxin interferences on the recovery of total AFs (5 μg kg−1, n = 3)
| Interference |
Concentration (μg kg−1) |
Recovery (%) |
| ZEN |
5 |
95.13 |
| OTA |
5 |
94.92 |
| DON |
5 |
98.73 |
| Mixture |
Total |
94.11 |
3.6 Analytical parameters
The proposed method was validated in terms of linearity, limit of detection, and intra-day and inter-day precisions. Samples for the construction of the calibration curve were prepared by spiking an appropriate amount of the total AF stock solution (with final concentrations of 0.05, 0.1, 0.5, 5, 20, 50, 100, 250, 400 and 500 μg L−1) to the non-contaminated pistachio samples and subjecting this to the proposed DLLME-D-SPE procedure following the enhanced fluorescence measurements. Under optimum experimental conditions, the calibration curve was linear over the concentration range of 0.05–500 μg L−1 with a calibration equation of Y = 87.61C + 4.71 and correlation coefficient (R2) of 0.9984. Furthermore, the sensitivity of the method for each individual aflatoxin was also measured by analyses of non-contaminated pistachio samples spiked with each aflatoxin with a corresponding concentration in samples containing total AFs. Thus, samples containing 0.02, 0.04, 0.2, 2, 8, 20, 40, 100, 160, and 200 μg L−1 for AFB1 or AFG1 and samples containing 0.005, 0.01, 0.05, 0.5, 2, 5, 10, 25, 40, and 50 for AFB2 or AFG2 were analyzed. The calibration equations of Y = 38.93C + 3.77 (R2 of 0.9982), Y = 279.80C + 8.49 (R2 of 0.9992), Y = 49.43C − 21.96 (R2 of 0.9993) and Y = 236.22C − 9.99 (R2 of 0.9991) were obtained for AFB1, AFB2, AFG1 and AFG2 respectively. The limit of detection (LOD = 3.3Sb/m, where Sb is the standard deviation of ten replicate measurements of a blank solution and m is the slope of the calibration curve) was found to be 21 ng L−1. The precision of the method was evaluated as RSD% through investigation of the intra-day and inter-day variations. The intra-day precision was evaluated using five replicate measurements of two spiked samples with concentrations of 2 and 200 μg L−1 in the same day and the inter-day precision was evaluated using five replicate measurements of spiked samples at the same concentration levels in five consecutive days. The results which are summarized in Table 2 indicate the good precision of the proposed method. The adsorption capacity of the adsorbent was also determined by the static method. For this purpose, 60 mg of the hydrophobic adsorbent was equilibrated with 18 mL of a solution containing the dispersed analyte after the DLLME step, at different concentration levels at the optimum conditions. After 10 min, the mixture was magnetically decanted and the supernatant was analyzed. The results showed that the amount of analytes adsorbed per mass unit of the adsorbent was increased with increasing concentration of the total AFs and then reached a plateau value (adsorption capacity value), which represents the saturation of the active surface of the hydrophobic adsorbent. The maximum adsorption capacity of the adsorbent for the total AFs was found to be 531 μg g−1.
Table 2 The characteristic data for the determination of total AFs by the proposed method
| Parameters |
Value |
| For 2 μg L−1 of total AFs. For 200 μg L−1 of total AFs. Sb is the standard deviation for ten blank measurements and m is the slope of the calibration curve. |
| Dynamic range (μg L−1) |
0.05–500 |
| Correlation coefficient (R2) |
0.9993 |
| Intra-day precision (RSD%, n = 5) |
3.3a |
| 2.3b |
| Inter-day precision (RSD%, n = 5) |
4.6a |
| 3.4b |
| Limit of detection (3.3Sb/mc, ng L−1) |
21 |
3.7 Real sample analysis
To evaluate the applicability of the proposed method in real matrices, it was applied to the determination of AFs in pistachio samples. Recovery studies were carried out by spiking the samples with different amounts of total AFs and the obtained results are summarized in Table 3. The acceptable recoveries in the range of 91.6% to 99.6% demonstrate that the matrix of the pistachio sample did not affect the extraction efficiency. Further examination of the accuracy was performed by comparison of the results obtained from the proposed method and the AOAC standard method (IAC-HPLC-FL) no. 999.07:2000, chap. 49.2.29 (ref. 21) for the determination of AFs in five contaminated pistachio samples. The results are summarized in Table 4. The statistical t-test analysis of the results showed that there are no significant differences between the data obtained by the two methods at a 95% confidence level. Furthermore, a comparison of the analytical characteristics obtained by the proposed method and some other reported methods for the determination of AFs is presented in Table 5. As can be seen, the proposed method has distinct advantages in terms of a low detection limit, wide linear range, ease of operation and simplicity.
Table 3 Determination of the total AFs in pistachio samples (n = 3, ±SD)
| Sample |
Spiked (μg kg−1) |
Found (μg kg−1) |
Recovery (%) |
| Not detected. |
| Sample 1 |
0 |
2.21 ± 0.41 |
— |
| 10 |
11.44 ± 1.63 |
93.69 |
| 50 |
50.74 ± 2.81 |
97.18 |
| 300 |
300.12 ± 2.89 |
99.32 |
| Sample 2 |
0 |
NDa |
— |
| 10 |
9.16 ± 1.78 |
91.67 |
| 50 |
47.66 ± 2.73 |
95.32 |
| 300 |
298.81 ± 2.85 |
99.60 |
| Sample 3 |
0 |
ND |
— |
| 10 |
9.39 ± 2.19 |
93.91 |
| 50 |
46.73 ± 1.97 |
93.46 |
| 300 |
294.32 ± 2.63 |
98.11 |
Table 4 Comparison of AF analyses (mean ± SD, n = 3) in contaminated pistachio samples by the proposed and standard IAC-HPLC-FD methods
| Sample |
Proposed method |
HPLC-FD-IACa |
| AFs (μg kg−1) |
AFs (μg kg−1) |
| HPLC analysis by AOAC standard method. |
| Sample 1 |
2.21 ± 0.41 |
2.33 ± 0.13 |
| Sample 2 |
2.78 ± 0.29 |
2.52 ± 0.30 |
| Sample 3 |
1.98 ± 0.43 |
2.12 ± 0.37 |
| Sample 4 |
3.55 ± 0.29 |
3.32 ± 0.41 |
| Sample 5 |
2.41 ± 0.24 |
2.55 ± 0.22 |
Table 5 Comparison of the proposed method with some previously reported methods for the determination of total AFs
| Method |
Analyte |
Matrix |
Linear range (μg kg−1) |
LOD (μg kg−1) |
Ref. |
| High performance liquid chromatography-mass spectrometry. Ultra-high performance liquid chromatography-tandem mass spectrometry. ng mL−1. |
| HPLC-IAC |
B1, G1 |
Pistachio |
0.12–8 |
87.7 |
2 |
| HPLC-IAC |
B2, G2 |
Pistachio |
0.06–4 |
87.7 |
2 |
| HPLC-MSa |
B1, B2, G1, G2 |
Leaves |
0.04–50 |
0.01–0.03 |
29 |
| UHPLC-MS/MSb |
B1 |
Seeds |
0.3–10 |
0.09 |
30 |
| UHPLC-MS/MS |
B2, G1, G2 |
Seeds |
0.6–20 |
0.11–0.22 |
30 |
| DLLME-HPLC-FD |
B1, G1 |
Cereals |
0.1–20c |
0.06–0.17 |
27 |
| DLLME-HPLC-FD |
B2, G2 |
Cereals |
0.025–5c |
0.01–0.04 |
27 |
| DLLME-D-SPE-fluorimetry |
B1, B2, G1, G2 |
Pistachio |
0.05–500 |
0.021 |
This work |
4 Conclusion
In this study, a two-step extraction technique namely, DLLME coupled with magnetic nanoparticle based VA-D-SPE followed by surfactant enhanced spectrofluorimetric detection was developed for the extraction of total AFs in pistachio samples. The proposed method demonstrates that an organic solvent with a lower density than water can be used in DLLME and it can be retrieved without involving any additional handling procedure and apparatus by the application of hydrophobic magnetic nanoparticles. The method has many advantages including simplicity of extraction, low organic solvent consumption, excellent enrichment in a short period of time, good repeatability and reproducibility for the determination of AFs, low cost and high accuracy. The good recoveries obtained for real samples and the inherent sensitivity and selectivity of the spectrofluorimetric method showed that the present method is effectively applicable for the determination of AFs in pistachio samples.
References
- K. Perényi, A. Lásztity and S. Pusztai, Microchem. J., 2007, 85, 149–156 CrossRef PubMed.
- R. Ghali, I. Belouaer, S. Hdiri, H. Ghorbel, K. Maaroufi and A. Hedilli, J. Food Compos. Anal., 2009, 22, 751–755 CrossRef CAS PubMed.
- A. Saidi, M. Mirzaei and S. Zeinali, Chemom. Intell. Lab. Syst., 2013, 127, 29–34 CrossRef CAS PubMed.
- E. Calleri, G. Marrubini, G. Brusotti, G. Massolini and G. Caccialanza, J. Pharm. Biomed. Anal., 2007, 44, 396–403 CrossRef CAS PubMed.
- S. M. Herzallah, Food Chem., 2009, 114, 1141–1146 CrossRef CAS PubMed.
- Q.-H. He, Y. Xu, D. Wang, M. Kang, Z.-B. Huang and Y.-P. Li, Food Chem., 2012, 134, 507–512 CrossRef CAS PubMed.
- L. Wang, Z. Wang, W. Gao, J. Chen, M. Yang, Y. Kuang, L. Huang and S. Chen, Food Chem., 2013, 138, 1048–1054 CrossRef CAS PubMed.
- Y. Nonaka, K. Saito, N. Hanioka, S. Narimatsu and H. Kataoka, J. Chromatogr. A, 2009, 1216, 4416–4422 CrossRef CAS PubMed.
- L. E. Edinboro and H. T. Karnes, J. Chromatogr. A, 2005, 1083, 127–132 CrossRef CAS PubMed.
- L. Fang, H. Chen, X. Ying and J.-M. Lin, Talanta, 2011, 84, 216–222 CrossRef CAS PubMed.
- F.-Y. Yu, A. V. Gribas, M. M. Vdovenko and I. Y. Sakharov, Talanta, 2013, 107, 25–29 CrossRef CAS PubMed.
- C. N. Rossi, C. R. Takabayashi, M. A. Ono, G. H. Saito, E. N. Itano, O. Kawamura, E. Y. Hirooka and E. Y. S. Ono, Food Chem., 2012, 132, 2211–2216 CrossRef CAS PubMed.
- I. Y. Goryacheva, M. A. Karagusheva, C. V. Peteghem, L. Sibanda and S. D. Saeger, Food Control, 2009, 20, 802–806 CrossRef CAS PubMed.
- M. Rezaee, Y. Assadi, M.-R. Milani Hosseini, E. Aghaee, F. Ahmadi and S. Berijani, J. Chromatogr. A, 2006, 1116, 1–9 CrossRef CAS PubMed.
- M. Rezaee, Y. Yamini and M. Faraji, J. Chromatogr. A, 2010, 1217, 2342–2357 CrossRef CAS PubMed.
- D. Ge and H. K. Lee, J. Chromatogr. A, 2013, 1317, 217–222 CrossRef CAS PubMed.
- M. Shamsipur, N. Fattahi, Y. Assadi, M. Sadeghi and K. Sharafi, Talanta, 2014, 130, 26–32 CrossRef CAS PubMed.
- L. Guo and H. K. Lee, J. Chromatogr. A, 2013, 1300, 24–30 CrossRef CAS PubMed.
- D. Djozan, M. A. Farajzadeh, S. M. Sorouraddin and T. Baheri, J. Chromatogr. A, 2012, 1248, 24–31 CrossRef CAS PubMed.
- A. Zgoła-Grześkowiak and T. Grześkowiak, J. Chromatogr. A, 2012, 1251, 40–47 CrossRef PubMed.
- AOAC International, Natural toxins. Official methods of analysis, current through revision 1, Maryland, 18th edn, 2006 Search PubMed.
- T. Gong, D. Yang, J. Hu, W. Yang, C. Wang and J. Q. Lu, Colloids Surf., A, 2009, 339, 232–239 CrossRef CAS PubMed.
- J. Liang, H. Li, J. Yan and W. Hou, Energy Fuels, 2014, 28, 6172–6178 CrossRef CAS.
- N. Wu, L. Fu, M. Su, M. Aslam, K. C. Wong and V. P. Dravid, Nano Lett., 2004, 4, 383–386 CrossRef CAS.
- M. Appell and W. B. Bosma, J. Lumin., 2011, 131, 2330–2334 CrossRef CAS PubMed.
- J. Hashemi, G. A. Kram and N. Alizadeh, Talanta, 2008, 75, 1075–1081 CrossRef CAS PubMed.
- L. Campone, A. L. Piccinelli, R. Celano and L. Rastrelli, J. Chromatogr. A, 2011, 1218, 7648–7654 CrossRef CAS PubMed.
- M. Hashemi, N. Jahanshahi and A. Habibi, Desalination, 2012, 288, 93–97 CrossRef CAS PubMed.
- M. Alcaide-Molina, J. Ruiz-Jiménez, J. M. Mata-Granados and M. D. Luque de Castro, J. Chromatogr. A, 2009, 1216, 1115–1125 CrossRef CAS PubMed.
- Z. Han, Y. Zheng, L. Luan, Z. Cai, Y. Ren and Y. Wu, Anal. Chim. Acta, 2010, 664, 165–171 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra11484a |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.