
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recovery and reuse of homogeneous palladium catalysts via organic solvent nanofiltration: application in the synthesis of AZD4625†
Hui
Xiao
 *a,
William R. F.
Goundry
a,
Rhys
Griffiths
a,
Yanyue
Feng
b and
Staffan
Karlsson
b
*a,
William R. F.
Goundry
a,
Rhys
Griffiths
a,
Yanyue
Feng
b and
Staffan
Karlsson
b
aEarly Chemical Development, Pharmaceutical Sciences, Biopharmaceuticals R&D, AstraZeneca, Macclesfield SK10 2NA, UK. E-mail: hui.xiao4@astrazeneca.com
bEarly Chemical Development, Pharmaceutical Sciences, Biopharmaceuticals R&D, AstraZeneca Gothenburg, SE-431 83 Mölndal, Sweden
First published on 28th February 2025
Abstract
Homogeneous catalysts are frequently used in the pharmaceutical industry but suffer from problematic separation from the reaction mixture and subsequent reuse. As an alternative to traditional separation methods like distillation and extraction, organic solvent nanofiltration (OSN) shows great potential to address the challenge of efficiently recovering and reusing homogeneous catalysts, without high energy usage and cumbersome biphasic separation. Here, we demonstrate the effective recovery of homogeneous palladium catalysts from the reaction mixture using commercial OSN membranes in a real pharmaceutical manufacturing case study to synthesize AZD4625, without altering the existing catalyst/ligand system. Despite the inherent challenges, the recovered catalyst and ligand were successfully reused up to five times, maintaining high conversion of over 90%. Furthermore, life cycle assessment shows that the sustainability of the process could be further enhanced by using greener bio-derived solvents and implementing solvent recovery to reduce solvent consumption.
Green foundation1. This work demonstrates the application of organic solvent nanofiltration (OSN) for the efficient recovery and reuse of a homogeneous palladium catalyst in the synthesis of AZD4625. Conducted under industrially relevant conditions with high catalyst/product concentrations and the green solvent 2-MeTHF, this study highlights its practicality and environmentally friendly nature.2. Using OSN membrane technology, the catalyst was successfully reused for up to five cycles with high conversion of over 90%. Life cycle assessment (LCA) indicates that the OSN process using bio-derived 2-MeTHF has a carbon footprint comparable to the adsorption method using CUNO filters. 3. Further sustainability gains can be achieved by integrating a solvent recovery unit to reduce solvent consumption of the OSN diafiltration process. Additionally, future research can extend the findings from this study to other pharmaceutical syntheses. |
1. Introduction
Homogeneous catalysts are widely used in the pharmaceutical and chemical industries due to their high reaction efficiency and selectivity for key transformations such as cross-coupling and asymmetric hydrogenation.1 However, separating homogeneous catalysts is inherently challenging because they exist in the same phase as the product, unlike heterogeneous catalysts, which can be easily removed by filtration.2 The presence of toxic precious metals in these catalysts, such as palladium (Pd), ruthenium, and rhodium, makes separation essential in the pharmaceutical industry, as strict regulatory limits on residual metal levels in final drug products must be met to ensure patient safety.3 Furthermore, the high cost of these metals and ligands provides a strong incentive for their recycling and reuse, as it can reduce costs and improve the sustainability of industrial processes.4,5 Therefore, developing efficient methods for the recovery and reuse of homogeneous catalysts is of critical importance.Traditional separation methods, such as distillation, extraction, and adsorption, have been explored, but unfortunately, no single method can solve all separation challenges.6–8 For example, distillation requires phase changes and high energy input, making it unsuitable for heat-sensitive catalysts and products.1 The application of extraction necessitates that the catalyst dissolves in one phase while the product goes into another, resulting in cumbersome biphasic operations.2 Adsorption using scavengers is popular but its efficiency depends on the type of scavengers and is not suitable if the products are also adsorbed or if scavengers leak into the products as new impurities.1 As an alternative, organic solvent nanofiltration (OSN) can selectively separate catalysts from the product without phase transition and biphasic operation, making homogeneous catalyst recovery easier and greener.4,9 Additionally, the absence of thermal transitions makes OSN quite suitable for the recovery of heat-sensitive catalysts and products.10
OSN is a membrane-based process used for separating molecules (150–1000 Da) in organic solvents.10 For successful catalyst and product separation using OSN, the membrane should have as high as possible rejection towards the catalyst (ideally 100%) and as low as possible rejection towards the product.4,10 The high rejection of the catalyst could prevent the catalyst/ligand from permeating into the product side, thus reducing potential impurities.4 Conversely, a low product rejection typically indicates that a low volume of diafiltration solvent is required to achieve the desired product yield.11 To meet these requirements, many strategies have been proposed by researchers. One approach is to enhance the selectivity of the membrane by applying novel membrane materials12,13 or modifying the membrane itself.14 Besides the commercial polydimethylsiloxane (PDMS)-based and polyimide-based membranes, new polymeric membrane made of poly(ether ether ketone)15 and crosslinked polybenzimidazole16 have been investigated for the recovery of homogeneous catalysts. Another important category of polymeric membrane, thin film composite membranes, prepared via interfacial polymerization also shows great potential for OSN applications especially in polar solvents.13,17–20 Modified ceramic membranes via Grignard reactions have expanded their applications in both polar and non-polar solvents, demonstrating their effectiveness in catalyst recovery.14,21,22 In addition, OSN membranes made of new materials such as graphene oxide,23 nanoporous graphene24 and covalent organic framework12,25 are promising candidates for OSN applications. Although these material innovations are very promising, challenges such as membrane performance under industrially relevant conditions and manufacturability at scale need to be addressed to bridge gaps for large-scale industrial applications.26
Another common strategy to improve the membrane selectivity is to increase the catalyst rejection by enlarging the catalyst or ligand.27–31 This can be achieved by enlarging the catalyst size through anchoring it to soluble supports, such as dendrimers27,28 and soluble polymers,29 or by combining the catalyst with larger ligands.30 However, the enlargement strategy may compromise catalytic activity, and the additional steps required for catalyst synthesis can increase costs and add complexity to the process. Thus, making such modifications is not always practical, especially in the pharmaceutical industry, since any modifications to existing catalyst/ligand systems in drug manufacturing must undergo rigorous validation and approval processes by regulatory authorities.
Additionally, several studies have demonstrated catalyst recovery using commercial membranes.31–35 For example, Wen et al.35 recently achieved successful recovery of homogeneous photocatalyst tetrabutylammonium decatungstate (TBADT) (>99%) using a commercial membrane (Solsep NF030306), and a continuous-flow system was demonstrated by coupling a microflow photoreactor with the OSN-based catalyst recovery unit. However, the model reaction in their study involved TBADT which has a molecular weight of 3320 Da, more than 10 times larger than that of the product (238 Da). Such a large molecular weight difference between the catalyst and product is not often obtained in homogeneous catalyst reactions. Similar model reactions with small molecular size products were often used to demonstrate catalyst recovery using OSN,32–34 with product rejection/recovery frequently neglected although it is crucial for industrial processes.4 Therefore, we believe it is meaningful to demonstrate the application of OSN for catalyst recovery and reuse in a case study conducted under real industrial conditions, including commonly used sizes of catalysts/ligands and products, as well as much higher catalyst and product concentrations.
In recent years, 2-methyltetrahydrofuran (2-MeTHF) has gained significant attention as a green alternative to tetrahydrofuran in organometallic chemistry and biocatalysis, owing to its derivation from renewable resources such as corncobs and sugarcane bagasse.36 However, the use of OSN in model reactions involving 2-MeTHF remains limited. For example, Bayrakdar et al. demonstrated the potential of OSN for separating gold and platinum catalysts from diluted reaction mixtures in 2-MeTHF.34,37,38 While their work achieved good flux and catalyst/product selectivity, the reaction solvents for these gold-catalysed34,38 and platinum-catalysed model reactions37 were not 2-MeTHF, which requires an additional solvent exchange step for the diafiltration experiments.
Herein, we report the successful separation of Pd homogeneous catalysts from products using commercial membranes in one of the key steps to synthesize AZD4625. AZD4625 is a potent and selective inhibitor of KRASG12C, which is a high-value intractable oncology drug target.39 The reaction scheme of this case study is shown Fig. 1. Stage 1 is a Pd-catalyzed Buchwald–Hartwig amination reaction in 2-MeTHF, followed by a hydrolysis reaction in Stage 2.
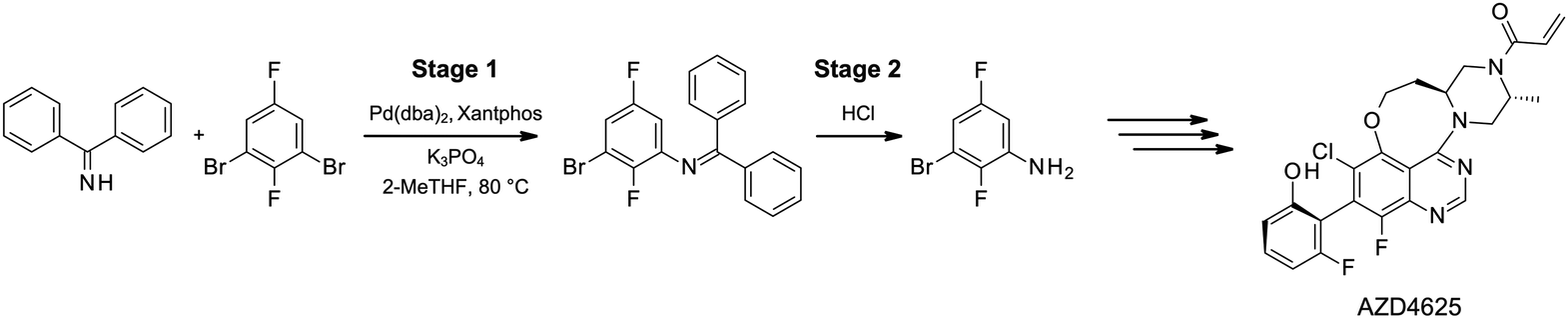 | ||
| Fig. 1 Reaction scheme of the membrane case study: AZD4625 Stage 1 and Stage 2, and the structure of the final product AZD4625. | ||
This case study differs from previous work in three key aspects. First, we deliberately choose not to modify the original reaction conditions (e.g., by enlarging the catalyst or ligand), ensuring compatibility with existing processes. The Pd(dba)2/Xantphos catalyst/ligand system, with molecular weights of 575 and 578.6 g mol−1, respectively, reflects molecular weights commonly used in industrial processes. Second, our OSN experiments are conducted under industrially relevant conditions, including significantly higher concentrations than those typically investigated in the literature. Third, 2-MeTHF is used directly in the reaction without requiring additional solvent exchange steps. Our work here demonstrates the use of the green solvent 2-MeTHF in an industrially relevant case study and provides valuable insights into the performance of OSN membranes under more realistic conditions.
Furthermore, the reusability of the recovered catalyst/ligand is investigated, and a simple cradle-to-gate life cycle analysis is conducted to evaluate the sustainability of the OSN membrane process compared to the traditional adsorption method.
2. Experimental
2.1 Materials
The reagents benzophenone imine (Sigma-Aldrich, UK), 1,3-dibromo-2,5-difluorobenzene (Fluorochem UK), potassium phosphate tribasic (Sigma-Aldrich, UK), palladium(0) bis(dibenzylideneacetone) (Pd(dba)2, Sigma-Aldrich, UK), Xantphos (Sigma-Aldrich, UK) and 2-MeTHF (VWR, UK) were purchased and used without further purification. Deionized water was collected from a Milli-Q® IQ-7003 ultrapure/pure water system (Millipore, UK). The commercially available OSN membranes selected were the Borsig series (oNF-1, oNF-2, oNF-3), the Evonik PuraMem series (Selective, Performance, and Flux), and the SolSep NF10206, all purchased separately from Borsig GmbH (Germany), Evonik Operations GmbH (Germany), and SolSep BV (Netherland).2.2 AZD4625 Stage 1 and 2 procedures
A typical procedure for AZD4626 Stage 1 and 2 is as follows: the jacketed vessel with an overhead stirrer, fitted with a reflux condenser is purged with nitrogen three times. Then, potassium phosphate tribasic (2 equiv., 83 mmol), Xantphos (0.02 equiv., 0.83 mmol), Pd(dba)2 (0.015 equiv., 0.62 mmol), benzophenone imine (1 equiv., 41 mmol), 1,3-dibromo-2,5-difluorobenzene (1.05 equiv., 43 mmol), 2-MeTHF (10 mL g−1) are added to the vessel. After dissolving the components by stirring, the vessel is heated to 80 °C under nitrogen atmosphere. After a 24-hour reaction, the vessel is cooled to room temperature and deionized water (10 mL g−1) is added to quench the reaction. After phase separation, the bottom aqueous layer is removed. The crude product in the organic layer is used as such in the following step (Stage 2). 2 M HCl (10 mL g−1) is added to the organic layer and stirred for 7 hours at room temperature. The organic layer is then directly subjected for further membrane experiments.2.3 Membrane performance characterization
The membrane screening experiments were conducted on a MiniMem cross-flow membrane system (PS Prozesstechnik GmbH, Switzerland) at room temperature (Fig. 2). All the membranes were immersed in 2-MeTHF for more than 2 hours before tests. The membrane disc, with an effective membrane area of 33 cm2, was fitted into the membrane cell (Fig. S2†). The initial test was operated in pure solvent (2-MeTHF) at 20 bar until the stable flux was achieved. Subsequently, the feed solution was changed to the reaction mixture (ten times dilution with 2-MeTHF) for the membrane screening test. The weight of the permeance over time was monitored using a scale. The samples of retentate and permeate were collected at 20 bar. The concentration of the product was analyzed using high-performance liquid chromatography-mass spectrometry (HPLC-MS). The palladium and phosphorus concentrations, which were utilized to represent the catalyst and ligand concentration, were determined by inductively coupled plasma optical emission spectrometry (ICP-OES). The membrane rejection and permeance were calculated by eqn (1) and (2), | (1) |
 | (2) |
 | ||
| Fig. 2 Flowchart of the Minimem cross-flow system with an AutoMem system for diafiltration. | ||
2.4 Catalyst recovery and reuse
After the reactions of AZD4625 Stage 1 and Stage 2, the reaction mixture (after aqueous work-up) was directly served as the feed solution for diafiltration. During the diafiltration process, the diafiltration solvent (2-MeTHF) was added to the feed vessel at the same rate as the permeate leaving the membrane cell. This process was controlled by an AutoMem system (PS Prozesstechnik GmbH, Switzerland) in conjunction with the MiniMem system (Fig. 2). Following diafiltration with a specified amount of diafiltration solvents, the permeate containing the product was collected. The retentate was also collected for the catalyst reuse experiments: the retained catalyst and ligand were added into a jacketed reaction vessel containing the same quantity of starting materials, but no additional catalyst or ligand, for another batch of reaction. Each reaction was sampled regularly, and the conversion was calculated using HPLC-MS analysis.2.5 Analytical techniques
HPLC-MS was used to determine the concentration of reactants and products on a Waters ACQUITY UPLC H-Class PLUS System, fitted with a ACQUITY QDa Detector and ACQUITY UPLC BEH C18 Column. The conversion of the limiting substrate (benzophenone imine) to the product was calculated by comparing the area of the individual characteristic peaks [conversion = areaproduct/(areaproduct + areasubstrate)].ICP-OES was used to determine the concentration of palladium and phosphorus in the feed, permeate and retentate. Samples of 0.2 mL feed, permeate, and retentate were initially digested in 6 mL of nitric acid/hydrochloric acid (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) using a ThermoFisher UltraWAVE microwave digestion system. Subsequently, each sample was diluted to 20 mL in centrifuge tubes with deionized water. Analysis was performed using ICP-OES on a ThermoFisher iCAP Pro XP Duo with a prepFast autosampler. The results were compared against a calibration curve generated from standard samples containing 0.01 ppm, 0.05 ppm, 0.5 ppm, and 5 ppm of palladium and phosphorus.
:1 v/v) using a ThermoFisher UltraWAVE microwave digestion system. Subsequently, each sample was diluted to 20 mL in centrifuge tubes with deionized water. Analysis was performed using ICP-OES on a ThermoFisher iCAP Pro XP Duo with a prepFast autosampler. The results were compared against a calibration curve generated from standard samples containing 0.01 ppm, 0.05 ppm, 0.5 ppm, and 5 ppm of palladium and phosphorus.
3. Results and discussion
3.1 Membrane screening
Membrane screening experiments were conducted to select suitable membranes for the catalyst/product separation in AZD4625 Stage 1 and 2. We first tried to remove the Pd catalyst directly after the Stage 1. Besides solvent compatibility, the molecular weight cut-off (MWCO) of a membrane should ideally fall between the size of the catalyst (575 g mol−1) and the Stage 1 product (373 g mol−1). However, due to the complexity of solvent-solute-membrane interactions, MWCO data by the manufacturer is not always reliable.40 In addition, the availability of GMP-qualified membrane modules for potential scale-up experiments is a key consideration in our selection process. Therefore, we selected seven commercially available membranes to ensure practical implementation and reproducibility both at laboratory and pilot scale. The seven membranes were screened with the Stage 1 reaction mixture (ten times dilution with 2-MeTHF) as feed solution, and their performance data at operating pressure of 20 bar were summarized in Table 1. As mentioned before, a high catalyst rejection is a prerequisite for a successful catalyst/product separation. The Borsig oNF-1, oNF-2, and Evonik Puramem® Performance were three possible candidates for the catalyst removal in terms of high catalyst rejection (>98.3%) (Table 1, entries 1, 2 and 5). However, their high Stage 1 product rejection (59.4% to 89.3%) rendered the separation difficult.| Entry | Membrane type | MWCO | Permeance (L m−2 h−1 bar−1) | Pd(dba)2 rejection | Xantphos rejection | Stage 1 product rejection | Stage 2 product rejection |
|---|---|---|---|---|---|---|---|
| The values represent averages, with detailed data presented in Table S3.† | |||||||
| 1 | Borsig oNF-1 | 600 | 3.9 | 100.0% | 99.6% | 82.8% | 38.6% |
| 2 | Borsig oNF-2 | 350 | 4.3 | 98.5% | 97.2% | 59.4% | 28.3% |
| 3 | Borsig oNF-3 | 900 | 9.5 | 95.1% | 91.0% | 48.5% | — |
| 4 | Evonik Puramem® Selective | — | 0.7 | 96.9% | 97.0% | 89.1% | — |
| 5 | Evonik Puramem® Performance | — | 1.8 | 98.3% | 97.3% | 89.3% | 56.3% |
| 6 | Evonik Puramem® Flux | — | 6.9 | 96.5% | 95.5% | 63.7% | — |
| 7 | Solsep NF10206 | — | 0.9 | 93.2% | 92.9% | 92.5% | — |
Since the Stage 2 product has much smaller molecular weight (208 g mol−1) compared to the Stage 1 product (372 g mol−1), we believed that the rejection of the Stage 2 product would be lower. Instead of separating the catalyst after Stage 1, we attempted the catalyst/product separation after a telescoped reaction to Stage 2. Fortunately, we found that for the three best membranes in Stage 1 (Table 1, entries 1, 2 and 5), the Stage 2 product rejection is only half of the Stage 1 product rejection, verifying that molecular size is a critical factor affecting the membrane rejection. As shown in Table 1, the Borsig oNF-1 membrane has almost perfect catalyst rejection and 38.6% rejection towards the Stage 2 product. In contrast, the Borsig oNF-2 membrane has a slightly lower catalyst rejection of 98.5% but also lower product rejection of 28.3%.
The better rejection of the oNF-1 membrane was unexpected since it has a larger MWCO of 600 Da compared to the oNF-2 membrane (350 Da). This observation is also similar to former gold catalyst recovery studies using Borsig membranes.34,38 This suggests that solute transport through the Borsig oNF series membrane is not solely determined by solute size or pore size, ie., size exclusion mechanism. It is believed that the interactions between solute, solvent, and membrane significantly affect OSN membrane performance.4 Zeidler et al.41 found that for PDMS-based membranes, solute rejection increased when the affinity of the solute to the solvent was stronger than its affinity to the membrane. In our case, both Borsig oNF-1 and oNF-2 membranes are PDMS-based, thus we hypothesize the difference in membrane affinity to the solutes affects the rejection. The oNF-1 membrane has a slightly more hydrophobic surface,42 resulting in lower affinity for the polar solute (Stage 2 product) compared to the oNF-2 membrane. This reduced affinity leads to poor dissolution of the solutes into the oNF-1 membrane material, which primarily governs the transport of solutes through the membrane. In other words, the solutes have a stronger tendency to remain in the solvent phase rather than permeating through the membrane. As a result, higher solute rejection is observed with the oNF-1 membrane.
Considering the similar high catalyst rejection and low product rejection, both Borsig oNF-1 and oNF-2 are selected for further catalyst/product separation experiments. Notably, both membranes have a high permeance of 3.9–4.3 L m−2 h−1 bar−1. The operating pressure was also optimized for both membranes (Fig. S3†). For both membranes, the Stage 2 product rejection increased slightly as the pressure increased from 10 bar to 40 bar. In contrast, the catalyst/ligand rejection slightly increased from 10 bar to 20 bar and then reached a plateau with further pressure increases to 40 bar. Therefore, 20 bar was selected for further experiments due to the best catalyst rejection and relatively low product rejection.
3.2 Catalyst recovery from the undiluted reaction mixture
After identifying suitable membrane and operation conditions, we explored the catalyst/product separation using undiluted reaction mixtures. Although the catalyst separation in Stage 2 is much more efficient than in Stage 1 using Borsig oNF-1 membrane (Fig. 3a), we still could not achieve complete separation in a single step due to imperfect product rejection. Therefore, diafiltration is required to wash the product out of the membrane cell by using fresh solvents. Using the rejection data in Fig. 3a, we calculated the catalyst retention and product recovery with varying diavolumes (DVs, the volume of diafiltration solvents relative to the original feed solution) using eqn (3):34| Retention (%) = 100 × e−N×(1−Ri) | (3) |
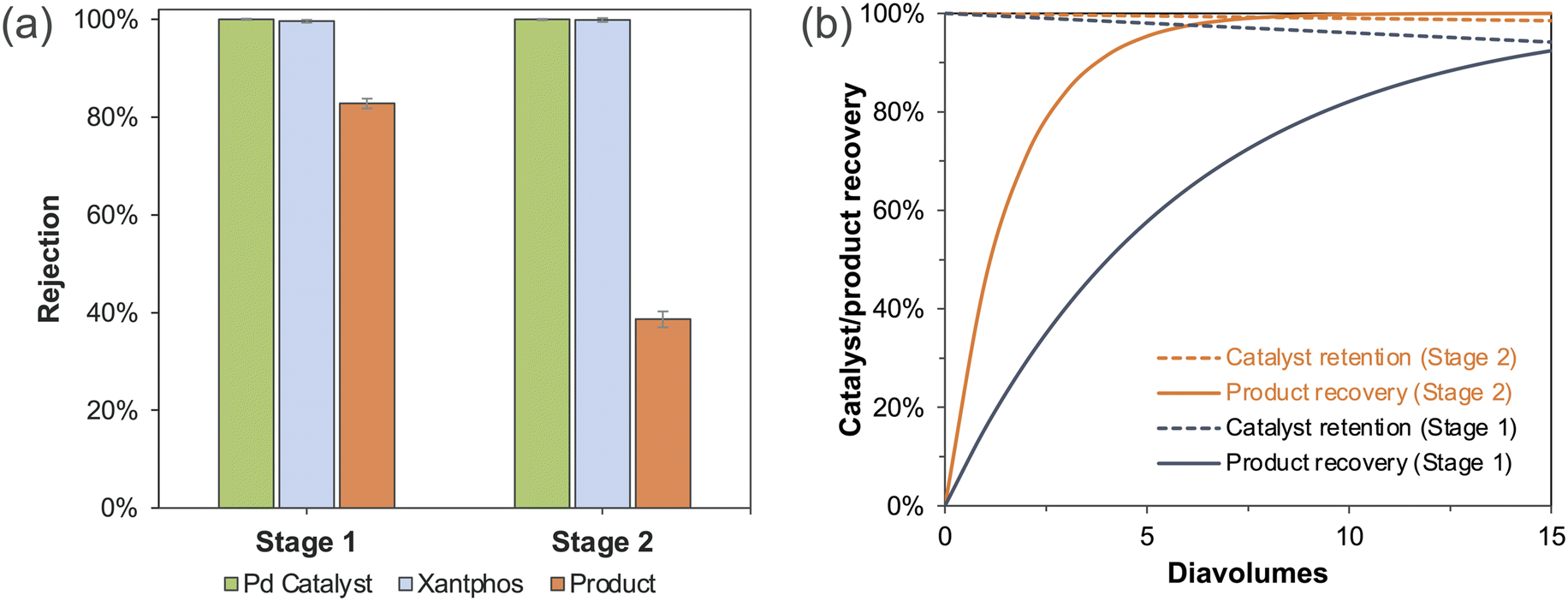 | ||
| Fig. 3 (a) The rejection of Borsig oNF-1 (600 Da) towards catalyst and product in Stage 1 vs. Stage 2, (b) the comparison of the calculated catalyst retention and product recovery during diafiltration in Stage 1 vs. Stage 2. | ||
Borsig oNF-1 membrane was first used to separate the catalyst and product in Stage 2 reaction mixture, which consisted of about 150 g L−1 Stage 2 product and 900 ppm Pd. 6 DVs of fresh solvent was used to achieve higher product recovery. As a result, 98.1% products were recovered and 91.7% of catalyst/ligands were retained, demonstrating successful separation using OSN. Also, the Pd level, which was 900 ppm in the feed vessel, decreased to only 12 ppm in the permeate. However, we noticed the catalyst retention was much lower than expected, considering the perfect catalyst/ligand rejection (100%/99.6%) observed in our membrane screening experiments. We speculated a decrease in catalyst rejection, due to the tenfold concentrated feed solution, could be the reason for the decrease in catalyst recovery. Further experiments confirmed our speculation and showed that the catalyst and ligand rejection at the concentrated solution decreased to 98.0%, 97.3%, respectively. Comparable separation results were also achieved by using Borsig oNF-2 membrane (Table S4†). After 5 DVs diafiltration, 97.2% product recovery and 90.4% catalyst retention were attained.
3.3 Reuse of the recovered catalyst and ligand
After a successful recovery of the catalyst/ligand, we further explored the possibility of reusing the recovered catalyst/ligand for repeated reactions. The experimental setup for catalyst reuse was depicted in Fig. 4. In this setup, the reaction and OSN separation here were separate steps. After the reaction and phase separation were completed, the reaction mixture was sent to the OSN unit for catalyst/product separation. The recovered catalyst/ligand was then sent back to the reactor with the same molar quantity of starting materials added, without any catalyst/ligand replenishment. A new batch of reaction was initiated, and the reaction mixture was subsequently subjected to the OSN diafiltration for catalyst recovery. The catalyst reuse experiment continued until a significant drop in conversion was observed. | ||
| Fig. 4 Schematic of the catalyst recovery and reuse: the recovered catalyst/ligand is added to the same quantity of starting materials as the first cycle for further reactions and subsequent OSN diafiltration. | ||
For the study of recycling/reuse of the Pd catalyst, in the first cycle, we used 1.5 mol% catalyst and 2.0 mol% ligand, which were the recommended loading in our large-scale manufacturing. The conversion of Stage 1 was monitored over time to demonstrate the catalyst performance. As shown in Fig. 5a, the catalyst maintained similar conversion in the first two cycles, and slightly decreased in the following three cycles. The catalyst experienced an obvious performance drop in the 6th cycle. The conversion after 24 hours (Fig. 5b) demonstrates that the catalyst maintained conversion of >90% for up to 5 cycles.
 | ||
| Fig. 5 (a) Stage 1 conversion with time in each reaction cycle and (b) Stage 1 conversion after 24 h in each reaction cycle. | ||
For the diafiltration experiments, the Borsig oNF-1 membrane was used, and the same membrane coupon was reused for all the diafiltration experiments. Table 2 summarizes the product recovery and catalyst/ligand retention in each cycle. An excessively large volume of diafiltration solvent (6–8 DVs) was used to achieve a high product recovery, which can help reduce side-reactions where product reacts with starting materials. In the first cycle, 98.1% product was recovered, and 91.7% catalyst was retained. With the increase of diafiltration volumes in the following cycles, the product recovery increased, while the catalyst and ligand retention decreased. Besides Borsig oNF-1 membrane, Borsig oNF-2 membrane was also evaluated for catalyst reuse experiments, the catalyst was successfully reused for up to 4 times with a high conversion >90% (Table S4†).
| Cycle | Conversion in 24 h (%) | Number of DVs | Product recovery (%) | Catalyst retention (%) | Ligand retention (%) |
|---|---|---|---|---|---|
| 1 | 99.3 | 6.0 | 98.1 | 91.7 | 88.9 |
| 2 | 99.0 | 5.8 | 97.4 | 88.7 | 89.5 |
| 3 | 96.8 | 7.1 | 98.7 | 85.5 | 85.0 |
| 4 | 94.0 | 7.8 | 99.1 | 78.6 | 82.1 |
| 5 | 90.1 | 8.1 | 99.2 | 74.1 | 82.1 |
| 6 | 65.5 |
To understand the reason for the gradual decrease in conversion in the catalyst reuse experiment, we measured the content of the catalyst and ligand in each cycle. Fig. 6a shows that the content of catalyst/ligand, normalized to the first cycle, dropped dramatically to only 16% in the 6th reaction cycle. Thus, we believe the loss of the catalyst/ligand could be the cause of the decline in conversion. We also converted the catalyst content to mol%, and the conversion vs. catalyst mol% data in Fig. 6b showed that a catalyst content of 0.5 mol% was essential for achieving a high conversion of over 90%. Additionally, we observed a gradual accumulation of impurities over successive reaction cycles in the HPLC-MS data (Fig. S4a†). These impurities can be attributed to the reaction between the Stage 2 product in the recovered catalyst/ligand and the starting material of Stage 1 (Fig. S4b†). Although we have used excessive solvent washes to achieve a high product yield of 98–99%, there was still 1–2% of the product that carried over to the next reaction with the recovered catalyst/ligand. This side reaction was not particularly noticeable in the second cycle, possibly due to the presence of sufficient catalysts, but became severe in the next three cycles as the catalyst content decreases (Fig. S4a†).
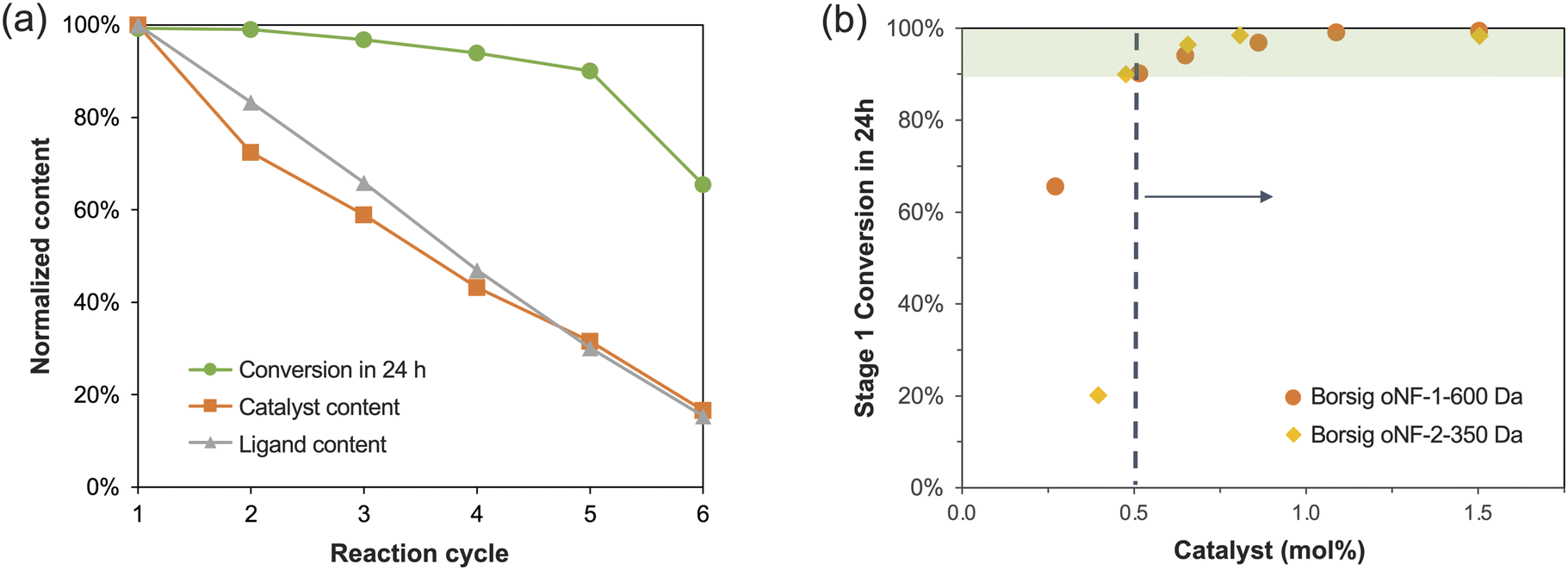 | ||
| Fig. 6 (a) The normalized content of catalyst and ligand in each reaction cycle during the catalyst reuse experiment using the Borsig oNF-1-600 Da membrane. (b) Stage 1 conversion and the catalyst mol% in each reaction cycle. | ||
We further analyzed the origin of the catalyst loss and found that the primary cause was the formation of Pd black, which was removed during the extractive work-up. The inevitable Pd black formation is a typical reason for the deactivation of Pd homogenous catalysts.32,43 This observation is also consistent with our experimental findings: a significant amount of Pd black formed and adhered to the reactor's inner wall. Besides the degradation during the reaction, catalyst degradation may also happen during the diafiltration process, due to the exposure to air. The 31P NMR results showed the ligand Xantphos, which contributes to catalyst stability, was oxidized to form Xantphos mono-oxide and Xantphos bis-oxide in air (detailed experiments in Note S2 and Fig. S5†). The ratio of unoxidized catalyst complex also decreased gradually with time (Table S5†). However, we did not observe any significant difference between the solution with and without the membrane, indicating that the degradation process is not related to the membrane itself. Membrane adsorption is also a potential source of catalyst loss. To minimize this effect, we used the same membrane disc throughout the diafiltration experiments and thoroughly washed it after each experiment by pumping a specific volume of fresh solvent through it. As a result, catalyst adsorption by the membrane was reduced to an acceptable level, accounting for 3.6% to 5.3% of the catalyst in the feed during each diafiltration cycle.
It worth noting that the permeate after each cycle diafiltration mainly consists of a mixture of the Stage 2 product, the by-product benzophenone, and dba ligand from the Pd(dba)2 catalyst, as verified by the HPLC-MS spectra (Fig. S6 and Fig. S7†). Consequently, product isolation is necessary. Since the composition of the permeate is quite similar to that of the reaction mixture after Pd removal using CUNO filter adsorption, we can refer to our previous manufacturing process (this has been validated by AstraZeneca manufacturing department). Isolation was achieved by performing a solvent swap from 2-MeTHF to isopropyl acetate under reduced pressure, followed by the addition of hydrochloric acid to form a crystalline hydrochloride salt of the main product, which precipitated from the solution while the by-product benzophenone and dba ligand remained in the filtrate.
In addition, we calculated the Pd content relative to the product in the permeate (without product isolation) as an indicator of product purity after diafiltration (Table S6†). After the first cycle of diafiltration, the Pd content was 484 μg Pd g−1 product. A slight increase in Pd content was observed across subsequent cycles, which can be attributed to the decreased yields in Stage 1 and Stage 2, as well as the increased diafiltration volumes used with each cycle. Although the Pd content is higher than the International Conference on Harmonization (ICH) limits for final drug products,3 it is important to note that the isolation process via crystallization can further purify the product and significantly reduce Pd levels. Also, higher Pd content is typically acceptable in the early stages of synthesis, as in our study, because Pd levels are expected to reach acceptable limits after multiple subsequent crystallisations of downstream intermediates.6
Membrane stability is a critical factor for the large-scale membrane applications. As mentioned earlier, we used the same membrane coupon for the diafiltration experiments. To compare membrane performance in each cycle, we calculated the average rejection using eqn (3) as a reference (Fig. 7a). From the first cycle to the fifth cycle, the average catalyst rejection decreased from 98.6% to 96.3%, while Xantphos rejection declined slightly from 98.0% to 97.6%. The slightly decrease in catalyst/ligand rejection might be attributed to the membrane fouling in the high concentration of the reaction mixture, with up to 150 g L−1 of product during diafiltration. However, we did not observe the flux decrease with time, due to the decreased feed concentration during diafiltration. Also, the membrane permeance before each diafiltration experiment in the pure solvent 2-MeTHF increased slightly from the first to the fifth diafiltration cycle (Fig. S8†). This indicates that our initial assumption of membrane fouling over time was incorrect, although we observed reaction mixture accumulated on the membrane surface after the fifth diafiltration (Fig. 7a inset). The increased solvent flux indicates the precipitation on the membrane might have been cleaned by pure solvent wash before each diafiltration experiment.
 | ||
| Fig. 7 (a) The average catalyst and product rejection in each diafiltration cycle. Inset: photo of the Borsig oNF-1 membrane coupon after five cycles of catalyst reuse experiments. (b) The rejection of Stage 2 product at varying product concentrations. The feed solution was prepared by diluting the Stage 2 reaction mixture with 2-MeTHF to the desired concentration. | ||
Instead, we tend to attribute the slight decrease in catalyst/ligand rejection to catalyst degradation. This assumption is also consistent with literature reports on OSN applied to homogenous catalyst recovery.44,45 For example, Nair et al. observed a sharp drop in ruthenium catalyst rejection from 98.8% to 76.8%, which they attributed to the decomposition of the catalyst complex, thereby lowering catalyst rejection.44 However, we did not observe such a significant decrease in catalyst rejection in our experiments. One possible explanation is that the Borsig oNF-1 membrane demonstrated consistently high rejection for both the catalyst and ligand separately. The rejection of the catalyst and ligand individually was comparable to the rejection of the catalyst/ligand mixture (Table S7†), suggesting that the membrane maintains good separation performance even if Xantphos/Pd(dba)2 dissociates into free Xantphos and Pd(dba)2. Thus, we believe that the catalyst complex may decompose into smaller fragments (not catalyst or ligand themselves), which pass through the membrane and contribute to the slight reduction in catalyst rejection.
Meanwhile, we observed the average product rejection slightly increased. To understand the reason for the rejection increase, we further measured the effects of product concentration on the membrane rejection. As shown in Fig. 7b, product rejection dropped significantly at low concentrations (<30 g L−1) and continued decreasing to a plateau as the concentration increased. The rejection increased by up to 18.0% when the product concentration decreased from 128.6 g L−1 to 1.5 g L−1. Thus, we believe the lower product concentration due to the conversion drop could be the main reason for the increase in product rejection. Despite the slight decrease in membrane performance, it is worth noting that the recovered catalyst/ligand maintained high conversion for up to five cycles.
3.4 Improving sustainability
The successful catalyst reuse experiments can significantly reduce the catalyst/ligand consumption in large-scale manufacturing. However, we used 6–8 DVs of fresh 2-MeTHF to achieve a high product yield, which inevitably increases the process mass intensity (PMI, defined as the total mass of materials used to produce a specified mass of product) (Table S2†). In contrast, the original method to remove the Pd catalyst in Stage 1 was using CUNO filters based on adsorption. This method could result in higher solid waste generation due to the single-use nature of the filters and prevents the direct reuse of the catalyst.To compare the environmental impacts of the membrane process and the non-membrane process (i.e., adsorption by CUNO filters), we conducted a cradle-to-gate life cycle assessment to calculate carbon emissions.46 To simplify the model, we only compared the two processes in terms of the global warming potential (kg CO2e per kg limiting material [LM]) of the catalyst/ligand and solvents (Fig. 8, detailed methods are included in Note S1†). The manufacturing and disposal of the OSN membrane module and CUNO filters were not considered here due to lack of data. Therefore, our calculation provides a lower band on the true impact of the two processes.
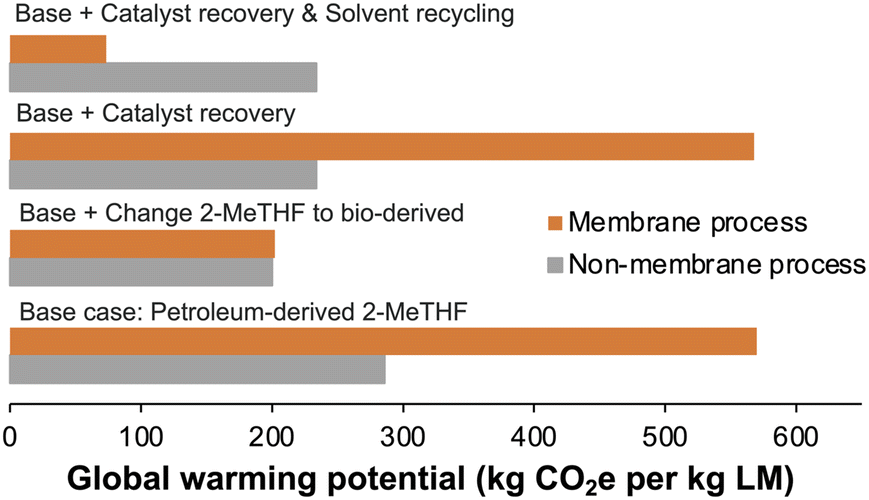 | ||
| Fig. 8 The global warming potential (GWP) comparison between the membrane process (orange bar) and the non-membrane process (adsorption by CUNO filters, grey bar) in different scenarios. The GWP includes carbon emissions related to the catalyst/ligand and solvent. The base scenario uses petroleum-based 2-MeTHF, without catalyst recovery and solvent recycling. The next case refers to the base case, but the solvent is replaced with bio-derived 2-MeTHF. The third case refers to the base case, but catalyst recovery (80%) is added for the CUNO filter process. Scenario 4 refers to the base case but both catalyst recovery (80%) and solvent recycling (95%) are added. | ||
Firstly, we noticed the source of the solvents is a critical factor. If bio-derived 2-MeTHF is used to replace the petroleum-derived 2-MeTHF, the carbon footprint of the membrane process significantly decreases and becomes comparable to that of non-membrane process (Fig. 8), as both the manufacturing and incineration of petroleum-based 2-MeTHF incur much higher carbon footprints. Secondly, the treatment of the recovered Pd in CUNO filters affects the overall carbon footprints of the non-membrane process. Unlike the membrane process, the recovered Pd by CUNO filters cannot be reused directly in reactions. Instead, it must be transported to the catalyst manufacturer for reprocessing and only Pd is recovered. If we do not consider the carbon emissions from the transportation of CUNO filters and Pd reprocessing, Pd recovery can improve the sustainability of the CUNO filter process (Fig. 8). Thirdly, integrating a solvent recovery unit with the membrane process can significantly enhance its sustainability by reducing overall solvent consumption. As shown in Fig. 8, recycling 95% of the diafiltration solvent could greatly decrease the carbon footprint of the membrane process, making it greener than the non-membrane process. Meanwhile, solvent recycling can substantially reduce the PMI of the membrane process to 25.4, compared to 31.9 for the non-membrane process (Table S2†). Overall, to increase the sustainability of the membrane process, both green bio-derived solvents and efficient solvent recovery are vital.
4. Conclusions
Without altering the existing catalyst/ligand system, we successfully demonstrated the recovery and reuse of Pd homogeneous catalysts using commercial OSN membranes in a case study from the pharmaceutical manufacture of AZD4625. Membrane screening experiments identified Borsig oNF-1 and oNF-2 as the best membranes for catalyst/product separation, in terms of their high catalyst rejection and low product rejection. Compared with direct catalyst/product separation after the Pd-catalyzed Buchwald–Hartwig reaction (Stage 1) in 2-MeTHF, we found that separation after Stage 2 could save up to 10 DVs of fresh solvent, due to much lower product rejection in Stage 2. The Borsig oNF-1 membrane maintained high performance during the catalyst reuse experiment, and the non-destructive recovered catalyst/ligand were reused up to five times with a high conversion of over 90%. The preliminary life cycle assessment calculations, in terms of the catalyst/ligand and solvent usage, demonstrated that catalyst recovery and reuse using OSN could be much greener than the non-membrane process using CUNO filters, if a bio-derived solvent is used or a solvent recovery unit is added to reduce the solvent consumption of the diafiltration process. This study demonstrates OSN could be a more sustainable and efficient approach to homogenous catalyst recovery and reuse, which holds great potential for broader application in the pharmaceutical industry.Author contributions
Hui Xiao: conceptualization; data curation; investigation; methodology; formal analysis; writing – original draft. William R. F. Goundry: supervision; funding acquisition; writing – review and editing. Rhys Griffiths: methodology; data curation; writing – review and editing. Staffan Karlsson: supervision; funding acquisition; writing – review and editing. Yanyue Feng: writing – review and editing.Data availability
The data supporting the findings of this study are available within the article and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has received funding from the European Union's Horizon Europe Framework Programme (HORIZON) under grant agreement No. 101057668. The work of UK-based Associated Partners has been funded by UK Research and Innovation (UKRI) under the UK government's Horizon Europe funding guarantee (No. 10039890). The authors would like to thank Dr Elizabeth M. Dauncey, Dr Matthew Tatton, Dr Qing Yu for their guidance of the chemical synthesis experiments. We thank Robert Peeling for the contribution of the process scheme (Fig. S1†). We also appreciate Miriam Turner, and Chloë Smithers for the insightful discussion on the life cycle assessment calculation.References
- D. J. Cole-Hamilton, Science, 2003, 299, 1702–1706 CrossRef CAS PubMed.
- T. A. Fassbach, J.-M. Ji, A. J. Vorholt and W. Leitner, ACS Catal., 2024, 14, 7289–7298 CrossRef CAS.
- ICH, International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) guideline Q3D (R2) on elemental impurities – Step 5, https://www.ema.europa.eu/en/documents/scientific-guideline/international-conference-harmonisation-technical-requirements-registration-pharmaceuticals-human-use-ich-q3d-elemental-impurities-step-5-revision-2_en.pdf, (accessed 03/12/2024).
- H. Xiao, Y. Feng, W. R. F. Goundry and S. Karlsson, Org. Process Res. Dev., 2024, 28, 891–923 CrossRef CAS PubMed.
- S. Cotty, J. Jeon, J. Elbert, V. S. Jeyaraj, A. V. Mironenko and X. Su, Sci. Adv., 2022, 8, eade3094 CrossRef CAS PubMed.
- M. Economidou, N. Mistry, K. M. P. Wheelhouse and D. M. Lindsay, Org. Process Res. Dev., 2023, 27, 1585–1615 CrossRef CAS.
- I. Vural Gürsel, T. Noël, Q. Wang and V. Hessel, Green Chem., 2015, 17, 2012–2026 RSC.
- G. Ignacz, A. K. Beke, V. Toth and G. Szekely, Nat. Energy, 2024 DOI:10.1038/s41560-024-01668-7.
- M. Janssen, C. Müller and D. Vogt, Green Chem., 2011, 13, 2247–2257 RSC.
- P. Marchetti, M. F. Jimenez Solomon, G. Székely and A. G. Livingston, Chem. Rev., 2014, 114, 10735–10806 CrossRef CAS PubMed.
- G. Székely, J. Bandarra, W. Heggie, B. Sellergren and F. C. Ferreira, J. Membr. Sci., 2011, 381, 21–33 CrossRef.
- H. Yang, J. Xu, H. Cao, J. Wu and D. Zhao, Nat. Commun., 2023, 14, 2726 CrossRef CAS PubMed.
- B. Alhazmi, G. Ignacz, M. Di Vincenzo, M. N. Hedhili, G. Szekely and S. P. Nunes, Nat. Commun., 2024, 15, 7151 CrossRef CAS PubMed.
- D. Ormerod, M. Dorbec, E. Merkul, N. Kaval, N. Lefèvre, S. Hostyn, L. Eykens, J. Lievens, S. Sergeyev and B. U. W. Maes, Org. Process Res. Dev., 2018, 22, 1509–1517 CrossRef CAS.
- L. Peeva, J. Arbour and A. Livingston, Org. Process Res. Dev., 2013, 17, 967–975 CrossRef CAS.
- P. Kisszékelyi, S. Nagy, B. Tóth, B. Zeller, L. Hegedűs, B. Mátravölgyi, A. Grün, T. Németh, P. Huszthy and J. Kupai, Periodica Polytech., Chem. Eng., 2018, 62, 489–496 Search PubMed.
- S. Karan, Z. Jiang and A. G. Livingston, Science, 2015, 348, 1347–1351 CrossRef CAS PubMed.
- T. Huang, B. A. Moosa, P. Hoang, J. Liu, S. Chisca, G. Zhang, M. AlYami, N. M. Khashab and S. P. Nunes, Nat. Commun., 2020, 11, 5882 CrossRef CAS PubMed.
- J.-B. Li, C.-Y. Zhu, H.-N. Li, J.-H. Xin, H.-Y. Fan, C. Zhang, J. Wu, L. Zhang, H.-C. Yang and Z.-K. Xu, J. Membr. Sci., 2024, 704, 122841 CrossRef CAS.
- S. Liu, F. Yang, J. Zhou, Y. Peng, E. Wang, J. Song and B. Su, J. Membr. Sci., 2023, 687, 122052 CrossRef CAS.
- S. R. Hosseinabadi, K. Wyns, V. Meynen, R. Carleer, P. Adriaensens, A. Buekenhoudt and B. Van der Bruggen, J. Membr. Sci., 2014, 454, 496–504 CrossRef.
- D. Ormerod, N. Lefevre, M. Dorbec, I. Eyskens, P. Vloemans, K. Duyssens, V. Diez de la Torre, N. Kaval, E. Merkul, S. Sergeyev and B. U. W. Maes, Org. Process Res. Dev., 2016, 20, 911–920 CrossRef CAS.
- Q. Yang, Y. Su, C. Chi, C. T. Cherian, K. Huang, V. G. Kravets, F. C. Wang, J. C. Zhang, A. Pratt, A. N. Grigorenko, F. Guinea, A. K. Geim and R. R. Nair, Nat. Mater., 2017, 16, 1198–1202 CrossRef CAS PubMed.
- J. Kang, Y. Ko, J. P. Kim, J. Y. Kim, J. Kim, O. Kwon, K. C. Kim and D. W. Kim, Nat. Commun., 2023, 14, 901 CrossRef CAS PubMed.
- L. Cao, C. Chen, S. An, T. Xu, X. Liu, Z. Li, I. C. Chen, J. Miao, G. Li, Y. Han and Z. Lai, J. Am. Chem. Soc., 2024, 146, 21989–21998 CrossRef PubMed.
- J. R. McCutcheon and M. S. Mauter, Science, 2023, 380, 242–244 CrossRef CAS PubMed.
- S. A. Chavan, W. Maes, L. E. M. Gevers, J. Wahlen, I. F. J. Vankelecom, P. A. Jacobs, W. Dehaen and D. E. De Vos, Chem. – Eur. J., 2005, 11, 6754–6762 CrossRef CAS PubMed.
- A. Krupková, K. Kubátová, L. Č. Šťastná, P. Cuřínová, M. Müllerová, J. Karban, J. Čermák and T. Strašák, Catalysts, 2021, 11, 1317 CrossRef.
- C. Matassa, D. Ormerod, U. T. Bornscheuer, M. Höhne and Y. Satyawali, Process Biochem., 2019, 80, 17–25 CrossRef CAS.
- D. Schoeps, V. Sashuk, K. Ebert and H. Plenio, Organometallics, 2009, 28, 3922–3927 CrossRef CAS.
- P. Kisszekelyi, R. Hardian, H. Vovusha, B. Chen, X. Zeng, U. Schwingenschlögl, J. Kupai and G. Szekely, ChemSusChem, 2020, 13, 3127–3136 CrossRef CAS PubMed.
- H.-t. Wong, C. J. Pink, F. C. Ferreira and A. G. Livingston, Green Chem., 2006, 8, 373–379 RSC.
- P. van der Gryp, A. Barnard, J.-P. Cronje, D. de Vlieger, S. Marx and H. C. M. Vosloo, J. Membr. Sci., 2010, 353, 70–77 CrossRef CAS.
- T. A. C. A. Bayrakdar, F. Nahra, O. Zugazua, L. Eykens, D. Ormerod and S. P. Nolan, Green Chem., 2020, 22, 2598–2604 RSC.
- Z. Wen, D. Pintossi, M. Nuño and T. Noël, Nat. Commun., 2022, 13, 6147 CrossRef CAS PubMed.
- S. Monticelli, L. Castoldi, I. Murgia, R. Senatore, E. Mazzeo, J. Wackerlig, E. Urban, T. Langer and V. Pace, Monatsh. Chem., 2017, 148, 37–48 CrossRef CAS PubMed.
- T. A. C. A. Bayrakdar, B. P. Maliszewski, F. Nahra, D. Ormerod and S. P. Nolan, ChemSusChem, 2021, 14, 3810–3814 CrossRef CAS PubMed.
- T. A. C. A. Bayrakdar, F. Nahra, D. Ormerod and S. P. Nolan, J. Chem. Technol. Biotechnol., 2021, 96, 3371–3377 CrossRef.
- J. G. Kettle, S. K. Bagal, S. Bickerton, M. S. Bodnarchuk, S. Boyd, J. Breed, R. J. Carbajo, D. J. Cassar, A. Chakraborty, S. Cosulich, I. Cumming, M. Davies, N. L. Davies, A. Eatherton, L. Evans, L. Feron, S. Fillery, E. S. Gleave, F. W. Goldberg, L. Hanson, S. Harlfinger, M. Howard, R. Howells, A. Jackson, P. Kemmitt, G. Lamont, S. Lamont, H. J. Lewis, L. Liu, M. J. Niedbala, C. Phillips, R. Polanski, P. Raubo, G. Robb, D. M. Robinson, S. Ross, M. G. Sanders, M. Tonge, R. Whiteley, S. Wilkinson, J. Yang and W. Zhang, J. Med. Chem., 2022, 65, 6940–6952 CrossRef CAS PubMed.
- R. Verbeke, I. Nulens, M. Thijs, M. Lenaerts, M. Bastin, C. Van Goethem, G. Koeckelberghs and I. F. J. Vankelecom, J. Membr. Sci., 2023, 677, 121595 CrossRef CAS.
- S. Zeidler, U. Kätzel and P. Kreis, J. Membr. Sci., 2013, 429, 295–303 CrossRef CAS.
- C. G. Lopresto, S. Darvishmanesh, A. Ehsanzadeh, A. Amelio, S. Mazinani, R. Ramazani, V. Calabrò and B. Van der Bruggen, Biofuels, Bioprod. Biorefin., 2017, 11, 307–324 CrossRef CAS.
- M. Tromp, J. R. A. Sietsma, J. A. van Bokhoven, G. P. F. van Strijdonck, R. J. van Haaren, A. M. J. van der Eerden, P. W. N. M. van Leeuwen and D. C. Koningsberger, Chem. Commun., 2003, 128–129 RSC.
- D. Nair, H.-T. Wong, S. Han, I. F. J. Vankelecom, L. S. White, A. G. Livingston and A. T. Boam, Org. Process Res. Dev., 2009, 13, 863–869 CrossRef CAS.
- J. M. Dreimann, M. Skiborowski, A. Behr and A. J. Vorholt, ChemCatChem, 2016, 8, 3330–3333 CrossRef CAS.
- D. F. Akl, D. Poier, S. C. D’Angelo, T. P. Araújo, V. Tulus, O. V. Safonova, S. Mitchell, R. Marti, G. Guillén-Gosálbez and J. Pérez-Ramírez, Green Chem., 2022, 24, 6879–6888 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc06334a |
| This journal is © The Royal Society of Chemistry 2025 |