
Tensile-strain-driven interstitial Ru doping structure on an FeCoP/FF electrode accelerates the reaction kinetics of water electrolysis†
Lu
Zhan
ab,
Yanru
Liu
 ac,
Guizhong
Zhou
*b,
Kang
Liu
ac,
Yunmei
Du
*ab and
Lei
Wang
ac
ac,
Guizhong
Zhou
*b,
Kang
Liu
ac,
Yunmei
Du
*ab and
Lei
Wang
ac
aKey Laboratory of Eco-chemical Engineering, Ministry of Education, International Science and Technology Cooperation Base of Eco-chemical Engineering and Green Manufacturing, Qingdao University of Science and Technology, Qingdao, People's Republic of China
bShandong Engineering Research Center for Marine Environment Corrosion and Safety Protection, College of Environment and Safety Engineering, Qingdao University of Science and Technology, Qingdao, People's Republic of China. E-mail: zhougz@126.com; duyunmeiqust@163.com
cCollege of Chemistry and Molecular Engineering, Qingdao University of Science and Technology, Key Laboratory of Optic-electric Sensing and Analytical Chemistry of Life Science, Taishan Scholar Advantage and Characteristic Discipline Team of Eco Chemical Process and Technology, Qingdao, People's Republic of China
First published on 7th February 2025
Abstract
Ru atoms with a large radius are often doped into a lattice as substituents due to reaction kinetics and thermodynamics limitations. Therefore, overcoming kinetic resistance to realize interstitial Ru doping in phosphides and establishing the internal relationship of its electrocatalytic performance are challenging. Considering that the tensile strain induced by a quenching-induced huge temperature difference can provide the possibility of interstitial doping, an interstitially Ru-doped FeCoP/FF electrode with 2.32% tensile strain was innovatively constructed by a strain-driven interstitial Ru doping strategy. As expected, the Ru-FeCoP/FF∥Ru-FeCoP/FF electrolyzer needs a cell voltage of only 1.64 V to deliver 1 A cm−2. Notably, the tensile strain and interstitially doped Ru synergistically promote the movement of electrons from Fe and Co sites to P and plummeting of the activation energy (Ea), thus accelerating the reaction kinetics of HER and OER. Overall, this work provides new ideas for designing an Ru interstitially doped electrode and optimizing the HER and OER kinetics.
Green foundation1. This work advances the field of green chemistry through electrocatalytic total water decomposition for hydrogen production and solar panel testing.2. This work developed two kinds of Ru gap doped OER and HER electrocatalyst, and the gas production effect was good under power supplied by solar panels. One of the electrocatalysts has been successfully written up and published in the Journal of Colloid and Interface Science. 3. First of all, the rate and output of hydrogen generation should be increased by improving the catalyst, and then the emission of CO2 should be reduced by controlling the temperature of phosphorization. |
1. Introduction
Electrochemical water splitting requires efficient and stable electrocatalysts that can significantly reduce the overpotential of the two half-reactions, namely the hydrogen evolution reaction (HER) on the cathode and the oxygen evolution reaction (OER) on the anode.1–7 Currently, to improve the conductivity and intrinsic activity of phosphide, a suitable proportion of precious metals is used for doping. Among them, as the most promising active material among noble metal materials, ruthenium (Ru) shows special advantages in electrochemical water splitting due to its unique electronic structure and strong hydrogen adsorption capacity.8–10 Therefore, the introduction of Ru dopant is of great significance for optimizing the structure and function of electrode materials.11 Currently, Ru dopants are mainly incorporated into the catalyst by solvothermal methods, electrodeposition, etching methods, and other. Moreover, due to the large atomic radius of Ru, it is more inclined to replace the main metal atoms in the lattice during the formation of the crystal, thereby optimizing the electrocatalytic reaction kinetics.12–15 Wang et al. pointed out that, when a single atom of Ru is doped into the 2H-MoS2 lattice by replacing an Mo atom, it results in more active centers and some vacancies, thereby improving the HER performance of Ru@2H-MoS2.16 Jang et al. found that Ru atoms substituted some of the Co and Fe atoms doped into the lattice gap of CoFeP nano-frames (RCFP/NF) to adjust the electronic structure, so that the energy band center of RCFP/NF moved down, and the adsorption free energy of the best adsorbate (OH*/H*) was adjusted.17 However, Ru doped into the lattice in the form of substitution may not be conducive to the stability of the crystal structure and the exposure of active sites due to its large atomic radius.18Interestingly, researchers have found that dopants can also be incorporated into the interstitial sites of the lattice, effectively regulating electrical conductivity, and reducing the d-band center.19–21 Notably, this unique interstitial doping can effectively avoid lattice damage and atomic overflow. In particular, non-metallic atoms (such as P, B, or C) are more advantageous in interstitial doping due to their smaller atomic radius. Song et al. successfully prepared P interstitially doped P-NiFe ANs-400 materials using a hydrothermal method under the action of low-temperature thermal reduction and atomic diffusion. The interstitially doped P atoms share the charge around Fe and active Ni sites and optimize the free energy of OER intermediates.22 Mao et al. introduced B into the lattice of Pt by a simple one-pot method. The interstitially doped B atom not only inhibits the formation of OH and O species at the active site but also prevents the oxidation of the indicated Pt atom.23 Zhang et al. doped C atoms into the lattice of PdMo diene by thermal decomposition. C doping can optimize the oxygen binding in the electrochemical process, reduce the operating potential for oxidation/dissolution of the metal, and improve the catalytic performance of PdMo diene in terms of activity and durability.24 However, unlike non-metallic atoms, transition metal atoms show great potential in further optimizing the structure and performance of catalysts due to their rich unfilled d-orbitals that can be used as electrophilic sites.25–27 Significantly, transition metal interstitial dopants have been shown to improve the structural stability and reversibility of related materials in the fields of photocatalysis and batteries. However, their role in the field of electrocatalysis has not yet been developed.28 Moreover, it is difficult for transition metal atoms with a large atomic radius (especially Ru) to spontaneously incorporate into tightly arranged lattice gaps during the doping process, due to the dual limitations of reaction kinetics and thermodynamics.29–31 Therefore, inducing the interstitial doping of Ru and determining the electrocatalytic mechanism of this unique form of doping are difficult and provide the focus for the current electrode design. External factors (doping, reaction temperature, electromagnetic field, etching, etc.) can induce lattice expansion to produce a tensile strain effect. This effect not only increases the lattice spacing but also regulates the coordination degree, d-band center, and valence electron configuration of the crystal.32 This provides a convenient way for the dopant to enter the lattice gap position. Hence, the use of the strain effect to drive metal atoms for interstitial doping shows important research potential.
Based on this, an interstitially Ru doped-FeCoP/FF electrode with 2.32% tensile strain was innovatively constructed by a strain-driven interstitial Ru doping strategy. Interestingly, this work cleverly utilizes the ‘thermal expansion and cold contraction’ mechanism of the lattice, caused by the huge temperature difference resulting from quenching, to prompt Fe2P and CoP crystals to produce a certain tensile strain. Stimulated by the stretched lattice, a small amount of Ru atoms is spontaneously embedded into the interstitial void of the phosphide crystal. Notably, density functional theory (DFT) and activation energy (Ea) tests reveal that the tensile strain and interstitially doped Ru synergistically promote the movement of electrons from Fe and Co sites to P and plummeting of the activation energy, thus accelerating the reaction kinetics of HER and OER. Moreover, an SCN− poisoning experiment further reveals that Ru dopants serve as the real HER active species. As expected, the well-designed Ru-FeCoP/FF requires only 0.158 V and 0.390 V to achieve an industrial-grade current density of 1 A cm−2 for HER and OER processes, respectively. Notably, the Ru-FeCoP/FF∥Ru-FeCoP/FF electrolyzer needs a cell voltage of only 1.64 V to yield a current density of 1 A cm−2 for total water decomposition, which is much better than that of the Pt/C-NF∥RuO2-NF benchmark electrolyzer. In summary, a strain-driven interstitial Ru doping strategy was proposed for the first time in the preparation of phosphide electrodes. Simultaneously, this work provides a new idea for constructing an Ru interstitially doped electrode and optimizing the HER and OER kinetics.
2. Experimental section
2.1. Chemicals
Ruthenium chloride (RuCl3·XH2O, 35–42%) and cobalt nitrate hexahydrate (Co (NO3)2·6H2O, 99%) were purchased from Aladdin Inc., China. Potassium hydroxide (KOH, 99%) and alcohol were obtained from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). Commercial Pt/C (20 wt%) was acquired from Johnson Matthey. All the chemicals were used without further purification.2.2 Synthesis of CoFe2O4/FF
Firstly, foamed iron (2 × 2 cm2, 1 mm) was ultrasonically cleaned in acetone and ethanol for 20 min to remove excess impurities. Then, the foamed iron was transferred to 55 mL of deionized water solution with 0.873 g of Co (NO3)2·6H2O, stirred for 30 min, and then stood at room temperature for 5 h. The target product was placed in an oven at 60 °C until it was dry. Finally, the intermediate sample CoFe2O4/FF was obtained.2.3 Synthesis of Ru-FeCoP/FF
The intermediate sample CoFe2O4/FF was phosphatized in a tube furnace at 350 °C for 2 h, and the sample was quickly taken out when the tube furnace had cooled to 200 °C. The sample was immersed in a pre-prepared 2 mg mL−1 iced RuCl3 solution (10 mg of RuCl3·XH2O was added to 5 mL of deionized water for ultrasonic mixing) for 30 min. The target product was washed with deionized water and placed in an oven at 60 °C until dry to obtain a sample of the final product, Ru-FeCoP/FF. (The iced RuCl3 solution in the experiment was treated with liquid nitrogen.)2.4 Synthesis of FeCoP/FF-Q
The intermediate sample CoFe2O4/FF was phosphatized in a tube furnace at 350 °C for 2 h, and the sample was quickly taken out when the tube furnace had cooled to 200 °C. The sample was immersed for 30 min in liquid nitrogen that had been prepared in advance. The target product was washed with deionized water and placed in an oven at 60 °C until it was dry to obtain the FeCoP/FF sample.2.5 Synthesis of FeCoP/FF-C
The intermediate sample CoFe2O4/FF was phosphated in a tube furnace at 350 °C for 2 h and naturally cooled to room temperature in a tube furnace. The sample was immersed for 30 min in liquid nitrogen that had been prepared in advance. The target product was washed with deionized water and placed in an oven at 60 °C until it was dry to obtain the FeCoP/FF sample.2.6 Synthesis of Ru-FeCoP/FF-X (20, 25, 35, 40)
The experimental method is similar to the synthesis method for Ru-FeCoP/FF. Ru-FeCoP/FF-X (20, 25, 35, 40) can be obtained by adjusting the immersion time in RuCl3 solution to x min (x = 20, 25, 35, 40). Other experimental steps remain unchanged.2.7 Materials characterizations
To characterize the morphology of the catalysts and analyze their structure, scanning electron microscopy (SEM) was conducted with a Zeiss Sigma 500. A transmission electron microscope (TEM; JEM-2100F) and high-resolution TEM (HRTEM) were used to further analyze the special morphology of Ru-FeCoP/FF and its comparison catalyst samples. Powder X-ray diffraction (XRD) was used to analyze phase composition and crystal structure with a Rigaku D-MAX 2500/PC, and the measurement was carried out under the operation of Cu NT radiation at a 10 min−1 scanning rate. X-ray photoelectron spectroscopy (XPS; Thermo ESCALAB 250Xi) was used to obtain the elemental composition, molecular structure, and chemical states of the sample surface.2.8 Electrochemical measurements
All electrochemical measurements were performed in an electrochemical workstation (Gamry Instruments Reference 2000), in an alkaline solution of 1.0 M KOH at room temperature. The HER made use of a conventionally simple system of three electrodes. The synthesized catalysts supported by carbon paper acted as the working electrode. The carbon paper was ultrasonicated with ethanol and deionized water three times before use. A carbon rod served as the counter electrode, and a mercuric oxide electrode (Hg/HgO) was used as the reference electrode. The Ru-FeCoP/FF electrode with a loading of 0.223 g of Ru-FeCoP powder, calculated from the mass difference of Fe foam before and after the reaction, was directly used as a working electrode for electrochemical (HER and OER) tests. In this work, all potentials measured were adjusted with reversible hydrogen electrodes (RHE), calculated with the relevant formula E(RHE) = E(Hg/HgO) + 0.098 V + 0.0592 × pH.All of the electrochemical properties were tested using linear sweep voltammograms (LSVs), and the LSV data curves were obtained at a sweep rate of 5 mV s−1. To test the rate of charge transfer of the catalysts, the electrochemical impedance spectroscopy (EIS) measurement was implemented in a frequency window ranging from 0.1 to 105 Hz, which applied a −1.219 V voltage to the HER tests. Cyclic voltammetry (CV) tests were measured in the non-faradaic section, and in this work, the scanning speeds of CV were set from 40 to 200 mV s−1 with a tolerance of 40 mV s−1.
3. Results and discussion
As shown in Fig. S1,† CoFe2O4 micro-flowers were grown on the FF substrate by a simple liquid-phase precipitation method. At the same time, Fig. S2† shows that the XRD diffraction peak of the CoFe2O4/FF precursor matches well with the CoFe2O4 phase, confirming the formation of the CoFe2O4 phase. Then, the FeCoP/FF-Q electrode was obtained by the phosphorization–liquid nitrogen quenching method. Fig. S3a–c† show that the FeCoP/FF-Q electrode possesses a micro-flower structure composed of nanosheets. As shown in Fig. S3d,† the crystal planes of CoP species on the FeCoP nanosheets indicate that the FeCoP/FF-Q electrode is composed of Fe2P and CoP phases. Interestingly, the lattice fringes belonging to Fe2P and CoP phases in the FeCoP/FF-Q electrode undergo significant expansion, indicating that quenching induces a certain strain in the electrode material. To further explore the specific strain parameters in FeCoP nanosheets induced by quenching treatment, the well-designed FeCoP/FF-Q electrode and the FeCoP/FF-C counterpart synthesized by natural cooling after phosphorization were tested by geometric phase analysis (GPA). As anticipated, the strain distribution (exy) diagram (Fig. 1a) of FeCoP/FF-Q shows that its lattice region is mainly red pixels, indicating that the huge temperature difference induced by liquid nitrogen quenching promotes the tensile strain of the FeCoP crystal. Additionally, the related strain histogram shows that FeCoP/FF-Q has a tensile strain with a strain coefficient of 2.05%. Unlike FeCoP/FF-Q, the FeCoP/FF-C counterpart with the same micro-flower structure (Fig. S4a and b†) shows no obvious strain in the lattice region due to the slow cooling process (Fig. 1b). Moreover, the above phenomenon confirms that quenching treatment can successfully introduce tensile strain into the phosphide lattice, laying the foundation for subsequent Ru interstitial doping. Finally, the well-designed Ru-FeCoP/FF electrode was obtained by impregnating FeCoP/FF-Q into Ru solution. Fig. 1c–e illustrate that Ru-FeCoP/FF still maintains a micro-flower structure composed of ultrathin nanosheets. Fig. S4c† shows that the FeCoP/FF electrode is a cyan–black sheet. Fig. 1f shows that there are obvious lattice fringes in the Ru-FeCoP nanosheets, belonging to the (110) crystal plane of Fe2P species, and the (011) and (112) crystal planes of CoP species, respectively, meaning that the Ru-FeCoP/FF electrode is composed of Fe2P and CoP phases. Notably, the crystal lattices of both Fe2P and CoP phases are significantly expanded due to the Ru doping. To confirm the doping position of Ru atoms, a high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) test was conducted.33–35 As shown in Fig. 1g and h, Ru dopants are successfully incorporated into the interstitial void of Fe2P and CoP lattices, confirming that the obvious tensile strain induced by quenching can indeed promote the interstitial doping of Ru atoms. Furthermore, the element mapping result (Fig. 1i) confirms the uniform distribution of Co, Fe, P, and Ru elements on Ru-FeCoP nanosheets. Simultaneously, Fig. S5† discloses that the content of Ru is only 1.51%. | ||
| Fig. 1 (a and b) GPA (geometric phase analysis) test of FeCoP/FF-Q and FeCoP/FF-C electrodes. (c and d) SEM images of Ru-FeCoP/FF. (e) TEM image of powders on the surface of the Ru-FeCoP/FF electrode. (f) HRTEM images of powders on the surface of the Ru-FeCoP/FF electrode. (g and h) HAADF-STEM images of powders on the surface of the Ru-FeCoP/FF electrode. (i) EDX elemental mappings of powders on the surface of the Ru-FeCoP/FF electrode. | ||
In addition, to further explore the effect of quenching treatment on the doping position of Ru in the electrode, the FeCoP/FF-C counterpart was immersed in Ru solution at room temperature for 15 min to obtain the Ru-FeCoP/FF-C comparison sample. As displayed in Fig. S6a–c,† Ru-FeCoP/FF-C still has the morphology of micro-flowers. Moreover, Fig. S6d† shows that Ru-FeCoP/FF-C has non-expanding lattice fringes belonging to the Fe2P and the CoP species, meaning that Ru atoms in Ru-FeCoP/FF-C are not incorporated into the lattice of Fe2P and CoP phases but adsorbed on the surface of phosphides. Interestingly, the GPA result of the well-constructed Ru-FeCoP/FF electrode shows that the Ru interstitially doped-FeCoP nanosheets still have a tensile strain of 2.32% (Fig. S7a†). The slightly increased tensile strain coefficient further reveals the existence of interstitial Ru dopants in FeCoP nanosheets. The GPA result of Ru-FeCoP/FF-C shows that Ru-FeCoP nanosheets still show no obvious strain (Fig. S7b†), further revealing that the Ru atoms in Ru-FeCoP/FF-C are difficult to incorporate into the lattice of Fe2P and CoP phases due to the lack of quenching treatment and affects the crystal structure of the phosphides. Simultaneously, Table S1† shows that the Ru content in the Ru-FeCoP nanosheets collected by the Ru-FeCoP/FF electrode is 1.82%, while the Ru content in the Ru-FeCoP-C nanosheets from the Ru-FeCoP-C electrode is only 0.51%, further suggesting that the tensile strain induced by quenching treatment can prompt more Ru atoms to be interstitially doped into the phosphide, and the lack of strain effect can only cause a small amount of Ru atoms to physically deposit on the surface of the phosphide.
The composition and electronic configuration of Ru-FeCoP/FF were further evaluated using X-ray diffraction (XRD) and X-ray photoelectron spectroscopy (XPS). As displayed in Fig. 2a, Ru-FeCoP/FF, FeCoP/FF-Q and Ru-FeCoP/FF-C show obvious diffraction peaks belonging to Fe2P (PDF#76-0089) and CoP (PDF#29-0497) phases. Notably, the diffraction peaks belonging to the phosphide in the Ru-FeCoP/FF-C electrode do not shift significantly compared with the standard cards of Fe2P and CoP, proving that there is no strain in the crystal structure of Fe2P or CoP in Ru-FeCoP/FF-C. The peaks belonging to phosphides in Ru-FeCoP/FF and FeCoP/FF-Q are shifted to the left compared with that in the Ru-FeCoP/FF-C electrode, again revealing that Ru atoms were doped into the lattice gap of Fe2P and CoP phases due to the tensile strain induced by the quenching treatment. Fig. 2b illustrates the signal peaks of Co, Fe, Ru, and P elements in Ru-FeCoP/FF, confirming the elemental composition of Ru-FeCoP/FF. The Co 2p spectrum (Fig. 2c) of Ru-FeCoP/FF displays peaks at 778.8 eV and 793.4 eV belonging to Co–P species, indicating the formation of the CoP phase. Other peaks at 781.7 eV and 797.7 eV, 787.2 and 803.2 eV are attributed to the Co–O bond and satellite peaks, respectively. As shown in Fig. 2d, the peaks at 711.8 eV and 724.8 eV in Ru-FeCoP/FF are assigned to Fe–P species, confirming the formation of the Fe2P phase. Moreover, the peaks of Ru-FeCoP/FF in Fig. 2e at 127.8 and 128.7 eV belong to P–M (M = Co, Fe, Ru) species, while the peaks at 132.4 and 133.7 eV belong to the P–O species. As shown in Fig. 2f, the Ru 3p spectrum of Ru-FeCoP/FF has two sets of peaks. The peaks at 462.9 eV and 486.1 eV belong to the Ru–P species, while the peaks at 466.5 eV and 488.1 eV belong to the Ru–O species. Significantly, the peaks belonging to the Co–P and Fe–P bonds in Ru-FeCoP/FF show a positive shift compared with those in the FeCoP/FF electrode, while the peak of P–M shows a negative move. This implies that Co and Fe in Ru-FeCoP/FF carried more positive charges, and P carried more electrons. In addition, compared with the reported Ru2P,36 the peak of the Ru–P bond in Ru-FeCoP/FF also shifts negatively, indicating that the interstitially doped Ru atoms carry some electrons. The above results confirm that the interstitial Ru dopants greatly optimize the electronic configuration of the Fe2P and CoP phases.
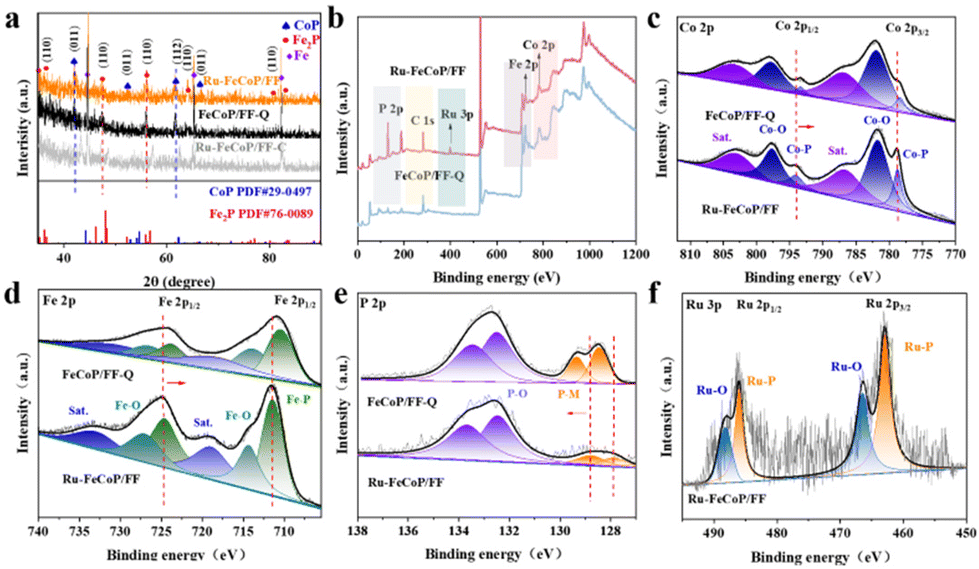 | ||
| Fig. 2 (a) XRD patterns of Ru-FeCoP/FF, FeCoP/FF-Q and Ru-FeCoP/FF-C electrodes. (b) XPS surveys of Ru-FeCoP/FF and FeCoP/FF-Q electrodes. High-resolution spectra of (c) Co 2p, (d) Fe 2p, (e) P 2p, (f) Ru 3p of Ru-FeCoP/FF and FeCoP/FF-Q electrodes. | ||
Notably, Co and Fe atoms with more positive charges are conducive to the formation of Co(Fe)–OOH species with high OER activity.
Ru with more electrons can also be more stable during the electrocatalytic process. Moreover, a stable Ru dopant with optimized H* adsorption ability promotes alkaline HER kinetics. Interestingly, compared with the Ru-FeCoP/FF-C counterpart (Fig. S8†), the peak belonging to the M–P (M = Co, Fe) species in Ru-FeCoP/FF has a positive shift. At the same time, the peaks belonging to the P–M and Ru–P species in Ru-FeCoP/FF shift negatively. The above phenomena reveal that interstitial Ru dopants and appropriate lattice strain can effectively optimize the electronic configuration of phosphides. Notably, the peaks belonging to the Ru-P species in the Ru-FeCoP/FF-C counterpart are significantly weakened in comparison with that in Ru-FeCoP/FF, further proving that Ru-FeCoP/FF-C contains only a small amount of Ru element due to the lack of tensile strain.
Based on the above characterization, the well-designed Ru-FeCoP/FF and other comparison electrodes were tested for alkaline HER at room temperature using a conventional three-electrode system.37–39 As shown in Fig. 3a, the Ru-FeCoP/FF electrode requires overpotentials of only 5 mV and 158 mV at 10 mA cm−2 and 1 A cm−2 for alkaline HER, much lower than for Ru-FeCoP/FF-C (17 mV, 213 mV) or FeCoP/FF (36 mV, 272 mV), implying that Ru-FeCoP/FF shows exceptional HER activity. Its excellent HER activity is mainly due to the tensile strain effect, interstitial Ru dopants with outstanding H* adsorption ability, and the optimized electron configuration. Moreover, the voltages of the Ru-FeCoP/FF electrode at 10 mA cm−2 and 1 A cm−2 are 0.1 and 0.78 times those of Pt/C-FF (46 mV, 201 mV), respectively, confirming that the Ru-FeCoP/FF electrode shows alkaline HER activity beyond that of the Pt-based electrode. Fig. 3b shows that the order of the Tafel slope is Ru-FeCoP/FF (36.2 mV dec−1) < Pt/C-FF (78.3 mV dec−1) < Ru-FeCoP/FF-C (81.6 mV dec−1) < FeCoP/FF-Q (96.4 mV dec−1), indicating that the Ru-FeCoP/FF electrode exhibits speedy HER kinetics. This is inseparable from the unique physical structure and electronic configuration of Ru-FeCoP/FF. The Ru-FeCoP/FF electrode has the smallest Rct value (0.74 Ω) among all the as-synthesized samples (Fig. S9a and Table S2†), which are lower than those of Ru-FeCoP/FF-C (1.44 Ω) or FeCoP/FF-Q (2.17 Ω), indicating that the Ru-FeCoP/FF electrode has the fastest charge transfer speed, stemming from the optimal electronic structure induced by the interstitial Ru dopants. As the ECSA value is proportional to the value of the double-layer capacitance (Cdl), a cyclic voltammetry (CV) test (Fig. S10†) was performed to obtain the corresponding Cdl value. As exhibited in Fig. 3c, the Cdl value of the Ru-FeCoP/FF electrode is as high as 82.09 mF cm−2, confirming that the Ru-FeCoP/FF electrode has a large ECSA, due to its unique interstitial Ru doping and distinct lattice tensile strain. Meanwhile, Fig. S11† shows that the Ru-FeCoP/FF electrode still maintains the morphology of micro-flower clusters after HER, meaning that the Ru-FeCoP/FF electrode exhibits excellent structural stability. As shown in from Fig. S12,† the Ru species in the Ru-FeCoP/FF electrode are still stable in the phosphide, as the charge redistribution promotes the interstitial Ru atoms to be in a more stable electron-rich state. However, the peaks of M–P (M = Ru, Co, Fe) species in the Ru-FeCoP/FF electrode did not change significantly, indicating that the chemical composition of the Ru-FeCoP/FF electrode was also very stable during the HER process. Besides HER performance, the OER test of Ru-FeCoP/FF and other counterparts was also conducted under the same conditions. As shown in Fig. 3d, the Ru-FeCoP/FF electrode requires overpotentials of only 160 mV, 290 mV, and 390 mV to reach current densities of 50 mA cm−2, 100 mA cm−2, and 1 A cm−2, respectively, which are 0.31, 0.53, and 0.51 times those of RuO2-FF (510 mV, 550 mV, 770 mV). This confirms that the Ru-FeCoP/FF electrode shows superior alkaline OER performance, mainly due to the charge redistribution of Fe and Co driven by the trace interstitial Ru doping. Fig. 3e shows that the Ru-FeCoP/FF electrode has an ultra-low Tafel slope (68.1 mV dec−1). The Ru-FeCoP/FF electrode also has the smallest Rct value (0.69 Ω), lower than those of Ru-FeCoP/FF-C (1.52 Ω) or FeCoP/FF-Q (2.33 Ω), revealing that the Ru-FeCoP/FF electrode shows fast charge transfer ability during the OER process (Fig. S9b and Table S2†). As shown in Fig. 3f and S13,† the Cdl value of the Ru-FeCoP/FF electrode for OER is as high as 17.09 mF cm−2, derived from its unique lattice tensile strain that promotes the exposure of more Fe and Co active species. Fig. S14† displays no significant change in the morphology of the Ru-FeCoP/FF electrode after OER. However, the XPS spectrum after OER (Fig. S15†) shows that the peaks belonging to P–M (M = Fe, Co, Ru) bonds in Ru-FeCoP/FF decreased significantly. In contrast, the peaks belonging to M–O (M = Fe and Co) bonds increased significantly, revealing the formation of M–OOH (M = Fe and Co) active phases with high OER activity. To prove the high utilization of Ru species by the Ru-FeCoP/FF catalyst, we performed an ICP test (Table S1†), and calculated the mass activity of Ru (or Pt) in Ru-FeCoP/FF, Ru-FeCoP/FF-C, Pt/C-FF and RuO2, respectively. Surprisingly, Fig. 3g displays that the well-designed Ru-FeCoP/FF catalyst shows ultra-high mass activity (594.8 mA cm−2 mg−1Ru for HER) at 50 mV, which is much higher than that of the Ru-FeCoP/FF-C counterpart (313.3 mA cm−2 mg−1Ru) or the Pt/C-FF catalyst (120.1 mA cm−2 mg−1Pt) for HER. The Ru-FeCoP/FF catalyst exhibits ultrahigh mass activity (601.7 mA cm−2 mg−1Ru for OER) at 350 mV, which is much higher than that of the Ru-FeCoP/FF-C catalyst (186.3 mA cm−2 mg−1Ru) or the Ru2O-FF catalyst (42.6 mA cm−2 mg−1Ru) for OER (Fig. 3h). The above phenomenon reveals that the utilization rate of Ru species in the well-constructed Ru-FeCoP/FF catalyst is much higher than that of Ru in Ru-FeCoP/FF-C or that of Pt in Pt/C-FF. This further confirms the high utilization of Ru species in Ru-FeCoP/FF. Fig. 3i shows that the current density of Ru-FeCoP/FF for HER and OER remains ∼49 mA cm−2 after 50 h. Fig. 3j shows that the alkaline HER performance of the Ru-FeCoP/FF electrode exceeded those of most recently reported noble-metal-based electrodes.
 | ||
| Fig. 3 (a) HER polarization curves and (b) HER corresponding Tafel slopes of Ru-FeCoP/FF, FeCoP/FF-Q, Ru-FeCoP/FF-C and Pt/C-FF for HER in 1.0 M KOH. (c) The Cdl values calculated according to the current density at different scan rates of HER. (d) OER polarization curves and (e) OER corresponding Tafel slopes of Ru-FeCoP/FF, FeCoP/FF-Q, Ru-FeCoP/FF-C and Pt/C-FF for HER in 1.0 M KOH. (f) The Cdl values calculated according to the current density at different scan rates of HER. (g) Mass activity of Ru-FeCoP/FF, Ru-FeCoP/FF-C and Pt/C-FF for HER in 1.0 M KOH. (h) Mass activity of Ru-FeCoP/FF, Ru-FeCoP/FF-C and Pt/C-FF for OER in 1.0 M KOH. (i) Chronoamperometry tests of Ru-FeCoP/FF for HER and OER. (j) Comparison of η10 and Tafel for HER with recently reported noble-metal-based electrocatalysts. (k and l) ECSA-normalized LSV curves of Ru-FeCoP/FF, Ru-FeCoP/FF-C, FeCoP/FF-Q for HER and OER. (m and n) a histogram showing the TOF for HER and OER of Ru-FeCoP/FF, Ru-FeCoP/FF-C, FeCoP/FF-Q. | ||
Moreover, we have listed the specific values of ECSA (Tables S4 and S5†) and plotted the ECSA-normalized LSV curve to confirm the intrinsic catalytic activity of Ru-FeCoP/FF. As shown in Fig. 3k and l, the voltage value of Ru-FeCoP/FF is 100 mV at a normalized ECSA current density of 0.1 mA cm−2, which is smaller than that of Ru-FeCoP/FF-C (169 mV) or FeCoP/FF-Q (197 mV), revealing that the Ru-FeCoP/FF catalyst shows exceptional intrinsic activity for HER. Moreover, the voltage value of Ru-FeCoP/FF is 340 mV at a normalized ECSA current density of 0.3 mA cm−2, which is smaller than that of Ru-FeCoP/FF-C (430 mV) or FeCoP/FF-Q (500 mV), revealing that the Ru-FeCoP/FF catalyst shows exceptional intrinsic activity for OER. To further evaluate the unit catalytic site activity of Ru-FeCoP/FF and comparison samples, the turnover frequency (TOF) value was calculated by a CV test with a scan rate of 100 mV s−1 in 1.0 M phosphate buffered saline (PBS). As all the as-prepared catalysts were loaded on carbon paper for performance testing, the CV test was also conducted on pure carbon paper, and it was deducted as the reaction background (Fig. S16a and S16c†). As expected, the TOF value of Ru-FeCoP/FF (0.91 s−1) at 100 mV is 2.8 and 13 times those of Ru-FeCoP/FF-C (0.32 s−1) and FeCoP/FF-Q (0.07 s−1), respectively, in HER (Fig. S16† and Fig. 3m). Moreover, the TOF value of Ru-FeCoP/FF (0.2 s−1) at 100 mV is 22 and 200 times those of Ru-FeCoP/FF-C (0.009 s−1) and FeCoP/FF-Q (0.001 s−1), respectively, in OER (Fig. S16† and Fig. 3n). This phenomenon proves that the active sites in the well-designed Ru-FeCoP/FF show ultra-high intrinsic activity.
To further explore the effect of interstitial Ru dopants on the charge density distribution at the heterointerface, the Fe2P/CoP model with 2.05% tensile strain, and the Ru-Fe2P/Ru-CoP model with interstitial Ru dopants and 2.32% tensile strain were established. Fig. 4a illustrates the difference in charge density at the Fe2P/CoP interface. The cyan region indicates charge accumulation, while the yellow region indicates charge depletion. The local charge density at the Fe2P/CoP interface is significantly increased, indicating that there is strong electronic interaction at the interface. For the Fe2P/CoP interface, the electrons around the Fe and Co atoms are transferred to the adjacent P sites, resulting in charge accumulation around the P atoms. As shown in Fig. 4b, the charge aggregation region at the Ru-Fe2P/Ru-CoP interface increases compared with the Fe2P-CoP interface, indicating that the introduction of Ru promotes a shift in the electron cloud from Fe and Co atoms to P atoms. The above results further indicate that the obvious charge redistribution is stimulated by the interstitial Ru atoms, consistent with the XPS results in Fig. 2. To further explore the effect of thermodynamic temperature on the intrinsic activity of Ru-FeCoP/FF, FeCoP/FF, and Ru-FeCoP/FF-C, alkaline HER and OER tests were carried out in the temperature range 298–328 K.40 As displayed in Fig. S17–20,† the electrocatalytic performance of Ru-FeCoP/FF, FeCoP/FF, and Ru-FeCoP/FF-C increases with an increase in the test temperature, indicating that an increase in thermodynamic temperature can accelerate the kinetic process of HER and OER. The activation energy (Ea) values of HER and OER are obtained by fitting the slope of the ‘log (j0) ∼ 1000/T′ curves (Fig. 4c and d), directly reflecting the intrinsic activity of HER and OER. Fig. 4e show that the Ea value of Ru-FeCoP/FF for HER is only 32.7 kJ mol−1, lower than those of FeCoP/FF-Q (53.1 kJ mol−1) or Ru-FeCoP/FF-C (40.2 kJ mol−1). For OER, the Ea value of Ru-FeCoP/FF is 35.8 kJ mol−1, lower than those of FeCoP/FF-Q (70.8 kJ mol−1) or Ru-FeCoP/FF-C (43.1 kJ mol−1). The above phenomena fundamentally reveal that the unique interstitial Ru doping and distinct lattice tensile strain can effectively promote a reduction in the HER and OER energy barrier, thereby promoting an improvement in their reaction kinetics. In addition, to reveal the important role of interstitial Ru dopant in Ru-FeCoP/FF in the HER process, a poisoning experiment with SCN− was conducted in this work. Subsequently, SCN−-poisoning experiments were also carried out to determine the role of Ru sites in Ru-FeCoP/FF during the alkaline HER process. After adding 9.6 g of KSCN to the 1 M KOH electrolyte, Fig. 4f shows that the HER performance of Ru-FeCoP/FF deteriorates significantly after 3 h, indicating that SCN− leads to significant degradation in HER performance by poisoning Ru sites in Ru-FeCoP/FF. This phenomenon confirms that the interstitial Ru dopant induced by the tensile strain effect is the real active species for alkaline HER.
 | ||
| Fig. 4 Atomic models with charge density difference plot of (a) FeCoP/FF-Q (Fe2P/CoP model), (b) Ru-CoP/FF (Ru-Fe2P/Ru-FeCoP model). (c and d) Arrhenius linear curves of FeCoP/FF-Q, Ru-FeCoP/FF and Ru-FeCoP/FF-C. (e) Histogram of Ea value for HER and OER. (f) LSV curves of Ru-FeCoP/FF and Ru-FeCoP/FF in KSCN solution. (g) Calculated free energy diagrams of the HER pathway for the Ru–Co dual site, Ru site, and Co site in the Ru-Fe2P/Ru-CoP model. (h and i) Free energy diagrams of the OER pathway for the Co site and Fe site in the Ru-Fe2P/Ru-CoP and Fe2P/CoP models. | ||
To more accurately reveal the catalytic mechanism and reaction kinetics of Ru-FeCoP/FF, this work was combined with DFT calculations to conduct an in-depth exploration. To further reveal the electrocatalytic mechanism of Ru-FeCoP/FF, we established the DFT model of Ru-Fe2P/Ru-CoP (Fig. 4a) and examined the electrocatalytic path for alkaline HER. It is known that alkaline HER has multiple reaction processes, mainly including the adsorption and dissociation of water molecules, the adsorption of H*, and the desorption of hydrogen. Considering the complex reaction steps of alkaline HER, the Gibbs free energy of the Ru site, Co site, and Ru–Co dual sites (i.e., Ru is responsible for adsorbing/dissociating water molecules, and Co is responsible for adsorbing/dissociating H* intermediates) in the Ru-Fe2P/Ru-CoP model were calculated. As displayed in Fig. 4g, the Gibbs free energy of adsorbed water for the Ru site (0.45 eV) is much higher than that for the Co site, indicating that the Ru site is the optimal site for adsorbing water molecules. Interestingly, the rate-determining steps of the Co site, Ru site and Ru–Co dual sites are all H2 desorption steps during the alkaline HER process. Notably, the energy barrier of the Ru–Co dual sites for the rate-determining step is −0.36 eV, which is much lower than those of the Ru site (−0.55 eV) or the Co site (−0.46 eV). Therefore, the Ru-Fe2P/Ru-CoP model follows the Ru–Co dual active sites mechanism during the alkaline HER process. Specifically, water molecules preferentially adsorb on the Ru site, and then the dissociated H* intermediate migrates to the adjacent Co site and releases H2 at the Co site. In addition, the OER process was also simulated in this work. The OER process involves four main processes, namely OH* → *OH, *OH → *O, *O → *OOH and *OOH → O2. As shown in Fig. 4h and i, the rate-determining step (RDS) of the Ru-Fe2P/Ru-CoP and Fe2P/CoP models in the OER process is the *OH → *O process. Obviously, the RDS barriers of Fe and Co sites in the original Fe2P/CoP model are 2.19 eV and 1.61 eV, respectively. Notably, the RDS barrier of the Fe and Co sites near the Ru dopants in the well-designed Ru-Fe2P/Ru-CoP model are 0.88 eV and 1.12 eV, respectively. The above results confirm that Fe atoms near the Ru dopants act as a real OER active site to accelerate oxygen evolution. Furthermore, the RDS barriers of Fe and Co sites in the Ru-Fe2P/Ru-CoP model are much lower than those of Fe and Co sites in the Fe2P/CoP model, indicating that the introduction of Ru reduces the RDS barrier of the Fe (Co) site and stimulates the intrinsic activity of the OER active site. Surprisingly, the Fe active site in the interstitially doped Ru-Fe2P/Ru-CoP model has a much lower RDS barrier than that of Fe in the Fe2P/CoP model, revealing that the interstitial Ru dopant can greatly optimize the OER reaction kinetics. In summary, the tensile-strain-driven interstitial Ru doping has an important influence on the reaction kinetics of HER and OER of phosphides. The interstitial Ru dopant is used as the HER active site, and the Ru-bonded Fe (or Co) atom is used as the high-efficiency OER active site.
Given the excellent alkaline HER and OER performance and durability of the Ru-FeCoP/FF electrode, this work further assembled an Ru-FeCoP/FF electrolytic cell for an overall water splitting (OWS) test. As shown in Fig. 5a, the Ru-FeCoP/FF electrodes require only 1.52 V and 1.64 V to achieve current densities of 100 mA cm−2 and 1 A cm−2. This is due to the Fe and Co atoms with more positive charges acting as OER active sites, and the interstitial Ru dopants with more electrons serving as HER active sites, synergistically expediting the HER and OER kinetics. Fig. 5b and Table S6† show that the voltage (E100) of the Ru-FeCoP/FF electrode at a current density of 100 mA cm−2 is better than those of most reported bifunctional electrodes. Notably, to further study the OWS efficiency, the Faraday efficiency (FE) was measured (Fig. 5c). As shown in Fig. 5d, the amount of H2 and O2 produced at a constant current density of 100 mA cm−2 is in good agreement with theoretical calculations. The formation rates of H2 and O2 are about 0.02 mmol min−1 and 0.01 mmol min−1, respectively. The ratio of H2 to O2 is close to 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, indicating that water is completely decomposed into hydrogen and oxygen. Surprisingly, the Faraday efficiencies of HER and OER are close to 99.8% and 99.4%, respectively. In addition, Fig. 5e demonstrates the remarkable stability of Ru-FeCoP/FF for OWS. Notably, under power supplied by solar panels, comparison images for gas production by the Ru-FeCoP/FF electrode and the pure FF electrode in the same test environment (Fig. 5f) confirm that the gas production effect of the well-designed Ru-FeCoP/FF electrode is ideal. These results indicate that Ru-FeCoP/FF shows ultra-high electrocatalytic efficiency and good application prospects.
:1, indicating that water is completely decomposed into hydrogen and oxygen. Surprisingly, the Faraday efficiencies of HER and OER are close to 99.8% and 99.4%, respectively. In addition, Fig. 5e demonstrates the remarkable stability of Ru-FeCoP/FF for OWS. Notably, under power supplied by solar panels, comparison images for gas production by the Ru-FeCoP/FF electrode and the pure FF electrode in the same test environment (Fig. 5f) confirm that the gas production effect of the well-designed Ru-FeCoP/FF electrode is ideal. These results indicate that Ru-FeCoP/FF shows ultra-high electrocatalytic efficiency and good application prospects.
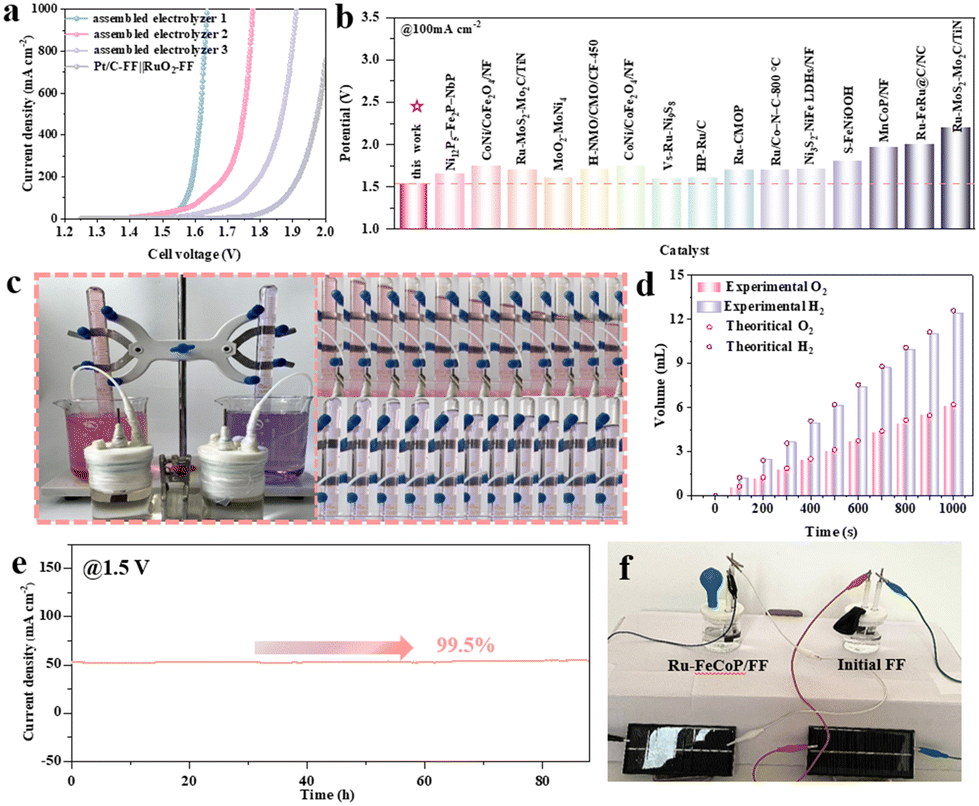 | ||
| Fig. 5 Overall water splitting tests in 1.0 M KOH. (a) LSV of Ru-FeCoP/FF∥Ru-FeCoP/FF (assembled electrolyzer 1), Ru-FeCoP/FF-C∥Ru-FeCoP/FF-C (assembled electrolyzer 2), FeCoP/FF-Q∥FeCoP/FF-Q (assembled electrolyzer 3), and RuO2-NF∥Pt/C-FF with iR-compensation. (b) Comparison of η100 for OWS with recently reported noble-metal-based electrocatalysts, (c) The amount of H2 and O2 varying with time in 1.0 M KOH and photographs of H2 and O2 collected at different time points. (d) The gas collection device for overall water splitting. (e) Stability tests of Ru-FeCoP/FF∥Ru-FeCoP/FF. (f) Comparison of gas production images with different solutions for power supplied by solar panels. | ||
In addition, to explore the effect of Ru soaking time after the phosphorization–liquid nitrogen quenching on the structure and properties of phosphides, the Ru soaking time (20 min, 25 min, 35 min, and 40 min) were changed to obtain Ru-FeCoP/FF-20, Ru-FeCoP/FF-25, Ru-FeCoP/FF-35 and Ru-FeCoP/FF-40. As shown in Fig. S21,† the Ru-FeCoP/FF-20 electrode has a smooth micro-flower structure. However, the Ru-FeCoP/FF-25 electrode (Fig. S23†) still has a micro-flower structure but with a rough surface. However, due to Ru soaking for a long time, the thickness of the nanosheets in Ru-FeCoP/FF-25 is greater than that in Ru-FeCoP/FF-20. As shown in Fig. S25 and S27,† too long an Ru soaking time leads to serious agglomeration of nanosheets in the Ru-FeCoP/FF-35 and Ru-FeCoP/FF-40 electrodes, which is not conducive to the exposure of active sites. Therefore, the Ru soaking time has an important regulatory effect on the physical structure of the phosphide electrode. Fig. S22, 24, 26, and 28† show that Ru-FeCoP/FF-20, Ru-FeCoP/FF-25, Ru-FeCoP/FF-35, and Ru-FeCoP/FF-40 display peaks belonging to CoP and Fe2P phases, indicating that the four comparison electrodes are composed of CoP and Fe2P phases. Moreover, the XRD pattern peaks of the CoP and Fe2P phases in Ru-FeCoP/FF-20, Ru-FeCoP/FF-25, Ru-FeCoP/FF-35, and Ru-FeCoP/FF-40 all shift to the left, confirming that the tensile strain is always present in all electrodes. Furthermore, the corresponding ICP test (Table S1†) reveals that the doping amount of Ru in the phosphide increases with extension of Ru soaking time. In addition, the HER and OER performances of Ru-FeCoP/FF-20, Ru-FeCoP/FF-25, Ru-FeCoP/FF-35, and Ru-FeCoP/FF-40 were also measured. Fig. S29a† shows that for the current density of Ru-FeCoP/FF-20, Ru-FeCoP/FF-25, and Ru-FeCoP/FF-35 to reach 1 A cm−2 for HER requires overpotentials of 223, 211, 243, and 178 mV, respectively, much higher than that of the Ru-FeCoP/FF electrode (158 mV). Similarly, Fig. S29b† shows that the OER overpotentials of Ru-FeCoP/FF-20, Ru-FeCoP/FF-25, Ru-FeCoP/FF-35, and Ru-FeCoP/FF-40 are 550, 490, 430, and 620 mV at a current density of 1 A cm−2, respectively, larger than that of Ru-FeCoP/FF (390 mV). The above phenomena confirm that the appropriate doping of Ru plays an important role in optimizing the performance of HER and OER. Fig. S29c and d† show that Ru-FeCoP/FF-20, Ru-FeCoP/FF-25, Ru-FeCoP/FF-35, and Ru-FeCoP/FF-40 have higher Tafel slopes for HER and OER, indicating that these four comparison samples have slower HER and OER reaction kinetics than Ru-FeCoP/FF. Fig. S30 and Table S3† show that the Rct values of HER and OER for Ru-FeCoP/FF-20 (1.18 Ω, 1.07 Ω), Ru-FeCoP/FF-25 (1.36 Ω, 1.29 Ω), Ru-FeCoP/FF-35 (2.08 Ω, 1.89 Ω) and Ru-FeCoP/FF-40 (4.68 Ω, 2.11 Ω) are higher than those of the Ru-FeCoP/FF electrode. As shown in Fig. S29e, f, S31 and 32,† the Cdl values of the Ru-FeCoP/FF-20, Ru-FeCoP/FF-25, Ru-FeCoP/FF-35 and Ru-FeCoP/FF-40 electrodes for HER and OER are lower than those of Ru-FeCoP/FF, due to the Ru soaking time greatly affecting their physical structure and the doping content of Ru in the electrode. In addition, Fig. S33† shows that Ru-FeCoP/FF-20, Ru-FeCoP/FF-25, Ru-FeCoP/FF-35, and Ru-FeCoP/FF-40 need cell voltages of 1.69, 1.75, 1.85, 1.91 V, respectively, to achieve a current density of 1 A cm−2, lower than that of Ru-FeCoP/FF (1.64 V). Therefore, the interstitial doping of trace Ru in the phosphide can be achieved under the premise of maintaining the morphology by using an appropriate Ru soaking time after the phosphorization–liquid nitrogen quenching treatment, leading to an enhancement in Ru utilization.
4. Conclusions
This work creatively developed a strain-driven interstitial Ru doping strategy to synthesize an interstitially Ru doped-FeCoP/FF electrode with 2.32% tensile strain. Significantly, we cleverly utilized the ‘thermal expansion and cold contraction’ mechanism of the lattice caused by the huge temperature difference from quenching to promote Fe2P and CoP crystals to produce a certain tensile strain. Stimulated by the stretched lattice, a small amount of Ru atoms is spontaneously embedded into the interstitial void of the phosphide crystal. As expected, the well-designed Ru-FeCoP/FF electrode requires voltages of only 0.158 V and 0.390 V to achieve 1 A cm−2 for HER and OER, respectively. Moreover, the Ru-FeCoP/FF∥Ru-FeCoP/FF electrolyzer needs a cell voltage of only 1.64 V to yield 1 A cm−2 for OWS, which is much better than that of Pt/C-FF∥RuO2-FF. Various characterizations reveal that the superior electrocatalytic activity and stability of Ru-FeCoP/FF mainly came from the tensile-strain-driven interstitial Ru doping, effectively promoting charge reset of the Ru-FeCoP/FF electrode. Notably, Co and Fe atoms with more positive charges are conducive to the formation of Co(Fe)–OOH species with high OER activity. Ru with more electrons can also be more stable during the electrocatalytic process. Moreover, a stable Ru dopant with optimized H* adsorption ability promotes alkaline HER kinetics. In addition, the tensile strain and interstitial Ru doping also greatly optimize the Ea values of HER and OER, thereby reducing the reaction energy barrier. In summary, this work provides new ideas for designing an Ru interstitially doped electrode with optimized HER and OER kinetics.Author contributions
Lu Zhan: investigation, methodology, software, formal analysis, validation, data curation, writing – original draft. Yanru Liu: validation. Guizhong Zhou: validation. Kang Liu: visualization. Yunmei Du: conceptualization, supervision. Lei Wang: investigation, formal analysis, resources. All authors discussed the results and commented on the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (52072197, 52302274), the 111 Project of China (Grant No. D20017), Natural Science Foundation of Shandong Province, China (ZR2022QE098), and Major Basic Research Program of Natural Science Foundation of Shandong Province under Grant (ZR2020ZD09), Youth Innovation Team of Colleges and Universities in Shandong Province (2023KJ313), Postdoctoral Innovation Project of Shandong Province (SDCX-ZG-20220307), Double-Hundred Talent Plan of Shandong Province (WST2020003). The authors would like to thank Fei Zhang from SCI-GO (https://www.sci-go.com) for the DFT calculation.References
- J. Luo, H. Han, X. Wang, X. Qiu, B. Liu, Y. Lai, X. Chen, R. Zhong, L. Wang and C. Wang, Appl. Catal., B, 2023, 328, 122495–122507 CrossRef CAS.
- Y. X. Zeng, J. Z. Xu, Y. Wang, S. Li, D. Y. Luan and X. W. Lou, Angew. Chem., 2022, 134, e202212031 CrossRef.
- X. Xu, C. Ye, D. Chao, B. Chen, H. Li, C. Tang, X. Zhong and S.-Z. Qiao, Adv. Mater., 2022, 34, 2108688 CrossRef CAS PubMed.
- M. Cao, K. Liu, Y. Song, C. Ma, Y. Lin, H. Li, K. Chen, J. Fu, H. Li, J. Luo, Y. Zhang, X. Zheng, J. Hu, M. Liu and J. Regulating, Energy Chem., 2022, 72, 125 CrossRef CAS.
- Q. Hong, Y. Wang, R. Wang, Z. Chen, H. Yang, K. Yu, Y. Liu, H. Huang, Z. Kang and P. W. Menezes, Small, 2023, 2206723 CrossRef CAS PubMed.
- Y. Lu, L. Zhou and S. Wang, Nano Res., 2023, 16, 1890–1912 CrossRef CAS.
- C. Li, H. Jang, S. Liu, M. G. Kim, L. Hou, X. Liu and J. Cho, Adv. Energy Mater., 2022, 12, 2200029 CrossRef CAS.
- H. Y. Gong, X. Liang, G. L. Sun, D. W. Li, X. J. Zheng, H. Shi, K. Zeng, G. C. Xu, Y. Li, R. Z. Yang and C. Z. Yuan, Rare Met., 2022, 41, 4034 CrossRef CAS.
- J. Wang, Y. Song, C. Zuo, R. Li, Y. Zhou, Y. Zhang and B. Wu, J. Colloid Interface Sci., 2022, 625, 722–733 CrossRef CAS PubMed.
- C. Tian, R. Liu and Y. Zhang, Ru-doped functional porous materials for electrocatalytic water splitting, Nano Res., 2024, 17, 982–1002 CrossRef CAS.
- M. Kuang, Y. Wang, W. Fang, H. Tan, M. Chen, J. Yao, C. Liu, J. Xu, K. Zhou and Q. Yan, Adv. Mater., 2020, 32, 2002189 CrossRef CAS PubMed.
- D. Y. Li, L. L. Liao, H. Q. Zhou, Y. Zhao, F. M. Cai, J. S. Zeng, F. Liu, H. Wu, D. S. Tang and F. Yu, Mater. Today Phys., 2021, 16, 100314 CrossRef CAS.
- S. Han, Y. Ma, Q. Yun, A.-L. Wang, Q. Zhu, H. Zhang, C. He, J. Xia, X. Meng, L. Gao, W. Cao and Q. Lu, Adv. Funct. Mater., 2022, 32, 2208760 CrossRef CAS.
- W. Wei, F. Guo, C. Wang, L. Wang, Z. Sheng, X. Wu, B. Cai and A. Eychmüller, Small, 2024, 20, 2310603 CrossRef CAS PubMed.
- C. Feng, M. Lv, J. Shao, H. Wu, W. Zhou, S. Qi, C. Deng, X. Chai, H. Yang, Q. Hu and C. He, Adv. Mater., 2023, 352305598 Search PubMed.
- J. Wang, W. Fang, Y. Hu, Y. Zhang, J. Dang, Y. Wu, B. Chen, H. Zhao and Z. Li, Appl. Catal., B, 2021, 341, 298120490 Search PubMed.
- K. Jang, H. Yoon, J. Hyoung, D. Pratama, C. Lee and D. Kim, Appl. Catal., B, 2024, 341, 123327 CrossRef CAS.
- X. Li, Z. Niu, M. Niu, J. Wang, D. Cao and X. Zeng, Small, 2024, 2311335 CrossRef CAS PubMed.
- H. Y. Gong, X. Liang, G. L. Sun, D. W. Li, X. J. Zheng, H. Shi, K. Zeng, G. C. Xu, Y. Li, R. Z. Yang and C. Z. Yuan, Rare Met., 2022, 41, 4034 CrossRef CAS.
- M. Ramadoss, Y. Chen, X. Chen, Z. Su, M. Karpuraranjith, D. Yang, M. A. Pandit and K. Muralidharan, J. Phys. Chem. C, 2021, 125, 20972–20979 CrossRef CAS.
- Y. Yang, R. Zou, J. Gan, Y. Wei, Z. Chen, X. Li, S. Admassie, Y. Liu and X. Peng, Green Chem., 2023, 25, 4104–4112 RSC.
- M. Song, D. Zhang, F. Miao, Y. Shi, N. Wang, L. R. Zhan and P. L. Wang, Appl. Catal., B, 2024, 342, 123376 CrossRef.
- Z. Mao, C. Liu, X. Zhang, Q. Qin, X. Li, H. Yang, F. Li, Q. Zhang, X. Zhang, J. Cai and W. Bin, ACS Catal., 2022, 128848–128856 Search PubMed.
- K. Zhang, Y. Guo, R. Wang, W. Zhan, Q. Li, R. He, T. Wu, C. Jin and M. He, ACS Energy Lett., 2022, 7, 3329–3336 CrossRef CAS.
- Q. Mao, S. Jiao, K. Ren, S. Wang, Y. Xu, Z. Wang, X. Li, L. Wang and H. Wang, Chem. Eng. J., 2021, 426, 131227 CrossRef CAS.
- P. Su, W. Fu, Z. Hu, J. Jing and M. Zhou, Appl. Catal., B, 2022, 313, 121457 CrossRef CAS.
- B. Gao, X. Du, Y. Zhao, W. Chen, S. Ding, C. Xiao, Z. Song and H. Jang, Chem. Eng. J., 2022, 433, 133768 CrossRef CAS.
- I. Kwon, I. Kwak, S. Ju, S. Kang, S. Han, Y. Park and J. Park, ACS Nano, 2020, 14, 12184–12194 CrossRef CAS PubMed.
- C. Fan, Z. Zang and X. Zhang, Int. J. Hydrogen Energy, 2024, 56, 1273–1283 CrossRef CAS.
- Y. Zhou, J. Zhang, H. Ren, Y. Pan, Y. Yan, F. Sun, X. Wang, S. Wang and J. Zhang, Appl. Catal., B, 2020, 268, 118467 CrossRef CAS.
- X. Lin, L. Chen and X. Zhong, Adv. Compos. Hybrid Mater., 2023, 6, 79 CrossRef CAS.
- Y. T. Yan, J. H. Lin, K. K. Huang, X. H. Zeng, L. Qiao, S. D. Liu, J. Cao, J. S. C. Jun, Y. S. Yamauchi and J. L. Qi, J. Am. Chem. Soc., 2023, 145, 24218–24229 CrossRef CAS PubMed.
- N. Zhou, L. Yu, R. Liu, Y. Fan, Q. Han and M. Chai, EcoEnergy, 2023, 1, 12 CrossRef.
- Y. Gu, L. Nie, J. Liu, Y. Yang, L. Zhao, Z. Lv, Q. Zhan and P. Lai, EcoEnergy, 2023, 1, 9 CrossRef.
- A. Kerner, V. Cohen, Z. Xu, R. Kirmani, Y. Park, P. Harvey, P. Murphy, C. Cawthorn, C. Giebink, M. Luther, K. Zhu, J. Berry, L. Kronik and P. Rand, Adv. Mater., 2023, 35, 2302206 CrossRef PubMed.
- Q. Yu, W. Yu, Y. Wang, J. He, Y. Chen, H. Yuan, R. Liu, J. Wang, S. Liu, J. Yu, H. Liu and W. Zhou, Small, 2023, 19, 2208045 CrossRef CAS PubMed.
- V. H. Hoa, M. Austeria, H. T. Dao, M. Mai and D. H. Kim, Appl. Catal., B, 2023, 327, 122467 CrossRef CAS.
- K. Jang, H. Yoon, S. Hyoung, S. Pratama, C. Lee and W. Kim, Appl. Catal., B, 2024, 341, 123327 CrossRef CAS.
- X. Mu, X. Gu, S. Dai, J. Chen, Y. Cui, Q. Chen, M. Yu, C. Chen, S. Liu and S. Mu, Energy Environ. Sci., 2022, 15, 4048–4057 RSC.
- Y. Du, W. Wang, H. Zhao, X. Jiang, Y. Liu, R. Chen, B. Yang and L. Wang, ChemCatChem, 2021, 13, 1362 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4gc06286h |
| This journal is © The Royal Society of Chemistry 2025 |