
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMorphological and mechanical characterization of high-strength sulfur composites prepared with variably-sized lignocellulose particles†
Moira K.
Lauer
a,
Zoe E.
Sanders
b,
Ashlyn D.
Smith
ab and
Rhett C.
Smith
 *a
*a
aDepartment of Chemistry, Clemson University, Clemson, South Carolina 29634, USA. E-mail: rhett@clemson.edu
bDepartment of Chemistry and Biology, Anderson University, Anderson, South Carolina 29621, USA
First published on 29th September 2021
Abstract
The extent to which lignocellulose biomass particle size influences the properties of biomass–sulfur composites prepared from these particles was evaluated. For this purpose several materials were prepared by the reaction of sulfur with peanut shell particles that had been fractionated into narrow size ranges using ASTM certified sieves. Eight particle size fractions with an upper cutoff range of 710 microns were thus used to prepare a series of eight composites PSS-1 to PSS-8. The use of biomass particles having defined size ranges allowed for a 36-fold faster reaction rate relative to the analogous reactions employing unfractionated biomass, while the resultant composites still maintained excellent strength characteristics. Composites prepared with smaller biomass particles exhibited the most uniform dispersion, yet similar ultimate strength characteristics were observed for most of the composites irrespective of the biomass particle size. The strength characteristics of these materials could be rationalized by the interplay of the dispersion of filler in the network versus the unfavorable interactions between the hydrophilic biomass filler and hydrophobic sulfur network. This work highlights the importance of quantifying filler effect for microscopically non-homogeneous composite materials and provides insight on simple strategies for drastically impacting the time and energy expenditures for biomass composite synthesis and resultant properties.
Introduction
Developing mechanically-improved and more environmentally compatible structural materials is imperative to advance society beyond the petrochemical era. Concrete, comprised largely of ordinary portland cement (OPC), is the most widely-utilized construction material worldwide, with current production levels of over four and a half billion metric tons per year.1 The production of concrete accounts for about 30% of global materials utilization and is responsible for approximately 8% of anthropogenic CO2 production.1 The production of CO2 in the manufacture of concrete is unavoidable as the formation of its key intermediate, cement clinker, is made by heating mined carbonate minerals to >1400 °C to form their respective oxides, a process that produces stoichiometric quantities of CO2. Cement production has approximately doubled in the last two decades and with an ever-increasing population and the rapid modernization of less developed countries, global concrete production is forecast to continue to increase.2 A key component of efforts to reduce CO2 emissions must be to decrease the amount of OPC used in structural applications by adopting new formulations that use less environmentally-deleterious components.3–6 Although significant progress has been made towards this aim, many improved cement formulations still produce large amounts of CO2 while ultimately generating a non-recyclable product with a limited operational lifetime. Furthermore, many recent efforts have focused on filler-type additives that, while lowering the amount of new clinker that must be made, often do not impart attributes to their composite concretes that would improve their eventual recyclability, a property essential for a circular lifecycle.7–10Our group has recently reported on various composite materials that have strength properties on par with or better than cement. These new composites are generally easily recycled, acid-resistant, and hydrophobic—characteristics that are possible due to the materials’ high sulfur contents.11–33 These high sulfur content materials (HSMs) are prepared using waste sulfur that is produced in megaton quantities as a byproduct of fossil fuel refining.34,35 Mechanistically, such HSMs are predominantly prepared by inverse vulcanization.36–41 Inverse vulcanization is the crosslinking of olefin-containing organic species in concert with thermally-induced homologation of excess elemental sulfur (Scheme 1). Inverse vulcanization can be up to 100% atom economical and proceeds at significantly lower temperatures (∼180 °C) than that required for the production of cement clinker. The sulfur catenates in HSMs can homologate when the material is reheated, allowing the material to flow for facile remolding, pouring and paving. The HSMs can thus be reprocessed over many cycles with negligible changes in mechanical properties. Beyond structural applications, HSMs also have utility in lithium–sulfur batteries,42–50 as IR transparent lenses,51–57 and as adsorbents in environmental remediation.58–64 As the potential application scope of this class of materials continues to expand, important research on improved processing, recycling and alternative synthetic avenues to HSMs have been developed, even expanding the substrate scope to previously inaccessible organic monomers.39,65–72
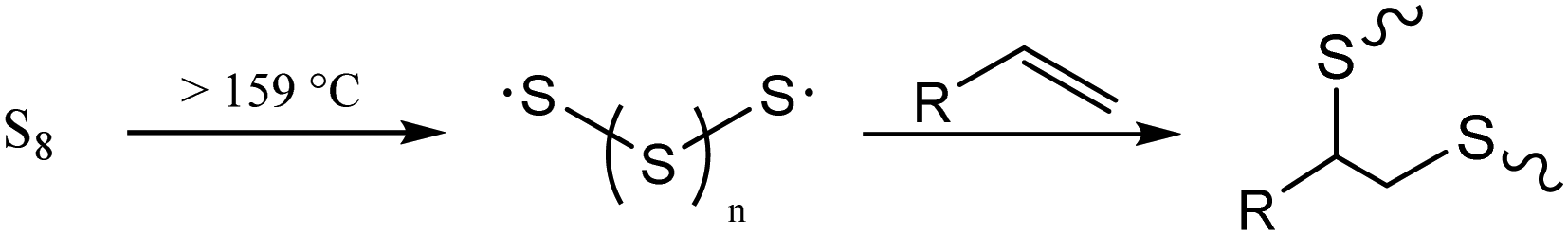 | ||
| Scheme 1 Thermal homologation of S8 and its subsequent reaction with an olefin. When the wt% of sulfur is greater than that of the organic species, the process is known as inverse vulcanization. | ||
Ideally HSMs will be as sustainably-produced as possible. To meet this goal the organic component to be reacted with waste sulfur must be renewably-sourced and preferably carbon negative to minimize or eliminate contributions to CO2 emission. Towards this end, researchers have demonstrated successful HSM production by reacting sulfur with fatty acids,25,31,33 terpenoids,19,21,57,73–75 starch,18,24 lignin,12,17,28,29 cellulose,13,19 and lignocellulosic biomass derivatives.16,17 Some of those biologically-derived materials do not possess the olefin functionalities needed for inverse vulcanization and therefore require derivatization or use of alternative reaction strategies. Derivatization, however, can detract from the atom economy and add to the economic/energetic cost of the process. Derivatizing biopolymers is also fraught with challenges due to biopolymer agglomeration or hornification during derivatization steps.76–86 Our previous work showed that if raw biomass is used as a precursor for the preparation of HSMs, the organic material must be mechanically milled to improve sulfur-organic surface area of contact and thus improve homogeneous dispersion of the material into resulting composites. Though lignocellulosic biomass is an especially attractive starting material, biomass faces a litany of well-known challenges to their commercial applications in competition with established petrochemical- and mineral-based materials. As a chemical feedstock, biomass can suffer from compositional changes depending on the source—even for the same species of origin—particle size/shape effects, moisture content variation, and inhomogeneity of samples in terms of the percent composition of various plant components (stems, leaves, seed hulls, etc.) and thus variable chemical composition. Our previous work with peanut shell composites, for example, revealed that the composites’ properties were strongly dependent on the peanut oil content of the peanut shell material employed in their preparation.16,17
The issues discussed above for biomass utilization necessitate assessing (1) the extent to which lignocellulose composite mechanical properties depend on particle size and (2) whether the chemical composition of the lignocellulosic feedstock varies on the basis of particle size due to the difference in grindability of divergent botanical structures of which the biomass is comprised. Chemical composition and particle morphology drastically impact particle dispersion and mechanical properties of composites prepared from cellulose nanocrystals, suggesting that these characteristics would also be important in lignocellulose–sulfur composites.87–90 Towards this aim, we have selected a model system of finely ground peanut shells as the biomass feedstock for the current study. Peanuts hold a fascinating place in the historical development of sustainable agriculture and chemistry dating to George Washington Carver's pioneering work on the use of peanuts as a nutrient-restoring plant for use in crop rotation-based land stewardship strategies.91–93 Later, peanut and soybean oil were some of the earliest feedstocks studied for preparation of what would later be called plastics, even predating the petrochemical plastics boom of the twentieth century. Peanut-based food products for human consumption are made primarily from the cotyledon (kernel) alone (Fig. 1), after it has been separated from the seed coat and shell components. Animal bedding and feed products can be made from some of the peanut shell waste, but the amount of shell waste available outpaces demand for these products, and smaller particle size fractions of peanut shells are not suitable for use in these applications. As a result, large quantities of bulk shell waste and small particle-size waste remain to be valorised. Peanuts remain a high production crop with annual production of about 44 Mt, of which over 11 Mt is in the form of shells that are not nutritionally valuable for human consumption.94 Peanut shells are primarily comprised of cellulose (52%) and lignin (35%) with ∼1 wt% being peanut oil.
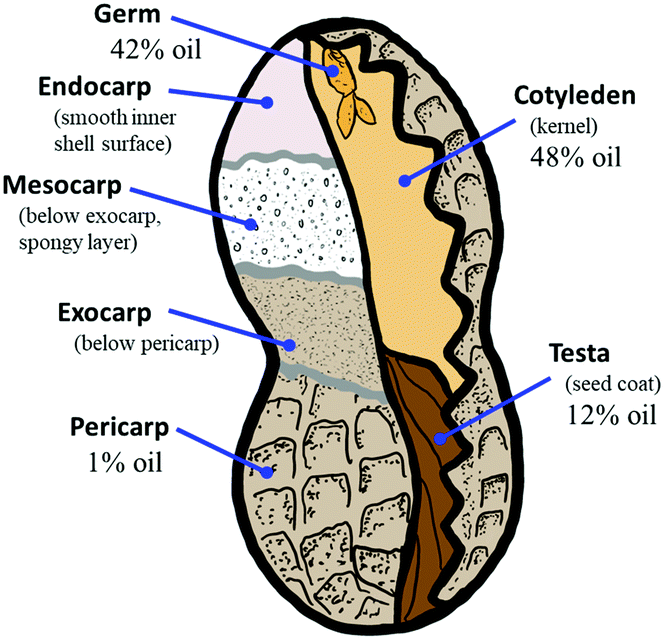 | ||
| Fig. 1 Botanical breakdown of a typical two-seed peanut legume with oil content for peanut-oil-containing components. Artist's rendering based on information accessed 4-24-21 at www.aboutpeanuts.com/peanut-facts/97-the-parts-and-composition-of-the-peanut. | ||
In previous work, we demonstrated several strategies for preparing composites from peanut shell and sulfur waste. The properties of these composites are competitive with traditional structural materials like OPC, suggesting their possible commercial applicability. Those studies demonstrated that high strength materials are possible due to the presence of a small amount of olefin-containing peanut oil (∼1% by weight) in the peanut shells, but only one particle size was used. The extent to which mechanical properties may also be attributable to a filler effect that depends on particle size of the lignocellulosic precursor thus remains unknown.95–102 Additionally, the material referred to as peanut shell waste can be further subdivided as being comprised of several botanical structures having different oil contents and therefore different potentials for crosslinking by inverse vulcanization (Fig. 1). It is conceivable that differences in the grindability of these botanical structures might influence their distribution by particle size in ground peanut shell materials. Herein, peanut shells were fractionated into eight different size ranges with an upper cutoff of 710 μm (Fig. 1 and Table 1) and the oil (olefin) content of each fraction was evaluated by 1H NMR spectrometric analysis. Each fraction was reacted with elemental sulfur to produce eight HSMs (PSS1–8). The extent to which particle size influences resultant composite mechanical properties were assessed by mechanical strength analysis, while morphology and thermal behavior were assessed by, differential scanning calorimetry (DSC), scanning electron microscopy element mapping by energy-dispersive X-ray analysis (SEM-EDX), and thermogravimetric analysis (TGA).
| Fraction code | Minimum size cutoff (μm)/ASTM size | Maximum size cutoff (μm)/ASTM size |
|---|---|---|
| a All particles passing the ASTM 200 sieve were retained in this sample. | ||
| PS-1 | 500/35 | 710/25 |
| PS-2 | 300/50 | 500/35 |
| PS-3 | 250/60 | 300/50 |
| PS-4 | 212/70 | 250/60 |
| PS-5 | 150/100 | 212/70 |
| PS-6 | 125/120 | 150/100 |
| PS-7 | 75/200 | 125/120 |
| PS-8 | NAa | 75/200 |
Results and discussion
Characterization and fractionation of biomass feedstock
The initial peanut shell material used for the current study was a finely-ground material (Fig. 2, unfractionated) from a facility that produces animal feed products from peanut shells. Animal feed production can only utilize a fraction of total peanut shells production and after processed into animal feed, there is a fraction remaining that is not usable for animal feed products due to the small particle size. This small particle size is, however, especially amenable to the target process of converting the shell residue into homogeneous composites. The industrial peanut shell product was therefore further fractionated using ASTM standard sieves beginning by removing particles larger than 710 μm, a portion that represented a negligible fraction of the total mass. The fractionation process was continued by collecting the material that would not pass through the 500 μm sieve to yield material with particles having dimensions of 500–710 μm (fraction PS-1). Sequentially smaller size ASTM sieves were employed according to the workflow provided in Table 1 to yield seven additional fractions from PS-2 through the smallest particle size fraction PS-8 (Fig. S1, ESI†). Optical micrographs of PS-X fractions (Fig. 2) demonstrate the range of sizes resulting from the fractionation process as well as revealing the irregular particle shapes present in the materials. | ||
| Fig. 2 Optical micrographs demonstrating the range of particle sizes (A) and irregular particle shapes (B) in peanut shell fractions PS-X. | ||
Due to inhomogeneity and small sample sizes analysed in DSC runs (∼5 mg), the magnitude and position of Tg values varied slightly for duplicate analysis of each PS-X fraction. The position of the Tg attributable to cellulose varied, on average, by 7 °C while the Tg values attributable to the lignin fraction of the peanut shell varied, on average, by 6 °C (Fig. S2–S8, ESI†). Unsurprisingly, all fractions of peanut shells had nearly indistinguishable infrared (IR) spectra (Fig. S9, ESI†) but somewhat different TGA results. All peanut shell fractions show an initial mass loss of ∼9 wt% due to adsorbed moisture in the range 25–120 °C (Fig. 3A). Due to increased surface area, smaller peanut shell particles began to decompose at slightly lower temperatures and much more rapidly than larger particle sizes while also burning more completely to give a lower char yield (i.e., less residue at 800 °C, Fig. 3B). Notable differences in thermal stability are observed only at temperatures in excess of 300 °C, as obviated in the derivative TGA inset shown in Fig. 3C. At the composite preparation temperature of 180 °C, there should thus be no particle size-dependant thermal degradation that could influence composite properties.
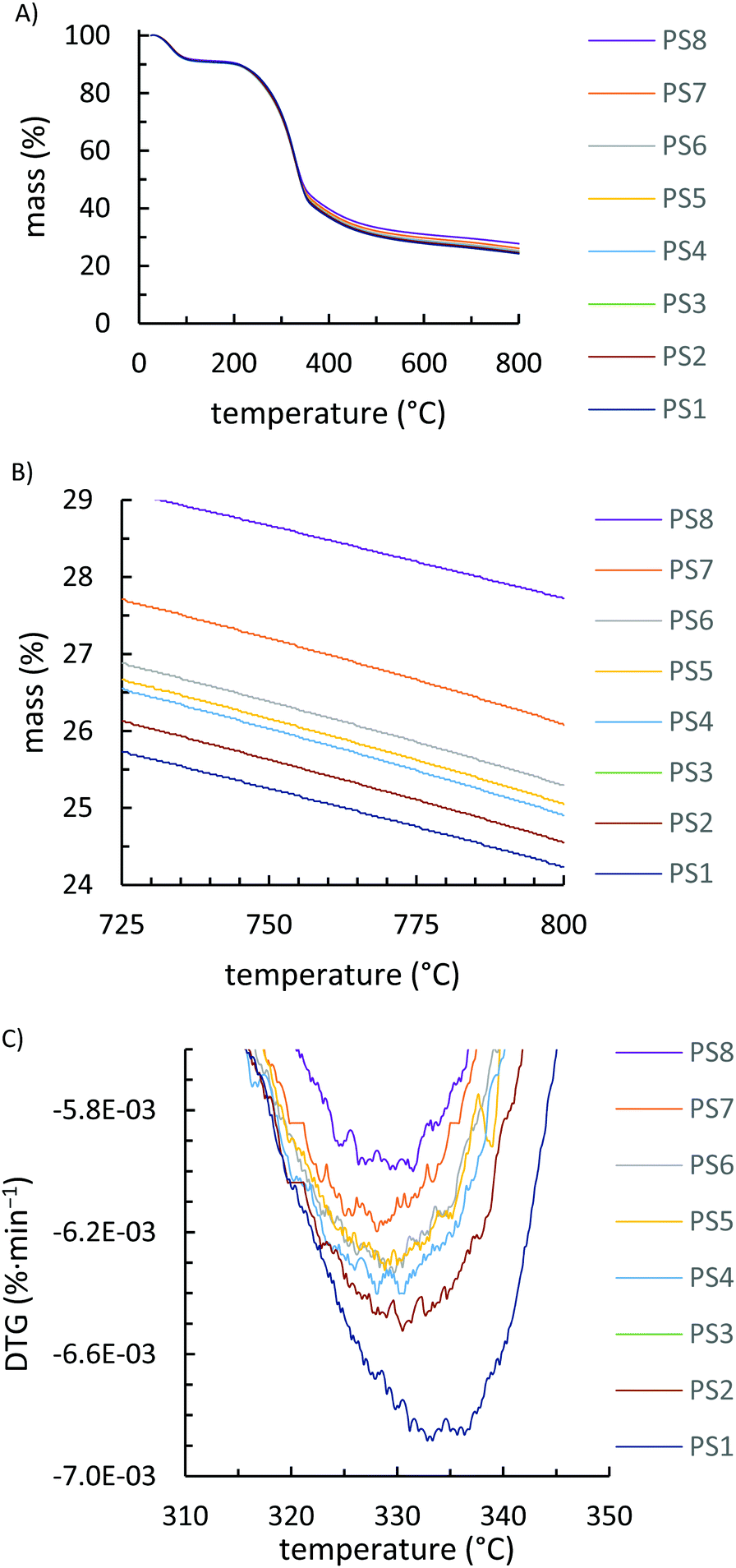 | ||
| Fig. 3 Thermogravimetric analysis of PS-X fractions from 25–800 °C (A), from 725–800 °C to emphasize the difference in residual mass (B), and the derivative TGA (DTG) curve over the primary decomposition step, emphasizing the more rapid and lower-temperature decomposition with smaller particle size (C). Note that PS-2 and PS-3 are coincident. | ||
Composite synthesis and chemical characterization
Each PS-X fraction (10 wt%) was mixed with sulfur (90 wt%) and the mixture was then heated at 180 °C while stirring until the mixture became visibly homogeneous to prepare eight inversely vulcanized materials PSS-X, where X corresponds to number of PS-X fraction used for the composite's preparation (Table 1). The feed composition of 10 wt% biomass and 90 wt% sulfur was selected to match that used to prepare previously-reported PS90, which was prepared from the unfractionated form of the same peanut shells used for the current study. PS90 also possessed compressional strength characteristics on par with that required for residential concrete construction and so represents a benchmark for comparing properties to composites prepared from fractionated peanut shells.While unfractionated peanut shells required three days of reaction to reach homogeneity in the preparation of PS90, fractionated shells were found to homogenize significantly more quickly during reaction, requiring only 2 h total reaction time. Although all eight of the reaction mixtures became homogenous at the reaction temperature, the two materials that were prepared with the largest particle-size fractions (PSS-1 and PSS-2) phase separated on cooling (Fig. S10, ESI†). Only homogeneous materials PSS-3 through PSS-8 will be further discussed. Cylinders of PSS-3–8 suitable for compressional analysis were prepared by remelting and pouring the molten material into silicone moulds (Fig. 4). In order to better understand the impact of filler morphology on ultimate composite properties, two control materials were prepared (Fig. 4B and C). The first control material was prepared to attempt to understand the impact of the peanut shells as filler on material properties. This was done by preparing a material containing no biomass and instead containing only sulfur and peanut oil in the same ratios as would be found in a material containing 10% peanut shell in which 1% of that mass was attributable to peanut oil. The second control material sought out to understand the impact of the peanut oil's incorporation into the lignocellulosic biomass as well as the impact of lignin–cellulose crosslinks. This material was consequently prepared with the same amount of peanut oil in the first control material but also containing a mixture of microcrystalline cellulose and alkali lignin in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio (the approximate ratio found in the peanut shell) as 10 wt% of the feed ratio. Both of these materials would not homogenize and showed drastic phase separation upon cooling in addition to a light-yellow colour indicative of (non-inversely vulcanized) orthorhombic sulfur. The inability for these materials to homogenize emphasizes the importance of the native biomass structure in facilitating the inverse vulcanization reaction while uniformly dispersing the filler.
:2 ratio (the approximate ratio found in the peanut shell) as 10 wt% of the feed ratio. Both of these materials would not homogenize and showed drastic phase separation upon cooling in addition to a light-yellow colour indicative of (non-inversely vulcanized) orthorhombic sulfur. The inability for these materials to homogenize emphasizes the importance of the native biomass structure in facilitating the inverse vulcanization reaction while uniformly dispersing the filler.
 | ||
| Fig. 4 Photos of compression test cylinders (A) and identically-prepared control materials made by reaction of sulfur and peanut oil (B) or sulfur, peanut oil, lignin, and cellulose (C) in the same ratios present in peanut shells used to prepare PSS-3–8. Control materials are heterogeneous and within minutes of cooling begin to rapidly re-develop the lighter yellow colour typical of orthorhombic sulfur, while the brown-orange colour of PSS-X composites persists for over 16 days (aging time before analysis herein). | ||
Because metastable polymeric sulfur has the potential to slowly revert to S8 over time when present in a crosslinked network, all materials were cured at room temperature for sixteen days before any mechanical, thermal, or morphological analysis was performed to ensure the probing of end-use structure and properties.13,16,17,103
HSMs can vary in the extent to which monomer feed sulfur is incorporated into their structures. Elemental analysis revealed that this holds for PSS3–8 as well, with the composition of each materials containing somewhat less than 90% of sulfur (∼81–88 wt%, Fig. 5A). A plot of particle size versus percent incorporation of sulfur revealed a definite trend in sulfur incorporation as a function of particle size, with composites prepared from the smallest and largest particles incorporating the most sulfur (86–88 wt%) and the intermediary composite PSS-5 incorporating the least sulfur (∼81 wt%, Fig. 4). To better understand this trend, extraction of composites PSS-X was undertaken utilizing CS2. Orthorhombic sulfur—sulfur that is not covalently linked to peanut shell molecules—is quite soluble in CS2, while peanut shell and covalently-attached sulfur is completely insoluble (as confirmed by elemental microanalysis of extracts). Fractionation of the material with CS2 paired with elemental analysis results therefore allows for the determination of how much of the sulfur present in each material is free or covalently bound to peanut shells. The fraction of each material that was soluble in CS2 varied predictably with the amount of sulfur that was incorporated into the material and, with the exception of PSS-8, the trend in total incorporated sulfur tracked with the amount of sulfur covalently bound (Fig. 5B). Materials PSS-4, PSS-5, and PSS-6 were effectively unable to crosslink with sulfur resulting in the largest discrepancy between feed ratio and the amount of incorporated sulfur whereas PSS-3, PSS-7, and PSS-8 were able to crosslink some amount of sulfur resulting in good agreement between the feed ratio and amount of sulfur incorporated into the material. The outlier, PSS-8 was able to distribute peanut shell particles most effectively through the sulfur network (from SEM analysis, vide infra), likely due to the low viscosity of the melt resulting from the small particle size. These data indicate that homogenization of organic species with few crosslinkable sites may be benefited by using as small a particle size as possible to provide good dispersion in the material.
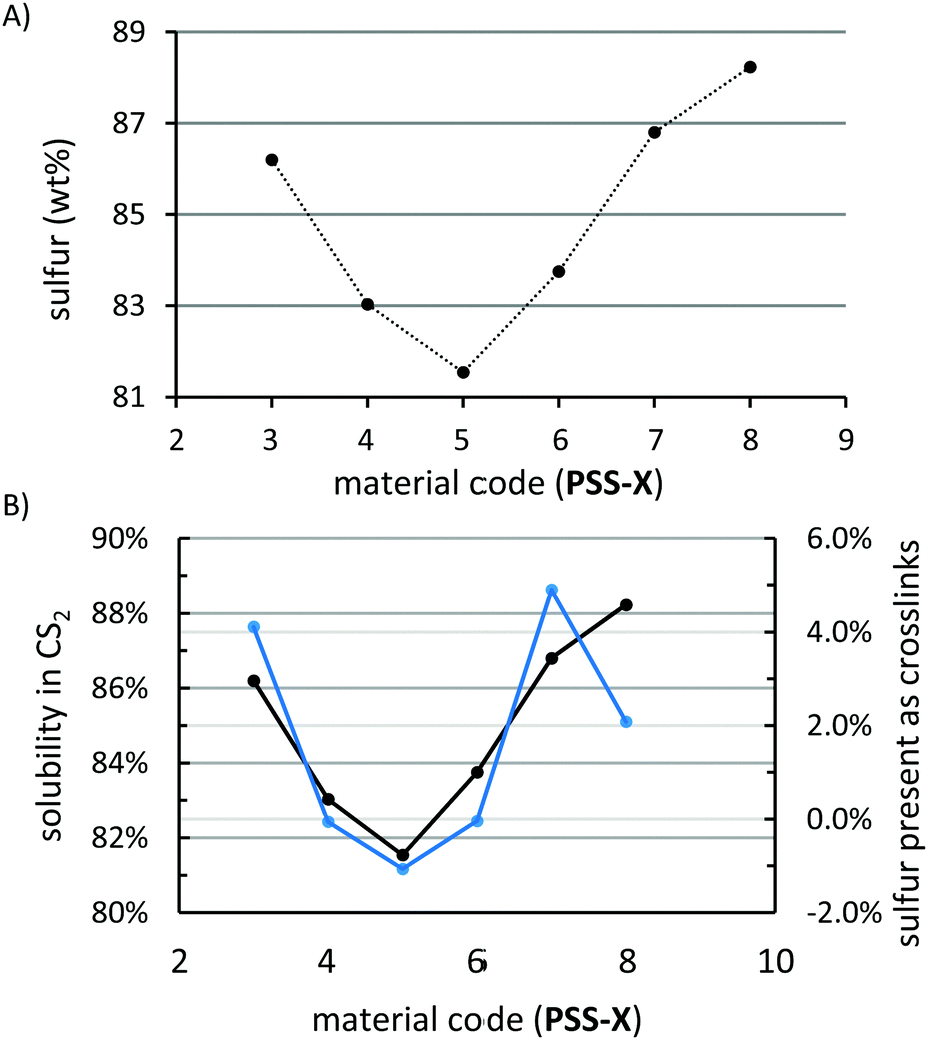 | ||
| Fig. 5 Chemical characterization of PSS-X materials showing the wt% of the material that was determined to be sulfur by elemental microanalysis (A), the amount of orthorhombic sulfur that was determined to be present in the material by CS2 fractionation studies (B, left axis, black trace) and the amount of sulfur that was determined to be present in the composite as crosslinked sulfur (the difference between the amount of sulfur present and the amount of orthorhombic sulfur identified) (B, right axis, blue trace). | ||
Morphological characteristics of biomass–sulfur composites
To gain further insight into the morphology of the materials, the composites were examined by scanning electron microscopy element mapping by energy-dispersive X-ray analysis (SEM-EDX, Fig. 6). Whereas composites prepared from smaller particle-size biomass showed homogeneous distribution of organic material throughout the HSM (i.e., PSS-7 and PSS-8, Fig. 6I–L), organic particle agglomeration became more pronounced for the materials prepared with larger particle size precursors, as evident from the images of PSS-3 and PSS-4 (Fig. 6A–D). Such trends in particle dispersion as a function of their size is a common phenomenon in composite formation that can often be traced to differences in viscosity.104 In the current case, the increased melt viscosity is likewise evident when pouring molten samples of composites with larger particle size precursors. | ||
| Fig. 6 SEM-EDX images of PSS-X showing the EM image (black and white: A, C, E, G, I and K) and the carbon element mapping (blue images: B, D, F, H, J and L) for PSS-3 (A and B), PSS-4 (C and D), PSS-5 (E and F), PSS-6 (G and H), PSS-7 (I and J), and PSS-8 (K and L). | ||
Thermal analysis revealed little in the differences between composites. TGA analysis of PSS-X showed a similar trend as observed for PS-X but on a smaller magnitude commensurate with the lower quantity of organics in the composites (Fig. S11, ESI†). Results obtained by DSC showed nearly identical curves for all of the materials except that PSS-3 showed a consistent (triplicate analysis) cold crystallization exotherm at ∼20 °C (Fig. S12–S17, ESI†). This can be attributed to the poor dispersion of peanut shells in this material producing a more rigid network in local areas, hindering crystallization on cooling, thus resulting in partial recrystallization upon reheating. The materials also showed a linear trend in crystallization temperature with the exception of PSS-4 that showed a surprisingly high crystallization temperature consistent with the lack of crosslinking observed (see also Table S3 and Fig. S18, ESI†).
Composite mechanical strength and correlation with botanical composition/fat content
Compressive strength measurements were conducted in an attempt to find a correlation between strength and particle size. Previously reported PS90, prepared from 90 wt% sulfur and unfractionated peanut shells, exhibited an impressive compressive strength of 21.3 ± 1.2 MPa.16,17,103 A higher average compressive strength was observed for all of the PSS-X composites that were prepared from fractions of the same peanut shells, but the compressive strength was less reproducible from sample-to-sample for PSS-X compared to PS90, as reflected in the larger standard deviations shown for these data in Table 2 (all measurements were done in triplicate). Unfortunately, even with replicate analysis, most materials had such a large standard deviation that the materials could not be differentiated in terms of compressional strength as a function of particle size.| Material | Strengtha (MPa) | n | wt% of hexane soluble in corresponding PS-X (%) |
|---|---|---|---|
| a Material ultimate strength under a compressional force. b The number of replicates performed to obtain the strength and standard deviation. c The material prepared in our previous manuscript from unfractionated peanut shells and heated for three days. | |||
| PSS-3 | 29.2 ± 6.3 | 4 | 1.10 |
| PSS-4 | 30.6 ± 0.9 | 3 | 0.00 |
| PSS-5 | 28.0 ± 3.2 | 4 | 0.50 |
| PSS-6 | 30.3 ± 10.2 | 5 | 0.04 |
| PSS-7 | 24.3 ± 4.4 | 4 | 0.54 |
| PSS-8 | 25.4 ± 10.6 | 8 | 0.05 |
| PS90 | 21.3 ± 1.2 | 3 | 1.60 |
The effect of fillers on a material's mechanical properties can generally be described by two primary considerations: (1) the compatibilization between the filler and the matrix, and (2) the dispersion of the filler in the matrix.101,104 For the PSS-X series of materials, the compatibility is not favourable due to the highly hydrophilic nature of the peanut shells juxtaposed with the hydrophobic nature of sulfur. On this basis, maximizing surface area (i.e., using smaller particle size filler) should be unfavourable for the development of strength properties. This is in contrast to the trend seen with particle dispersion where better dispersion was generally observed with smaller particle size. These two counterbalancing trends are likely responsible for the relatively similar strength properties across the series.
Based on the results from CS2 extractions, it may have been anticipated that PSS-3, PSS-7, and PSS-8 would have the greatest strength due to the peanut shell not only acting as a filler but also crosslinking with sulfur. Such a supposition is also borne out in strength trends of particle-free HSMs, wherein mechanical strength generally increases with increased covalent cross-linking. Such a trend was not observed in the case of composites PSS-X, however. The strengths of materials on either extreme of particle size were not as reproducible as are strengths of particle-free HSMs. The underlying cause for this behaviour in PSS-3 was traced to significant agglomeration of organic material observed in the SEM images. In contrast, the dispersion of particles in PSS-8 was much higher, though unfavourable sulfur-biomass interface effects are maximized among members of the series, so that PSS-8 also shows diminished strength. The other materials exhibited some extent of sulfur crosslinking, and consequently samples PSS-7 had a relatively reproducible strength yet provided a relatively low average compressional strength value. The reproducibility can be attributed to the uniform dispersion of organic particles through the sulfur matrix—the best seen throughout the series—providing consistent samples for analysis. The source of low strength can again be rationalized by the high surface area of the very small particles.
The materials that exhibited very little sulfur crosslinking—PSS-4, PSS-5, and PSS-6—produced surprisingly high average strength values. In these cases, the peanut shells are primarily acting as a filler with low contribution to a crosslinked network to stabilize polymeric sulfur. In this morphological regime, larger particle size produces the most reproducible results indicating minimization of surface area was more beneficial than the uniform dispersion of peanut shell.
The property variability and potential relationship to the complex interplay of surface area and size effects motivated us to quantify the amount of peanut oil in each of the peanut shell fractions. By stirring the peanut shells in hexane overnight, the peanut oil was extracted from the shell fractions, isolated, and quantified by 1H NMR spectrometry with an internal standard (2,3,4,5,6-pentafluorobenzaldehyde). This fractionation process resulted in removal of peanut oil from the shells (Table 2). The amount of oil extractable from the peanut shell samples varied across the series. Unfortunately, the presence of unidentified compounds other than peanut oil that produced resonances in the NMR spectrum made it difficult to make any conclusive deductions from these data for most of the samples (Fig. S19 and S20, ESI†). The 1H NMR data did, however, help to explain the wide variability of PSS-6 sample compressive strengths. In agreement with the CS2-extraction study that indicated that PSS-6 crosslinked a negligible amount of sulfur, PSS-6 also contained trivial quantities of peanut oil or any other hexane-soluble species. These data suggest that PSS-6 features the poorest compatibility/covalent crosslinking between peanut shell particles and sulfur among all of the composites studied. All materials possessed considerably less hexane soluble material than unfractionated peanut shell, a result that made practical sense but seemed counterintuitive considering the PSS-X series exhibited slightly better strength characteristics than PS90. This result may be explained by the difference in requisite heating times. The longer heating times required for PS90 (72 h versus 2 h for PSS-X) likely results in some destruction of the native peanut shell structure which we have previously shown to be disadvantageous in cellulose systems.19 As previously noted, attempts to prepare composites analogous to PS90 by heating sulfur with unfractionated peanut shells for shorter times led to visibly heterogeneous materials, so a direct comparison of PSS-X to that hypothetical direct control material is not possible. Fractionation thus proved to be successful in reducing reaction time, thereby preserving the native structure of the lignocellulosic components, and reducing energy requirements for composite preparation, but at the expense of material homogenization and reproducibility of material properties.
Conclusions
This work demonstrates the ability to attain high strength materials from unfractionated, unmodified biomass after heating to a relatively low temperature for only a few hours using raw biomass that has been fractionated by a simple mechanical sifting process. All prepared materials possessed compressional strength characteristics on par with residential Portland cement. Opposing trends in the minimization of surface area and the more uniform dispersion of biomass filler provides rationale for similarity of strength between all of the materials in the series. Fractionation of biomass into narrow particle size distributions did, however, prove to be beneficial for significantly decreasing reaction times and thus energy costs for achieving the high-strength materials.Experimental
General considerations
Fourier transform infrared spectra were obtained using an IR instrument (Shimadzu IRAffinity-1S) with an ATR attachment. Scans were collected over the range 400–4000 cm−1 at ambient temperature with a resolution of 8. TGA was recorded (Mettler Toledo TGA 2 STARe System) over the range 20–800 °C with a heating rate of 10 °C min−1 under a flow of N2 (100 mL min−1). Each measurement was acquired in duplicate and presented results represent an average value. DSC was acquired (Mettler Toledo DSC 3 STARe System) over the range 60 to 150 °C with a heating rate of 5 °C min−1 under a flow of N2 (200 mL min−1). Each DSC measurement was carried out over three heat–cool cycles. Each measurement was acquired in triplicate to ensure consistent results were obtained.Carbon disulfide extractions were performed by suspending 0.3 g of finely ground material (measured to 0.0001 g) in 20 mL of CS2, allowing the solid to settle for 30 minutes, pipetting off the supernatant into a separate vial, and adding another 20 mL of CS2. This process was repeated an additional 3 times so that a total of 5 washes was performed. The residual CS2 was evaporated under a flow of N2 and each vial was weighed to determine the fraction that was soluble (collected as supernatant) or insoluble (remained in the initial vial).
Compressional analysis was performed on a Mark-10 ES30 test stand equipped with a M3-200 force gauge (1 kN maximum force with 1 N resolution) with an applied force rate of 3–4 N s−1. Compression cylinders were cast from silicone resin moulds (Smooth-On Oomoo 30 tin-cure) with diameters of approximately 6 mm and heights of approximately 10 mm. Samples were manually sanded to ensure uniform dimensions and measured with a digital calliper with 0.01 mm resolution. Compressional analysis was initially performed three times; however, more samples were analysed when the standard deviation was greater than four MPa. The number of replicates was increased until the standard deviation was consistent with additional runs. The number of replicates for each sample is reported in Table 2.
SEM was acquired on a Schottky Field Emission Scanning Electron Microscope SU5000 operating in variable pressure mode with an accelerating voltage of 15 keV.
Materials and methods
Ground peanut shells were obtained from Golden Peanut and Tree Nuts (Product ES). Sulfur was obtained from Alfa Aesar. Carbon disulfide was obtained from Beantown Chemical. 2,3,4,5,6-Pentafluorobenzaldehyde was obtained from Acros Organics. All reagents were used as received unless otherwise specified.General composite synthesis
PSS-X materials were prepared by combining 9.0 g of sulfur and 1.1 g of fractionated peanut shells in a glass vial equipped with a Teflon coated stir bar. The vials were placed in an oil bath set to 180 °C for two hours. Specific reaction conditions can be found in the ESI.†Hexane extraction and NMR analysis
Peanut shell powder (8 g) was suspended in hexane (150 mL) and stirred at room temperature for 24 h. The suspension was filtered, and the hexane was removed from the filtrate under reduced pressure. The yield of material was determined as a first approximation of peanut oil in each fraction. A solution of 2,3,4,5,6-pentaflourobenzaldehyde (PFBA) in CDCl3 was prepared by serial dilution. First, 0.4657 g of PFBA was dissolved in 9.5337 g of CDCl3, 2.0118 g of this solution was then dissolved in 18.0001 g of CDCl3 to bring the final concentration of the solution to 4.6817 mg of PFBA per g of CDCl3. Approximately one gram of this solution was used to dissolve the residue for each hexane soluble fraction and then this solution was transferred to an NMR tube for analysis. External standards were also prepared by dissolving 10, 20, or 30 mg of peanut oil into 1 g of CDCl3.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding for this project from the National Science Foundation (CHE-1708844) is gratefully acknowledged.Notes and references
- K. L. Scrivener, V. M. John and E. M. Gartner, Cem. Concr. Res., 2018, 114, 2–26 CrossRef CAS.
- R. M. Andrew, Earth Syst. Sci. Data, 2018, 10, 195–217 CrossRef.
- K. L. Scrivener, V. M. John and E. M. Gartner, Eco-efficient cements: Potential, economically viable solutions for a low-CO2 cement-based materials industry, Paris, 2016 Search PubMed.
- S. Shirani, A. Cuesta, A. Morales-Cantero, A. G. De la Torre, M. P. Olbinado and M. A. G. Aranda, Cem. Concr. Res., 2021, 147, 106499 CrossRef CAS.
- R. Zhao, L. Zhang, G. Fan, Y. Chen, G. Huang, H. Zhang, J. Zhu and X. Guan, Cem. Concr. Res., 2021, 144, 106420 CrossRef CAS.
- Y. Briki, F. Avet, M. Zajac, P. Bowen, M. B. Haha and K. Scrivener, Cem. Concr. Res., 2021, 146, 106477 CrossRef CAS.
- A. Al-Hamrani, M. Kucukvar, W. Alnahhal, E. Mahdi and N. C. Onat, Materials, 2021, 14, 351 CrossRef CAS PubMed.
- F. Blomsma and G. Brennan, J. Ind. Ecol., 2017, 21, 603–614 CrossRef.
- J. Deschamps, B. Simon, A. Tagnit-Hamou and B. Amor, J. Cleaner Prod., 2018, 185, 14–22 CrossRef CAS.
- Y. Yu, D. M. Yazan, S. Bhochhibhoya and L. Volker, J. Clean. Prod., 2021, 293, 126083 CrossRef.
- T. Thiounn, M. K. Lauer, M. S. Bedford, R. C. Smith and A. G. Tennyson, RSC Adv., 2018, 8, 39074–39082 RSC.
- M. S. Karunarathna, M. K. Lauer, T. Thiounn, R. C. Smith and A. G. Tennyson, J. Mater. Chem. A, 2019, 7, 15683–15690 RSC.
- M. K. Lauer, T. A. Estrada-Mendoza, C. D. McMillen, G. Chumanov, A. G. Tennyson and R. C. Smith, Adv. Sustainable Syst., 2019, 3, 1900062 CrossRef CAS.
- M. S. Karunarathna, M. K. Lauer and R. C. Smith, J. Mater. Chem. A, 2020, 8, 20318–20322 RSC.
- M. S. Karunarathna, M. K. Lauer, A. G. Tennyson and R. C. Smith, Polym. Chem., 2020, 11, 1621–1628 RSC.
- M. K. Lauer, M. S. Karunarathna, A. Tennyson and R. C. Smith, Mater. Adv., 2020, 1, 590–594 RSC.
- M. K. Lauer, M. S. Karunarathna, A. G. Tennyson and R. C. Smith, Mater. Adv., 2020, 1, 2271–2278 RSC.
- M. K. Lauer and R. C. Smith, Comprehensive Reviews in Food Science and Food Safety, 2020, pp. 1–53, DOI:10.1111/1541-4337.12627.
- M. K. Lauer, A. G. Tennyson and R. C. Smith, ACS Appl. Polym. Mater., 2020, 2, 3761–3765 CrossRef CAS.
- C. V. Lopez, M. S. Karunarathna, M. K. Lauer, C. P. Maladeniya, T. Thiounn, E. D. Ackley and R. C. Smith, J. Polym. Sci., 2020, 58, 2259–2266 CrossRef CAS.
- C. P. Maladeniya, M. S. Karunarathna, M. K. Lauer, C. V. Lopez, T. Thiounn and R. C. Smith, Mater. Adv., 2020, 1, 1665–1674 RSC.
- T. Thiounn, M. S. Karunarathna, L. M. Slann, M. K. Lauer and R. C. Smith, J. Polym. Sci., 2020, 58, 1347–1364 CrossRef CAS.
- T. Thiounn, M. K. Lauer, M. S. Karunarathna, A. G. Tennyson and R. C. Smith, Sustainable Chem., 2020, 1, 183–197 CrossRef.
- M. K. Lauer, A. G. Tennyson and R. C. Smith, Mater. Adv., 2021, 2, 2391–2397 RSC.
- A. D. Smith, T. Thiounn, E. W. Lyles, E. K. Kibler, R. C. Smith and A. G. Tennyson, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 1704–1710 CrossRef CAS.
- T. Thiounn, A. G. Tennyson and R. C. Smith, RSC Adv., 2019, 9, 31460–31465 RSC.
- T. Thiounn and R. C. Smith, J. Poly. Sci., 2020, 58, 1347–1364 CrossRef CAS.
- M. S. Karunarathna and R. C. Smith, Sustainability, 2020, 12, 734–748 CrossRef CAS.
- M. S. Karunarathna, A. G. Tennyson and R. C. Smith, J. Mater. Chem. A, 2020, 8, 548–553 RSC.
- C. V. Lopez, C. P. Maladeniya and R. C. Smith, Electrochemistry, 2020, 1, 226–259 Search PubMed.
- A. D. Smith, C. D. McMillin, R. C. Smith and A. G. Tennyson, J. Polym. Sci., 2020, 58, 438–445 CrossRef CAS.
- A. D. Smith, R. C. Smith and A. G. Tennyson, Sustain. Chem. Pharm., 2020, 16, 100249 CrossRef.
- A. D. Smith, R. C. Smith and A. G. Tennyson, Sustain. Chem., 2020, 1, 209–237 CrossRef.
- X. Zhang, Y. Tang, S. Qu, J. Da and Z. Hao, ACS Catal., 2015, 5, 1053–1067 CrossRef CAS.
- A. Demirbas, H. Alidrisi and M. A. Balubaid, Pet. Sci. Technol., 2015, 33, 93–101 CrossRef CAS.
- W. J. Chung, J. J. Griebel, E. T. Kim, H. Yoon, A. G. Simmonds, H. J. Ji, P. T. Dirlam, R. S. Glass, J. J. Wie, N. A. Nguyen, B. W. Guralnick, J. Park, A. Somogyi, P. Theato, M. E. Mackay, Y.-E. Sung, K. Char and J. Pyun, Nat. Chem., 2013, 5, 518–524 CrossRef CAS PubMed.
- Y. Zhang, R. S. Glass, K. Char and J. Pyun, Polym. Chem., 2019, 10, 4078–4105 RSC.
- T. S. Kleine, R. S. Glass, D. L. Lichtenberger, M. E. MacKay, K. Char, R. A. Norwood and J. Pyun, ACS Macro Lett., 2020, 9, 245–259 CrossRef CAS.
- P. Yan, W. Zhao, B. Zhang, L. Jiang, S. Petcher, J. A. Smith, D. J. Parker, A. I. Cooper, J. Lei and T. Hasell, Angew. Chem., Int. Ed., 2020, 59, 13371–13378 CrossRef CAS PubMed.
- M. J. H. Worthington, R. L. Kucera and J. M. Chalker, Green Chem., 2017, 19, 2748–2761 RSC.
- J. M. Chalker, M. J. H. Worthington, N. A. Lundquist and L. J. Esdaile, Top. Curr. Chem., 2019, 377, 1–27 CrossRef CAS PubMed.
- A. G. Simmonds, J. J. Griebel, J. Park, K. R. Kim, W. J. Chung, V. P. Oleshko, J. Kim, E. T. Kim, R. S. Glass, C. L. Soles, Y.-E. Sung, K. Char and J. Pyun, ACS Macro Lett., 2014, 3, 229–232 CrossRef CAS.
- I. Gomez, O. Leonet, J. A. Blazquez and D. Mecerreyes, ChemSusChem, 2016, 9, 3419–3425 CrossRef CAS PubMed.
- A. Hoefling, D. T. Nguyen, Y. J. Lee, S.-W. Song and P. Theato, Mater. Chem. Front., 2017, 1, 1818–1822 RSC.
- S. Zeng, L. Li, L. Xie, D. Zhao, N. Wang and S. Chen, ChemSusChem, 2017, 10, 3378–3386 CrossRef CAS PubMed.
- Y. Zhang, J. J. Griebel, P. T. Dirlam, N. A. Nguyen, R. S. Glass, M. E. MacKay, K. Char and J. Pyun, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 107–116 CrossRef CAS.
- P. Dong, K. S. Han, J.-I. Lee, X. Zhang, Y. Cha and M.-K. Song, ACS Appl. Mater. Interfaces, 2018, 10, 29565–29573 CrossRef CAS PubMed.
- Q. Jiang, Y. Li, X. Zhao, P. Xiong, X. Yu, Y. Xu and L. Chen, J. Mater. Chem. A, 2018, 6, 17977–17981 RSC.
- F. Zhao, Y. Li and W. Feng, Small Methods, 2018, 2, 1–34 CAS.
- T. S. Sahu, S. Choi, P. Jaumaux, J. Zhang, C. Wang, D. Zhou and G. Wang, Polyhedron, 2019, 162, 147–154 CrossRef CAS.
- J. J. Griebel, S. Namnabat, E. T. Kim, R. Himmelhuber, D. H. Moronta, W. J. Chung, A. G. Simmonds, K.-J. Kim, J. van der Laan, N. A. Nguyen, E. L. Dereniak, M. E. MacKay, K. Char, R. S. Glass, R. A. Norwood and J. Pyun, Adv. Mater., 2014, 26, 3014–3018 CrossRef CAS PubMed.
- S. Namnabat, J. J. Gabriel, J. Pyun and R. A. Norwood, Proc. SPIE, 2014, 8983, 89830D CrossRef.
- S. Namnabat, J. J. Gabriel, J. Pyun, R. A. Norwood, E. L. Dereniak and J. van der Laan, Proc. SPIE, 2014, 9070, 90702H Search PubMed.
- J. J. Griebel, N. A. Nguyen, S. Namnabat, L. E. Anderson, R. S. Glass, R. A. Norwood, M. E. MacKay, K. Char and J. Pyun, ACS Macro Lett., 2015, 4, 862–866 CrossRef CAS.
- L. E. Anderson, T. S. Kleine, Y. Zhang, D. D. Phan, S. Namnabat, E. A. LaVilla, K. M. Konopka, L. Ruiz Diaz, M. S. Manchester, J. Schwiegerling, R. S. Glass, M. E. Mackay, K. Char, R. A. Norwood and J. Pyun, ACS Macro Lett., 2017, 6, 500–504 CrossRef CAS.
- T. S. Kleine, T. Lee, K. J. Carothers, M. O. Hamilton, L. E. Anderson, L. Ruiz Diaz, N. P. Lyons, K. R. Coasey, W. O. Parker, Jr., L. Borghi, M. E. MacKay, K. Char, R. S. Glass, D. L. Lichtenberger, R. A. Norwood and J. Pyun, Angew. Chem., Int. Ed., 2019, 58, 17656–17660 CrossRef CAS PubMed.
- J. Kuwabara, K. Oi, M. M. Watanabe, T. Fukuda and T. Kanbara, ACS Appl. Polym. Mater., 2020, 2, 5173–5178 CrossRef CAS.
- T. Hasell, D. J. Parker, H. A. Jones, T. McAllister and S. M. Howdle, Chem. Commun., 2016, 52, 5383–5386 RSC.
- W. Thielke Michael, A. Bultema Lindsey, D. Brauer Daniel, P. Theato, B. Richter and M. Fischer, Polymers, 2016, 8(7), 266 CrossRef PubMed.
- D. J. Parker, H. A. Jones, S. Petcher, L. Cervini, J. M. Griffin, R. Akhtar and T. Hasell, J. Mater. Chem. A, 2017, 5, 11682–11692 RSC.
- A. M. Abraham, S. V. Kumar and S. M. Alhassan, Chem. Eng. J., 2018, 332, 1–7 CrossRef CAS.
- H.-K. Lin, Y.-S. Lai and Y.-L. Liu, ACS Sustainable Chem. Eng., 2019, 7, 4515–4522 CrossRef CAS.
- Y. Chen, A. Yasin, Y. Zhang, X. Zan, Y. Liu and L. Zhang, Materials, 2020, 13, 632 CrossRef CAS PubMed.
- A. D. Tikoalu, N. A. Lundquist and J. M. Chalker, Adv. Sustainable Syst., 2020, 4, 1900111 CrossRef CAS.
- C. R. Westerman and C. L. Jenkins, Macromolecules, 2018, 51, 7233–7238 CrossRef CAS.
- C. Herrera, K. J. Ysinga and C. L. Jenkins, ACS Appl. Mater. Interfaces, 2019, 11, 35312–35318 CrossRef CAS PubMed.
- K. Orme, A. H. Fistrovich and C. L. Jenkins, Macromolecules, 2020, 53, 9353–9361 CrossRef CAS.
- X. Wu, J. A. Smith, S. Petcher, B. Zhang, D. J. Parker, J. M. Griffin and T. Hasell, Nat. Commun., 2019, 10, 10035–10044 Search PubMed.
- B. Zhang, S. Petcher and T. Hasell, Chem. Commun., 2019, 55, 10681–10684 RSC.
- B. Zhang, H. Gao, P. Yan, S. Petcher and T. Hasell, Mater. Chem. Front., 2020, 4, 669–675 RSC.
- N. A. Lundquist, A. D. Tikoalu, M. J. H. Worthington, R. Shapter, S. J. Tonkin, F. Stojcevski, M. Mann, C. T. Gibson, J. R. Gascooke, A. Karton, L. C. Henderson, L. J. Esdaile and J. M. Chalker, Chem. – Eur. J., 2020, 26, 10035–10044 CrossRef CAS PubMed.
- S. J. Tonkin, C. T. Gibson, J. A. Campbell, D. A. Lewis, A. Karton, T. Hasell and J. M. Chalker, Chem. Sci., 2020, 11, 5537–5546 RSC.
- M. P. Crockett, A. M. Evans, M. J. H. Worthington, I. S. Albuquerque, A. D. Slattery, C. T. Gibson, J. A. Campbell, D. A. Lewis, G. J. L. Bernardes and J. M. Chalker, Angew. Chem., Int. Ed., 2016, 55, 1714–1718 CrossRef CAS PubMed.
- F. Wu, S. Chen, V. Srot, Y. Huang, S. K. Sinha, P. A. van Aken, J. Maier and Y. Yu, Adv. Mater., 2018, 30(13), 1706643 CrossRef PubMed.
- J. A. Smith, S. J. Green, S. Petcher, D. J. Parker, B. Zhang, M. J. H. Worthington, X. Wu, C. A. Kelly, T. Baker, C. T. Gibson, J. A. Campbell, D. A. Lewis, M. J. Jenkins, H. Willcock, J. M. Chalker and T. Hasell, Chem. – Eur. J., 2019, 25, 10433–10440 CrossRef CAS PubMed.
- J. E. M. Ballesteros, V. dos Santos, G. Marmol, M. Frias and J. Fiorelli, Cellulose, 2017, 24, 2275–2286 CrossRef CAS.
- R. P. Da Fonseca, J. C. Rocha and M. Cheriaf, Materials, 2021, 14, 155 CrossRef CAS PubMed.
- S. R. Ferreira, F. d. A. Silva, P. R. L. Lima and R. D. Toledo Filho, Constr. Build. Mater., 2017, 139, 551–561 CrossRef CAS.
- H. Fu, W. Gao, B. Wang, J. Zeng, Z. Cheng, J. Xu and K. Chen, Cellulose, 2020, 27, 1327–1340 CrossRef CAS.
- C. Gong, J.-p. Ni, C. Tian and Z.-h. Su, Int. J. Biol. Macromol., 2021, 172, 573–579 CrossRef CAS PubMed.
- C.-H. Ham, H. J. Youn and H. L. Lee, BioResources, 2020, 15, 9197–9211 CAS.
- M. N. F. Norrrahim, H. Ariffin, M. A. Hassan, N. A. Ibrahim, W. M. Z. W. Yunus and H. Nishida, Int. J. Nanotechnol., 2019, 16, 668–679 CrossRef CAS.
- P. Posada, J. Velasquez-Cock, C. Gomez-Hoyos, A. M. Serpa Guerra, S. V. Lyulin, J. M. Kenny, P. Ganan, C. Castro and R. Zuluaga, Cellulose, 2020, 27, 10649–10670 CrossRef CAS.
- A. Santmarti, T. Tammelin and K.-Y. Lee, Carbohydr. Polym., 2020, 250, 116870 CrossRef CAS PubMed.
- V. Vilchez, E. Dieckmann, T. Tammelin, C. Cheeseman and K.-Y. Lee, ACS Sustainable Chem. Eng., 2020, 8, 14263–14267 CrossRef CAS.
- C. Palange, M. A. Johns, D. J. Scurr, J. S. Phipps and S. J. Eichhorn, Cellulose, 2019, 26, 9645–9659 CrossRef CAS.
- X. Yang, E. Bakaic, T. Hoare and E. D. Cranston, Biomacromolecules, 2013, 14, 4447–4455 CrossRef CAS PubMed.
- M. S. Reid, M. Villalobos and E. D. Cranston, Langmuir, 2017, 33, 1583–1598 CrossRef CAS PubMed.
- S. J. Eichhorn, A. Dufresne, M. Aranguren, N. E. Marcovich, J. R. Capadona, S. J. Rowan, C. Weder, W. Thielemans, M. Toman, S. Renneckar, W. Gindl, S. Veigel, J. Keckes, H. Yano, K. Abe, M. Nogi, A. N. Nakagaito, A. Mangalam, J. Simonsen, A. S. Benight, A. Bismarck, L. A. Berglund and T. Peijs, J. Mater. Sci., 2010, 45, 1–33 CrossRef CAS.
- M. Mariano, N. El Kissi and A. Dufresne, J. Polym. Sci., Part B: Polym. Phys., 2014, 52, 791–806 CrossRef CAS.
- C. W. Wright, J. Chem. Educ., 1946, 23, 268–270 CrossRef CAS.
- S. P. Massie, Chemistry, 1970, 43, 18–21 CAS.
- O. Zavgorodnya, J. L. Shamshina, P. Berton and R. D. Rogers, ACS Symp. Ser., 2017, 1250, 17–33 CrossRef CAS.
- M.-A. Perea-Moreno, F. Manzano-Agugliaro, Q. Hernandez-Escobedo and A.-J. Perea-Moreno, Sustainability, 2018, 10, 3254 CrossRef CAS.
- K.-H. Kim, J. L. Ong and O. Okuno, J. Prosthet. Dent., 2002, 87, 642–649 CrossRef CAS PubMed.
- S. Lin-Gibson, L. Sung, A. M. Forster, H. Hu, Y. Cheng and N. J. Lin, Acta Biomater., 2009, 5, 2084–2094 CrossRef CAS PubMed.
- T. Sakai, H. Li, T. Abe, S. Yamaguchi and S. Imazato, Dent. Mater., 2021, 37, 168–174 CrossRef CAS PubMed.
- J. D. Satterthwaite, A. Maisuria, K. Vogel and D. C. Watts, Dent. Mater., 2012, 28, 609–614 CrossRef CAS PubMed.
- G. E. Padawer and N. Beecher, Polym. Eng. Sci., 1970, 10, 185–192 CrossRef CAS.
- J. M. Adams, Clay Miner., 1993, 28, 509–530 CrossRef CAS.
- Additives for Plastics Handbook, ed. J. Murphy, 2nd edn, 2001 Search PubMed.
- Z.-L. He, L.-N. Chen, L. Zhang, H.-Y. Ren, M.-D. Xu and Y.-W. Lou, J. Appl. Polym. Sci., 2020, 137, 49010 CrossRef CAS.
- M. K. Lauer, M. S. Karunarathna, A. G. Tennyson and R. C. Smith, Mater. Adv., 2020, 1, 590–594 RSC.
- S. Samal, S. Kim and H. Kim, J. Am. Ceram. Soc., 2012, 95, 1595–1603 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Instrumentation, synthetic details, DSC curves, TGA curves, FTIR data, NMR spectral data, and material images. See DOI: 10.1039/d1ma00689d |
| This journal is © The Royal Society of Chemistry 2021 |