
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHarnessing photo/electro-catalytic activity via nano-junctions in ternary nanocomposites for clean energy
Ekta
Rani
 *a,
Parisa
Talebi
a,
Wei
Cao
a,
Marko
Huttula
ab and
Harishchandra
Singh
a
*a,
Parisa
Talebi
a,
Wei
Cao
a,
Marko
Huttula
ab and
Harishchandra
Singh
a
aNano and Molecular Systems Research Unit, University of Oulu, FIN-90014, Finland. E-mail: ades.ekta@gmail.com
bSchool of Materials Science and Engineering, Henan University of Science and Technology, Luoyang 471023, Henan, China
First published on 20th October 2020
Abstract
Though solar energy availability is predicted for centuries, the diurnal and asymmetrical nature of the sun across the globe presents significant challenges in terms of harvesting sunlight. Photo/electro-catalysis, currently believed to be the bottleneck, promises a potential solution to these challenges along with a green and sustainable environment. This review aims to provide the current highlights on the application of inorganic-semiconductor-based ternary nanocomposites for H2 production and pollutant removal. Various engineering strategies employing integration of 2D materials, 1D nanorods, and/or 0D nanoparticles with inorganic semiconductors to create multiple nano-junctions have been developed for the excellent photocatalytic activity. Following a succinct description of the latest progress in photocatalysts, a discussion on the importance of ternary electrocatalysts in the field of next-generation supercapacitors has been included. Finally, the authors’ perspectives are considered briefly, including future developments and critical technical challenges in the ever-growing field of photo/electro-catalysis.
1. Introduction
The unprecedented industrial growth, uncontrolled environmental pollution, and the current energy scenario pose a significant challenge in realizing a carbon-neutral society. The key to this challenge is to expand sustainable and environmentally benign energy systems to avert the impending climate change. Thus, tangible steps are required to have a greener era, wherein one is expected to rely on a renewable and clean energy source that leaves limited and regulated environmental damage. Solar-light-driven energy conversion, environmental purification, and storage technologies via photo- and electrocatalysts have drawn tremendous attention. Solar energy is an inexhaustible, clean, and green resource, along with the fact that the sunlight reaching the Earth's surface on an hourly basis surpasses the annual global energy consumption.1,2Since the demonstration of water splitting via TiO2 by employing ultraviolet (UV) excitation light by Fujishima and Honda in 1972,3 photocatalytic technology incorporating inexhaustible solar energy-driven water splitting and degradation of pollutants has been contemplated as one of the most promising approaches to solve the global energy and environmental crisis. Solar water splitting involving hydrogen production via sunlight is indisputably one of the most imperative approaches to produce clean energy. The produced hydrogen can release energy via direct combustion or in a hydrogen fuel cell, with the only by-product being water, whereas degradation of pollutants tackles another major global issue as a significant amount of pollutants are discharged into lakes, rivers, and groundwater due to rapid industrialization. It is known that around 1.8 billion people consume contaminated water. Thus, these harmful compounds must be turned into harmless compounds, which can be realized by exploiting photocatalysts to absorb light and produce active radicals that degrade the pollutants via redox processes.
For an active photocatalyst, various parameters such as excitation wavelength, suitable bandgaps, band positions with adequate driving potential, and charge carrier separation play a significant role. Since photocatalytic H2 production and pollutant degradation hinge on the sun and photocatalyst, not consumed during the process, this is possibly a low-cost and environmentally friendly approach for energy production and pollution abatement. However, though solar energy availability is predicted for centuries, the diurnal and asymmetrical nature of this source across the globe presents significant challenges in terms of harvesting, storage, and utilization. Furthermore, the large bandgap of a commonly used active semiconductor such as TiO2 (∼3.2 eV for anatase) requires an excitation wavelength that falls in the UV region. The UV spectral regime consists of less than ∼5% of the total sunlight (solar light consists of ∼5% UV, ∼45% visible, and ∼52% infrared) incident at the Earth's surface. In contrast, the photocorrosion of the visible light active CdS (∼2.42 eV) catalyst is considered as the Achilles heel of CdS. So far, the research has suggested that a solar-driven energy conversion efficiency of ∼10% under optimal operational conditions could allow the photocatalytic technology to be economically viable for hydrogen production and environmental enrichment.4,5 However, the current water-splitting technologies using particulate homogeneous photocatalysts on the laboratory scale are only ∼1% efficient.6 This necessitates the idea of developing highly stable and efficient photocatalytic systems covering a broad range of sunlight to achieve the required performance.
Another efficient and practical way to harness sunlight is to convert it into electricity using photovoltaics.7 However, the sun's diurnal nature brings out the importance of energy storage systems in electronics, automotive, aerospace, and stationary markets to counterbalance its variability.8,9 Furthermore, with the persistent demands in miniaturization of electronic devices due to the addition of portable technology into our daily life, there are serious struggles to manufacture small but powerful devices. Among various possible energy storage systems, supercapacitors have enormous potential owing to their superior power density, fast charge/discharge rate, and long cycle life compared to conventional capacitors and batteries. The most widely used electrode materials are carbon, conducting polymers, and transition metal oxides for active supercapacitors. However, these materials do have their drawbacks. For example, graphite has low specific capacitances, and conducting polymers have poor cyclability. Among various available transition metal oxide materials, Co3O4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10 and MnO211 have attracted much attention as a pseudocapacitive electrode material because of their high theoretical capacitance of 3560 F g−1 and 1233 F g−1, respectively, along with their excellent redox activity, cost-effectiveness, and environmentally friendly nature. However, the performances are often compromised in terms of actual capacity and cycling stability, which originate from the limited redox kinetics for ion diffusion, electron transfer rates, and poor structural stability.12 The novel material design needs to be implemented to transcend these limitations to achieve unique properties such as an abundant specific surface area and high electrical conductivity to simultaneously improve both the capacitive performance and the stability of the electrode material.
10 and MnO211 have attracted much attention as a pseudocapacitive electrode material because of their high theoretical capacitance of 3560 F g−1 and 1233 F g−1, respectively, along with their excellent redox activity, cost-effectiveness, and environmentally friendly nature. However, the performances are often compromised in terms of actual capacity and cycling stability, which originate from the limited redox kinetics for ion diffusion, electron transfer rates, and poor structural stability.12 The novel material design needs to be implemented to transcend these limitations to achieve unique properties such as an abundant specific surface area and high electrical conductivity to simultaneously improve both the capacitive performance and the stability of the electrode material.
With the research being pioneered in the “conventional catalysts”, particularly homogeneous and binary catalysts, the development of “unconventional ternary catalysts” has been stimulated. The idea of harnessing an active system ensemble effect with two different components in one-pot is conceptualized as a “ternary system”. The application of the ternary system has been noticed in the field of biosciences.13 The Nobel Prize (Chemistry, 2004) winning discovery of “ubiquitin-mediated protein degradation” by Aaron Ciechanover, Avram Hershko, and Irwin Rose exhibits one of the most pertinent examples of nature's ternary enzyme system.14 An attempt to master the “nature-like ternary system”, the availability of highly sophisticated growth techniques and atomic-nano scale characterization tools have inspired scientists to develop highly efficient catalytic systems. Ternary photo/electrocatalysts involve active catalysts with electron transfer mediators such as 2D materials, 1D nanorods (NRs), and 0D nanoparticles (NPs). These heterostructures combine materials with distinctly different physical and chemical properties to yield a unique hybrid nano-system with multiple nano-junctions giving rise to tunable and enhanced properties that may not be attainable otherwise. Although the concept of using a ternary catalyst is sprouting, it has demonstrated its invaluable implications in terms of efficiency and stability. A substantial number of apposite reports utilizing various ternary photo/electrocatalysts have drastically increased in a decade; however, these reports remain scattered in the literature.
Furthermore, we would like to emphasize here that although several review articles have been published on catalysts along with some recent reviews on photocatalysts15,16 and electrocataylysts;17 however, none is related to inorganic semiconductor-based ternary systems. A recent extensive review on the photocatalytic activity of heterogeneous nanocomposites has also been reported; however, it primarily focused on the one-step reaction in binary nanocomposites, which produced H2.18 Another review article discussing the application of the ternary system for the degradation of pollutants has been reported; however, it only involves tungsten element-based ternary systems.19 This review article, on the other hand, focuses on the photo/electro-catalytic properties of semiconductor-based ternary nanocomposites, which have not been the topic of review so far.
We aim to propose the current advancements in ternary catalysts in a decade, their current status, and possible future directions. The article is divided into two sections, “ternary photocatalyst” and “ternary electrocatalyst”, highlighting the importance of multiple nano-junctions in these integrated systems comprising various components of distinct functionalities. In the end, we present our perspective on the current research situation, the challenge in this field, and where this field is headed.
2. Ternary photocatalyst
Based on the excellent and different properties of various nanostructures due to the confinement in different dimensions and surface to volume ratio, an active photocatalyst can be well integrated with (i) 2D materials that show excellent mobility of charge carriers and a high surface area, (ii) 1D NRs that show high electrical conductivity and directional flow of charge carriers due to the confinement in the radial direction and large aspect ratio, and (iii) 0D NPs with varying size to tune the excitation wavelength.2.1. Solar water splitting
In the last decade, a substantial amount of work has been carried out on the integration of 2D materials,20–30 1D NRs,31–34 0D NPs,35–43 and Ag NP–graphene blends,44–46 with active catalysts, wherein these materials act as electron collectors/transporters to enhance the overall hydrogen evolution rate (HER). This review will instead focus on selected case studies that relate to the physicochemical aspects of different photocatalysts. Furthermore, sacrificial reagents such as electron donors or hole scavengers have been employed to enhance the overall activity. It is to be noted that the unit of the HER will be μmol h−1 g−1 throughout the article unless noted otherwise.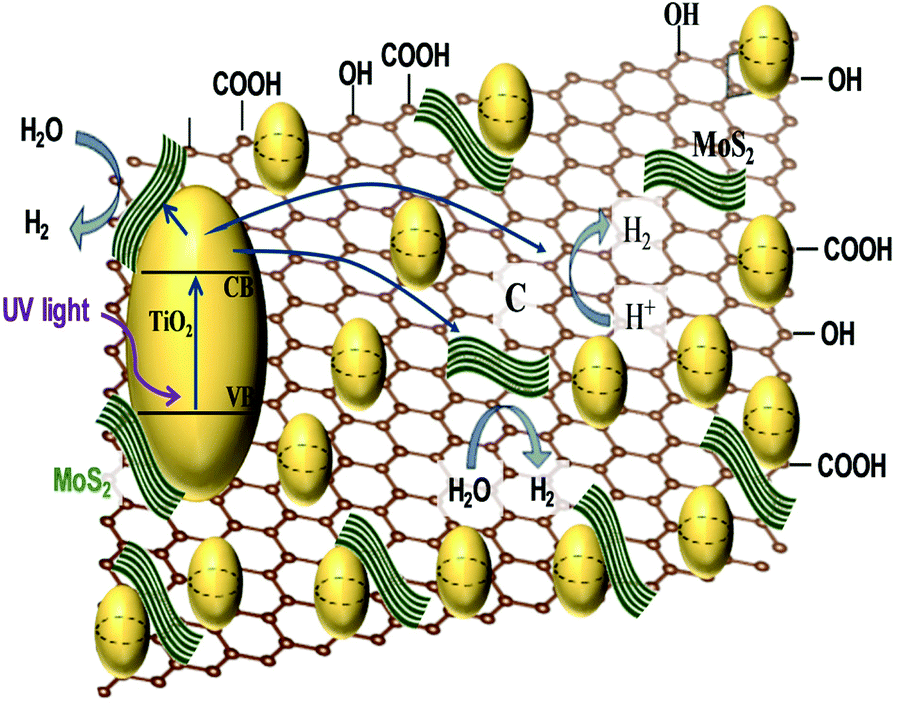 | ||
| Fig. 1 Schematic illustration of photocatalytic activity of the TiO2/MoS2/graphene nanocomposite. | ||
In another report, Ye et al.21 have reported the 3D porous hierarchical architecture (Fig. 2a) of CdS NPs (size ∼ 10 nm) decorated on MoS2/reduced graphene oxide (rGO) synthesized by a solution-chemistry method, as evidenced by scanning electron microscopy (SEM) and HRTEM (Fig. 2b & c). For different contents of the MoS2/rGO (MG) cocatalyst, 1:2 molar ratio of the M:G exhibited the highest activity, which is attributed to the uniform dispersion of MoS2 on rGO.47 Among various wt% of the MG cocatalyst, CdS/2.0 wt% MG annealed at 573 K showed the highest HER of ∼6100 (AQE: 20.6%) and ∼9000 (AQE: 28.1%), in Na2S–Na2SO3 and lactic acid solution (Fig. 2d–f), respectively, @420 nm. Furthermore, the observed trend of the HER of CdS/MG (9000) > CdS/M (6400) > CdS/G (200) > CdS (150) > MG (0) suggests highest activity of the ternary nanocomposite. The authors believed that a lower annealing temperature gives low crystallization, whereas too high temperature (673 K) leads to the agglomeration of CdS NPs and non-uniform dispersion of MG on the surface of CdS, thus, not facilitating the charge transfer. The observed remarkable HER in the lactic acid solution is attributed to the abundance of H+ ions facilitating higher production. No significant loss in the activity was noted after five recycles. The observed higher activity of ternary nanocomposites is attributed to the nanosized MG with more active edges, which shorten the electron transfer path, as evidenced by the transient photocurrent measurements (Fig. 2g). Thus, the formation of a ternary nanocomposite would enhance the catalytic performance of CdS and make it more stable.
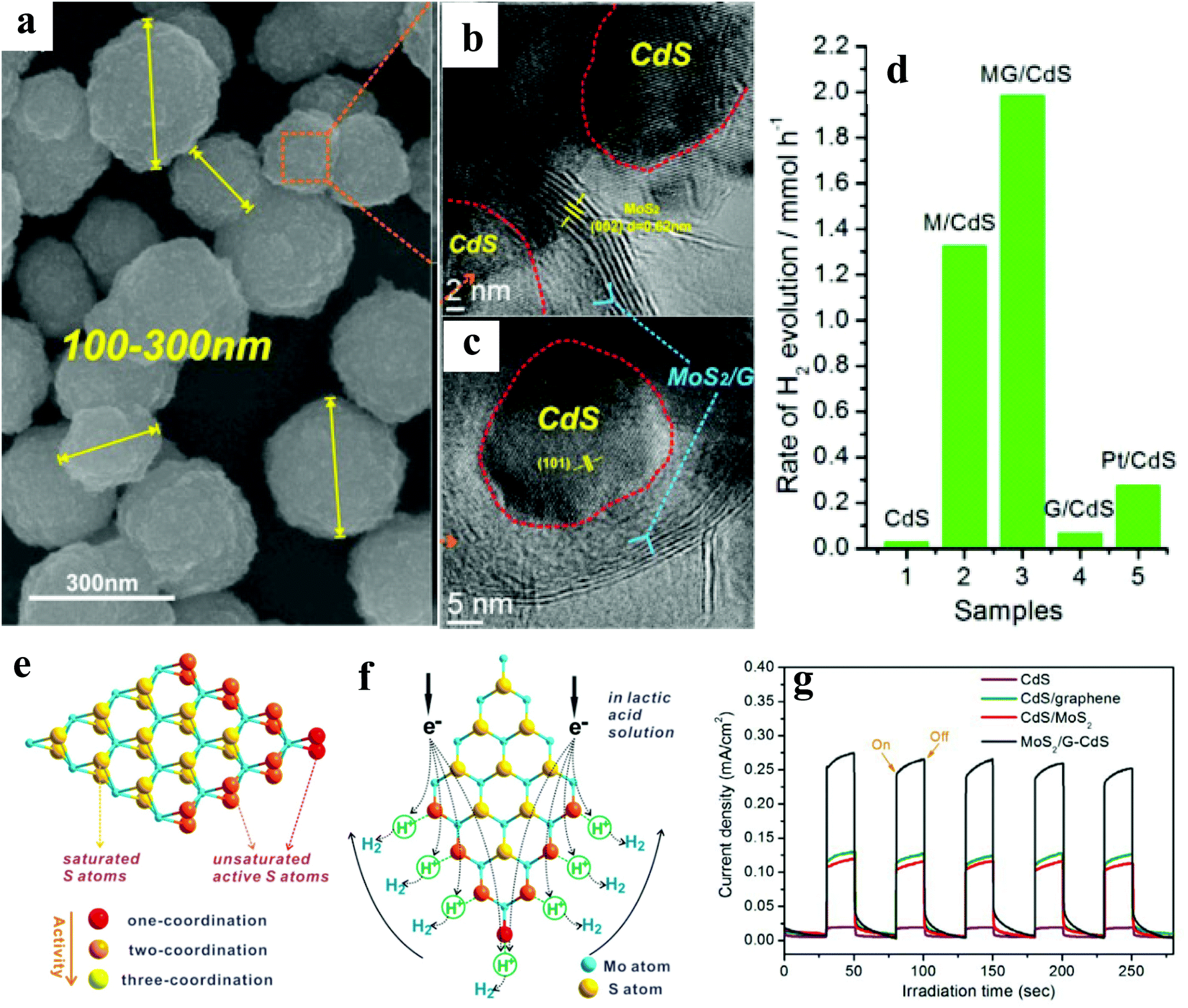 | ||
| Fig. 2 (a) SEM image of CdS/MG nanocomposites after annealing at 573 K for 2 h under an Ar atmosphere, (b) and (c) HRTEM images of the CdS/MG nanocomposite. (d) Photocatalytic H2 production activities of CdS, CdS/M, optimized CdS/MG, CdS/G, and Pt/CdS (0.5 wt%) in lactic acid solution, (e) schematic illustration of the microstructure of MoS2, and (f) its cocatalytic mechanism of H2 generation. (g) Transient photocurrent responses of pristine CdS and its binary/ternary nanocomposite under simulated irradiation Copyright 2014, American Chemical Society. Reprinted from ref. 21 with permission. | ||
Comparison of the HER reported by Ye et al.21 and Li and Xu et al.23 suggests that CdS NPs decorated on the MG ternary nanocomposite show a remarkable H2 evolution rate of ∼9000 (ref. 21) and ∼19000 (ref. 23) in lactic acid solution @420 nm. Furthermore, Xu et al.23 have reported the highest HER of ∼23200 with an AQE of 65.8% for CdS NR-NPs decorated onto MG. Although both the reports have used the same components, however, different results have been observed. The observed enhanced efficiency by Li and Xu et al.23 may be attributed to the smaller sized nanocomposite with more specific area leading to more active sites and the combination of CdS NP-NR morphology. Further comparison with Xiang et al.27 suggests that the observed lower activity of CdS NRs/WS2/G (∼1842) as compared to CdS NRs/MoS2/G (∼4000)23 may be attributed to the use of the Na2S–Na2SO3 sacrificial agent instead of lactic acid, as both WS2 and MoS2 have shown similar cocatalytic activity.48
Qiao et al.22 integrated Zn0.5Cd0.5S (ZCS) NPs within NiS NPs and rGO to form the ternary nanocomposite. The authors synthesized a series of NiS/ZCS/rGO nanocomposites containing different wt% and mol% of rGO and NiS by a co-precipitation-hydrothermal treatment. TEM and HRTEM imaging show NiS NPs (∼5–10 nm) decorated onto ZCS NPs (∼50 nm), which were uniformly dispersed on rGO nanosheets. The HER of the as-prepared samples was measured in Na2S–Na2SO3 solution. The results suggest that 0.25 wt% of rGO and 3 mol% of NiS show the highest activity of 7514 μmol h−1 g−1 with an AQE of 31.1%@420 nm. The authors proposed that the higher concentration of NiS, leading to the high loading amount of NiS NPs, may shield the incident light, thus, suppressing the photocatalytic activity. The detected HER from the optimized NiS/ZCS/rGO (7514) exceeds that of the bare ZCS (2228), ZCS/rGO binary (4118), and the ZCS/NiS binary (4790) nanocomposite and is in corroboration with the observed highest photocurrent intensity. Ternary nanocomposite's higher activity has been attributed to the formation of a p–n junction between p-type NiS NPs and n-type ZCS NPs, which is crucial for the charge transfer, as evidenced via Mott–Schottky plots. The authors have not reported stability studies.
In another report, Min and Xu et al.26 integrated ZCS NRs with MoS2/rGO, wherein ZCS NRs (diameter ∼ 100 nm) were synthesized by a solvothermal method followed by a dual reduction reaction to produce the ternary nanocomposite. TEM and HRTEM imaging showed the MoS2 location at interfacial contact formed between the ZCS and rGO. The ternary nanocomposites were synthesized with varying wt% MoS2 and rGO and found that 1.5 wt% MoS2 and 3.0 wt% rGO showed the best photocatalytic activity measured in a lactic acid solution. The observed HER from the ternary ZCS NRs@MG (77000) is much higher than that of pristine ZCS NRs (933) and is in corroboration with the observed negligible photoluminescence (PL) intensity and higher photocurrent response suggesting hampered charge recombination and efficient charge transfer in ZCS@MG, respectively. The nanocomposite's stability was noted for 25 h, and the decrease in the HER after 25 h was ascribed to the consumption of lactic acid in the solution.
The NiS/ZCS/rGO nanocomposite gave a HER yield of ∼7514 μmol h−1 g−1 with an AQE of 31.1%,22 whereas ZCS NRs@MG gave 77000 μmol h−1 g−1. Although HER yield is higher for ZCS NRs@MG;26 however, the authors have not mentioned the AQE, making the comparison difficult. The ZCS NRs@MG nanocomposite was found to be stable for 25 h,26 whereas the stability of the NiS/ZCS/rGO nanocomposite is not reported. Interestingly, in the graphene-based ternary nanocomposite, the highest AQE of 65.8% has been observed by employing CdS NR-NPs decorated onto MG.23 Furthermore, whether it is TiO2, CdS or ZCS, the photocatalytic mechanism of a semiconductor integrated with rGO and MoS2 can be summarized as the semiconductor-based catalyst generating electron–hole pairs. The photogenerated electrons in the conduction band (CB) of the semiconductors will be transferred to the edge of MoS2 due to the formation of intimate contact at the nano-junction followed by a reduction of the adsorbed H+ to H2 under the cocatalytic activity of unsaturated active S atoms (Fig. 2e & f). Electrons on the inactive MoS2 basal planes can be transferred to the edge of MoS2 through the rGO sheets followed by H2 evolution. This mechanism contrasts with that reported for NiS/ZCS/rGO,22 wherein the photogenerated electrons of ZCS are injected into the rGO and the lower graphene/graphene redox potential to reduce H+ to H2. The left-behind photogenerated holes in the valence band (VB) of ZCS will be transferred to NiS due to the inner electric field created in the p–n junction.
:nZnO = 83.81:10.55:5.64. The photocatalytic activity was measured in an aqueous solution of Na2S–Na2SO3 using an Xe lamp. The observed HER of ∼6000 μmol h−1 g−1 is much higher than that of bare CdS (120). Based on the Z-scheme charge-carrier transport, the authors have interpreted that in the ZnO–CdS@Cd ternary nanocomposite, effective and rapid recombination of the photoexcited electrons from the CB of ZnO with the holes from the VB of CdS is a key step to prolong the lifetime of the photoexcited electrons in the CB of CdS and holes in the VB of ZnO. The presence of a metal Cd core acting as an effective channel is certainly beneficial for such recombination. The photocatalyst was found to be stable for four recycles. The authors have not mentioned the AQE.
In another report, Amirav et al.32 have reported the Pt tipped CdSe@CdS ternary nanocomposite with a remarkable AQE of 100%. CdSe NPs were prepared by the solution chemistry method followed by the growth of CdS NRs, including the injection of CdSe NPs into these NRs. The photo-deposition of Pt NPs was carried out to realize the final nanocomposite. TEM and high angle annular dark field micrographs show CdSe NPs embedded asymmetrically within a CdS NR (diameter ∼ 2.3 nm) and a single Pt NP placed at the end of the NR (Fig. 3). The authors have examined the photocatalytic activity of Pt tipped CdSe@CdS with a hydroxyl anion/radical redox couple operating as a shuttle to relay the holes. A typical sample containing ∼6 × 1014, Pt tipped 50 nm long CdS NRs embedded with CdSe NPs, isopropyl alcohol (electron donor), and KOH gave rise to a HER of 142000 μmol h−1 g−1 corresponding to an AQE of 100% at pH of 15@455 nm excitation. Furthermore, the AQE > 90% was observed for numerous samples with varying CdSe NP size (2.3–2.7 nm) and CdS NR length (45–65 nm). The synthesized nanocomposite showed astounding stability of 44 h. The observation of an exponential decrease in the activity with the decrease in the pH (15 to 8) is supported by the hypothesis that high pH is required to shift the VB of CdS for the oxidation of hydroxyl ions,49 which enable a crossover between the VB maximum and the redox potential for hydroxyl anions and radicals. As a result, the hydroxyl anion–radical redox couple can be employed as a shuttle to efficiently relay the hole from CdSe NPs to the scavenger. The authors have proposed that this architecture facilitates localization of holes, which are three-dimensionally confined to the CdSe. At the same time, delocalized electrons are transferred to the Pt tip via CdS NRs for the reduction of H+ to H2. This design enabled efficient long-lasting charge carriers’ separation, thus, extending their availability for redox reactions. It is noteworthy that the system had a single Pt reduction catalyst. The authors interpreted that the observed decreased AQE of 58.5% for two Pt NPs can be correlated with the system's ability to mediate hole transfer on the CdS NR surface, a process that might be highly sensitive to the ligand coverage. Based on a previous report,50 the authors have proposed that for a multi-electron reaction system, the final photocatalyst should only include a single cocatalytic site per each segment of the semiconductor capable of light excitation.
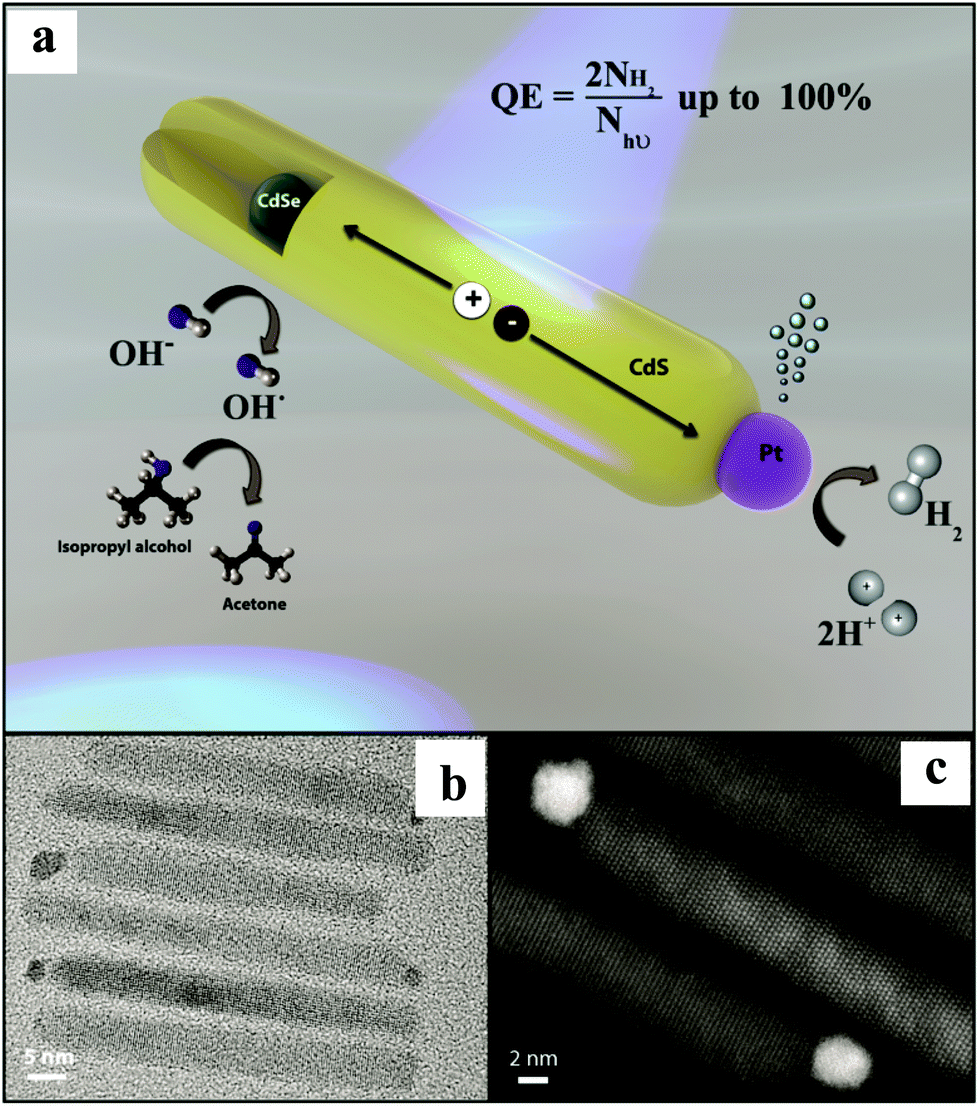 | ||
| Fig. 3 (a) Illustration of the NP photocatalyst depicting the photocatalytic cycle. (b) TEM and (c) high-resolution HAADF micrographs of a few representative Pt tipped CdSe@CdS rods. Copyright 2016, American Chemical Society. Reprinted from ref. 32 with permission. | ||
In a recent report, Burda et al.34 have reported the HER in the MoS2-stratified CdS-Cu2−xS ternary nanocomposite (Fig. 4) with an optimal 3.42 wt% of MoS2. The fascinating epitaxial vertical growth of 2D MoS2 on 1D CdS-Cu2−xS NRs (diameter ∼ 51 nm, length ∼ 987 nm) was carried out using sulfur powder dissolved in the oleyl amine precursor. The photocatalytic activity of the stratified screw-shaped MoS2@1D CdS-Cu2−xS ternary nanocomposite was measured in an aqueous solution of Na2S–Na2SO3. The detected highest HER with a MoS2-stratified CdS-Cu2−xS ternary system (14184) as compared to bare CdS (146), and the CdS-Cu2−xS binary system (1121) is in corroboration with the observed higher transient photocurrent response and weaker PL emission. However, the corresponding AQE is ∼1%@400 nm. The authors have proposed that the photogenerated electrons of Cu2−xS can transfer to the CB of CdS and MoS2; however, they mainly accumulate in the MoS2 due to the stronger driving force between Cu2−xS and MoS2. The electrons could migrate to the surface of MoS2 for the reduction of H+ ions. Furthermore, the mobile CuI ions of Cu2−xS can intercalate within the MoS2 basal plane due to nano-junction formation, thus making a CuI@MoS2 basal plane catalytically active. The stratified ternary nanocomposite was found to be stable during the cycling runs.
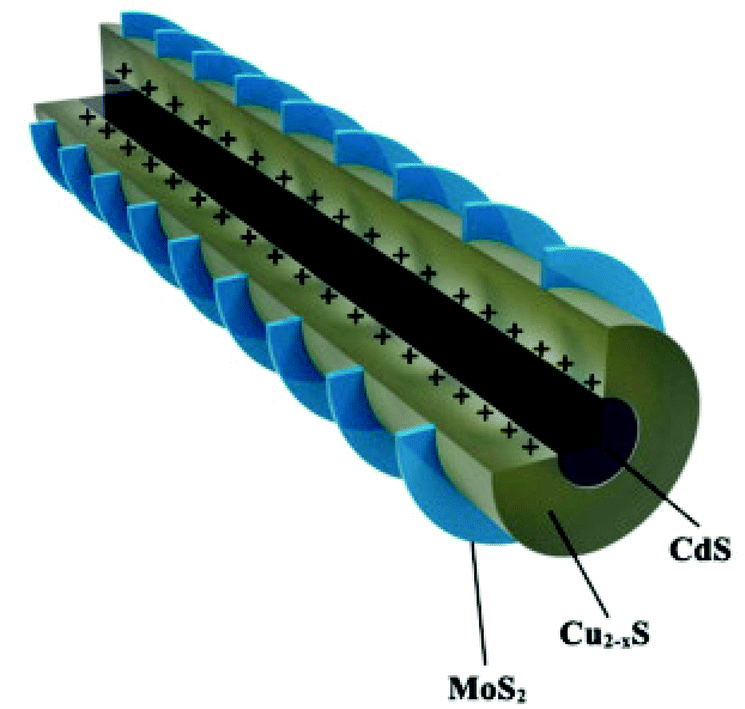 | ||
| Fig. 4 Schematic illustration of the crystal structure of heterointerfaces. Copyright 2020, American Chemical Society. Reprinted from ref. 34 with permission. | ||
It is interesting to note that the Pt tipped CdSe@CdS ternary nanocomposite with a remarkable AQE of 100% has been reported.32 However, this ternary nanocomposite involves Pt, an expensive component, making it less plausible for the large-scale applications. Furthermore, CdS is used as an active photocatalyst in the two potential reports,31,32 whereas the role of CdS in the overall photocatalytic activity is not very clear in the MoS2-stratified CdS-Cu2−xS ternary nanocomposite.34 For both graphene and 1D nanorod-based ternary nanocomposites, CdS-based nanocomposites have been shown to have the highest AQE.
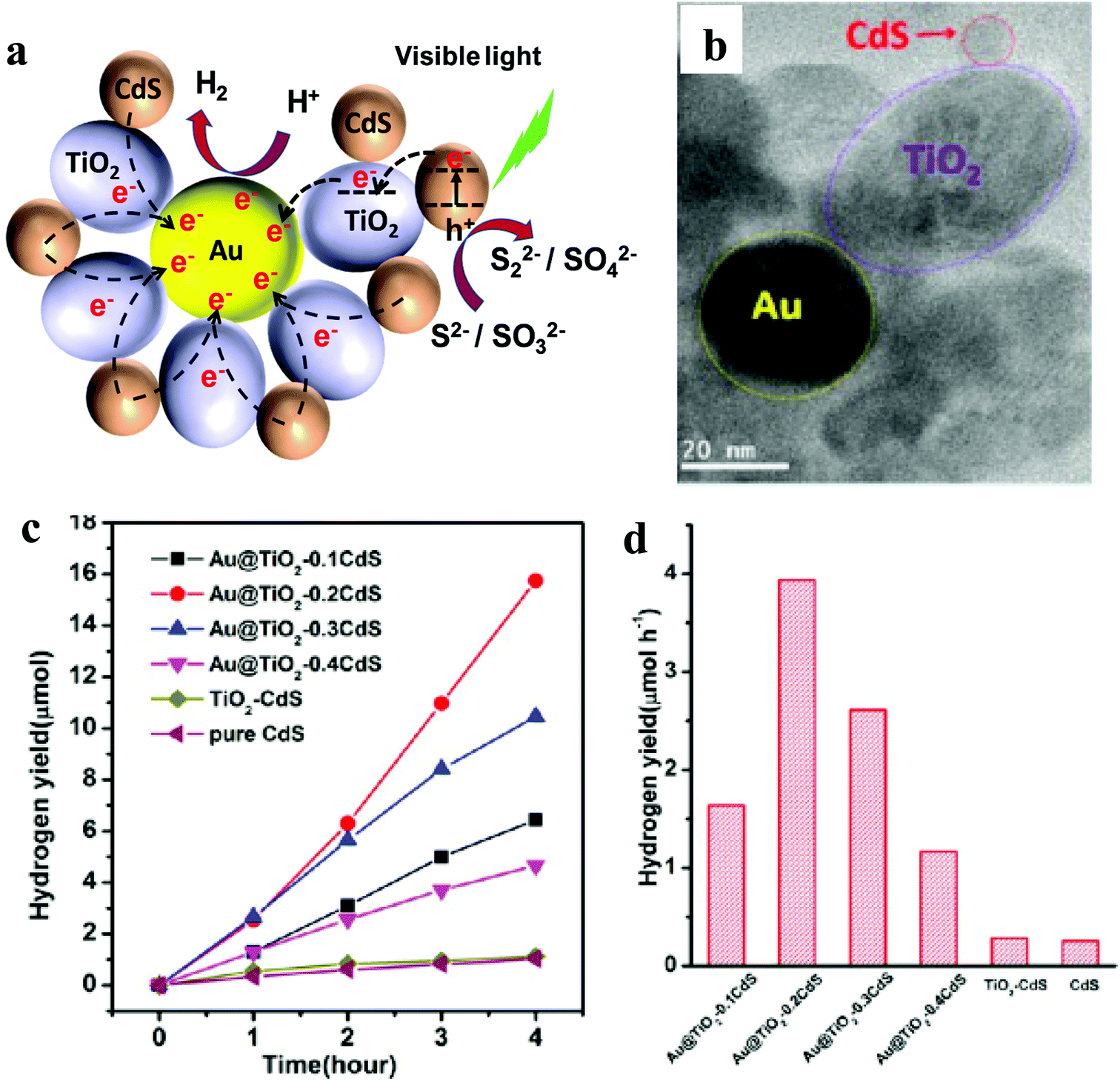 | ||
| Fig. 5 (a) Schematic illustration of photocatalytic activity of the Au@TiO2–CdS nanocomposite, (b) TEM, (c) plots of the photocatalytic HER versus visible light irradiation time and (d) HER for pristine CdS, binary TiO2–CdS and various ternary nanocomposites. Fig. 4b–d: Copyright 2013, American Chemical Society. Reprinted from ref. 35 with permission. | ||
In another report, Liu et al.36 prepared a CdS/Au/ZnO ternary nanocomposite, wherein a hydrothermal process was used to grow wurtzite ZnO (∼5 μm) followed by photo-deposition of Au NPs (10–20 nm) and subsequent deposition of hexagonal CdS NPs (∼50 nm) via chemical bath deposition. Interestingly, SEM imaging showed the final morphology of ZnO as nanosheets, which is attributed to the agglomeration of flower-like shaped crystals. The SAED pattern showed the single-crystalline nature of the nanosheet. The photocatalytic activity was measured in an aqueous solution of Na2S–Na2SO3. Compared with the HER of CdS/ZnO (134), CdS/Au/ZnO (608) showed remarkably improved photocatalytic activity under UV irradiation. The authors performed a comparative test in order to prove that the Au NPs strengthen the interfacial charge-carrier transfer between CdS (holes) and ZnO (electrons) via the Z-scheme by studying the photocatalytic activity of Au/CdS/ZnO (HER: 197). The photogenerated electrons and holes remaining in the CB CdS and VB ZnO, respectively, can be involved effectively in photocatalytic reactions for hydrogen evolution.
Zhang et al.39 have synthesized the g-C3N4/Pt/TiO2 ternary nanocomposite (Fig. 6) through a facile two-step synthetic methodology, involving photo-deposition of Pt NPs on the surface of anatase TiO2 NRs (diameter ∼ 30–80 nm and lengths of 2–20 μm) and subsequent growth of g-C3N4 (CN) layers, as evidenced by electron microscopy imaging. The TiO2 NRs loaded with Pt NPs are uniformly wrapped with the porous fluffy CN layers. The photocatalytic performance was evaluated in an aqueous solution of 17 vol% triethanolamine sacrificial reagent. Under visible light excitation, TiO2 and Pt/TiO2 did not show any H2 as expected. The observed higher HER of ∼893 (CN/Pt/TiO2) compared to 715 (CN/TiO2) is further supported with the observed higher photocurrent response and longer fluorescence lifetime in the CN/Pt/TiO2 ternary nanocomposite (2.60 ns) compared to CN/TiO2 binary (2.50 ns), Pt/TiO2 binary (0.38 ns), and bare TiO2 (0.36 ns). Furthermore, the electrochemical impedance spectra (EIS) curve radius of all electrodes showed the smallest radius for CN/Pt/TiO2, further suggesting the more efficient separation of electron–hole pairs and faster interfacial charge transfer. It is further shown that CN/Pt/TiO2 can simultaneously produce H2 by water splitting and degrade rhodamine B (RhB) in the same reaction system. The authors have described that under visible light irradiation, some of the photogenerated electrons in CN tend to transfer to the CB of TiO2via Pt NPs, whereas some of them will reduce H+ at the CN CB. At the same time, triethanolamine will consume h+ in the CN VB, while O2 is reduced to O2− at the TiO2 CB, which will further degrade RhB.
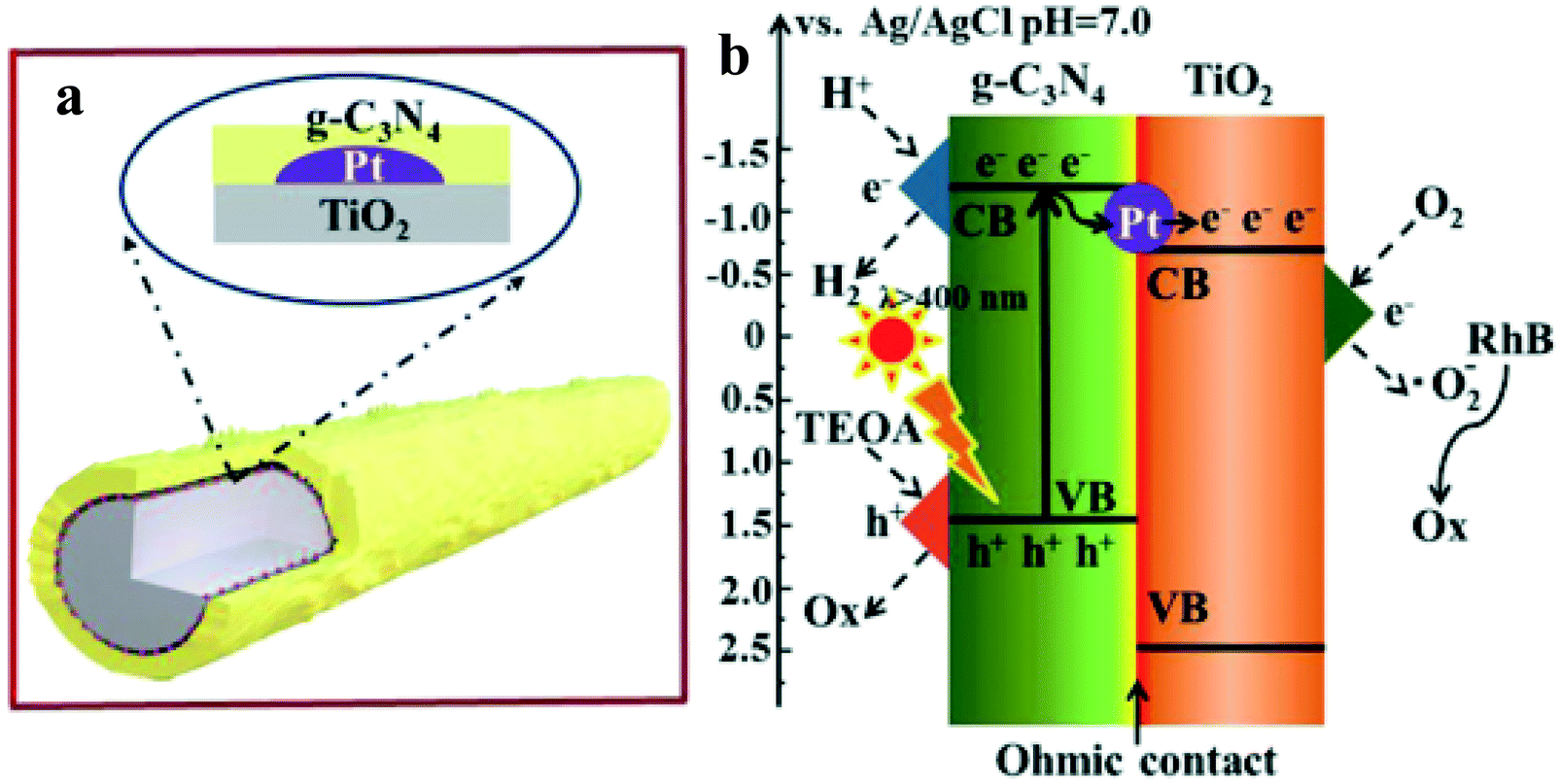 | ||
| Fig. 6 Schematic structure (a) and the photogenerated electron–hole transfer pathways (b) inside the CN/Pt/TiO2 composite under visible light irradiation. Copyright 2019, Royal Society of Chemistry. Reprinted from ref. 39 with permission. | ||
It is important to note here that none of the reports mentioned AQE, making it difficult to compare these three systems. However, based on HER yield, Au@TiO2–CdS has shown the highest activity (1970 μmol h−1 g−1). Besides, these reports again suggest that the ternary nanocomposite involving CdS shows the highest activity, which is similar to that observed for graphene and 1D-NR-based ternary nanocomposites.
:GO (TG), which is 5:1 and 10:1, denoted as 5 and 10, respectively. SEM and TEM imaging suggest smaller Ag NPs decorated on spherical and oval TiO2 NPs (diameter ∼ several to tens of nm), which are randomly distributed over GO's surface. Among various concentrations of AgNO3, the highest absorption was obtained for A15TG(5) and A10TG(10) nanocomposites with the smallest bandgap of ∼1.63 eV and 1.82 eV, respectively, as evidenced by UV-vis spectroscopy, thus indicating a required optimum concentration of Ag for the nanocomposite. The HER was measured in 20% methanol aqueous solution. The observed HER of 705.6 and 427.2 μmol g−1 h−1 with an AQE of 26.2% and 15.8% for A15TG(5) and A10TG(10), respectively, suggests that A15TG(5) is the optimal nanocomposite. The authors have not disused the probable mechanism for H2 production, and they have not mentioned the photocatalyst's stability. It is to be noted that the authors have discussed various trapping experiments to explain the degradation mechanism of pollutants in the water, which in turn increases the H2 production activity.
In short, CdS combined with cocatalysts has shown the best HER yield and high stability. Observed higher yield could be due to the intrinsic high visible light absorption of pristine CdS (bandgap ∼ 2.4 eV) and synergistic effect of the ternary nanocomposite. Simultaneously, the photocorrosion of the visible light active pristine CdS catalyst (∼2.4 eV) is considered the Achilles heel of pristine CdS, whereas the stability has been found to be increased in ternary nanocomposites. This suggests that the formed ternary nanocomposites have enhanced photocatalytic efficiency and increased overall stability.
2.2. Degradation of pollutants (3500)
A substantial amount of work has been published on integrating 2D materials,51–67 0D NPs,68–83 and Ag NP–graphene blends84–89 with an active catalyst for pollutant removal. This review will focus on selected case studies that relate to the degradation of various pollutants such as alcohol, methylene blue (MB), methyl orange (MO), Congo red (CR), and RhB.Chen et al.53 have studied 2D porous g-C3N4/nitrogen-doped GO/MoS2 (CN/NrGO/MoS2) synthesized by the hydrothermal method. Detailed HRTEM (Fig. 7a & b) imaging shows a distinguished and coherent interface among the CNNS, NrGO, and MoS2, indicating the formation of nano-junctions, resulting in better separation of photoinduced charge carriers and more efficient electron transfer within the nanocomposite structure. The photocatalytic oxidation of MB and the reduction of Cr(VI) were simultaneously carried out at a pH value of 3, containing 30 mg of catalyst mixed with aqueous 10 mg L−1 MB solution and 10 mg L−1 K2Cr2O7. The kinetic rate constant (k, min−1) for the oxidation of MB (0.0338) and the reduction of Cr (VI: 0.0157) is, respectively, enhanced by 141 and 265% when the binary photocatalyst of CN/N-doped partially rGO was replaced with the CN/NrGO/MoS2 (Fig. 7c & d). The authors have attributed the enhanced photocatalytic activity to the formation of nano-junctions and efficient light absorption. The observed significant enhancement is further supported with drastic quenching of PL emission (Fig. 7e) in the ternary nanocomposite, implying an enhanced separation of the photogenerated carrier. The photocurrent density of CNNS/NrGO/MoS2 reaches 27.76 μA cm−2, which is 8.14, 3.06, 1.46, and 10.93-fold higher than those of bulk CN (3.41 μA cm−2), CNNS, CNNS/N-doped partially rGO, and MoS2, respectively (Fig. 7f). Moreover, no significant photocurrent changes were observed within 1400 s of illumination, indicating that the nano-junctions are stable. The authors have proposed that photogenerated electrons in the CN CB can be transferred easily to MoS2 sheets via NrGO, where photoreduction reactions occur. Simultaneously, NrGO interlayers also provide pathways for photogenerated holes at the VB of MoS2 to reach the VB of CN for photooxidation reactions.
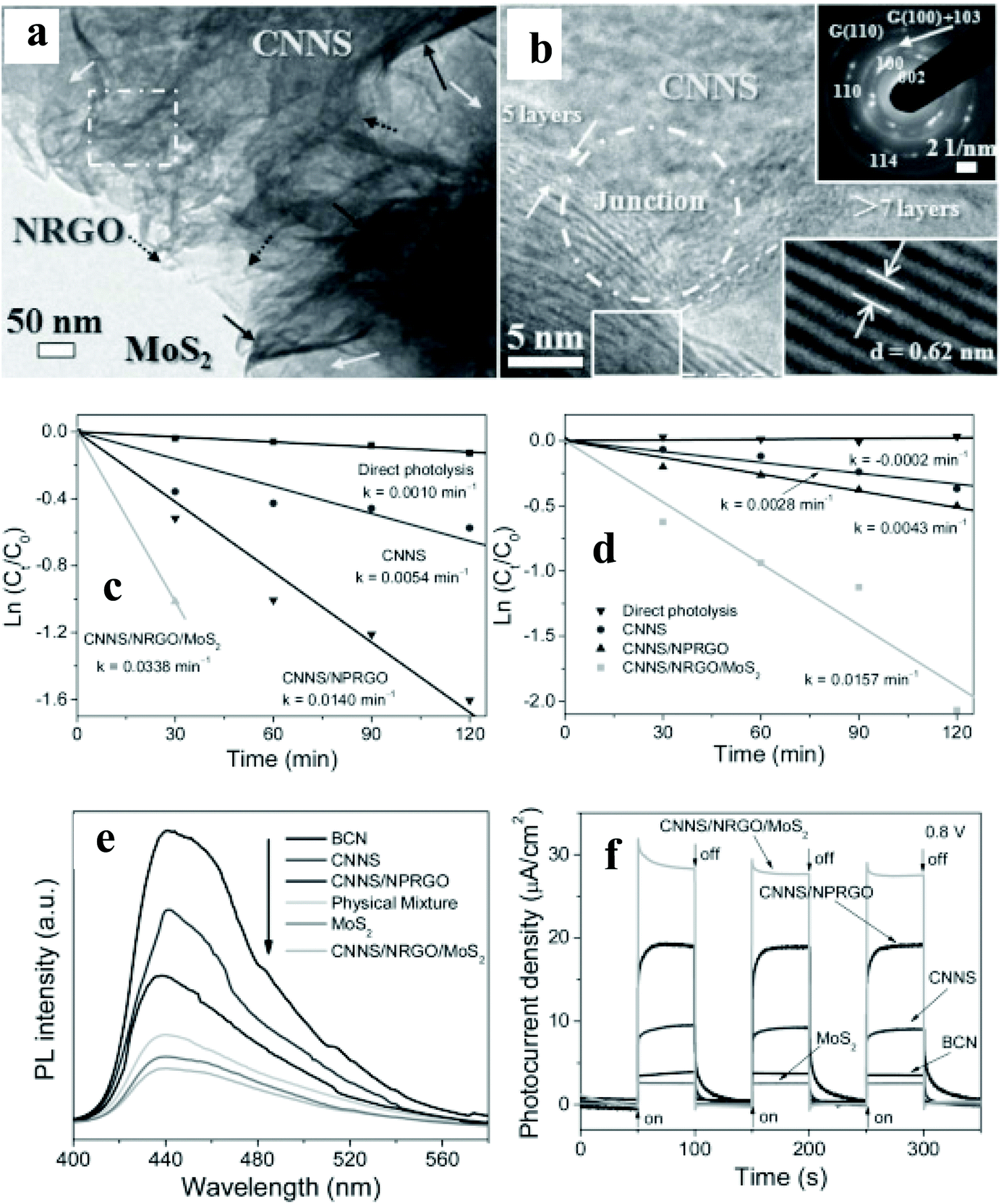 | ||
| Fig. 7 (a and b) TEM images, SAED pattern, and HRTEM images of CNNS/NRGO/MoS2 nanojunctions, (c and d) ln(Ct/C0) of MB and Cr(VI) versus time for different photoactive materials under simulated sunlight irradiation, (e) PL spectra and (f) transient photocurrent density versus Na2SO4 electrolyte. Copyright 2013, Wiley Online Library. Reprinted from ref. 53 with permission. | ||
In another report, Lu and Liang et al.54 have studied MB degradation by using a CN/BiOI/GO composite with 10 mg L−1 MB solution and 10 mg catalyst@300 W Xe lamp. For the preparation of g-C3N4/BiOI/GO, hydrothermally prepared g-C3N4/BiOI and GO were ultrasonicated, followed by drying at 60 °C for 6 h. TEM and HRTEM imaging demonstrated the deposition of g-C3N4/BiOI on the surface of GO. The kinetic rate constant for the photodegradation of MB was found to be ∼0.2334 min−1 for g-C3N4/BiOI/GO, which is ∼6 times higher than that reported by Chen et al.53 (0.0338 min−1@150 W Xe lamp). The authors have interpreted that the key issue for photocatalyst performance enhancement is electron–hole separation rather than adsorption capacity. The studied photocatalyst was found to be stable for five recycles.
In another report by Chen et al.,59 the Bi2WO6/MoS2/rGO (BMG) ternary nanocomposite synthesized via a two-step hydrothermal process has been reported for the photoreduction of Cr(VI). TEM and HRTEM imaging showed anchoring of MoS2 nanosheets (∼100 nm) and orthorhombic B NPs (∼2–15 nm) on rGO uniformly. The photocatalytic activity was determined at λ = 540 nm by decomposing K2Cr2O7 (10 mg L−1) aqueous solution containing 30 mg of the photocatalyst in 0.02 mL lactic acid solution. The BMG ternary nanocomposite with 7.5 wt% MG showed the highest activity with k ∼ 0.05513 min−1, which is in corroboration with the observed higher photocurrent response and negligible PL emission in the same. The photocatalyst was found to be stable for four recycles. This observed rate constant is much higher than that observed by Chen et al.53 (0.0157 min−1), which could be due to the use of lactic acid, monochromatic excitation source, and different active catalysts leading to a difference in the interaction at the nano-junctions.
Tian et al.62 have reported an effective strategy to synthesize a noble metal-free Bi2WO6@MoS2/rGO ternary nanocomposite using a two-step hydrothermal method with a visible light harvester to degrade RhB. The as-prepared Bi2WO6 (BWO) showed a clew-like microsphere structure with an average diameter of ∼4 μm. Among various ternary nanocomposites prepared, BWO/MG with 1.0% MG, containing 5% G showed the best performance. The catalyst's photocatalytic activity was measured in an aqueous solution of RhB (10 mg L−1) and 20 mg of photocatalyst@300 W Xe arc lamp. The observed kinetic rate constant of ∼0.01157 min−1 for Bi2WO6@MoS2/rGO is much higher than ∼0.00118 min−1 (BWO), suggesting that the ternary heterojunction showed the highest photodegradation rate. The authors have proposed that the photoexcited electrons on the CB of MoS2 can transfer to the CB of BWO via rGO, which can react with O2 to produce O2˙−. These oxidative species play a vital role in the oxidative degradation of organics.
Lee et al.67 have reported that CN hybridized with CdS NPs and rGO using a facile chemical method for the photodegradation of RhB and CR dyes under visible light and UV irradiation. HRTEM imaging suggests decorating hexagonal CdS NPs (∼40 nm) over the entire CN sheets, which are further dispersed onto rGO. The photocatalytic activity toward photodegradation of a 4 mg L−1 solution of RhB and a 20 mg L−1 solution of CR was measured with 100 mg photocatalyst. For RhB, the rate constant was found in the order of 0.02192 (CN/CdS/rGO) > 0.01049 (CN/rGO) > 0.00931 (rGO/CdS) > 0.00643 CN/CdS > 0.00177 (CN), whereas, for CR, the rate constant was found in the order of 0.02437 (CN/CdS/rGO) > 0.01480 (CN/rGO) > 0.00556 (rGO/CdS) > 0.00406 CN/CdS > 0.00191 (CN). The observed higher activity of CN/CdS/rGO is further corroborated with the decreased PL emission and the larger surface area of the CN/CdS/rGO (70.42 m2 g−1), resulting in rapid adsorption of dye onto the surface of the photocatalyst leading to effective degradation of organic pollutants. Post photodegradation, the total organic carbon concentration was measured to confirm the dye molecules’ complete degradation. Due to the large electron mobility of the rGO, the authors suggested that captured photoelectrons effectively transported from g-C3N4 to CdS and then to rGO. The captured electrons were utilized for the formation of O2˙− and OH˙ radicals, which were further used for degradation.
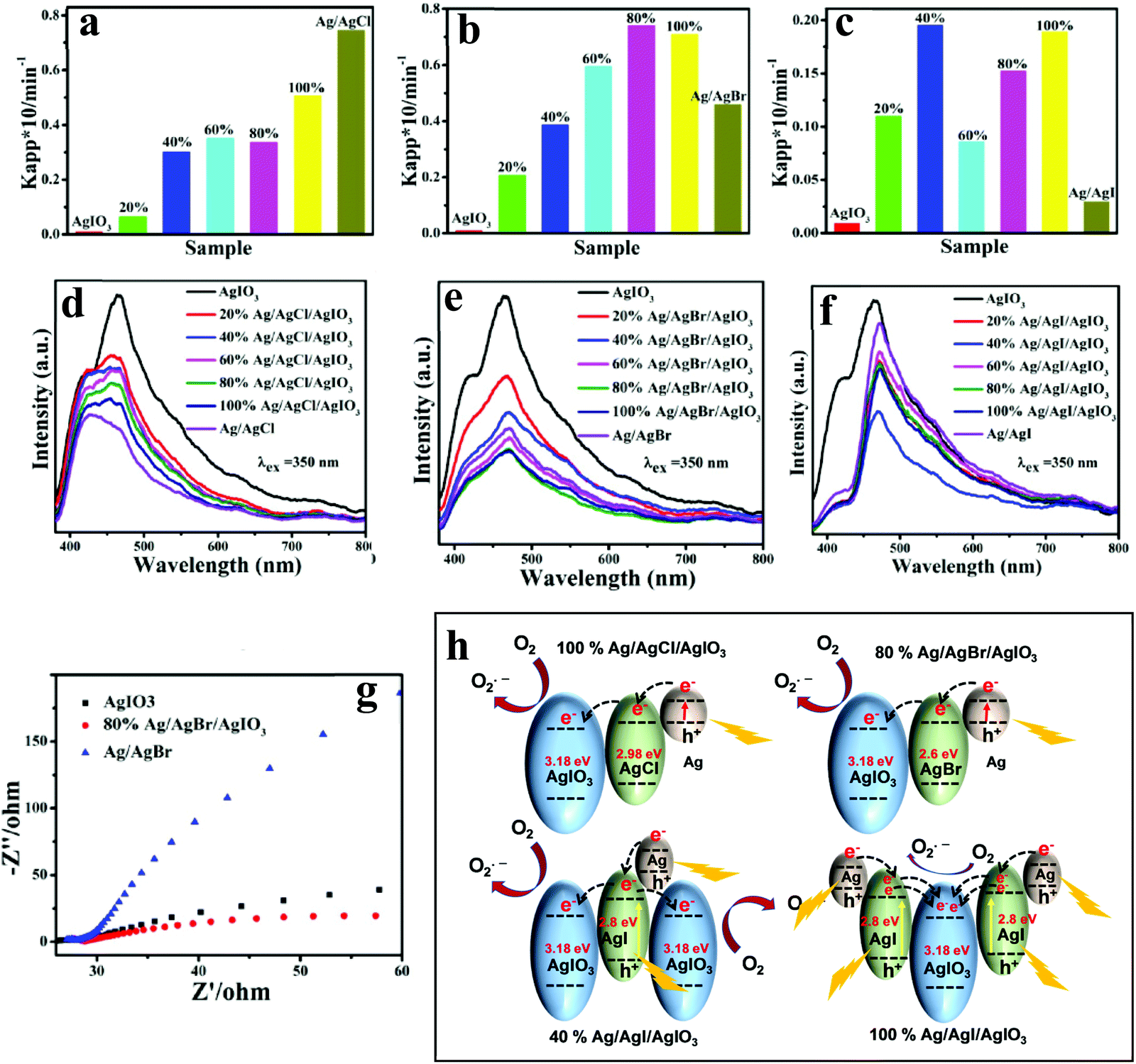 | ||
| Fig. 8 Apparent rate constants for the photodegradation of MO over AgIO3, Ag/AgCl, and Ag/AgCl/AgIO3 composites (a); AgIO3, Ag/AgBr, and Ag/AgBr/AgIO3 (b); AgIO3, Ag/AgI, and Ag/AgI/AgIO3 (c) under the irradiation of visible light, corresponding (d–f) PL spectra under an excitation of 350 nm, and (g) EIS Nyquist plot of AgIO3, 80% Ag/AgBr/AgIO3, and Ag/AgBr. (h) Schematic illustration of different charge-transfer mechanisms of 100% Ag/AgCl/AgIO3, 80% Ag/AgBr/AgIO3, 40% Ag/AgI/AgIO3, and 100% Ag/AgI/AgIO3. Fig. 6a–g: Copyright 2016, American Chemical Society. Reprinted from ref. 75 with permission. | ||
Niu and Zeng et al.76 have studied the Ag/AgCl/SrTiO3 ternary nanocomposite grown using the precipitation reaction between AgNO3 and NaCl at ambient temperature, wherein Ag/AgCl NPs were immobilized on the surface of SrTiO3 nanocubes. RhB, MO, MB, phenol, and bisphenol A (BPA) mixed with 50 mg of photocatalyst were utilized as target contaminants (10 mg L−1) to assess the photocatalytic activities under visible light excitation. The optimized ternary nanocomposite with 21.6% mass ratio of Ag to SrTiO3 could degrade ∼96% RhB (30 min), 93% MO (40 min) and 96% MB (70 min). The degradation efficiency for phenol and BPA could reach 70% and 83% within 4 h, respectively. Based on these results, the authors suggested that the optimal Ag/AgCl/SrTiO3 was conducive to preventing the charge carrier recombination, as evidenced by PL emission quenching. The authors have proposed that the photogenerated electrons in Ag NPs could transfer to the CB of SrTiO3 and AgCl. Since the CB potentials of SrTiO3 (−0.86 eV) and AgCl (−0.06 eV) are much more negative than the potential of O2/O2˙− (0.046 eV), the electrons in the CB would reduce O2 to O2˙−, which would further oxidize the organic contaminants. Simultaneously, the photogenerated holes can migrate to the AgCl surface to oxidize Cl− to Cl˙, which can oxidize organic contaminants and get reduced to Cl− again. No significant changes in the photocatalytic degradation of RhB repeated five times suggest good stability.
Choi et al.81 have studied noble metal-free CdS/TiO2/WO3 for photodegradation of polyoxometalate (POM: PMo12O403−). P25 TiO2–WO3 was synthesized by the sol–gel method followed by hydrolysis of CdS and it was found that 1:1:1 molar ratio of CdS:TiO2:WO3 gave the best output. HRTEM imaging showed uniform decoration of CdS onto TiO2, which is uniformly dispersed on WO3. The visible-light photocatalytic activity was measured with 100 mg L−1 POM in the presence of methanol (electron donor) and t-butyl alcohol at pH of ∼2.5 to support the reduction of POM is indeed due to CB electrons. The observed activity increased in the order of TiO2–WO3 < CdS < CdS–WO3 ≈ CdS–TiO2 < CdS–TiO2–WO3. The authors have suggested that photogenerated electrons in CdS efficiently transfer to WO3via TiO2 because of the cascaded positioning of the CBs of three semiconductors, which is consistent with the observed significant photocurrent generation efficiency of the ternary nanocomposite. However, the authors have also mentioned that the ternary nanocomposite was not very stable, and its photocatalytic activity was gradually reduced with repeated uses. The authors have attributed the issue to the instability of CdS; this is in contrast to other reports,32 which established enhanced stability of CdS in the ternary nanocomposite.
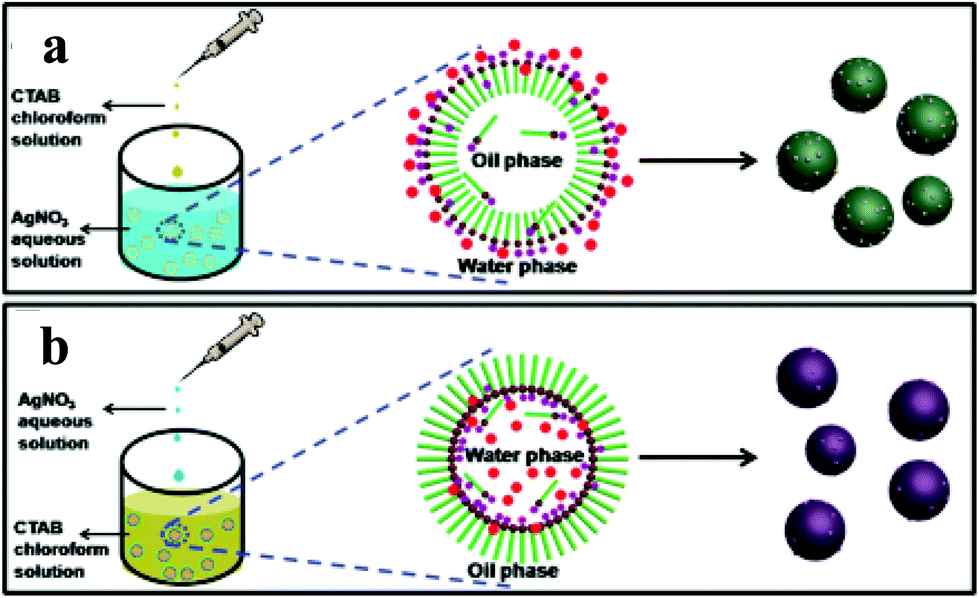 | ||
| Fig. 9 Schematic explanation for the one-pot surfactant-assisted synthesis of Ag/AgBr-based nanostructures via an oil/water (a) and a water/oil (b) microemulsion system. Copyright 2012, American Chemical Society. Reprinted from ref. 84 with permission. | ||
As an application, Chen et al.88 have reported a Ag/TiO2/rGO based reusable and highly sensitive surface-enhanced Raman scattering (SERS) substrate by implying the photocatalytic ability of TiO2 for the photodegradation of the adsorbed 4-aminothiophenol (ATP) molecules. The presence of Ag NPs onto TiO2 NPs decorated uniformly on wrinkled rGO was evidenced by TEM. A remarkable reduction in the detection limit of 4-ATP using ternary Ag/TiO2/rGO (10−14 M) as compared to the binary Ag/rGO (10−10 M)91 substrate was observed. The substrate cleaning was carried out via the photodegradation under UV illumination (30 min), and all characteristic peaks of 4-ATP were found to disappear. In the five cycles examination, the SERS spectrum of 4-ATP using the recycled substrate had almost the same intensity. This demonstrated that the presence of the TiO2 interlayer could restrain the enhancement effect of Ag nanoparticles on the Raman signals of rGO and could act as the photocatalyst for the surface cleaning to make the substrate reusable.
Degradation of various pollutants such as MB, Cr(IV), RhB, MO, and alcohol by employing ternary nanocomposites has shown better performance than their binary counterparts. Furthermore, a comparison of the different reports suggests that the highest kinetic rate constant for the photodegradation of MB was found to be ∼0.2334 min−1 by employing the g-CN/BiOI/GO ternary nanocomposite.54 The observed highest degradation rate constant for Cr(IV) is ∼0.05513 min−1 using Bi2WO6/MoS2/rGO.59 Lee et al.67 have reported an observed rate constant of ∼0.02192 for the degradation of RhB by employing CN/CdS/rGO. These studies suggest that in the case of degradation, GO plays an important role compared to 0D NPs, whereas not a single report of the 1D NR-based nanocomposite has been reported. Chen et al.,88 on the other hand, have observed 94% degradation of MO within 4 by employing Ag/AgBr/GO. The observed distinct enhancement of the catalytic activity upon the hybridization of GO is attributed to the smaller size of Ag/AgBr/GO and the excellent adsorptive capacity of Ag/AgBr/GO. This shows that the synergistic effect of graphene and 0D NPs can be combined to have high efficiency.
In summary, the lower overpotential for a redox reaction, hampered charge recombination and efficient charge transfer are the three crucial factors to enhance the photocatalytic activity. The superior activity of ternary nanocomposites can be attributed to the lower over-potential and synergistic effect of multiple nano-junctions. The formation of multiple nano-junctions leads to intimate contact between various nanocomposite components to facilitate efficient charge transfer among various components and thus enhanced separation of the photogenerated carriers. Although these are possible in binary nanocomposites, however, H2 generation or pollutant removal cannot be maximized because of either a low electron transfer rate or high overpotential, whereas, in the bare catalyst, fast recombination of charge carriers hampers the photocatalytic activity. Furthermore, the nano-size effect giving rise to more active sites for the adsorption of H+ ion or pollutants enhances the activity. The formation of ternary nanocomposites has additionally led to the increased stability of the active catalyst.
2.3. The photocatalytic mechanism in ternary nanocomposites
Several types of developed hetero-photocatalysts with different mechanisms,92 such as type-II, S-scheme, and Z-scheme, are discussed here, in brief, to bring out the similarity and difference in the photocatalytic mechanism of ternary and binary nanocomposites. For example, in the type-II binary photocatalyst, interparticle electron transfer in one direction and interparticle hole transfer in the opposite direction when two semiconductors are coupled together give rise to spatial separation of charge carriers. Although spatial separation of charge carrier type-II photocatalysts can hamper the charge recombination; however, the type-II system has its shortfalls. The improved charge-separation efficiency occurs at the cost of reduced redox ability, an unfavorable condition for the photocatalytic reaction. This is because photogenerated electrons pile up in the CB of semiconductors with weak reduction potential. In contrast, photogenerated holes gather in the VB of semiconductors with weak oxidation potential, which may weaken the overall driving force. The S-scheme binary heterojunction,93 on the other hand, is composed of reduction and oxidation photocatalysts with staggered band structures, wherein the significant photogenerated electrons and holes are reserved in the CB and VB of reduction and oxidation photocatalysts, respectively. In contrast, the inefficient photogenerated charge carriers are recombined. This whole process, thus, introduces a strong redox potential and stronger driving force for the photocatalytic reaction. In comparison, the Z-scheme involves the generation of electron–hole pairs in semiconductor (1), followed by the injection of these electrons into the coupled semiconductor (2) for hydrogen evolution, whereas VB holes of the semiconductor (1) are usually involved in a water oxidation reaction.94It is to be noted that this clear distinction of various mechanisms is made for binary nanocomposites, wherein both the components used are semiconductors and usually take part in the generation of charge carriers (type-II, S-scheme). Whereas when it comes to ternary nanocomposites, three different components are involved with mainly one active component (i.e., semiconductor to generate charge carriers upon light irradiation) and two supportive (cocatalyst) components. Due to (uni- or bi-) directional charge flow from the active catalyst towards cocatalyst/s and vice versa, it is tricky to recognize the specific type of mechanism in these ternary nanocomposites, a mechanistic fact which is in contrast to that of binary nanocomposites. Although a few of the reported studies on ternary nanocomposites have titled the mechanism type (such as the Z-scheme36), the underlying mechanism in most of the reported potential systems has been conversed without any specific type21,23,32 due to the complexity of charge transfer among the aforesaid three components.
2.4. Ternary electrocatalyst
A few reports have been published on the integration of noble metal NPs and carbon nanotube/graphene with active electrocatalysts for their application in supercapacitors.95–102Wang et al.99 have reported an N-doped carbon nanotube (NCT)/Au NP/MnO2-based thin film supercapacitor with ultrafast charge/discharge. TEM imaging shows the as-prepared MnO2 thin films with distributed Au NPs covering the surface of NCT (Fig. 10a–c). A three-electrode system is used to evaluate the electrochemical performances of NCTs/Au/MnO2 and NCT/MnO2 electrodes. The achieved gravimetric capacitance of ∼1106 and ∼740 F g−1 at a current density of 5 A g−1 with a capacity retention of 103% and 75% over 5000 cycles with NCT/Au/MnO2 and NCT/MnO2 electrodes, respectively, shows better performance of the ternary nanocomposite. The observed enhanced performance is attributed to three-dimensional scaffolds of NCT, providing a larger surface area and shorter ion diffusion. Furthermore, Au NPs distributed on MnO2 thin layers can provide abundant Au/MnO2 interfaces for rapid electron transport, thus, enhancing the conductivity of MnO2 without sacrificing its inherent electrochemical performance. To further explore the electrochemical properties of the NCTs/Au/MnO2 in practical application, the authors have studied a solid-state flexible symmetrical supercapacitor (SSC: NCTs/Au/MnO2//NCTs/Au/MnO2) and asymmetrical supercapacitor (ASC: NCTs/Au/MnO2//Fe2O3 nanorods) prototype device. The observed symmetrical discharge–charge curves have been attributed to the good capacitive behavior of the SSC and ASC devices (Fig. 10d–g). Furthermore, the SSC device exhibited a large energy density of 51 W h kg−1 (Fig. 10e), whereas the ASC device exhibited ultrafast charge/discharge (10 V s−1). The superior cycling performance for SSC (93%) and ASC (97%) devices over 5000 cycles has been noted. The SSC and ASC device's excellent rate performance was attributed to the lower resistance and faster charge transport, which is supported by nearly no semicircle in the high-frequency region in the Nyquist plots, indicating an ultrasmall transfer resistance ensuring fast charge transfer between the electrolyte and electrodes.
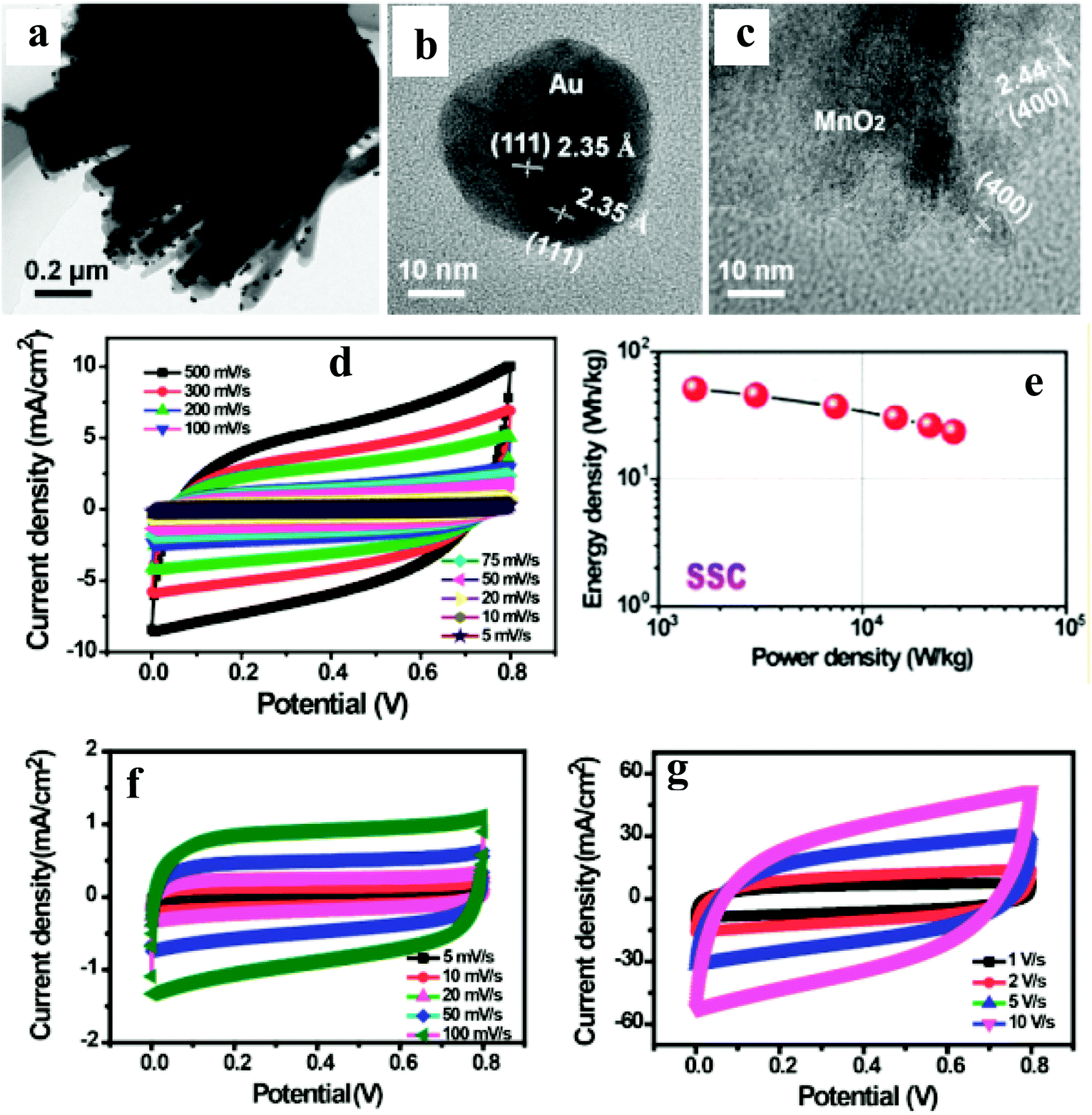 | ||
| Fig. 10 (a) Low-magnification TEM image of NCTs/ANPDM nanocomposite, (b and c) TEM images of Au NPs covered by the MnO2 and MnO2 besides the Au NPs on the surface of the NCTs, respectively. (d) CV curves of the assembled solid-state SSC device collected at different scan rates, (e) Ragone plots of the solid-state SSC device, and (f and g) CV curves of the assembled solid-state ASC device collected at different scan rates. Copyright 2016, American Chemical Society. Reprinted from ref. 98 with permission. | ||
In another report, Li and Liu et al.100 have synthesized a 3D macroporous structure of Ag NPs decorated on MnO2/sulfonated graphene (SG) by a hydrothermal process. SEM and TEM imaging demonstrated the hollow SG nanobeads with a well-ordered 3D porous structure. The average diameter of the hollow beads is ∼300 nm. SEM imaging further showed that MnO2 NPs with Ag NPs (∼8 nm) were deposited on SG's surface. A three-electrode system has evaluated the electrochemical performances of Ag-MnO2/SG, MnO2/SG, and MnO2 electrodes. The cyclic voltammetry (CV) curves at a 5 mV s−1 scan rate show that the 3D Ag-MnO2/SG electrode possesses enhanced charge storage ability compared to bare MnO2 and the MnO2/SG binary nanocomposite. A retained quasi-rectangular shaped CV curve of the Ag-MnO2/SG electrode at a high scan rate of 100 mV s−1 indicates fast charge propagation capability and facile ion transport. The maximum specific capacitance for Ag-MnO2/SG could reach 537.0 F g−1, which is higher than that of the MnO2/SG (508.8 F g−1) and MnO2 (172.26 F g−1) at a current density of 0.5 A g−1. Furthermore, the observed capacity retention of 99.72% for the Ag-MnO2/SG electrode after 5000 cycles is much higher than that of bare MnO2 (77.36%). From the Nyquist plots, the authors have inferred that the Ag-MnO2/SG shows the minimum charge transfer resistance due to the formation of intimate contact at the interface. The superior performances of Ag-MnO2/SG are attributed to (i) the interconnected 3D porous SG networks, which can reduce the diffusion distance for the electrolyte and accumulating more electrolyte ions, improving the rate performance and capacity, and (ii) improved conductivity due to Ag NPs.
Lan and Wang et al.101 have synthesized Ag NP embedded Co3O4 nanosheets on a three-dimensional graphene (3DG) network (Ag/Co3O4/3DG) by a hydrothermal method. The lack of the D-peak in the Raman spectra has been attributed to fewer defects in 3DG, giving rise to excellent conductivity, a desirable factor for current collectors in supercapacitors. The electrochemical performance was measured in a three-electrode configuration. Observed clear redox peaks in the CV curves were attributed to the conversion between different cobalt oxidation states. At a current density of 1 mA cm−2, the galvanostatic charge/discharge curves show that the charge and discharge times of bare Co3O4 foam and the Co3O4/3DG binary nanocomposite are shorter than those of the Ag/Co3O4/3DG hybrid. The areal-specific capacitance (Ca) was calculated to be 303.9 mF cm−2 (Ag/Co3O4/3DG), 145.9 (Co3O4/3DG), and 6.7 (Co3O4 foam), which is attributed to the enhanced conductivity in the ternary nanocomposite due to the formation of an ohmic contact. Furthermore, the areal capacitance retention of ∼120% after 104 cycles indicates a strong cycling performance. The galvanostatic charge/discharge curves of the Ag/Co3O4/3DG hybrid are found to be highly linear and symmetrical without an obvious IR drop, indicating a rapid IV response and excellent electrochemical reversibility. The coulombic efficiency of the Ag/Co3O4/3DG hybrid is above 97% during cycles. As displayed, the Nyquist plot of the Ag/Co3O4/3DG hybrid exhibits a negligible semicircular shape in the high-frequency range, which is related to the faradaic charge transfer resistance suggesting that the electrode possesses effective interfacial charge transfer efficiency. The authors have further the electrochemical properties of the two-electrode configuration for practical applications. The two-electrode configuration exhibited the Ca of 53.3 mF cm−2 at 1 mA cm−2. After 5000 cycles, the areal capacitance reaches 49.1 mF cm−2 and remains unchanged up to 10000 cycles.
In short, superior electro-catalytic activity in ternary nanocomposites, i.e., good electric conductivity, excellent specific capacitance, power performance, and good cyclic stability, can be attributed to fast electrode kinetics for facilitating fast electron transport from the electrode/electrolyte interface and improved conductivity due to the presence of NCT/graphene and Ag NPs.
3. Conclusions
With increasing attention toward the carbon-neutral society, the solar energy resource dominating the fossil-based energy resource is receiving heightened attention as a potentially widespread approach to achieve sustainable and green energy production. The possible energy production with sunlight striking earth in 1 h is much more than the entire year’ energy consumed. However, the sun's diurnal nature necessitates the idea of harnessing solar energy for conversion and storage cost-effectively by employing photo/electrocatalysts. However, the conventional (homogeneous) catalyst appears far-fetched, demanding special efforts to develop unconventional (heterogeneous) catalysts. Nature has provided the existential proof of heterogeneous catalysts, with the hydrogenase enzymes operating at the thermodynamic potential for production of H2 from H2O, and with the O2 evolving complex of photosystem II producing O2 from H2O in an energy-efficient fashion.Photo/electrocatalysts have been a part of our society for decades with an aim to achieve clean and green sustainable energy systems. Photocatalysis draws inspiration from natural photosynthesis and promises a direct route of photo-to-chemical energy conversion/storage and environmental pollutant removal, whereas photovoltaics employs photo-to-electrical energy conversion, which is stored in batteries/supercapacitors involving electro-catalysis. What has been grouped in this article represents the latest progress in the application of ternary nanocomposites. For convenience, we have categorized the photocatalytic section into four types: 2D materials, 1D nanorods, 0D nanoparticles, and 0D nanoparticle–2D material blends. Additionally, we have discussed Ag nanoparticles–graphene blend-based electrocatalyst, although there are only a few reports for the same. The formations of novel materials with multi nano-junctions have shed light on energy storage capabilities. We believe that the underlying mechanisms discussed can be of great inspiration to study and improve a large class of catalytic reactions.
Photocatalytic activity involving solar water splitting and pollutants’ degradation has been measured employing mainly hydrothermally grown particulate ternary nanocomposites. Sacrificial agents such as Na2S–Na2SO3 and lactic acid solution have been noticed to show a huge difference in the photocatalytic activity. Mainly, TiO2, CdS, and ZnCdS semiconductor-based ternary nanocomposites have been explored and shown enhanced activity compared to their pristine and peers under simulated UV and visible light excitation. It is particularly interesting to note that the apparent quantum efficiency of 100% has already been achieved by employing Pt tipped CdSe@CdS. However, this report includes an expensive Pt element, which makes it impossible for large scale applications in the future. It is evident from the current research that the CdS-based ternary nanocomposite comprising graphene shows AQE values greater than 60%; thus, it holds promise as a next-generation photocatalyst in terms of scalability, stability, and selectivity. Ternary nanocomposites have shown higher stability than their pristine and peers; for example, bare CdS is well known for its shown lesser stability, whereas CdS-based ternary nanocomposites have been proved to be highly stable. The observed higher activity in the ternary systems has been referred to the nano-size effect leading to more active sites and intimate contact between individual components, which can effectively suppress recombination and facilitate charge separation and transfer. Many visible light-responsive materials have been successfully implemented as H2 evolution photocatalysts and pollutants’ degradation due to the relaxation of thermodynamic limitations on photocatalyst materials in Z-scheme systems.
Ternary electrocatalysts involving noble metal nanoparticles and active carbon materials such as carbon nanotubes and graphene have been shown to give better cycling behavior and high energy density and power density. This enhancement has been attributed to three-dimensional scaffolds for binder-free electrodes, larger surface area, and shorter ion diffusion along with noble metal nanoparticle/active electrocatalyst interfaces, which provide direct and stable pathways for rapid electron transport, thus enhancing the conductivity. Thus, the electro-catalytic property can greatly be improved without sacrificing the inherent electrochemical performance. A three-electrode-based-prototype device has already been brought into the light, which has surpassed the perforce of many other electrocatalysts.
Although significant improvements have been achieved by employing ternary nanocomposites, challenges remain that need to be addressed to achieve benchmark efficiency. In the current scenario, the challenges that limit its development are by far connected to low cost, and requisite activity, selectivity, and durability. The challenges nonetheless provide a fertile field for research, to test different hypotheses and material design principles. For photocatalysts, compared with particle suspension systems, immobilized particulates can be more readily scaled up. In addition, the immobilization of photocatalysts on a conducting sub-layer can retain the material's intrinsic activity and show dramatic enhancement in photocatalytic performance.6 Thus, immobilization can provide a prototype device for the large-scale production of low-cost renewable energy resources and illustrate the importance of interfacial interaction, the kinetics of charge transport, adsorption of reactants, and transport of guest species to the active sites in the practical operation of such processes. In the literature, the photocatalytic activity has been mainly carried out using simulated sunlight. However, more experiments need to be carried out with solar light. Furthermore, experiments have been mainly carried out using UV and visible excitation; however, new active materials capable of harnessing IR or near-IR light are required as it covers a significant part of sunlight.
It is essential to mention here that even after decades of research, there is no standard method to compare the different reported efficiencies of photocatalysts in the literature. The amount of H2 produced can differ depending on the photocatalyst mass, reactor used, excitation source/power, and growth parameters.21,23 Therefore, the same photocatalyst can produce different values when used in different labs. We have made an attempt to compare different reports by comparing the rate of hydrogen in units of μmol h−1 g−1 and AQE. However, we would like to mention that hydrogen production and catalyst mass may not follow a linear relation, and often AQE is not measured with a standard protocol.103
Although encouraging results are being achieved; however, the field of a next-generation flexible supercapacitor is nascent. Great efforts are needed to go forward in the electrocatalyst technologies. A few of the challenges are to attain higher energy and higher-power density to fulfill our ever-increasing emery-dependency. Furthermore, advancements in ionic and electrical conductivity are indispensable for new-generation electrode materials at commercial levels. Thinner, lighter, smarter, or even transparent supercapacitors are needed for increasingly multifunctional consumer electronics. However, all this ends at one requirement; the energy storage mechanisms must be deeply understood for controlling interfacial reactions among various components of the electrode as well as between the electrode and electrolyte. A deeper level of understanding requires state-of-the-art in situ microscopic and spectroscopic studies to provide direct experimental proof of various reactions.
Despite significant progress towards high efficiency (HER) and high energy/power density (supercapacitors), most materials are far from the expected performance. Furthermore, the stringent requirements of multifunctionalities and industrial compatibility run far beyond the materials state-of-the-art. The current challenge in the field is the lack of rigorous control in designing decisive morphology for optimal performance on a large scale. In a broader perspective, ternary photo-/electro-catalysts are still in their developing stage. No human-made catalytic system, either homogeneous or heterogeneous, has yet been identified that shows performance even close to natural enzymatic systems. To use the solar source abundantly, it is indispensable to distinguish the functional catalyst and to develop a deep understanding of the mechanism of ternary nanocomposites. This necessitates the structure–property–function relationships in more detail, particularly in operando/in situ condition via recent Synchrotron advancement. The structure here can refer to the crystallographic crystal structure, morphology, and spectroscopic average and nanoscale local structure, for example, a cubic spinel crystal structure with different spectroscopic local structures such as octahedral and tetrahedral sites. Minute changes in spectroscopic signals can lead to enormous change in optical and electronic properties,104,105 which can affect material’ performances. For example, contrary to the current perspective of P25 with remarkable photocatalytic properties, rutile TiO2-based Ni–Ag-TiO2 has shown higher activity compared to the P25-based ternary system due to enormous change in optical and electronic properties.106 In another example, 1 at% doped Mn in lithium titanate showed ∼20% change in the electrochemical performance due to Mn2+ occupation in tetrahedral (8a) sites.107 Furthermore, various charge transfer pathways in such sophisticated designed systems such as mediator-induced interfacial charge transfer108,109 can be probed by synchrotron radiation-based technology as it has turned out to be an effective tool with remarkably high spatial/temporal-resolution,110,111 thanks to its energy tunability and high brilliance. Thus, the elusive contact mechanism between the active photocatalyst and co-catalyst can be revealed via simultaneous spectro-microscopic determinations empowered by synchrotron-based advanced methods for investigating the structure–property function relationships. This will bring all three characteristics to the same page to establish the underlying mechanism, which in turn will be useful to tune the material performance efficiently. A decisive role in tuning and optimizing the existing properties along with the control of their responsible key factors will bring us a step closer to the industrialization of such ternary systems for the greener environment. Overall, efforts inspired by a broad range of scientific areas such as materials science, chemistry, physics, chemical engineering, nanoscience, and nanotechnology will be highly desirable for future breakthroughs in the promotion of overall catalytic efficiency. The development of such catalysts would provide key enabling technology for a full solar energy conversion and storage system. One of the critical possible future directions is to combine experiments and theory to uncover the underlying catalytic mechanism in the ternary systems and the role of various nano-junctions. Besides, future work needs to focus on different facile synthetic approaches for constructing stable ternary materials with a highly active crystal surface and/or quantum size on a large scale. Clearly, better catalysts for the multi-electron transformations are needed along with the understanding of multiscale energy conversion processes in these ternary nanocomposites to promote fundamental research into practical applications. Notably, the era of photo/electro-catalytic reactions conducted with a prototype device is expected soon.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We acknowledge the European Regional Development Funding and the Oulu Council. The academy of Finland grants #311934 is acknowledged.References
- J. Barber, Chem. Soc. Rev., 2009, 38, 185–196 RSC
.
- N. S. Lewis, Science, 2007, 315, 798–801 CrossRef CAS
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS
- B. A. Pinaud, J. D. Benck, L. C. Seitz, A. J. Forman, Z. Chen, T. G. Deutsch, B. D. James, K. N. Baum, G. N. Baum, S. Ardo, H. Wang, E. Miller and T. F. Jaramillo, Energy Environ. Sci., 2013, 6, 1983–2002 RSC
- D. M. Fabian, S. Hu, N. Singh, F. A. Houle, T. Hisatomi, K. Domen, F. E. Osterloh and S. Ardo, Energy Environ. Sci., 2015, 8, 2825–2850 RSC
- Q. Wang, T. Hisatomi, Q. Jia, H. Tokudome, M. Zhong, C. Wang, Z. Pan, T. Takata, M. Nakabayashi, N. Shibata, Y. Li, I. D. Sharp, A. Kudo, T. Yamada and K. Domen, Nat. Mater., 2016, 15, 611–615 CrossRef CAS
- P. K. Nayak, S. Mahesh, H. J. Snaith and D. Cahen, Nat. Rev. Mater., 2019, 4, 269–285 CrossRef CAS
- Y. Zhou, C. H. Wang, W. Lu and L. Dai, Adv. Mater., 2020, 32, 1–24 Search PubMed
- J. Ding, W. Hu, E. Paek and D. Mitlin, Chem. Rev., 2018, 118, 6457–6498 CrossRef CAS
- J. H. Kwak, Y.-W. Lee and J. H. Bang, Mater. Lett., 2013, 110, 237–240 CrossRef CAS
- R. S. Kalubarme, H. S. Jadhav and C.-J. Park, Electrochim. Acta, 2013, 87, 457–465 CrossRef CAS
- W. Hong, J. Wang, Z. Li and S. Yang, J. Mater. Chem. A, 2015, 3, 2535–2540 RSC
- S. P. Sancheti, Urvashi, M. P. Shah and N. T. Patil, ACS Catal., 2020, 10, 3462–3489 CrossRef CAS
- A. Ciechanover, A. Hershko and I. Rose, J. Biol. Chem., 2006, 281, e32–e36 Search PubMed
- C. Xu, P. R. Anusuyadevi, C. Aymonier, R. Luque and S. Marre, Chem. Soc. Rev., 2019, 48, 3868 RSC
- J. You, Y. Guo, R. Guo and X. Liu, Chem. Eng. J., 2019, 373, 624–641 CrossRef CAS
- Z. Chang, J. Xu and X. Zhang, Adv. Eng. Mater., 2017, 7, 1700875 Search PubMed
- Z. Wang, C. Li and K. Domen, Chem. Soc. Rev., 2019, 48, 2109–2125 RSC
- J. Ke, M. A. Younis, Y. Kong, H. Zhou, J. Liu, L. Lei and Y. Hou, Nano-Micro Lett., 2018, 10, 69 CrossRef CAS
- Q. Xiang, J. Yu and M. Jaroniec, J. Am. Chem. Soc., 2012, 134, 6575–6578 CrossRef CAS
- K. Chang, Z. Mei, T. Wang, Q. Kang, S. Ouyang and J. Ye, ACS Nano, 2014, 8, 7078–7087 CrossRef CAS
- J. Zhang, L. Qi, J. Ran, J. Yu and S. Z. Qiao, Adv. Energy Mater., 2014, 4, 1301925 CrossRef
- M. Liu, F. Li, Z. Sun, L. Ma, L. Xu and Y. Wang, Chem. Commun., 2014, 50, 11004–11007 RSC
- W. Jiang, Y. Liu, R. Zong, Z. Li, W. Yao and Y. Zhu, J. Mater. Chem. A, 2015, 3, 18406–18412 RSC
- Y. J. Yuan, J. R. Tu, Z. J. Ye, D. Q. Chen, B. Hu, Y. W. Huang, T. T. Chen, D. P. Cao, Z. T. Yu and Z. G. Zou, Appl. Catal., B, 2016, 188, 13–22 CrossRef CAS
- S. N. Guo, Y. L. Min, J. C. Fan and Q. J. Xu, ACS Appl. Mater. Interfaces, 2016, 8, 2928–2934 CrossRef CAS
- Q. Xiang, F. Cheng and D. Lang, ChemSusChem, 2016, 9, 996–1002 CrossRef CAS
- S. Tonda, S. Kumar, Y. Gawli, M. Bhardwaj and S. Ogale, Int. J. Hydrogen Energy, 2017, 42, 5971–5984 CrossRef CAS
- M. Wang, P. Ju, J. Li, Y. Zhao, X. Han and Z. Hao, ACS Sustainable Chem. Eng., 2017, 5, 7878–7886 CrossRef CAS
- S. Chen, F. Li, T. Li and W. Cao, J. Colloid Interface Sci., 2019, 547, 50–59 CrossRef CAS
- X. Wang, G. Liu, L. Wang, Z. G. Chen, G. Q. Lu and H. M. Cheng, Adv. Energy Mater., 2012, 2, 42–46 CrossRef
- P. Kalisman, Y. Nakibli and L. Amirav, Nano Lett., 2016, 16, 1776–1781 CrossRef CAS
- Z. Yue, A. Liu, C. Zhang, J. Huang, M. Zhu, Y. Du and P. Yang, Appl. Catal., B, 2017, 201, 202–210 CrossRef CAS
- G. Liu, C. Kolodziej, R. Jin, S. Qi, Y. Lou, J. Chen, D. Jiang, Y. Zhao and C. Burda, ACS Nano, 2020, 14, 5468–5479 CrossRef CAS
- J. Fang, L. Xu, Z. Zhang, Y. Yuan, S. Cao, Z. Wang, L. Yin, Y. Liao and C. Xue, ACS Appl. Mater. Interfaces, 2013, 5, 8088–8092 CrossRef CAS
- Z. B. Yu, Y. P. Xie, G. Liu, G. Q. Lu, X. L. Ma and H. M. Cheng, J. Mater. Chem. A, 2013, 1, 2773–2776 RSC
- Z. Jiang, J. Pan, B. Wang and C. Li, Appl. Surf. Sci., 2018, 436, 519–526 CrossRef CAS
- H. Yu, W. Liu, X. Wang and F. Wang, Appl. Catal., B, 2018, 225, 415–423 CrossRef CAS
- H. Dou, Y. Qin, F. Pan, D. Long, X. Rao, G. Q. Xu and Y. Zhang, Catal. Sci. Technol., 2019, 9, 4898–4908 RSC
- J. Y. Do, R. K. Chava, Y. Il Kim, D. W. Cho and M. Kang, Appl. Surf. Sci., 2019, 494, 886–894 CrossRef CAS
- K. Maeda, A. Xiong, T. Yoshinaga, T. Ikeda, N. Sakamoto, T. Hisatomi, M. Takashima, D. Lu, M. Kanehara, T. Setoyama, T. Teranishi and K. Domen, Angew. Chem., Int. Ed., 2010, 49, 4096–4099 CrossRef CAS
- Y. Che, Q. Liu, B. Lu, J. Zhai, K. Wang and Z. Liu, Sci. Rep., 2020, 10, 721 CrossRef CAS
- D. Gao, W. Liu, Y. Xu, P. Wang, J. Fan and H. Yu, Appl. Catal., B, 2020, 260, 118190 CrossRef CAS
- Y. Yang, E. Liu, H. Dai, L. Kang, H. Wu, J. Fan, X. Hu and H. Liu, Int. J. Hydrogen Energy, 2014, 39, 7664–7671 CrossRef CAS
- F. J. Sheu and C. P. Cho, Int. J. Hydrogen Energy, 2017, 42, 17020–17029 CrossRef CAS
- F. J. Sheu and C. P. Cho, ChemistrySelect, 2018, 3, 354–362 CrossRef CAS
- K. Chang, D. Geng, X. Li, J. Yang, Y. Tang, M. Cai, R. Li and X. Sun, Adv. Energy Mater., 2013, 3, 839–844 CrossRef CAS
- W. Ho, J. C. Yu, J. Lin, J. Yu and P. Li, Langmuir, 2004, 20, 5865–5869 CrossRef CAS
- C. She, G. W. Bryant, A. Demortière, E. V. Shevchenko and M. Pelton, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 1–8 CrossRef
- Y. Nakibli, P. Kalisman and L. Amirav, J. Phys. Chem. Lett., 2015, 6, 2265–2268 CrossRef CAS
- N. Zhang, Y. Zhang, X. Pan, M. Q. Yang and Y. J. Xu, J. Phys. Chem. C, 2012, 116, 18023–18031 CrossRef CAS
- X. Liu, L. Pan, T. Lv and Z. Sun, J. Colloid Interface Sci., 2013, 394, 441–444 CrossRef CAS
- Y. Hou, Z. Wen, S. Cui, X. Guo and J. Chen, Adv. Mater., 2013, 25, 6291–6297 CrossRef CAS
- K. Dai, L. Lu, C. Liang, G. Zhu, Q. Liu, L. Geng and J. He, Dalton Trans., 2015, 44, 7903–7910 RSC
- A. Akhundi and A. Habibi-Yangjeh, Appl. Surf. Sci., 2015, 358, 261–269 CrossRef CAS
- N. Raghavan, S. Thangavel and G. Venugopal, Mater. Sci. Semicond. Process., 2015, 30, 321–329 CrossRef CAS
- W. K. Jo and N. Clament Sagaya Selvam, J. Hazard. Mater., 2015, 299, 462–470 CrossRef CAS
- D. B. Nimbalkar, H. H. Lo, P. V. R. K. Ramacharyulu and S. C. Ke, RSC Adv., 2016, 6, 31661–31667 RSC
- C. Zhang, G. Chen, C. Li, J. Sun, C. Lv, S. Fan and W. Xing, ACS Sustainable Chem. Eng., 2016, 4, 5936–5942 CrossRef CAS
- Q. Liu, Y. Guo, Z. Chen, Z. Zhang and X. Fang, Appl. Catal., B, 2016, 183, 231–241 CrossRef CAS
- C. Zhou, J. Yan, B. Chen, P. Li, X. Dong, F. Xi and J. Liu, RSC Adv., 2016, 6, 108955–108963 RSC
- M. Liu, X. Xue, S. Yu, X. Wang, X. Hu, H. Tian, H. Chen and W. Zheng, Sci. Rep., 2017, 7, 1–11 CrossRef
- L. Zhang, X. He, X. Xu, C. Liu, Y. Duan, L. Hou, Q. Zhou, C. Ma, X. Yang, R. Liu, F. Yang, L. Cui, C. Xu and Y. Li, Appl. Catal., B, 2017, 203, 1–8 CrossRef CAS
- M. Darvishi, M. Jamali-Paghaleh, F. Jamali-Paghaleh and J. Seyed-Yazdi, Mater. Res. Express, 2017, 4, 016501 CrossRef
- A. Ali, M. R. D. Biswas, Y. Areerob, D. C. T. Nguyen and W. C. Oh, J. Korean Ceram. Soc., 2018, 55, 381–391 CrossRef CAS
- Z. Xie, Y. Feng, F. Wang, D. Chen, Q. Zhang, Y. Zeng, W. Lv and G. Liu, Appl. Catal., B, 2018, 229, 96–104 CrossRef CAS
- R. C. Pawar, V. Khare and C. S. Lee, Dalton Trans., 2014, 43, 12514–12527 RSC
- L. Ye, J. Liu, C. Gong, L. Tian, T. Peng and L. Zan, ACS Catal., 2012, 2, 1677–1683 CrossRef CAS
- Q. Zhu, W. S. Wang, L. Lin, G. Q. Gao, H. L. Guo, H. Du and A. W. Xu, J. Phys. Chem. C, 2013, 117, 5894–5900 CrossRef CAS
- Y. Bu, Z. Chen and C. Sun, Appl. Catal., B, 2015, 179, 363–371 CrossRef CAS
- J. Li, Y. Xie, Y. Zhong and Y. Hu, J. Mater. Chem. A, 2015, 3, 5474–5481 RSC
- K. H. Leong, S. L. Liu, L. C. Sim, P. Saravanan, M. Jang and S. Ibrahim, Appl. Surf. Sci., 2015, 358, 370–376 CrossRef CAS
- L. Sun, W. Wu, Q. Tian, M. Lei, J. Liu, X. Xiao, X. Zheng, F. Ren and C. Jiang, ACS Sustainable Chem. Eng., 2016, 4, 1521–1530 CrossRef CAS
- S. Wu, X. Shen, G. Zhu, H. Zhou, Z. Ji, K. Chen and A. Yuan, Appl. Catal., B, 2016, 184, 328–336 CrossRef CAS
- C. Zeng, Y. Hu, Y. Guo, T. Zhang, F. Dong, Y. Zhang and H. Huang, ACS Sustainable Chem. Eng., 2016, 4, 3305–3315 CrossRef CAS
- S. F. Yang, C. G. Niu, D. W. Huang, H. Zhang, C. Liang and G. M. Zeng, Environ. Sci.: Nano, 2017, 4, 585–595 RSC
- Y. Xu, Q. Liu, C. Liu, Y. Zhai, M. Xie, L. Huang, H. Xu, H. Li and J. Jing, J. Colloid Interface Sci., 2018, 512, 555–566 CrossRef CAS
- Z. Zhang, X. Li, H. Chen, G. Shao, R. Zhang and H. Lu, Mater. Res. Express, 2018, 5, 015021 CrossRef
- Y. Zhen, J. Wang, F. Fu, W. Fu and Y. Liang, Nanomaterials, 2019, 9, 1054 CrossRef CAS
- X. H. Zou, S. W. Zhao, J. G. Zhang, H. L. Sun, Q. J. Pan and Y. R. Guo, Open Chem., 2019, 17, 779–787 CAS
- H. I. Kim, J. Kim, W. Kim and W. Choi, J. Phys. Chem. C, 2011, 115, 9797–9805 CrossRef CAS
- S. Zhang, T. Yu, H. Wen, R. Guo, J. Xu, R. Zhong, X. Liac and J. You, RSC Adv., 2020, 10, 16892 RSC
- A. I. Vaizoğullar, J. Inorg. Organomet. Polym. Mater., 2020, 30, 4129–4141 CrossRef
- M. Zhu, P. Chen and M. Liu, Langmuir, 2012, 28, 3385–3390 CrossRef CAS
- L. C. Sim, K. H. Leong, S. Ibrahim and P. Saravanan, J. Mater. Chem. A, 2014, 2, 5315–5322 RSC
- Y. Chen, W. Huang, D. He, Y. Situ and H. Huang, ACS Appl. Mater. Interfaces, 2014, 6, 14405–14414 CrossRef CAS
- J. Xue, S. Ma, Y. Zhou, Z. Zhang and M. He, ACS Appl. Mater. Interfaces, 2015, 7, 9630–9637 CrossRef CAS
- K. C. Hsu and D. H. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 27571–27579 CrossRef CAS
- K. H. Leong, L. C. Sim, D. Bahnemann, M. Jang, S. Ibrahim and P. Saravanan, APL Mater., 2015, 3, 104503 CrossRef
- N. Zhang, Y. Zhang, X. Pan, X. Fu, S. Liu and Y. J. Xu, J. Phys. Chem. C, 2011, 115, 23501–23511 CrossRef CAS
- K. C. Hsu and D. H. Chen, Nanoscale Res. Lett., 2014, 9, 1–9 CrossRef
- Q. Xu, L. Zhang, B. Cheng, J. Fan and J. Yu, Chem, 2020, 6, 1543–1559 CAS
- J. Fu, Q. Xu, J. Low, C. Jiang and J. Yu, Appl. Catal., B, 2019, 243, 556–565 CrossRef CAS
- Z. Qiang, X. Liu, F. Li, T. Li, M. Zhang, H. Singh, M. Huttula and W. Cao, Chem. Eng. J., 2021, 403, 126327 CrossRef CAS
- J. Yang and J. Y. Ying, Angew. Chem., Int. Ed., 2011, 50, 4637–4643 CrossRef CAS
- Y. Feng, H. Liu, P. Wang, F. Ye, Q. Tan and J. Yang, Sci. Rep., 2014, 4, 1–7 Search PubMed
- J. Kim, H. Ju, A. I. Inamdar, Y. Jo, J. Han, H. Kim and H. Im, Energy, 2014, 70, 473–477 CrossRef CAS
- W. Lan, Y. Sun, Y. Chen, J. Wang, G. Tang, W. Dou, Q. Su and E. Xie, RSC Adv., 2015, 5, 20878–20883 RSC
- Q. Lv, S. Wang, H. Sun, J. Luo, J. Xiao, J. W. Xiao, F. Xiao and S. Wang, Nano Lett., 2016, 16, 40–47 CrossRef CAS
- L. Liu, J. Zhao, W. H. Li, X. Y. Zhang, H. Z. Xu, K. Li, Y. Kai, L. S. Sun, C. Q. Li and F. Q. Liu, RSC Adv., 2016, 6, 94682–94686 RSC
- X. Wang, X. Zhang, W. Han, X. Mu, Y. Zhang, X. Zhao, Y. Chen, Z. Yang, Q. Su, E. Xie and W. Lan, Electrochim. Acta, 2017, 224, 260–268 CrossRef
- J. Li, Y. Sun, J. Wang, J. Tian, X. Zhang, H. Yang and B. Lin, J. Alloys Compd., 2018, 749, 783–793 CrossRef CAS
- H. Kisch and D. Bahnemann, J. Phys. Chem. Lett., 2015, 6, 1907–1910 CrossRef CAS
- E. Rani, A. A. Ingale, A. Chaturvedi, C. Kamal, D. M. Phase, M. P. Joshi, A. Chakrabarti, A. Banerjee and L. M. Kukreja, J. Raman Spectrosc., 2016, 47, 457–467 CrossRef CAS
- E. Rani, A. Ingale, D. M. Phase, A. Chaturvedi, C. Mukherjee, M. P. Joshi and L. M. Kukreja, Appl. Surf. Sci., 2017, 425, 1089–1094 CrossRef CAS
- P. M. Leukkunen, E. Rani, A. A. S. Devi, H. Singh, G. King, M. Alatalo, W. Cao and M. Huttula, Synergistic effect of Ni-Ag-rutile TiO2 ternary nanocomposite for efficient visible-light-driven photocatalytic activity, RSC Adv., 2020, 10, 36930 RSC
- H. Singh, M. Topsakal, K. Attenkofer, T. Wolf, M. Leskes, Y. Duan, F. Wang, J. Vinson, D. Lu and A. I. Frenkel, Phys. Rev. Mater., 2018, 2, 125403 CrossRef CAS
- K. Wang, J. Li and G. Zhang, ACS Appl. Mater. Interfaces, 2019, 1131, 27686 CrossRef
- X. Shi, S. Posysaev, M. Huttula, V. Pankratov, J. Hoszoeska, J. Dousse, F. Zeeshan, Y. Niu, A. Zakharov, T. Li, O. Miroshnichenko, M. Zhang, X. Wang, Z. Huang, S. Saukko, D. L. Gonzalez, S. van Dijken, M. Atatalo and W. Cao, Small, 2018, 14, 1–10 Search PubMed
- E. Paris, T. Sugimoto, T. Wakita, A. Barinov, K. Terashima, V. Kandyba, O. Proux, J. Kajitani, R. Higashinaka, T. D. Matsuda, Y. Aoki, T. Yokoya, T. Mizokawa and N. L. Saini, Phys. Rev. B: Condens. Matter Mater. Phys., 2017, 95, 035152 CrossRef
- H. Singh, D. Donetsky, J. Liu, K. Attenkofer, B. Cheng, J. R. Trelewicz, I. Lubomirsky, E. Stavitski and A. I. Frenkel, Rev. Sci. Instrum., 2018, 89, 045111 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2020 |