
Recent advances of dinuclear nickel- and palladium-complexes in homogeneous catalysis
Wentao
Xu
,
Muzi
Li
,
Liancheng
Qiao
and
Jin
Xie
 *
*
State Key Laboratory of Coordination Chemistry, Jiangsu Key Laboratory of Advanced Organic Materials, Chemistry and Biomedicine Innovation Center (ChemBIC), School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210023, P. R. China. E-mail: xie@nju.edu.cn
First published on 18th June 2020
Abstract
The development of dinuclear transition metal catalysis has gained considerable momentum in recent years. The synergistic interaction between two metals enables the development of dinuclear metal catalysts with high efficiency, robustness and uniqueness. In this highlight, we take the transition metals Ni and Pd as examples, and provide a current perspective of synthetic methodology development catalyzed by dinuclear Ni- and Pd-complexes in the past several years. The new catalytic reactivities of dinuclear Ni- and Pd-complexes are discussed.
 Wentao Xu | Wentao Xu was born in Hubei Province, China. He received his bachelor's degree from the Hefei University of Technology in 2014 and master's degree from the same university in 2017, under the supervision of Professor Huajian Xu. He is currently conducting his PhD studies under the supervision of Prof. Jin Xie and Prof. Chengjian Zhu at Nanjing University. His current research interest focuses on synergistic catalysis. |
 Muzi Li | Muzi Li was born in Jiangsu province, China, in 1998. Since 2017, he has been an undergraduate in Nanjing University. In 2019, he joined Prof. Xie's group, where his current research interest focuses on photochemistry. |
 Liancheng Qiao | Liancheng Qiao was born in Henan province, China, in 1998. Since 2016, he has been an undergraduate in Nanjing University. In 2018, he joined Prof. Xie's group, where his current research interest focuses on manganese chemistry. |
 Jin Xie | Jin Xie is currently a professor at Nanjing University. He was born in Chongqing, China, in 1985. He received his Bachelor's degree from Northeast Forestry University in 2008, and his PhD in 2013 from Nanjing University working under the direction of Prof. Chengjian Zhu. From 2014–2017, he was a postdoctoral research associate at Heidelberg University. In 2017, he returned to Nanjing University to start his independent career. His current research interests lie in bimetallic chemistry and radical chemistry. |
1. Introduction
In recent years, transition metal-catalyzed new chemical bond formation has gained great momentum in synthetic chemistry as it offers a new window to exploit unexplored chemical space.1 For example, in 2010, Pd-catalyzed C–C cross-coupling was identified in the Nobel Prize award to Heck, Negishi and Suzuki owing to their wide application of it in the concise construction of complex molecules.1e Among the transition metal catalysts, Pd and Ni are widely employed due to their high catalytic efficiency. However, to the best of our knowledge, most of these transition metal-catalyzed organic transformations are currently dominated by mononuclear metal catalyst precursors.2 Indeed, in nature a series of metalloenzymes contain binuclear or multinuclear metal centres (Scheme 1). For example, the urease from Bacillus pasteurii (Scheme 1b) contains dinuclear Ni, which is bridged by β-mercaptoethanol in the active site through the sulfur atom and chelates one Ni through a hydroxyl group.3 The synergistic interaction between two or more transition metals enables the development of metalloenzymes with high efficiency, robustness and exclusive selectivity. Inspired by the seminal work from Cotton and Liddle in metal–metal bonds,4 considerable interest have been dedicated to developing dinuclear metal-catalyzed novel organic transformations,5 exploiting the uncovered catalytic activity of Rh,6 Au,7 Mn,8etc.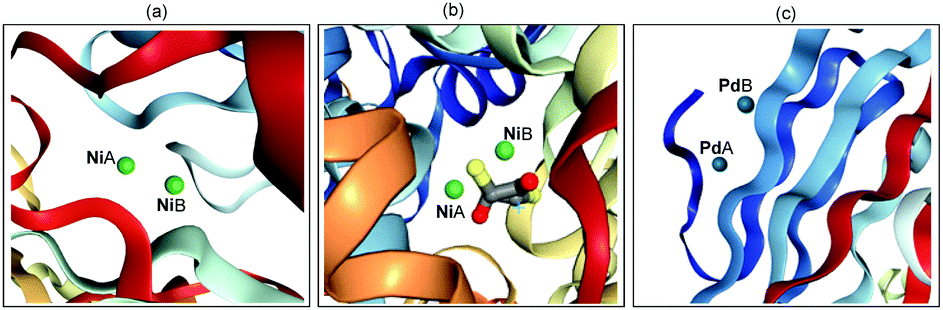 | ||
| Scheme 1 Selected dinuclear metallic enzymes: (a) Bacillus subtilis YXEP protein (PDB entry: 1YSJ), (b) urease from Bacillus pasteurii (PDB entry: 1UBP), (c) Aspergillus niger endoglucanase-palladium complex (PDB entry: 1KS4). | ||
In recent years, great progress has been made in dinuclear metal catalysis for organic synthesis. In this updated highlight, we mainly focus on discussing the recent achievements of dinuclear Ni- and Pd-catalysts involving metal–metal bonds during the last decade. Dinuclear metal catalysts or catalyst precursors9 (Scheme 2) manifest unique or better catalytic reactivity when compared to the corresponding mononuclear metal catalysts. Electronic unsymmetry in the transition state caused by the second synergistic metal centre is one key possibility. Previous reviews have focused on the synthesis of diverse dinuclear metal–metal complexes, but their synthetic applications as catalysts are still highly undeveloped.10,11 Due to the importance of dinuclear metal complexes in catalytic applications, we attempt to introduce historical and more recent cases in which the organic reactions are catalyzed by dinuclear Ni- and Pd-complexes. This highlight can provide a field of vision in dinuclear transition-metal complexes and attract more momentum to this area in near future.
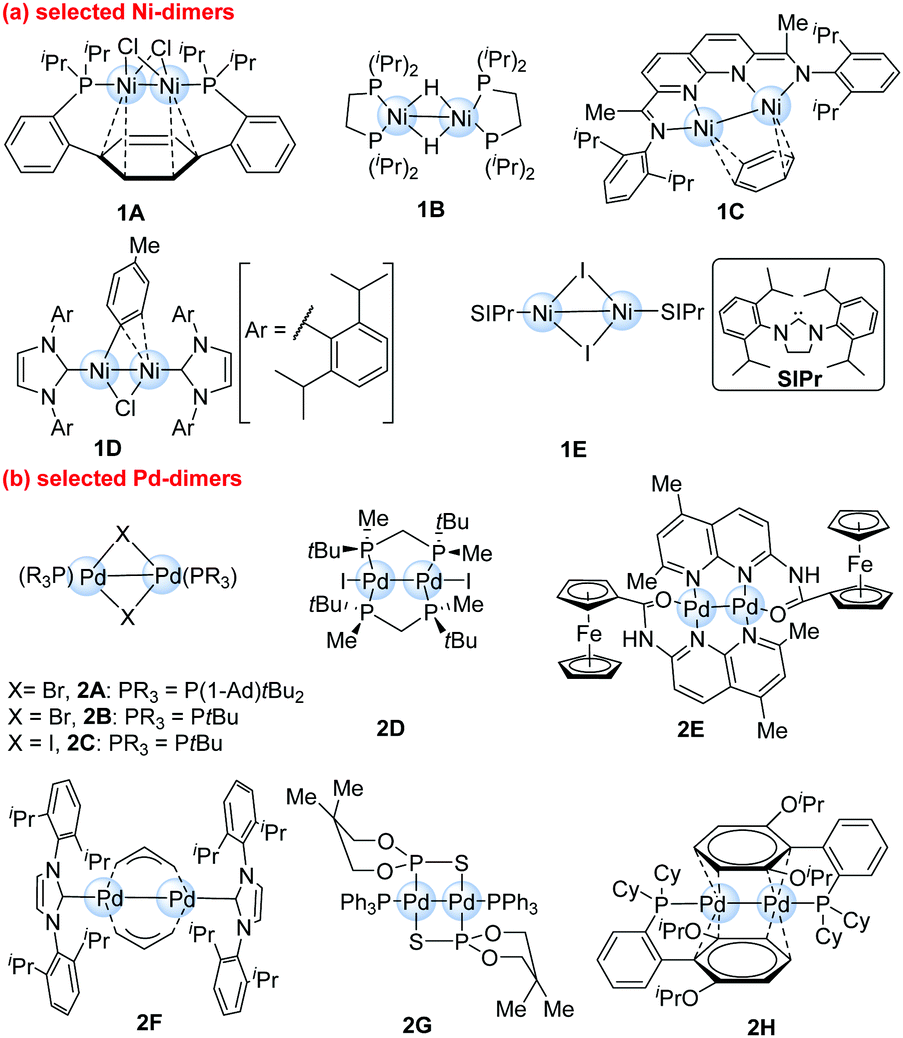 | ||
| Scheme 2 Selected examples of dinuclear Ni- and Pd-complexes with metal–metal bonds. | ||
2. Transformations catalyzed by dinuclear Ni-complexes
In 2010, Agapie and co-workers synthesized a bimetallic NiI–NiI complex (4) stabilized by metal–arene interactions (Scheme 3).12 The dinuclear Ni-complexes (1A), which can be obtained from a designed p-terphenyl diphosphine with Ni(COD)2 and NiCl2(dme), reacted with o,o′-biphenyldiyl Grignard reagents (3) to afford the isolable dinickel species (4). Treatment of the dinuclear nickel-complex (4) with excess CO at room temperature efficiently furnished fluorenones (5a), and the reaction of 4 with excess geminal dichloroalkanes afforded fluorene derivatives (5b). These early examples clearly demonstrate that dinuclear Ni-complex intermediates have promising possibilities in the construction of C–C bonds.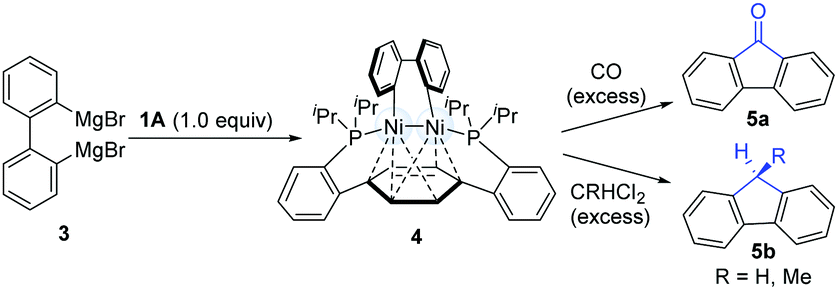 | ||
| Scheme 3 Examining the C–C coupling reactivity of a Ni-dimer complex. | ||
In 2011, Johnson's group successfully synthesized a dinuclear Ni-complex (7) via the C–C bond activation of relatively stable biphenylenes (6), avoiding the involvement of reactive Grignard reagents (Scheme 4).13 The use of a sterically hindered i-Pr3P and Ni(COD)2 together with biphenylene (6) allowed for the successful isolation of a dinuclear Ni-complex (7). Interestingly, a solution of 7 in pentane underwent reductive C–C coupling to generate a green Ni(I)–Ni(I) complex (9). The pathway involved the reductive elimination of the cis-disposed aryl groups at the formally Ni(III) centre in the intermediate 7, which directly resulted in a Ni(I)–Ni(I) complex without any subsequent rearrangements of the σ-bound ligands. In addition, a deuterium-labelling study also indicated that the intermediate 9 was an essential intermediate for the formation of the final coupling product (10). In general, the result was the dinuclear Ni-catalyzed dimerization of biphenylene (6) to produce tetraphenylene (10).
 | ||
| Scheme 4 Dinuclear Ni-catalyzed C–C activation. | ||
The catalytic hydrosilylation of alkynes can streamline the synthesis of silyl-substituted alkenes. To the best of our knowledge, this catalytic transformation is dominated by a mononuclear transition-metal catalyst. In 2012, Osakada and co-workers successfully synthesized dinickel(I) complexes (11) with bridging diphenylsilyl ligands from H2SiPh2 and Ni(cod)2 (Scheme 5).14 The reaction of 1,2-diphenylethyne with the dinuclear Ni-complex (11) generated a dialkenyldisilane (13) in 43% yield by intramolecular coupling of the dinickel structure, accompanied by the formation of an alkyne-coordinated Ni-complex (14).
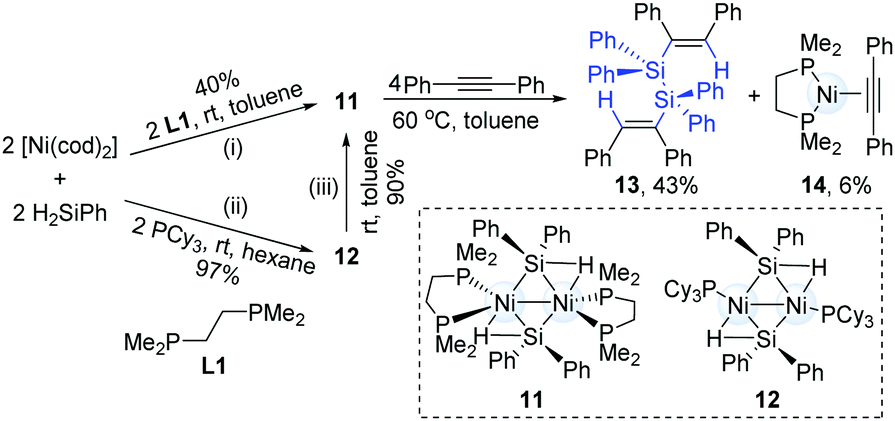 | ||
| Scheme 5 Catalytic hydrosilylation by a dinuclear nickel-complex. | ||
In 2015, Uyeda and co-workers reported the dinuclear Ni-catalyzed hydrosilylations of substrates containing alkenyl and carbonyl groups (Scheme 6).15 As can be seen in Scheme 6a, the activation model for mononuclear transition metals have been proposed to activate organosilanes via a Chalk–Harrod mechanism16 and a Tilley mechanism.17 However, the dinuclear [iPrNDI]Ni2 complex (NDI = naphthyridine-diimine) coordinated with secondary organosilanes (17) provides a complementary pathway to mononuclear Si–H oxidative addition (Scheme 10b). For aliphatic alkenes, such as 1-octene, anti-Markovnikov products (16a) were exclusively formed in a 77% yield, while styrenes yielded an approximately 1.1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of regioisomers (16b). The internal alkynes containing alkyl or aryl substituents were hydrosilylated in a syn fashion to yield (E)-alkene products (16c). In addition, aldehydes (16d) and ketones (16e) could be converted into the corresponding hydrosilylation products in high yields (88–97%). However, N,N-dimethylbenzamide (16f) was reductively deoxygenated to N,N-dimethylbenzylamine in a 76% yield instead of the hydrosilylation product. The control experiments demonstrated that the dinuclear Ni-complex catalyst (1C) was a much more reactive catalyst precursor than the mononuclear [bpy]Ni(COD) or [iPrDAD]Ni(COD) (iPrDAD = N1,N2-bis(2,6-diisopropylphenyl)ethane-1,2-diimine, yields: 93% vs. 0% for 16g).
:1 mixture of regioisomers (16b). The internal alkynes containing alkyl or aryl substituents were hydrosilylated in a syn fashion to yield (E)-alkene products (16c). In addition, aldehydes (16d) and ketones (16e) could be converted into the corresponding hydrosilylation products in high yields (88–97%). However, N,N-dimethylbenzamide (16f) was reductively deoxygenated to N,N-dimethylbenzylamine in a 76% yield instead of the hydrosilylation product. The control experiments demonstrated that the dinuclear Ni-complex catalyst (1C) was a much more reactive catalyst precursor than the mononuclear [bpy]Ni(COD) or [iPrDAD]Ni(COD) (iPrDAD = N1,N2-bis(2,6-diisopropylphenyl)ethane-1,2-diimine, yields: 93% vs. 0% for 16g).
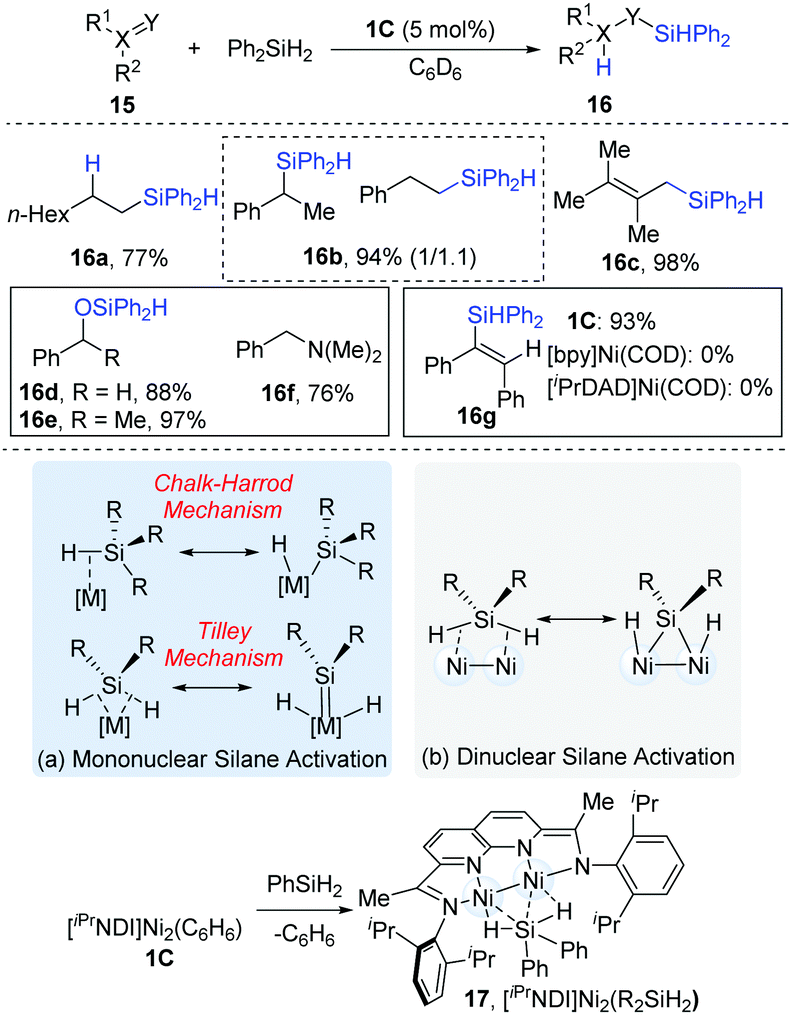 | ||
| Scheme 6 The reductive reaction of CO2 catalyzed by Et3SiH. | ||
Alongside their work in dinuclear nickel chemistry, Uyeda's group in 2015 reported a systematically comparative study of mono- and dinuclear nickel catalysts in the oligomerization of terminal alkynes (Scheme 7).18 When mononuclear nickel catalysts (19–21) were employed, they uniformly gave complex product mixtures, but the dinuclear catalyst (1C) was found to catalyze a rapid and selective cyclotrimerization of alkynes to form 1,2,4-trisubstituted arenes (18). Of note, the dinuclear catalytic system avoided the formation of the common 1,3,5-trisubstituted isomers. A wide range of terminal alkynes were found to be competent coupling partners in the cyclotrimerization. Interestingly, aromatic alkynes bearing electron-withdrawing groups (18c, 18d) underwent cyclotrimerization at a much faster rate (0.5 h vs. 8 h) than alkynes with electron-donating groups (18a, 18b). When 1,6-heptadiynes and propargyl ethers were used, the diarylalkane (18f) was obtained in a 94% yield in 15 min. In an effort to gain insights into the mechanistic pathway of this reaction, the key intermediate (23, R = SiMe2Ph), which could be formed via the reaction of the dinuclear Ni-catalyst and dimethylphenylsilylacetylene in C6D6, was isolated and successfully characterized. As shown in Scheme 7, the proposed mechanism starts with the dimerization of terminal alkynes to yield the intermediate 23, which undergoes selective cyclotrimerization to afford the desired products. The DFT calculation results illustrated that the electronic asymmetry, induced by the second Ni-centre in the dinuclear nickel-complex, could facilitate the formation of the lowest-energy transition state (25) and give 1,2,4-substituted arene products (18). It is thought that the selectivity of the cycloaddition is controlled by electronic bias in the π-system of the diene, caused by a secondary η2 interaction. In 2018, a new dinuclear digermane-catalyzed cyclotrimerization of terminal alkynes was developed to produce 1,2,4-triarylbenzenes with absolute regioselectivity by Sasamori's group.19
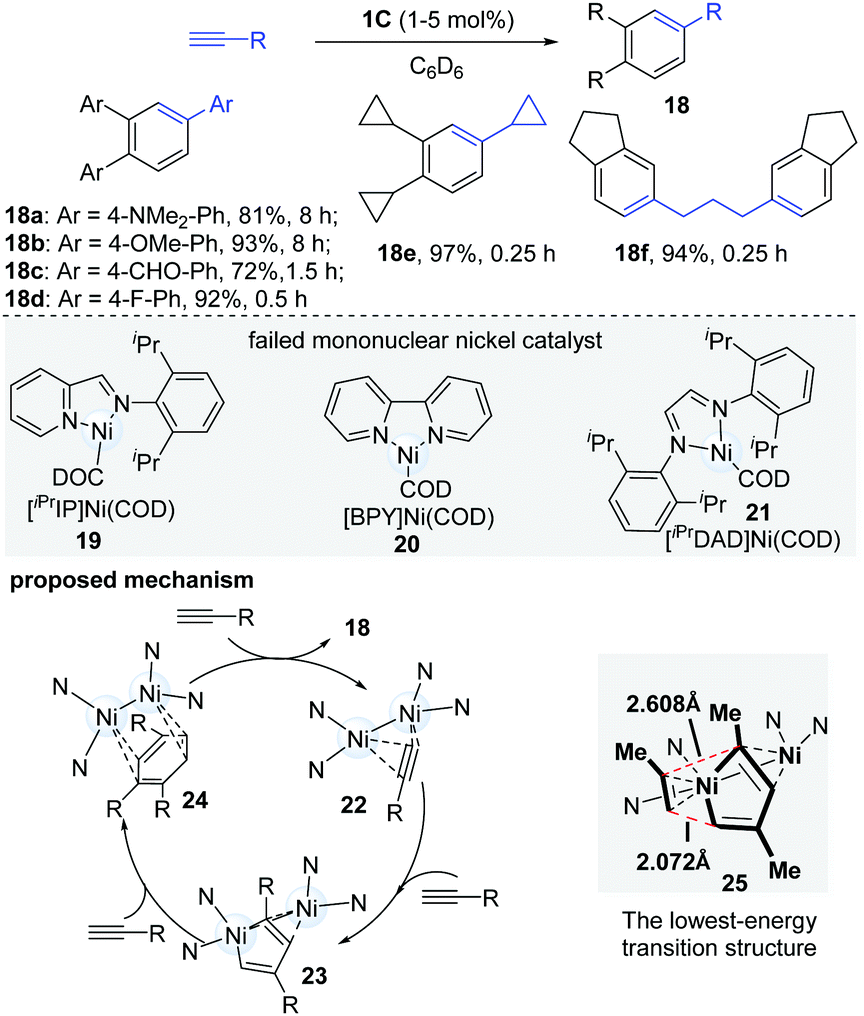 | ||
| Scheme 7 Dinuclear Ni-catalyzed alkyne cyclotrimerization. | ||
The highly reactive dinuclear nickel(I)-complexes (1D) bearing bulky N-heterocyclic carbene (NHC) ligands have been involved in the catalytic cycle of the Kumada–Tamao–Corriu cross-coupling of some aryl halides (Scheme 8).20 The reaction was performed at room temperature, where the less reactive aryl chlorides and fluorides are efficient coupling partners. During this catalytic process, the bridging halogen ligand may play an important role in maintaining the proximal positions of both the nickel centres. However, the other dinuclear Ni(I) species (26) might also play a key catalytic role due to the smaller steric hindrance during the oxidative addition of aryl halides.
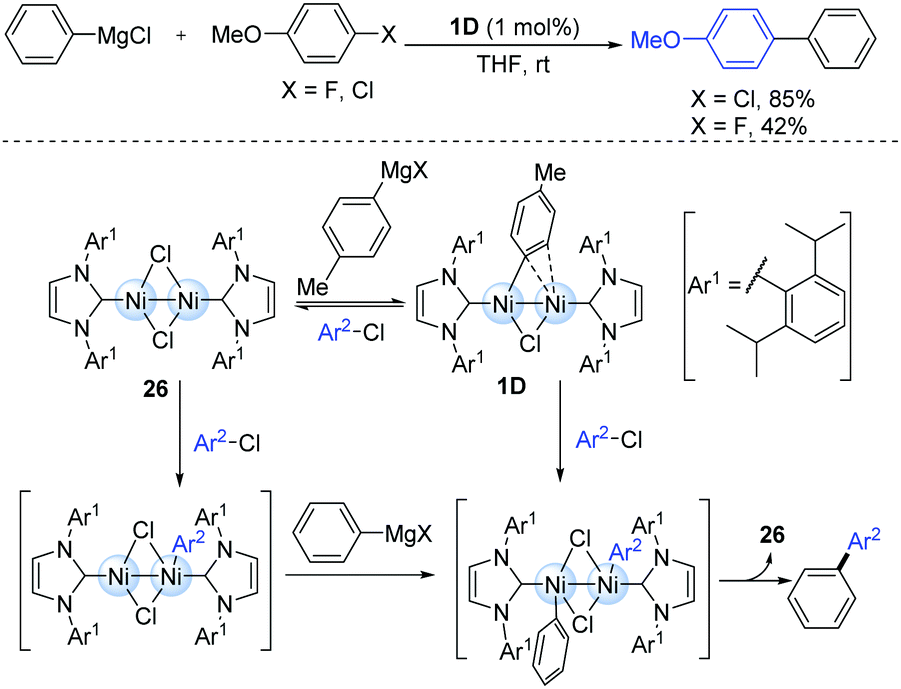 | ||
| Scheme 8 Kumada–Tamao–Corriu cross-coupling mediated by a Ni-dimer complex. | ||
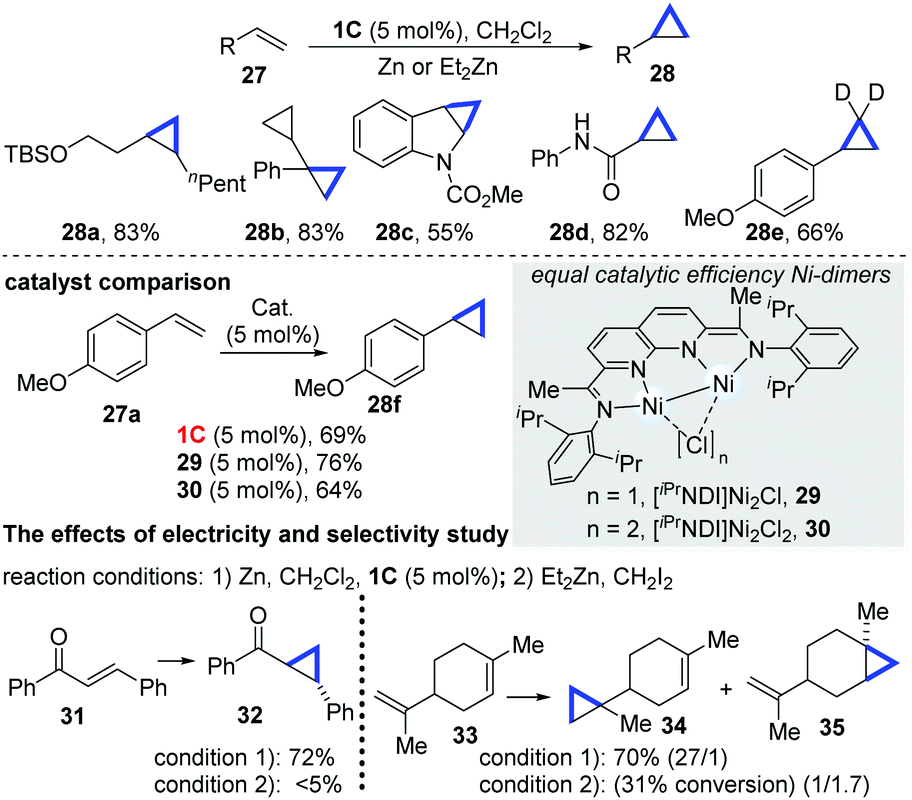
In 2016, Uyeda and co-workers subsequently developed an elegant dinuclear Ni-catalyzed cyclopropanation transformation of alkenes with dichloromethanes.21 Although the traditional cyclopropanation reactions of alkenes have been reported, they require preformed methylene equivalent compounds, such as diazoacetates,22 which are toxic and unstable, or carbenes that can be formed in situ from a dihalomethane–Zn powder combination.23 In this work by Uyeda's group, reductive cyclopropanation enabled by the dinuclear Ni-catalyst (1C) was achieved. The reaction required the formation of a population of unstable carbenes in situ from Zn and dihalomethanes. Importantly, NiI-dimers (1C, 29 and 30) have equal catalytic efficiency (28f), which are also much higher than that of common mononuclear Ni-complexes, such as [iPrIP]Ni(COD), [bpy]Ni(COD) and [iPrDAD]Ni(COD). A variety of acyclic (28a–b), carbo/heterocyclic alkenes (28c) and α,β-unsaturated carbonyl alkenes (28d) were found to be entirely compatible (Scheme 9). While using CD2Cl2, the deuterium-substituted cyclopropanes were obtained in a 66% yield (28e). In addition, the dinuclear Ni-catalyzed cyclopropanation of electron-deficient alkenes, such as chalcones, yielded trans-cyclopropanes in a 72% yield (32), while Simmons–Smith type Et2Zn reagents produced a low yield (<5%). For substrates such as limonene, containing several alkene units, it appeared that the dinuclear Ni-catalyzed cyclopropanation was sensitive to the steric effects, as the less sterically hindered alkene motif reacted preferentially (34), whereas the known Simmons–Smith cyclopropanation strategy afforded the products with an unsatisfying selectivity and low conversion.
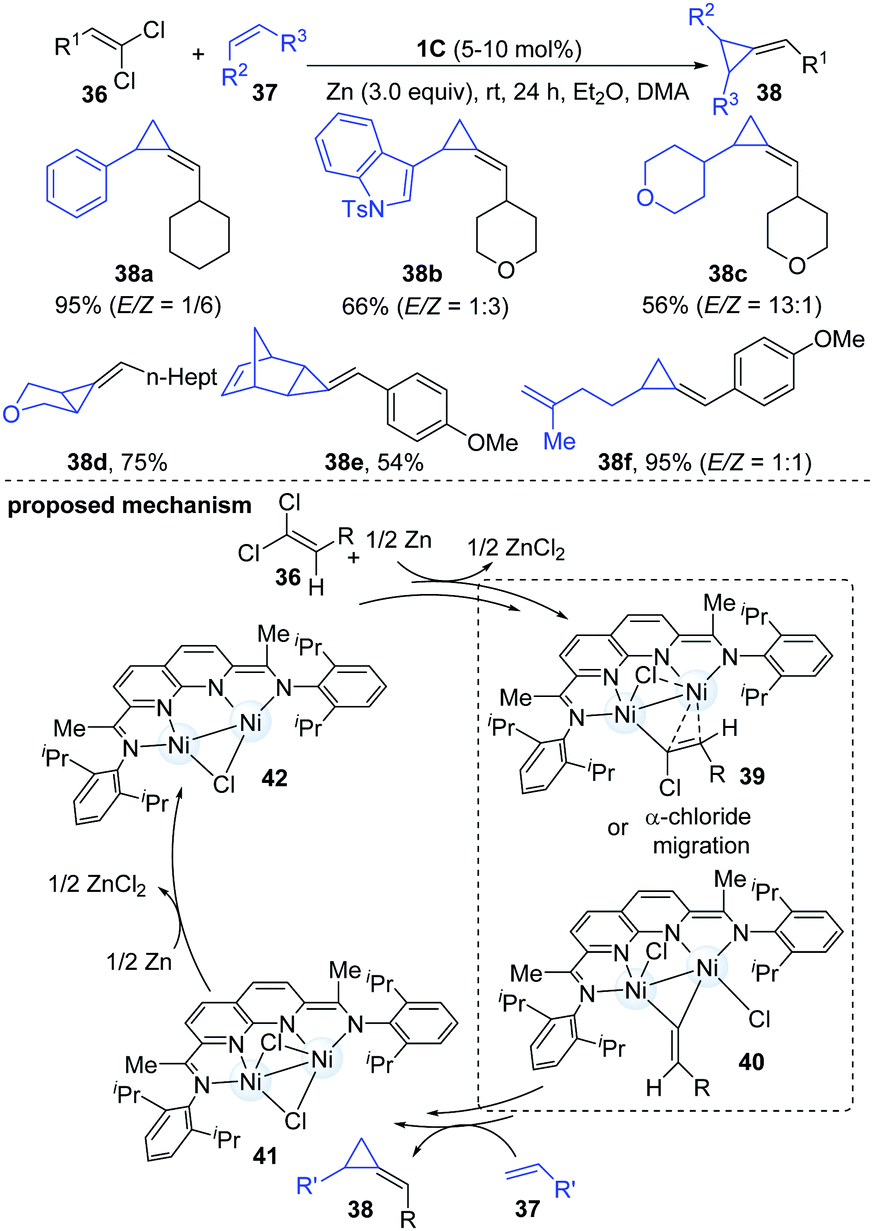
Both carbenes and vinylidenes (methylidene carbenes) are reactive organic intermediates. In a continuation of their ongoing interests in dinuclear Ni-catalyzed carbene chemistry, Uyeda and co-workers described a catalytic reductive methylenecyclopropanation reaction of simple olefins using 1,1-dichloroalkenes as vinylidene precursors.24 The dinuclear Ni-catalyst [iPrNDI]Ni2(C6H6) (1C) enabled the methylenecyclopropanation reaction in an almost quantitative yield. A variety of important functional groups could be well tolerated. The less hindered internal alkenes appeared to be better coupling partners than the relatively sterically hindered alkenes and furnished the corresponding cyclopropanated products (38d–e) in moderate to good yields. Interestingly, polyalkene substrates were monocyclopropanated at the less substituted alkene (38f). The authors proposed a two-electron oxidative pathway involving the addition of 1,1-dichloroalkene to the dinuclear Ni-complex to give a Ni2(1-chloroalkenyl)Cl intermediate (39) first. Then it encounters another alkene partner and forms the corresponding methylenecyclopropane product (40). Subsequently, the Ni2Cl2 complex (41) generated by vinylidene transfer undergoes a one-electron reduction to complete the catalytic cycle (Scheme 10).
 | ||
| Scheme 9 Dinuclear Ni-catalyzed cyclopropanation of olefins. | ||
 | ||
| Scheme 10 Catalytic reductive methylenecyclopropanation reaction. | ||
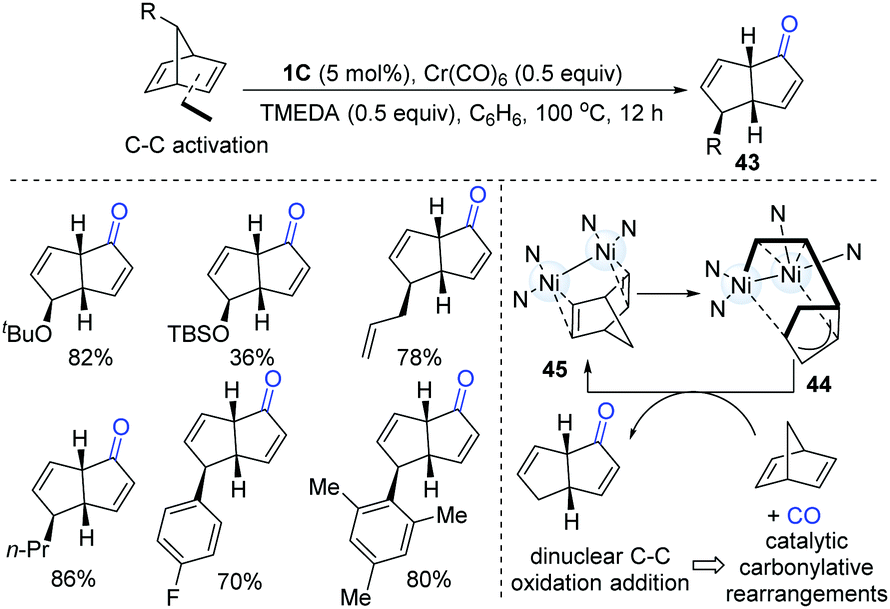
After gaining an understanding of the unique oxidative addition reactivity of dinuclear Ni-complexes, in 2017, Uyeda and co-workers discovered that dinuclear Ni-complexes could promote a rapid and reversible C–C oxidative addition of norbornadiene (Scheme 11).25 The intermediate (44) went through a migratory insertion-reductive elimination sequence to afford a bicyclo[3.3.0] product (43). The CO source was generated in situ from homoleptic metal carbonyl reagents, Cr(CO)6. This protocol is suitable for a wide range of alkyl, aryl and silyloxy-substituted norbornadienes.
 | ||
| Scheme 11 Dinuclear Ni-catalyzed carbonylative rearrangement of 7-substituted norbornadiene. | ||
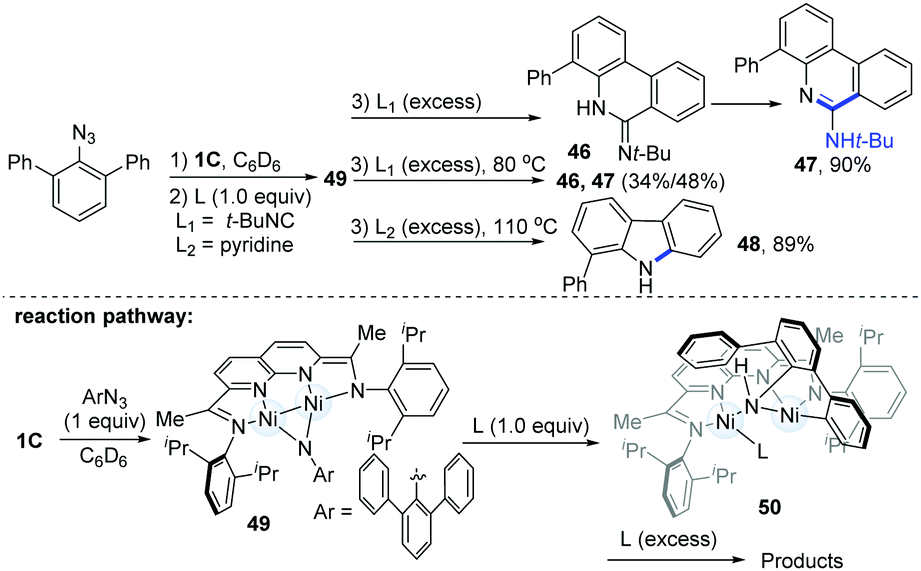
In the same year, Uyeda's group reported a tandem 1,2-addition of aryl azides and the C(sp2)–H activation pathway to produce phenanthridine (47) or carbazole (48) (Scheme 12).26 Treatment of the Ni-dimer (1C) with 10 equiv. of m-terphenyl azide led to a new paramagnetic species (49), which could undergo ortho C(sp2)–H bond addition to the Ni–(μ-NAr) bond with the addition of an external ligand to produce different final products (46–48). Usually, the employment of 1.0 equiv. of t-BuNC would lead to the formation of a C1-symmetric intermediate (50). However with excess t-BuNC, phenanthridin-6(5H)-imine (46) would be generated, and this was prone to form the more stable tautomeric phenanthridine product (47) in a 90% yield over 1 h at room temperature. Interestingly, if another 4.0 equiv. of pyridine were added as ligands, the dinuclear Ni-complexes (49) could finally afford the valuable phenylcarbazole (48) in an 89% yield.
 | ||
| Scheme 12 Dinuclear Ni-catalyzed ortho-C(sp2)–H oxidative addition. | ||
In 2017, Schoenebeck's group disclosed an elegant dinuclear Ni-catalyzed trifluoromethylselenolation of aryl halides (Scheme 13).27 In order to clarify the role and the reactivity of the dimer-NiI complex (1E), they synthesized an iodine- and a SeCF3-bridged dinuclear NiI intermediate (51). When the dinuclear Ni(I) intermediate (52) reacted with Ar–I, the corresponding aryl trifluoromethylselenolation products were successfully obtained. No conversion was observed with Ar–Br or Ar–Cl. No homocoupling biaryls were detected under these reactions, suggesting that the Ni(I)-complex did not simply serve as a catalyst precursor to Ni(0) species. The trifluoromethylselenolation protocol has very wide scope and can provide the desired products in excellent yields. Remarkably, when substrates containing F, Br and I substituents were used, the trifluoromethylselenolation exclusively occurred on the C–I bond and the other C–halogen bonds remained intact (51c). Of note, the use of dinuclear Ni-catalysts holds great advantages in organic synthesis because the well-established Ni(0) species can often promote oxidative addition of the ArSeCF3 products.
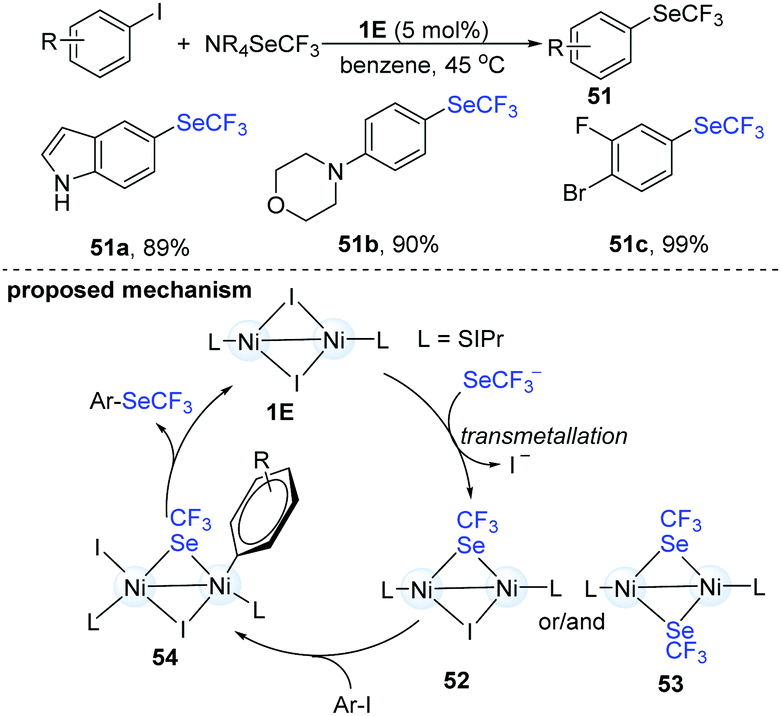 | ||
| Scheme 13 Dinuclear Ni-catalyzed trifluoromethylselenolation of aryl iodides. | ||
Based on the fact that benzo[h]quinoline ligands can stabilize Pd(III)–Pd(III),28 Diao's group successfully synthesized several kinds of high-valence dinuclear Ni-complexes from Ni(COD)2 and 10-bromobenzo[h]quinolone (Scheme 14).29 The addition of TDTT (1.0 equiv.) to 55 at −50 °C resulted in a high-valence intermediate (56) as a blue solid. The treatment of 56 with PhICl2 and TDTT gave rise to 58a and 58b in 90% and 75% yields, respectively. However, the introduction of bromide anions to 56 failed to generate 10-bromobenzo[h]quinolone (58c). In addition, the direct oxidation of 55 with PhNMe3Br3 at −80 °C in CD2Cl2 generated a dark green solution (57), which was then converted into 58c in a 190% yield (based on a 200% theoretical molar yield of 55).
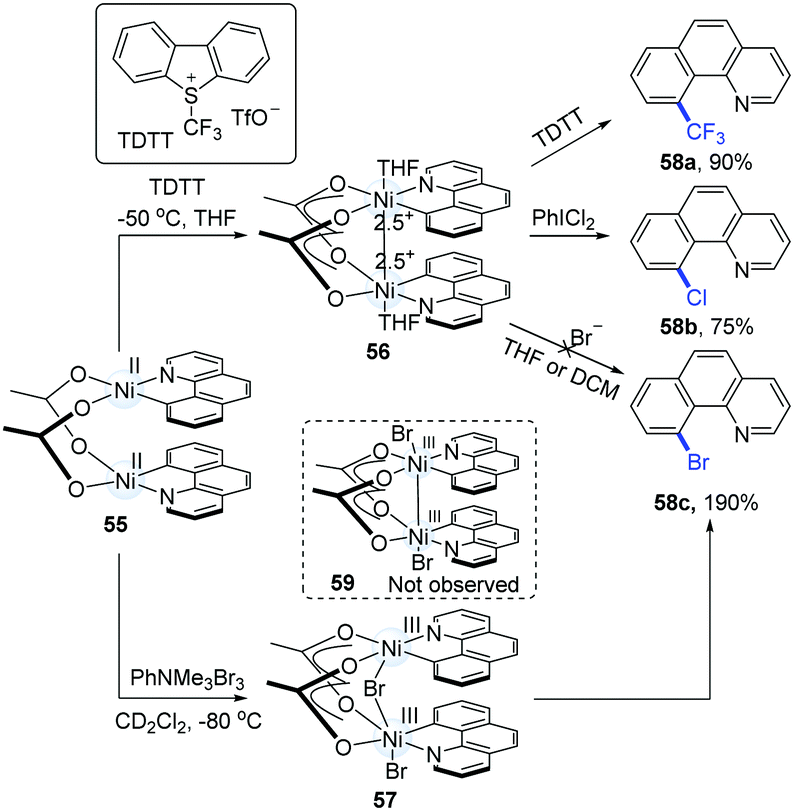 | ||
| Scheme 14 Mixed-valence and high-valence intermediates undergoing C–X reductive elimination. | ||
In 2018, Uyeda and co-workers used the same Ni-dimer [iPrNDI]Ni2(C6H6) (1C) to activate the C–C bond of strained three-membered ring substrates in vinyl aziridines and vinyl cyclopropanes (Scheme 15).30 They found that 1 mol% of the [iPrNDI]Ni2(C6H6) complex could efficiently convert vinyl cyclopropane into cyclopentene at room temperature, whereas the widely used mononuclear Ni-catalysts showed little catalytic efficiency. For phenyl-substituted vinyl cyclopropane (60), oxidative addition site-selectively occurred to the less hindered C–C bond, affording cyclopent-2-en-1-ylbenzene (61) as a single isomer in a 93% yield. Only the isomeric product (62) was obtained at high temperature (233 °C) in the absence of the dinuclear nickel catalyst (1C), which might be produced from a biradical intermediate via C–C bond homolysis at elevated temperature.
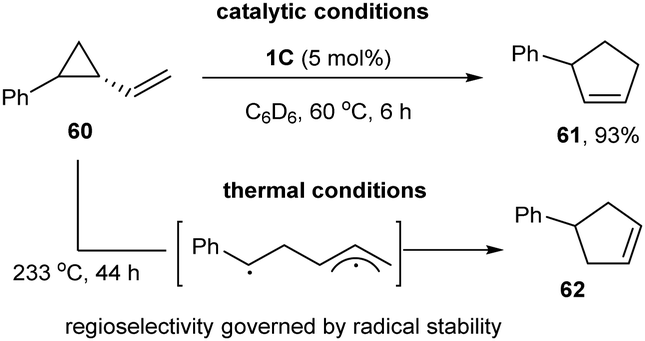 | ||
| Scheme 15 Dinuclear Ni-catalyzed ring-expansion of vinyl cyclopropane to cyclopentene. | ||
Despite the diversity of redox-based approaches to the formation of the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond, NN cross-coupling is generally a low-yield transformation. In 2018, the dinuclear Ni-catalyzed cross-coupling of aryl azides was achieved to afford synthetically useful organic chromophores in high yields under mild conditions (Scheme 16).31 The screening of Ni-complexes demonstrated that the [iPrIP]Ni(COD) catalyst (19) could furnish the NN coupling products in 13% yields via the formation of an azoarene π-complex (66); however, the stability of complex 66 blocks the catalytic turnover. In turn, the dimer-Ni catalyst bonds weakly with azoarene (67) and thus shows a highly efficient catalytic reactivity. Both electron-rich and electron-deficient substrates underwent NN coupling in moderate to high yields. Azoarenes derived from natural products (63a–c) were also found to be practical. Intramolecular NN coupling reaction (63d) and intermolecular NN polymerization (63e) have also been applied. Mechanistic studies with these were consistent with the initial formation of a μ-NAr intermediate, which then reacted with a second equiv. of the aryl azide to generate μ-N2Ar2 adduct. The mechanism begins with the 1,2-addition of aryl azides to the dinuclear Ni-complex (1C), immediately generating N2 gas accompanied by the formation of a new paramagnetic brown species (64). Upon the addition of another 1.0 equiv. of azides, the diamagnetic intermediate (65) can be formed. Finally, solvent-assisted ligand substitution gives the desired product azoarene (63), which was proposed as the rate-determining step.
N bond, NN cross-coupling is generally a low-yield transformation. In 2018, the dinuclear Ni-catalyzed cross-coupling of aryl azides was achieved to afford synthetically useful organic chromophores in high yields under mild conditions (Scheme 16).31 The screening of Ni-complexes demonstrated that the [iPrIP]Ni(COD) catalyst (19) could furnish the NN coupling products in 13% yields via the formation of an azoarene π-complex (66); however, the stability of complex 66 blocks the catalytic turnover. In turn, the dimer-Ni catalyst bonds weakly with azoarene (67) and thus shows a highly efficient catalytic reactivity. Both electron-rich and electron-deficient substrates underwent NN coupling in moderate to high yields. Azoarenes derived from natural products (63a–c) were also found to be practical. Intramolecular NN coupling reaction (63d) and intermolecular NN polymerization (63e) have also been applied. Mechanistic studies with these were consistent with the initial formation of a μ-NAr intermediate, which then reacted with a second equiv. of the aryl azide to generate μ-N2Ar2 adduct. The mechanism begins with the 1,2-addition of aryl azides to the dinuclear Ni-complex (1C), immediately generating N2 gas accompanied by the formation of a new paramagnetic brown species (64). Upon the addition of another 1.0 equiv. of azides, the diamagnetic intermediate (65) can be formed. Finally, solvent-assisted ligand substitution gives the desired product azoarene (63), which was proposed as the rate-determining step.
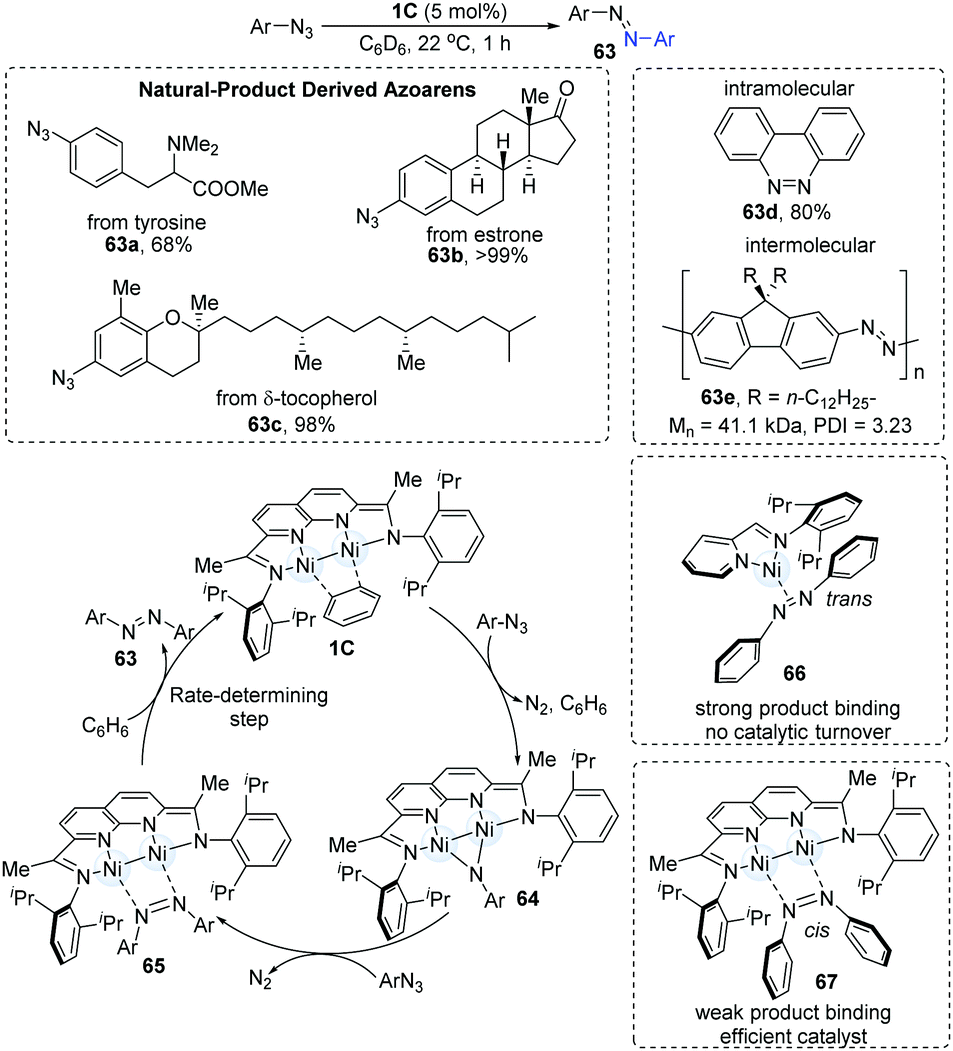 | ||
| Scheme 16 NN coupling catalyzed by Ni-dimer complex. | ||
Recently, when Martin's group routinely performed the nickel-catalyzed highly selective sp2 C–O functionalization of aryl esters, a Ni-dimer intermediate (69) was first successfully characterized (Scheme 17).32 The treatment of 1-naphthyl pivalate (68a) with [Ni(COD)2]/PCy3 could unexpectedly produce a dinuclear complex (69a) as a green air-sensitive solid in a 50% yield. Using [Ni(PCy3)2]2N2 instead of [Ni(COD)2] resulted in the formation of 69a in a quantitative yield. In addition, the π-extended aryl esters (68a–c) appeared to be more reactive for the formation of a dinuclear Ni complex than regular arenes (68d, 18% yield). Variable-temperature NMR spectroscopy indicated that the oxidative addition of a C–O bond to Ni(0)/PCy3 was facile, but was not the rate-determining step under the reaction conditions. Finally, based on the experimental and computational studies, a mechanism was proposed, as shown in Scheme 16b. The oxidative addition of arylpivalate can produce an off-cycle dinickel complex (69), which is in equilibrium with a mononickel Ni(II) complex (71) and Ni(0). A subsequent transmetalation step occurs with the mononickel complex. The resulting PivO–BPin is intercepted by fluoride ions to form insoluble fluoroborates, thus in favour of the catalyst turnover.
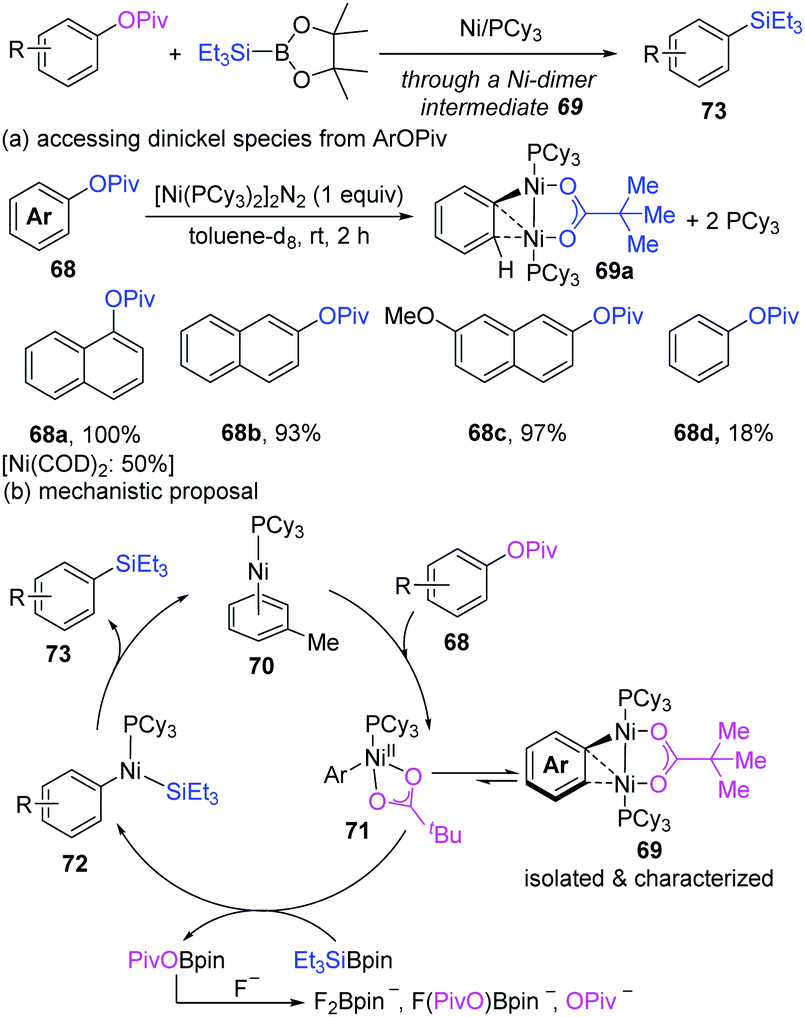 | ||
| Scheme 17 Intermediacy of Ni–Ni species in the sp2 C–O bond cleavage of aryl esters. | ||
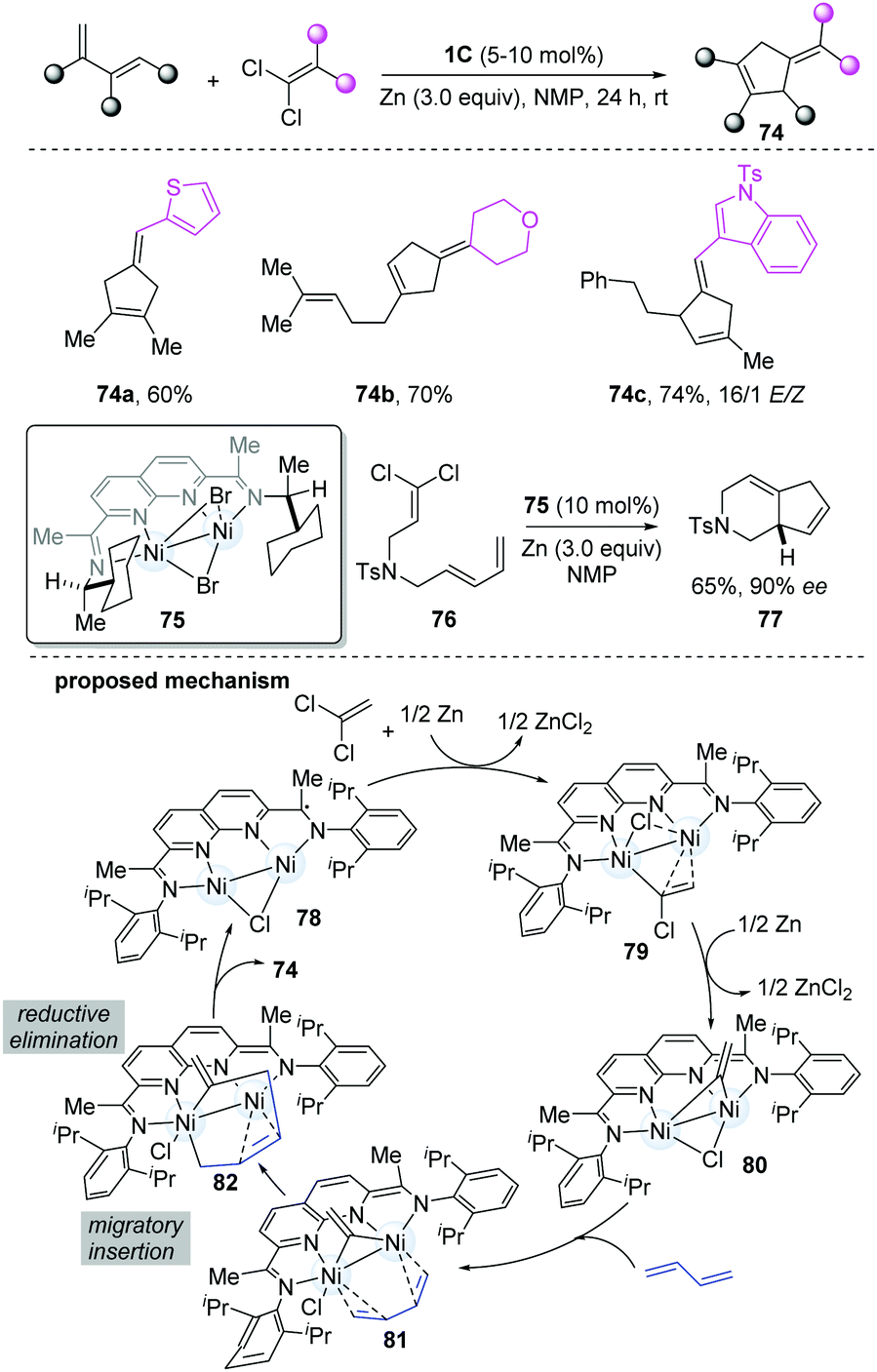 | ||
| Scheme 18 [4+1]-Cycloadditions of vinylidenes and dienes catalyzed by the dinickel catalyst. | ||
Based on the reported [2+1]-cycloaddition catalyzed by dinickel catalyst 1C, Uyeda's group developed a more challenging [4+1]-cycloaddition between a 1,3-diene and a C1 partner to construct five-membered rings,33 because of the competing [2+1]-cycloaddition. The steric profile of the catalyst was the key for the reaction. When a more-hindered functional group-substituted ligand was employed, such as isopropyl and cyclopentyl, the yield of [4+1]-cycloaddition products was much higher, without [2+1]-cycloaddition products. Under the optimal conditions, a series of substituted cyclopentenes were prepared (74a–c). When using a C2-symmetric chiral ligand-stabilized dinickel catalyst (75), high levels of asymmetric induction were achieved in the intramolecular cycloadditions (77). It is worth noting that Zn plays a crucial role in the formation of cycloadducts. The mechanism starts from oxidative addition of the 1,1-dichloroalkene by the [NDI]Ni2Cl, followed by one-electron reduction to form 79, which then undergoes a reductive process and C–Cl oxidative addition to give 80. Based on the DFT models, the Ni–Ni bond could be stabilized by the coordinating diene. Through migratory insertion, the metallacycle (82) resulted, which subsequently undergoes reductive elimination to give the final cycloadducts to finish the cycle (Scheme 18).
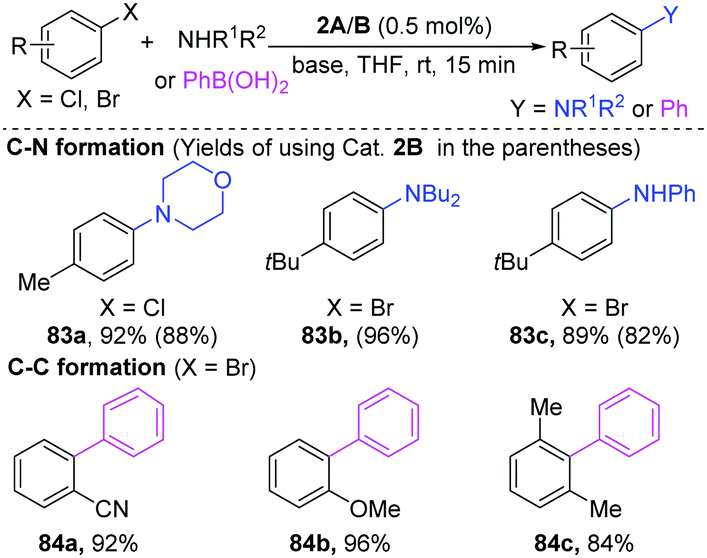 | ||
| Scheme 19 Dinuclear Pd-catalyzed Buchwald–Hartwig amination and Suzuki–Miyaura coupling. | ||
3. Transformations catalyzed by dinuclear Pd-complexes
Pd-Catalyzed cross-coupling has been extensively applied for the synthesis of pharmaceuticals and biologically important molecules.34 Compared to mononuclear palladium catalysts, the dinuclear Pd-complexes have the potential to share their redox ability, thus lowering the activation barriers. Consequently, research into dinuclear Pd-complexes is of great significance for the development of new reactions.35 The first dinuclear Pd(I)-complexes were successfully synthesized by Gelman and Meilakh in 1942,36 and the first dinuclear Pd(III) complex with the single bond between two trivalent palladium atoms was synthesized in 1998.28b During 1990s, Hartwig developed many useful transformations enabled by dinuclear-Pd complexes.37 In recent years, more and more elegant cases demonstrated that dinuclear palladium-complexes are indeed important reactive intermediates in organic transformations.35a,38In early 2002, Hartwig found that [PdBr(PR3)]2 was a superior catalyst for the C–N coupling of amines with aryl halides at room temperature (Scheme 19).39 Importantly, besides aryl bromides, the less reactive aryl chlorides are also good coupling partners (83a). When phenylboronic acids were employed for a Suzuki–Miyaura coupling, aryl bromides could give the Suzuki–Miyaura coupling products in high yields (84a–c). Preliminary mechanism studies demonstrated that oxidative addition would be the rate-limiting step for C–N coupling reactions.
Later, [PdBr-(PBut3)]2 was used as a catalyst for the α-arylation of esters (Scheme 20).40 Under the optimal conditions, primary (86a–b), secondary (86c–d) and tertiary esters (86e–h) could afford the corresponding α-arylation products with bench-stable Ar–Br in high yields (71–89%) with only 0.2–0.5 mol% catalyst loading, including heteroaryl bromides (86g–h).
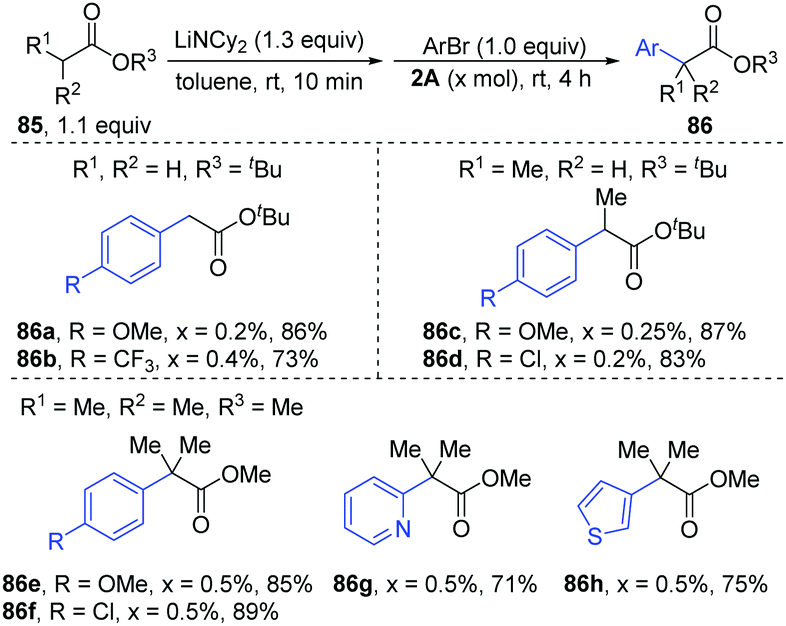 | ||
| Scheme 20 α-Arylation of esters catalyzed by dinuclear ([P(tBu)3]PdBr)2. | ||
The first example of chiral dinuclear palladium complexes and its potential application as a chiral catalyst was reported in 2009 by Imamoto's group.41 The combination of dinuclear Pd-complexes (2D) with silver triflate as additives exhibited high catalytic activity in the asymmetric ring-opening reactions of azabenzonorbornadienes with dimethylzinc (Scheme 21). Under the standard conditions, the desired products were obtained in good to excellent yields with high enantioselectivities (up to 99% ee), though the exception was the substrate containing a bulky substituent (88d).
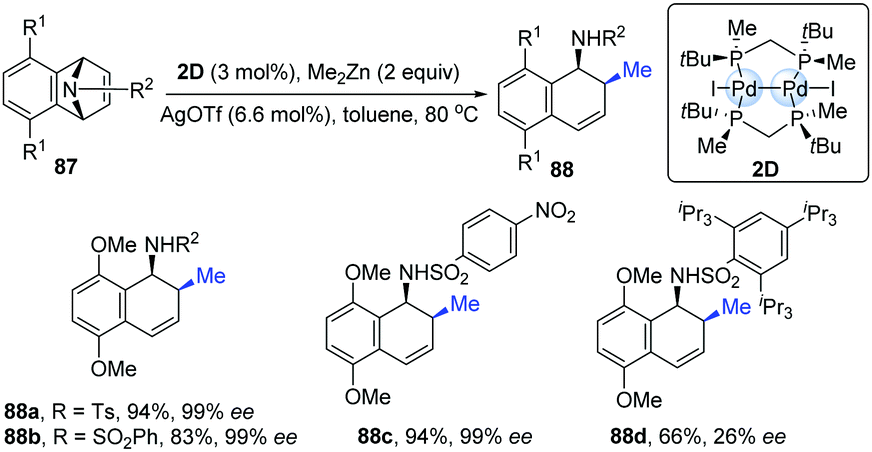 | ||
| Scheme 21 Ring-opening reactions catalyzed by dinuclear palladium complex with a chiral diphosphine ligand. | ||
In 2010, it was reported that dipalladium(I)-complex bridged by two [((5,7-dimethyl-1,8-naphthyridin-2-yl)-amino)carbonyl]ferrocene ligands (L) could exhibit the shortest Pd–Pd distance (2.3952 Å) among all dipalladium(I) compounds.42 The bond length between two metal centres may significantly influence its catalytic reactivity. Accordingly, a Pd-dimer catalyst (2E) was successfully employed in Suzuki–Miyaura and Heck cross-coupling reactions (Scheme 22). In general, electron-poor aryl bromides could smoothly achieve Suzuki cross-coupling, affording the corresponding products in higher yields than electron-rich aryl bromides. For electron-rich aryl chlorides, they gave moderate yields even with a higher catalyst loading. A variety of aryl bromides and styrenes could undergo the Heck coupling to afford the corresponding biaryl products in excellent yields and high stereoselectivity. In 2012, Schoenebeck and co-workers also developed a Suzuki cross-coupling by means of the Pd-dimer 2C.43
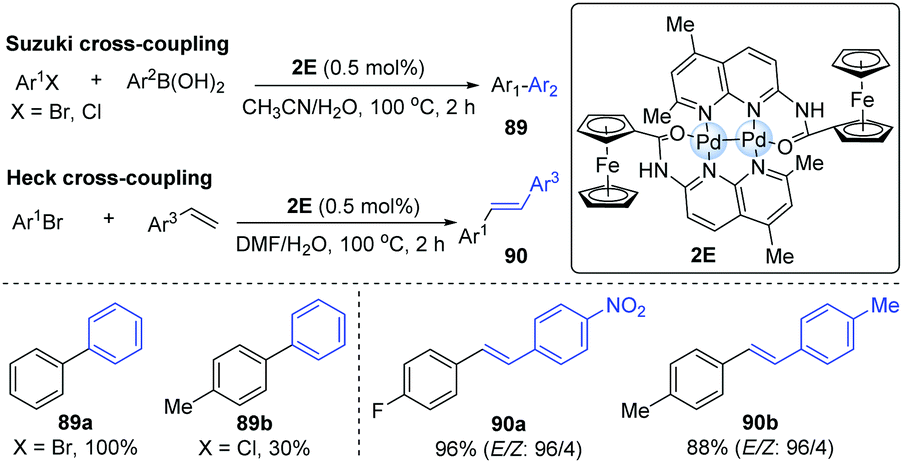 | ||
| Scheme 22 Dinuclear Pd-catalyzed Suzuki and Heck cross-coupling. | ||
In 2011, Hazari's group developed a new method for the mild activation of CO2 catalyzed by Pd(I)-bridging allyl dimers (Scheme 23).44 The catalytic reactivity of the dinuclear Pd-complex depended on the substituent on the allyl group and the ancillary ligand. Accordingly, a series of Pd-dimers was successfully synthesized (Scheme 23b). Among all the Pd-dimers (93), the allyl, 2-methylallyl and NHC (92d or 2F) ligands gave the best catalytic efficiency. When 2F was employed as the catalyst, the reaction afforded the carboxylation products of allylstannanes and allylboronates in satisfying yields (91a–c, Scheme 23a).
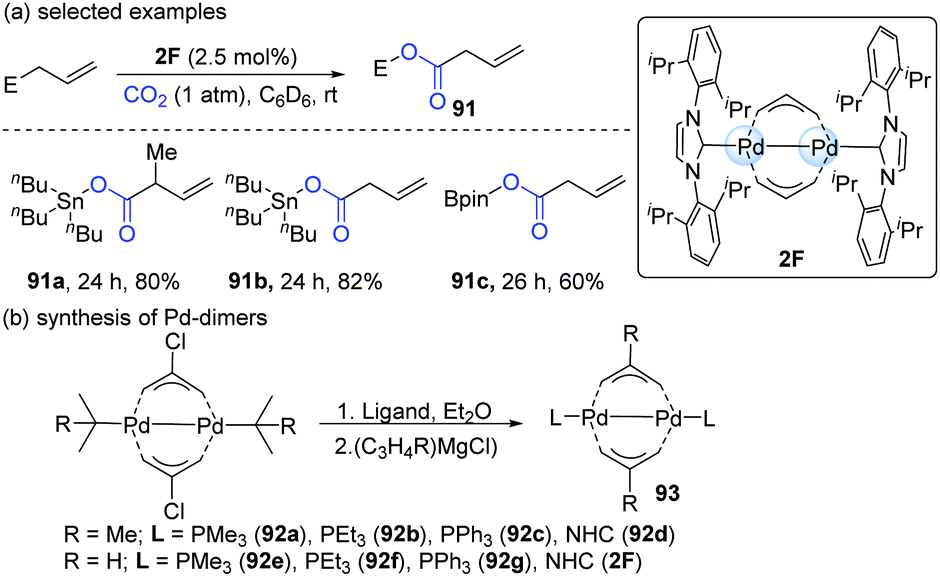 | ||
| Scheme 23 Carboxylation of different allyls with CO2 catalyzed by Pd-dimer. | ||
In general, phosphorylation reactions are generally limited to alkenes and alkynes, and the use of allenes has been mainly underdeveloped. In 2011, Swamy and co-workers applied an efficient and recoverable dinuclear palladium(I) catalyst [(OCH2CMe2CH2O)PSPd(PPh3)]2 (2G) for the selective phosphonylation/phosphanylation of allenes (Scheme 24).45 A large number of allenes and phosphonylating/phosphanylating agents were investigated. Importantly, it was found that the recovered dinuclear palladium(I) catalyst could keep the same catalytic activity with the new employed [Pd(PPh3)4], while other Pd catalysts (PdCl2, Pd(OAc)2, PdCl2(PPh3)2, Pd2(dba)3 and PdCl2(PhCN)2) were less efficient.
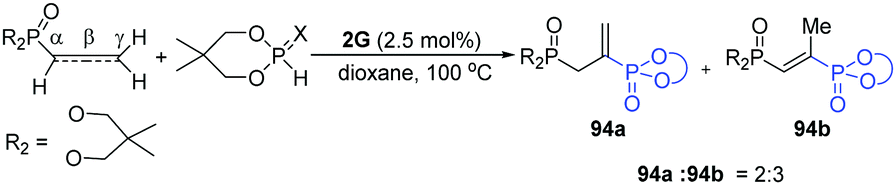 | ||
| Scheme 24 Dinuclear Pd-catalyzed phosphonylation of allenes. | ||
In 2013, Schoenebeck and co-workers reported an interesting dinuclear Pd-catalyzed Br/I halogen exchange of aryl iodides with NBu4Br (Scheme 25).46 Notably, the well-established mononuclear Pd(II)-complexes could not undergo the reductive elimination to give the desired product. In their initial test, the authors found that the addition of 1.0 equiv. of 2B to 10 equiv. of 9-iodoanthracene in THF for 3 h could produce 1.8 equiv. of 9-bromoanthracene (95a). The use of a catalytic amount of the dinuclear Pd-complex 2C (20 mol%) could achieve the I/Br exchange reaction of 9-iodoanthracenes in the presence of 10 equiv. of Bu4NBr (81%, 4 days); while slight heating of the mixture from room temperature to 35 °C significantly accelerated the reaction (61%, 20 h). However, the Pd-dimer (2B) was a less efficient catalyst and only a low yield (18–40%) was obtained, even with prolonging the reaction time. Upon the addition of an excess of Bu4NBr to Pd(I)–I-dimer (2C), the equilibrium shifted slightly towards the mixed Pd(I)–Br–I-dimer (96). Subsequent oxidative addition and reductive elimination produced the corresponding ArBr. Recently, C–I selective cross-coupling could be successfully finished by a cationic palladium trimer.47
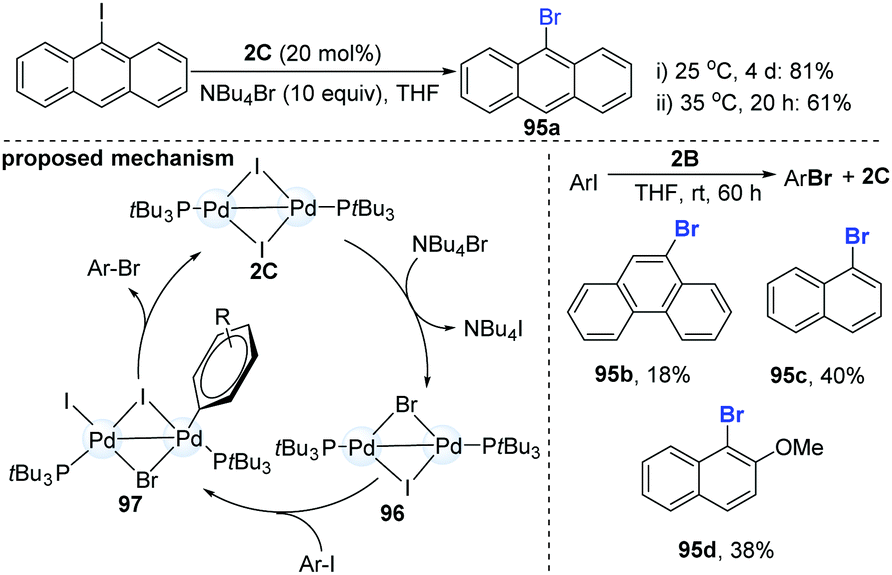 | ||
| Scheme 25 The I/Br exchange reaction of aromatic halide catalyzed by a Pd(I) dimer. | ||
Based on their success in dinuclear Pd-catalyzed halogen exchange,46 Schoenebeck's group hypothesized that strong nucleophiles might be easily converted into a reactive species. This could further expand the scope of the catalytic reaction (Scheme 26). Given the prevalence of SCF3 and SeCF3 motifs in biologically important molecules, these products are synthetically valuable. Consequently, Schoenebeck's group developed a highly efficient and operationally simple dinuclear Pd-catalyzed trifluoromethylthiolation of both aryl iodides and bromides with readily accessible (Me4N)SCF3 as a trifluoromethylthiolating reagent.48 Compared with the established mononuclear Pd-catalyzed similar transformation,49 the reaction with Pd(I)-dimers gave a much broader substrate scope (98). The versatile aldehyde, ketone, ester, ether, nitro, cyano and amine functional groups were found to be compatible. Mechanistic studies demonstrated that the formation of the SCF3-bridged Pd(I) dimer (102a) was thermodynamically favourable (ΔGrxn ≈ −21 kcal mol−1). Significantly, the authors succeeded to isolate and characterize the SCF3-bridged PdI dimer (102) by X-ray crystallography and 31P NMR analysis. In addition, the catalyst could be recycled from the reaction mixture, which afforded the product (98a) in an 88% yield for the second time (95% in the first catalytic cycle). A possible mechanism is shown in Scheme 25d. First, the reactive species 102a is generated via the exchange of I by an SCF3 anion at Pd(I). Then, the oxidative addition of ArI to a palladium dimer (103) followed by reductive elimination led to the formation of various ArSCF3 products. Later, similar coupling access to Ar–SeCF3 (99) was successfully achieved using (Me4N)SeCF3 as a nucleophile (Scheme 26b).50 In addition, the site-selective thiolation51 and selenolation52 of aryl halogens were also shown to be practical approaches under identical catalytic conditions (Scheme 26c and d). However, it is still difficult to know if Pd(0) species are involved or not in these catalytic coupling processes. Later, similar dinuclear catalytic platforms were applied in the synthesis of S-aryl phosphorothioates.53 In consideration of the less precious and more sustainable nickel, the NHC-derived dinuclear catalyst was synthesized. Predictably, this kind of dinuclear Ni(I) complex was successfully applied in the trifluoromethylselenolation of aryl iodides, which enriched the nickel chemistry.27
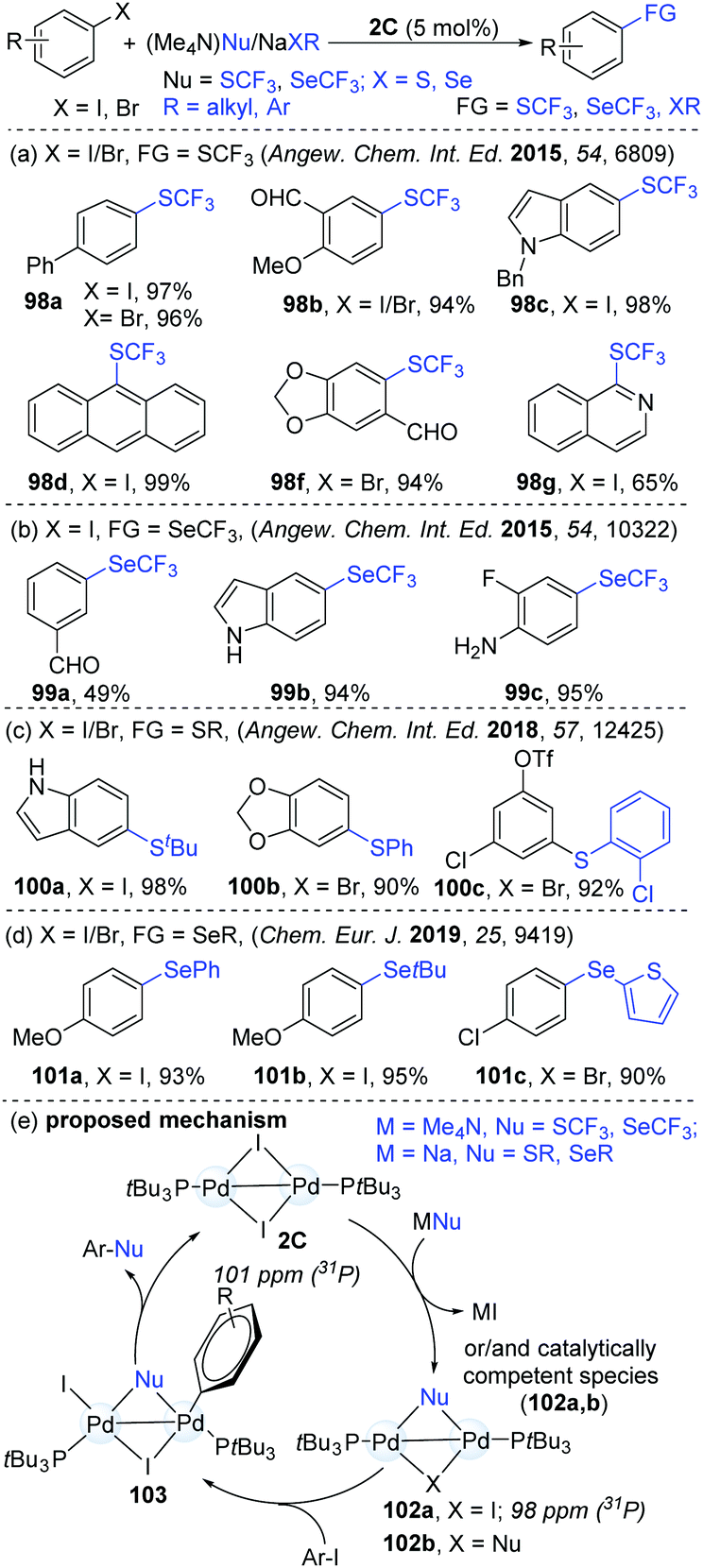 | ||
| Scheme 26 The efficient organic transformation of aromatic halides catalyzed by Pd(I) dimer. | ||
As part of their ongoing interests, Schoenebeck and co-workers recently developed a general dinuclear Pd-catalyzed chemoselective C(sp2)–C(sp2) coupling between aryl bromides and organometallics, keeping the versatile OTf functional group intact during the process (Scheme 27).54 Control of the selectivity generally appears in the oxidative addition steps, where the relative reactivity trend of the C–halogen bond is regarded as C–I > C–OTf ≈ C–Br > C–Cl.55 In general, the selectivity is controlled by several factors, such as the solvents, additives, ligands and substrates. Although a mononuclear metal catalyst shows a low catalytic reactivity and low selectivity in these couplings, the Pd-dimer46,48,50 could achieve good selectivity in Kumada coupling reactions. In addition, the dinuclear Pd-catalyst (2C) is also effective in Negishi couplings (104e and h). Soon, the first general chemo- and site-selective alkylation of C–Br bonds in the presence of C–OTf, C–Cl bonds was developed.56 Importantly, the reaction was completed very fast (≤5 min) under air conditions.
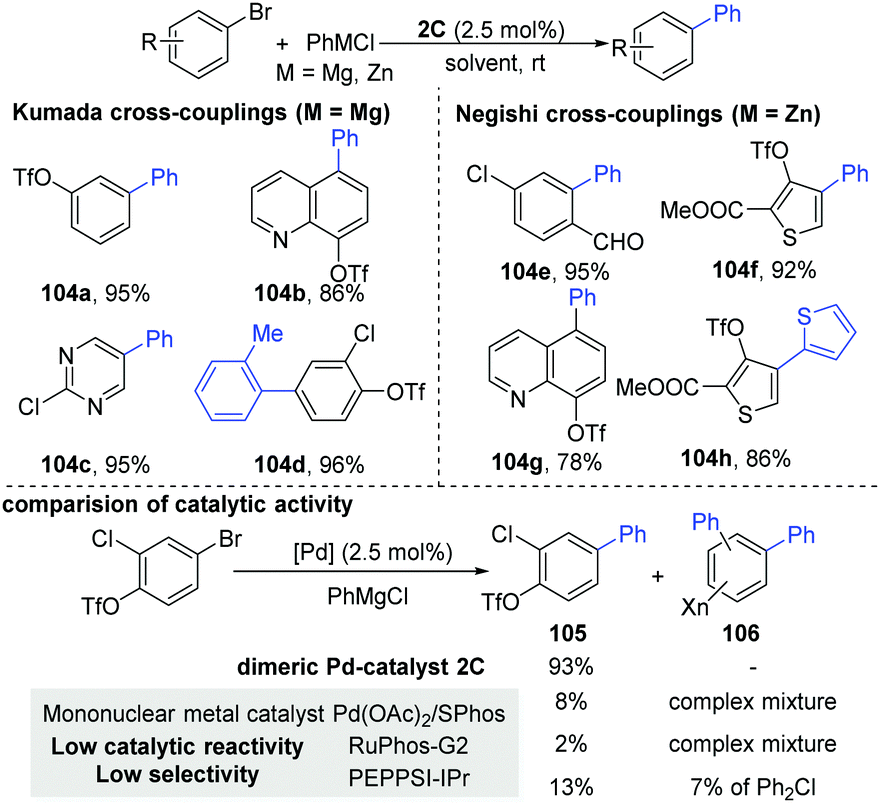 | ||
| Scheme 27 Dinuclear Pd-catalyzed Kumada and Negishi cross-coupling. | ||
In 2018, Schoenebeck's group developed a sequential strategy for the coupling of aryl halides with multiple competing coupling sites (–Br, –OTf, –Cl) catalyzed by the well-developed Pd-dimer 2C (Scheme 28A).57 Based on a previous work,58 they reasoned that the dinuclear entity acted as “Pd-ate” complexes in the polar media to preferentially react with C–OTf bonds over C–Cl bonds. As expected, selective C–OTf coupling over the C–Cl bond of a number of substrates was achieved in less than 10 min at room temperature in NMP. ortho-, meta- and para-substituted chlorophenyl triflates performed well (108a–c). Other functional groups, such as aldehyde (108d), fluorine (108e) and heterocyclic (108f) functional groups were also compatible. Under the same conditions, not only arylation (108g and 108h), but also the more challenging alkylation (primary and secondary alkyl groups) of diverse chlorophenyl triflates was successfully realized (108i and 108j). If no OTf motif was involved in the substrates, the aromatic C–Cl bond could efficiently undergo coupling with PhZnCl under an argon atmosphere (Scheme 28C). More importantly, the sequential selective functionalization of multifunctionalized aromatic halides in a one-pot process was developed. For instance, when the substrate contains two different reactivity sites, there are always three reaction models (I, Br and OTf: product 109a; II, OTf and Cl: product 109b; III, Br and Cl: product 109c). Taking product 109a as an example, one portion of organozincs in THF was added to bromoaryl triflate ti furnish a C–Br activation intermediate at first. Subsequently, when adding another portion of the coupling partners in NMP, the mixtures resulted in bis-functionalized arene 109a in a 66% total yield (model I). Using NMP as the solvent could achieve sequentially C–OTf and C–Cl functionalization (model II, 109b). For model III, a procedure analogous to model I was applied to deliver 109c in a 78% yield. Combining the results above, a very challenging sequential coupling of all three sites (–Br, –OTf and –Cl) was successfully developed in a one-pot three-steps process. The final functionalized products (109d–f) were obtained in good yields. Two years later, the selective functionalization of C–OSO2F (OFs) vs. C–Cl/C–Br in arenes was achieved by dinuclear-PdI (1C).59 As shown in Scheme 28B, the modular functionalization was realized in the sequence C–Br, then C–OFs, then C–Cl while using organozinc as a coupling partner (110a–c).
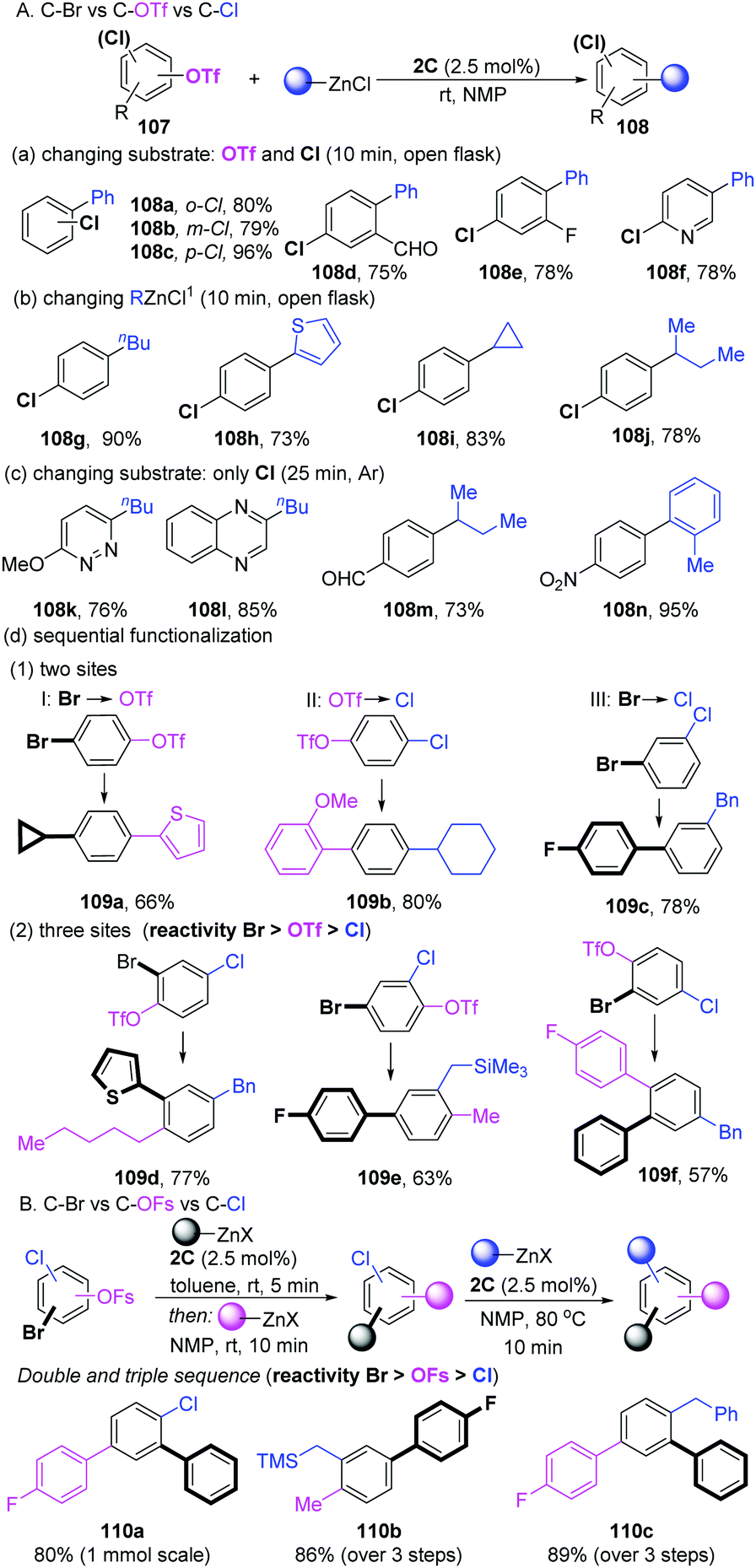 | ||
| Scheme 28 Sequential functionalization of arenes. | ||
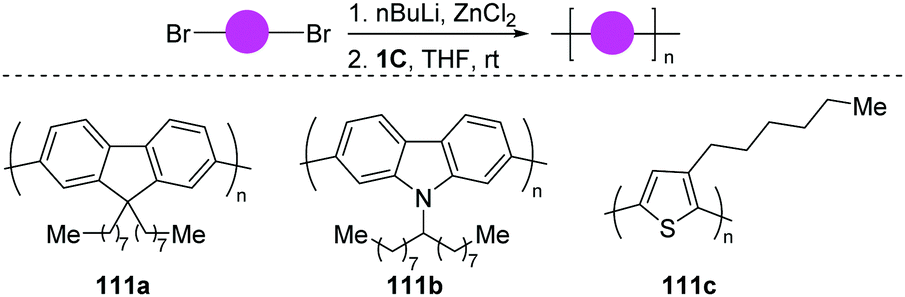
On the basis of the well-developed functionalization of aryl halides, the dinuclear-Pd catalyst (1C) shows a broad catalytic activity, independent of the steric or electronic characteristics of the particular substrate. In 2019, the same group disclosed an extremely rapid polymerization using dibromides as the monomer (Scheme 29).60 Polyfluorene, polycarbazole and polythiophene were efficiently accessible (111a–c). As to bromo-adamantylarenes, the same air-stable dinuclear-Pd catalyst (1C) enabled the selective ortho-functionalization.61
Besides the above dinuclear catalysts, which hold a metal–metal bond in the catalytic cycle, there are some dinuclear complexes that act as mono-metal catalyst precursors during the catalytic cycle according to the authors’ proposal. Generally, these complexes could efficiently catalyze the transformations, while the corresponding mono-metal catalysts did not work well. This part of the work is not discussed in this highlight.62–68 In addition, only the positive results have been reported in general and maybe a series of dinuclear metal complexes have low reactivity, which indicates that a deep mechanistic study into the dinuclear metal catalysis will be helpful and is urgently needed.
 | ||
| Scheme 29 Performance of the dinuclear-Pd catalyst in the polymerization. | ||
4. Conclusions and outlook
In recent years, many group-10 metal-dimer complexes have been successfully synthesized and increasing attention has been paid to their catalytic application in contemporary synthetic chemistry. In this highlight, the recent reports of organic transformations catalyzed by dinuclear Ni- and Pd-complexes during the past decade have been mainly discussed. In most cases, dinuclear transition-metal catalyst precursors hold much better catalytic reactivity than the corresponding mononuclear transition-metal catalysts and the examples cited herein show their unique catalytic reactivity. Their catalytic applications offer a powerful and complementary strategy to explore new chemical space, beyond what the well-established mononuclear transition-metal catalysis offers. Perhaps more importantly, it will stimulate chemists to seek to understand the relationship between the ligand effect, electronic structure and catalytic efficiency for dinuclear metal-complexes. Furthermore, based on understanding the effects of ligands, suitable chiral dinuclear catalysts (like 2D) may be promising for use in future asymmetric catalysis.Despite significant progress, there are still considerable opportunities and challenges in this field. The synthesis of new metal-dimer complexes and the exploitation of their synthetic application will still be of much interest to researchers. Currently, reaction development is outpacing the corresponding mechanism studies. A clear understanding of the reaction mechanisms will, in turn, help chemists to design and synthesize more efficient dinuclear transition-metal catalysts. Nonetheless, given the potential catalytic models of dinuclear transition-metal complexes in organic chemistry, we believe that this timely highlight will attract considerable enthusiasm in the near future.
Abbreviations
| COD | 1,5-Cyclooctadiene |
| dme | 1,2-Dimethoxyethane |
| iPrIP | N-(2,6-Diisopropylphenyl)-1-(pyridin-2-yl)methanimine |
| BPY | 2,2′-Dipyridine |
| iPrDAD | N 1,N2-Bis(2,6-diisopropylphenyl)ethane-1,2-diimine |
| iPrNDI | 1,1′-(1,8-Naphthyridine-2,7-diyl)bis(N-(2,6-diisopropylphenyl)ethan-1-imine) |
| NHC | 1,3-Bis(2,6-diisopropylphenyl)-1,3-dihydro-imidazol-2-ylidene |
| TDTT | 5-(Trifluoromethyl)-5H-dibenzo[b,d]thiophen-5-iumtrifluoromethanesulfonate |
| Mes | 1,3,5-Trimethyl-benzene |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Natural Science Foundation of China (21971108 and 21702098), the Fundamental Research Funds for the Central Universities (020514380214), the Natural Science Foundation of Jiangsu Province (Grant No. BK20190006), “Innovation & Entrepreneurship Talents Plan” of Jiangsu Province, “Jiangsu Six Peak Talent Project”, “1000-Youth Talents Plan” and start-up funding of Nanjing University.Notes and references
- (a) K. Ouyang, W. Hao, W. X. Zhang and Z. Xi, Chem. Rev., 2015, 115, 12045 CrossRef CAS PubMed; (b) J. Y. Corey, Chem. Rev., 2016, 116, 11291 CrossRef CAS PubMed; (c) Y. Yang, J. Lan and J. You, Chem. Rev., 2017, 117, 8787 CrossRef CAS PubMed; (d) Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247 CrossRef CAS PubMed; (e) J. R. Hummel, J. A. Boerth and J. A. Ellman, Chem. Rev., 2017, 117, 9163 CrossRef CAS PubMed; (f) J. B. Peng, F. P. Wu and X. F. Wu, Chem. Rev., 2019, 119, 2090 CrossRef CAS PubMed; (g) M. Caporali, L. Gonsalvi, A. Rossin and M. Peruzzini, Chem. Rev., 2010, 110, 4178 CrossRef CAS PubMed; (h) R. Skoda-Foldes and L. Kollar, Chem. Rev., 2003, 103, 4095 CrossRef PubMed; (i) A. Gansäuer and H. Bluhm, Chem. Rev., 2000, 100, 2771 CrossRef PubMed; (j) I. Ojima, M. Tzamarioudaki, Z. Li and R. J. Donovan, Chem. Rev., 1996, 96, 635 CrossRef CAS PubMed; (k) I. Nakamura and Y. Yamamoto, Chem. Rev., 2004, 104, 2127 CrossRef CAS PubMed.
- M. Mohadjer Beromi, A. Nova, D. Balcells, A. M. Brasacchio, G. W. Brudvig, L. M. Guard, N. Hazari and D. J. Vinyard, J. Am. Chem. Soc., 2017, 139, 922 CrossRef CAS PubMed.
- S. Benini, W. R. Rypniewski, K. S. Wilson, S. Ciurli and S. Mangani, J. Biol. Inorg. Chem., 1998, 3, 268 CrossRef CAS.
- (a) J. A. Bertrand, F. A. Cotton and W. A. Dollase, J. Am. Chem. Soc., 1963, 85, 1349 CrossRef CAS; (b) F. A. Cotton, Inorg. Chem., 1965, 4, 334 CrossRef CAS; (c) F. A. Cotton, Reactivity of Metal-Metal Bonds, American Chemical Society, 1981, ch. 1, vol. 155, p. 1 Search PubMed; (d) F. A. Cotton, Inorg. Chem., 1998, 37, 5710 CrossRef CAS; (e) F. A. Cotton, Acc. Chem. Res., 1969, 2, 240 CrossRef CAS; (f) F. A. Cotton, N. F. Curtis, C. B. Harris, B. F. Johnson, S. J. Lippard, J. T. Mague, W. R. Robinson and J. S. Wood, Science, 1964, 145, 1305 CrossRef CAS PubMed; (g) S. T. Liddle and D. P. Mills, Dalton Trans., 2009, 5592 RSC; (h) S. T. Liddle, Molecular Metal-Metal Bonds: Compounds, Synthesis, Properties, Wiley, 2015 Search PubMed.
- (a) C. Jones, Nat. Rev. Chem., 2017, 1, 0059 CrossRef CAS; (b) J. F. Berry, Acc. Chem. Res., 2016, 49, 27 CrossRef CAS PubMed.
- (a) M. P. Doyle and D. C. Forbes, Chem. Rev., 1998, 98, 911 CrossRef CAS PubMed; (b) H. M. L. Davies and J. R. Manning, Nature, 2008, 451, 417 CrossRef CAS PubMed; (c) M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Chem. Rev., 2010, 110, 704 CrossRef CAS PubMed; (d) J. Kwak, M. Kim and S. Chang, J. Am. Chem. Soc., 2011, 133, 3780 CrossRef CAS PubMed; (e) C. Qin, V. Boyarskikh, J. H. Hansen, K. I. Hardcastle, D. G. Musaev and H. M. Davies, J. Am. Chem. Soc., 2011, 133, 19198 CrossRef CAS PubMed; (f) A. DeAngelis, R. Panish and J. M. Fox, Acc. Chem. Res., 2015, 49, 115 CrossRef PubMed.
- (a) J. D. Basil, H. H. Murray, J. P. Fackler, J. Tocher, A. M. Mazany, B. Trzcinska-Bancroft, H. Knachel, D. Dudis, T. J. Delord and D. Marler, J. Am. Chem. Soc., 1985, 107, 6908 CrossRef CAS; (b) E. Tkatchouk, N. P. Mankad, D. Benitez, W. A. Goddard 3rd and F. D. Toste, J. Am. Chem. Soc., 2011, 133, 14293 CrossRef CAS PubMed; (c) A. Gomez-Suarez and S. P. Nolan, Angew. Chem., Int. Ed., 2012, 51, 8156 CrossRef CAS PubMed; (d) H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370 RSC; (e) D. S. Weinberger, M. Melaimi, C. E. Moore, A. L. Rheingold, G. Frenking, P. Jerabek and G. Bertrand, Angew. Chem., Int. Ed., 2013, 52, 8964 CrossRef CAS PubMed; (f) K. Liu, N. Li, Y. Ning, C. Zhu and J. Xie, Chem, 2019, 5, 2718 CrossRef CAS.
- (a) U. Flierler, M. Burzler, D. Leusser, J. Henn, H. Ott, H. Braunschweig and D. Stalke, Angew. Chem., Int. Ed., 2008, 47, 4321 CrossRef CAS PubMed; (b) P. Nuhant, M. S. Oderinde, J. Genovino, A. Juneau, Y. Gagne, C. Allais, G. M. Chinigo, C. Choi, N. W. Sach, L. Bernier, Y. M. Fobian, M. W. Bundesmann, B. Khunte, M. Frenette and O. O. Fadeyi, Angew. Chem., Int. Ed., 2017, 56, 15309 CrossRef CAS PubMed; (c) Z. Yan, X. A. Yuan, Y. Zhao, C. Zhu and J. Xie, Angew. Chem., Int. Ed., 2018, 57, 12906 CrossRef CAS PubMed; (d) D. Wang, J. Dong, W. Fan, X.-A. Yuan, J. Han and J. Xie, Angew. Chem., Int. Ed., 2020, 59, 8430 CrossRef CAS PubMed; (e) Y. Pang, G. Liu, C. Huang, X.-A. Yuan, W. Li and J. Xie, Angew. Chem., Int. Ed. DOI:10.1002/anie.202004950.
- A. Toledo, I. Funes-Ardoiz, F. Maseras and A. C. Albéniz, ACS Catal., 2018, 8, 7495 CrossRef CAS.
- (a) N. Hazari and D. P. Hruszkewycz, Chem. Soc. Rev., 2016, 45, 2871 RSC; (b) T. Murahashi and H. Kurosawa, Coord. Chem. Rev., 2002, 231, 207 CrossRef CAS; (c) I. G. Powers and C. Uyeda, ACS Catal., 2016, 7, 936 CrossRef.
- N. Xiong, Z. Guoxiang, S. Xiaolong and Z. Rong, Chin. J. Chem., 2020, 38, 185 CrossRef CAS.
- A. Velian, S. Lin, A. J. Miller, M. W. Day and T. Agapie, J. Am. Chem. Soc., 2010, 132, 6296 CrossRef CAS PubMed.
- R. Beck and S. A. Johnson, Chem. Commun., 2011, 47, 9233 RSC.
- M. Tanabe, R. Yumoto and K. Osakada, Chem. Commun., 2012, 48, 2125 RSC.
- T. J. Steiman and C. Uyeda, J. Am. Chem. Soc., 2015, 137, 6104 CrossRef CAS PubMed.
- A. J. Chalk and J. F. Harrod, J. Am. Chem. Soc., 1965, 87, 16 CrossRef CAS.
- P. B. Glaser and T. D. Tilley, J. Am. Chem. Soc., 2003, 125, 13640 CrossRef CAS PubMed.
- S. Pal and C. Uyeda, J. Am. Chem. Soc., 2015, 137, 8042 CrossRef CAS PubMed.
- T. Sugahara, J. D. Guo, T. Sasamori, S. Nagase and N. Tokitoh, Angew. Chem., Int. Ed., 2018, 57, 3499 CrossRef CAS PubMed.
- K. Matsubara, H. Yamamoto, S. Miyazaki, T. Inatomi, K. Nonaka, Y. Koga, Y. Yamada, L. F. Veiros and K. Kirchner, Organometallics, 2016, 36, 255 CrossRef.
- Y. Y. Zhou and C. Uyeda, Angew. Chem., Int. Ed., 2016, 55, 3171 CrossRef CAS PubMed.
- (a) B. Morandi and E. M. Carreira, Science, 2012, 335, 1471 CrossRef CAS PubMed; (b) S. A. Kunzi, J. M. Sarria Toro, T. den Hartog and P. Chen, Angew. Chem., Int. Ed., 2015, 54, 10670 CrossRef PubMed.
- H. E. Simmons and R. D. Smith, J. Am. Chem. Soc., 1958, 80, 5323 CrossRef CAS.
- S. Pal, Y. Y. Zhou and C. Uyeda, J. Am. Chem. Soc., 2017, 139, 11686 CrossRef CAS PubMed.
- D. R. Hartline, M. Zeller and C. Uyeda, J. Am. Chem. Soc., 2017, 139, 13672 CrossRef CAS PubMed.
- I. G. Powers, C. Kiattisewee, K. C. Mullane, E. J. Schelter and C. Uyeda, Chem. – Eur. J., 2017, 23, 7694 CrossRef CAS PubMed.
- A. B. Dürr, H. C. Fisher, I. Kalvet, K.-N. Truong and F. Schoenebeck, Angew. Chem., Int. Ed., 2017, 56, 13431 CrossRef PubMed.
- (a) D. C. Powers and T. Ritter, Acc. Chem. Res., 2012, 45, 840 CrossRef CAS PubMed; (b) F. A. Cotton, J. Gu, C. A. Murillo and D. J. Timmons, J. Am. Chem. Soc., 1998, 120, 13280 CrossRef CAS; (c) Y. Ye, N. D. Ball, J. W. Kampf and M. S. Sanford, J. Am. Chem. Soc., 2010, 132, 14682 CrossRef CAS PubMed.
- J. B. Diccianni, C. Hu and T. Diao, Angew. Chem., Int. Ed., 2017, 56, 3635 CrossRef CAS PubMed.
- H. R. Rounds, M. Zeller and C. Uyeda, Organometallics, 2018, 37, 545 CrossRef CAS.
- I. G. Powers, J. M. Andjaba, X. Luo, J. Mei and C. Uyeda, J. Am. Chem. Soc., 2018, 140, 4110 CrossRef CAS PubMed.
- R. J. Somerville, L. V. A. Hale, E. Gomez-Bengoa, J. Bures and R. Martin, J. Am. Chem. Soc., 2018, 140, 8771 CrossRef CAS PubMed.
- Y. Y. Zhou and C. Uyeda, Science, 2019, 363, 857 CrossRef CAS PubMed.
- J. Magano and J. R. Dunetz, Chem. Rev., 2011, 111, 2177 CrossRef CAS PubMed.
- (a) R. S. Paton and J. M. Brown, Angew. Chem., Int. Ed., 2012, 51, 10448 CrossRef CAS PubMed; (b) T. G. Saint-Denis, R.-Y. Zhu, G. Chen, Q.-F. Wu and J.-Q. Yu, Science, 2018, 359, 759 CrossRef CAS PubMed.
- A. Gelman and E. Meilakh, C. R. (Dokl.) Acad. Sci. URSS, 1942, 36, 171 Search PubMed.
- (a) F. Paul, J. Patt and J. F. Hartwig, J. Am. Chem. Soc., 1994, 116, 5969 CrossRef CAS; (b) J. F. Hartwig and F. Paul, J. Am. Chem. Soc., 1995, 117, 5373 CrossRef CAS; (c) M. S. Driver and J. F. Hartwig, J. Am. Chem. Soc., 1996, 118, 4206 CrossRef CAS; (d) F. Paul, J. Patt and J. F. Hartwig, Organometallics, 1995, 14, 3030 CrossRef CAS.
- (a) D. C. Powers, M. A. Geibel, J. E. Klein and T. Ritter, J. Am. Chem. Soc., 2009, 131, 17050 CrossRef CAS PubMed; (b) R. J. Pakula, M. Srebro-Hooper, C. G. Fry, H. J. Reich, J. Autschbach and J. F. Berry, Inorg. Chem., 2018, 57, 8046 CrossRef CAS PubMed; (c) J. N. Jaworski, S. D. McCann, I. A. Guzei and S. S. Stahl, Angew. Chem., Int. Ed., 2017, 56, 3605 CrossRef CAS PubMed; (d) J. Y. Wang, A. E. Strom and J. F. Hartwig, J. Am. Chem. Soc., 2018, 140, 7979 CrossRef CAS PubMed; (e) C. M. Palit, D. J. Graham, C.-H. Chen, B. M. Foxman and O. V. Ozerov, Chem. Commun., 2014, 50, 12840 RSC.
- J. P. Stambuli, R. Kuwano and J. F. Hartwig, Angew. Chem., Int. Ed., 2002, 41, 4746 CrossRef CAS PubMed.
- T. Hama and J. F. Hartwig, Org. Lett., 2008, 10, 1545 CrossRef CAS PubMed.
- T. Ogura, K. Yoshida, A. Yanagisawa and T. Imamoto, Org. Lett., 2009, 11, 2245 CrossRef CAS PubMed.
- R. K. Das, B. Saha, S. M. Rahaman and J. K. Bera, Chem. – Eur. J., 2010, 16, 14459 CrossRef CAS PubMed.
- F. Proutiere, M. Aufiero and F. Schoenebeck, J. Am. Chem. Soc., 2012, 134, 606 CrossRef CAS PubMed.
- (a) N. Hazari, D. Hruszkewycz and J. Wu, Synlett, 2011, 1793 CrossRef CAS; (b) D. P. Hruszkewycz, J. Wu, N. Hazari and C. D. Incarvito, J. Am. Chem. Soc., 2011, 133, 3280 CrossRef CAS PubMed.
- V. Srinivas, E. Balaraman, K. V. Sajna and K. C. Kumara Swamy, Eur. J. Org. Chem., 2011, 4222 CrossRef CAS.
- K. J. Bonney, F. Proutiere and F. Schoenebeck, Chem. Sci., 2013, 4, 4434 RSC.
- C. J. Diehl, T. Scattolin, U. Englert and F. Schoenebeck, Angew. Chem., Int. Ed., 2019, 58, 211 CrossRef CAS.
- G. Yin, I. Kalvet and F. Schoenebeck, Angew. Chem., Int. Ed., 2015, 54, 6809 CrossRef CAS.
- (a) G. Teverovskiy, D. S. Surry and S. L. Buchwald, Angew. Chem., Int. Ed., 2011, 50, 7312 CrossRef CAS; (b) C.-P. Zhang and D. A. Vicic, J. Am. Chem. Soc., 2011, 134, 183 CrossRef PubMed.
- M. Aufiero, T. Sperger, A. S. Tsang and F. Schoenebeck, Angew. Chem., Int. Ed., 2015, 54, 10322 CrossRef CAS PubMed.
- T. Scattolin, E. Senol, G. Yin, Q. Guo and F. Schoenebeck, Angew. Chem., Int. Ed., 2018, 57, 12425 CrossRef CAS PubMed.
- E. Senol, T. Scattolin and F. Schoenebeck, Chem. – Eur. J., 2019, 25, 9419 CrossRef CAS PubMed.
- X. Y. Chen, M. Pu, H. G. Cheng, T. Sperger and F. Schoenebeck, Angew. Chem., Int. Ed., 2019, 58, 11395 CrossRef CAS PubMed.
- I. Kalvet, G. Magnin and F. Schoenebeck, Angew. Chem., Int. Ed., 2017, 56, 1581 CrossRef CAS PubMed.
- (a) I. J. Fairlamb, Chem. Soc. Rev., 2007, 36, 1036 RSC; (b) J. Almond-Thynne, D. C. Blakemore, D. C. Pryde and A. C. Spivey, Chem. Sci., 2017, 8, 40 RSC.
- I. Kalvet, T. Sperger, T. Scattolin, G. Magnin and F. Schoenebeck, Angew. Chem., Int. Ed., 2017, 56, 7078 CrossRef CAS PubMed.
- S. T. Keaveney, G. Kundu and F. Schoenebeck, Angew. Chem., Int. Ed., 2018, 57, 12573 CrossRef CAS PubMed.
- F. Proutiere and F. Schoenebeck, Angew. Chem., Int. Ed., 2011, 123, 8342 CrossRef.
- M. Mendel, I. Kalvet, D. Hupperich, G. Magnin and F. Schoenebeck, Angew. Chem., Int. Ed., 2020, 59, 2115 CrossRef CAS PubMed.
- G. Magnin, J. Clifton and F. Schoenebeck, Angew. Chem., Int. Ed., 2019, 58, 10179 CrossRef CAS PubMed.
- I. Kalvet, K. Deckers, I. Funes-Ardoiz, G. Magnin, T. Sperger, M. Kremer and F. Schoenebeck, Angew. Chem., Int. Ed., 2020, 59, 7721 CrossRef CAS PubMed.
- A. N. Reyes-Sánchez, F. Cañavera-Buelvas, R. Barrios-Francisco, O. L. Cifuentes-Vaca, M. Flores-Alamo and J. J. García, Organometallics, 2011, 30, 3340 CrossRef.
- A. Arévalo, S. Ovando-Segovia, M. Flores-Alamo and J. J. García, Organometallics, 2013, 32, 2939 CrossRef.
- L. González-Sebastián, M. Flores-Alamo and J. J. García, Organometallics, 2013, 32, 7186 CrossRef.
- I. García-Ventura, M. Flores-Alamo and J. J. García, RSC Adv., 2016, 6, 101259 RSC.
- F. Schoenebeck, T. Sperger and C. Stirner, Synthesis, 2016, 115 CrossRef.
- M. Aufiero, T. Scattolin, F. Proutière and F. Schoenebeck, Organometallics, 2015, 34, 5191 CrossRef CAS.
- K. O. Kirlikovali, E. Cho, T. J. Downard, L. Grigoryan, Z. Han, S. Hong, D. Jung, J. C. Quintana, V. Reynoso, S. Ro, Y. Shen, K. Swartz, E. Ter Sahakyan, A. I. Wixtrom, B. Yoshida, A. L. Rheingold and A. M. Spokoyny, Dalton Trans., 2018, 47, 3684 RSC.
| This journal is © The Royal Society of Chemistry 2020 |