
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Copper(II) self-assembled clusters of bis((pyridin-2-yl)-1,2,4-triazol-3-yl)alkanes. Unusual rearrangement of ligands under reaction conditions†
Alexey
Gusev
 a,
Ivan
Nemec
bc,
Radovan
Herchel
b,
Victor
Shul'gin
a,
Irina
Ryush
a,
Michail
Kiskin
d,
Nickolay
Efimov
d,
Elena
Ugolkova
d,
Vadim
Minin
d,
Konstantin
Lyssenko
e,
Igor
Eremenko
d and
Wolfgang
Linert
*f
a,
Ivan
Nemec
bc,
Radovan
Herchel
b,
Victor
Shul'gin
a,
Irina
Ryush
a,
Michail
Kiskin
d,
Nickolay
Efimov
d,
Elena
Ugolkova
d,
Vadim
Minin
d,
Konstantin
Lyssenko
e,
Igor
Eremenko
d and
Wolfgang
Linert
*f
aGeneral and Physical Chemistry Department, V.I. Vernadsky Crimean Federal University, Acad. Vernadsky av. 4, Simferopol, 295007, Crimea. E-mail: galex0330@gmail.com
bDepartment of Inorganic Chemistry, Faculty of Science, Palacký University, 17. listopadu 1192/12, 771 46 Olomouc, Czech Republic
cRegional Centre of Advanced Technologies and Materials, Faculty of Science, Palacký University, 17. listopadu 12, Olomouc, Czech Republic
dN.S. Kurnakov Institute of General and Inorganic Chemistry, Russian Academy of Sciences, Moscow, 119991, Russia
eA. N. Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, 119991 Moscow, Russia
fInstitute of Applied Synthetic Chemistry, Vienna University of Technology, Getreidemarkt 9/163-AC, A-1060 Vienna, Austria. E-mail: wolfgang.linert@tuwien.ac.at
First published on 4th February 2019
Abstract
The reaction of two structurally related bridging ligands bis[5-(2-pyridyl)-1,2,4-triazole-3-yl]methane (H2L1) and bis[5-(2-pyridyl)-1,2,4-triazole-3-yl]ethane (H2L2) with copper(II) salts resulted in a surprising wide variety of complex structures [Cu2(H2L1)Cl2]Cl2·4CH3OH (1), [Cu4(L1)4]·4H2O (2), [Cu(H2L2)(ClO4)2] (3) and [Cu3(OH)Na2(L′)6](ClO4)·5H2O·C3H6O (4), where HL′ is 3,5-bis-(pyridin-2-yl)-1,2,4-triazole, which were structurally characterized by the X-ray diffraction method. Complexes 1 and 2 were prepared on the H2L1 basis and have binuclear and tetranuclear structures, respectively, demonstrating strong impact of the type of counter anion on the coordination mode of the ligand. In contrast, the reaction between Cu(ClO4)2 6H2O and H2L2 led to the preparation of mononuclear complex 3. The reaction of H2L2 with Cu(ClO4)2 under alkaline conditions led to oxidative rearrangement of the ligand and the homoleptic pentanuclear complex 4 with anionic ligand L′ was prepared. Magnetic properties were studied for compounds 1, 2 and 4 and for all of them the antiferromagnetic interactions between the Cu atoms were confirmed and analyzed by the spin Hamiltonian formalism. Furthermore, the occurrence of the antisymmetric exchange was confirmed in 4. The magnetic data analysis was supported by the X-band EPR measurements performed for complexes 1, 2 and 4.
1. Introduction
Current significant interest within supramolecular chemistry involves the design and synthesis of new ligand molecules to control the outcome of metal-assisted self-assembly processes.1 For this purpose, flexible ditopic ligands have attracted recent attention. Fine tuning of these ligand systems has resulted in the isolation of many structural types including grids, boxes, cylinders, various types of molecular polyhedra and helicates.2Significant progress has been achieved in the investigation of polynuclear complexes based on relatively simple bis(pyrazolyl-pyridine) bridging ligands and transition metal dications.3 M. Ward and coworkers have reported an extensive series of high-nuclearity cages based on self-assembly of the abovementioned ligands including an impressive number of polyhedral shapes of polynuclear complexes, e.g. M4L6 tetrahedra, M6L9 trigonal prisms, M8L12 cubes and ‘cuneane’, M12L18 truncated tetrahedra, and M16L24 tetra-capped truncated tetrahedra.4
Surprisingly, despite the attractive features of the nitrogen-rich triazole unit (e.g., multiple interaction modes, limited steric hindrance, moderate ligand field), the self-assembled structures have been much less studied for compounds with pyridyl-1,2,4-triazole ligands. It is noteworthy that the bis[5-(2-pyridyl)-1,2,4-triazole-3-yl]alkanes are excellent multidentate flexible ligands to construct supramolecular coordination complexes with various structures. They exhibit a strong chelate effect arising from the presence of the adjacent heterocyclic rings and furthermore, they can adopt various conformations due to large rotational flexibility of four aromatic rings, which may contribute to the preparation of complexes with interesting structures. Recent studies demonstrated that use of flexible alkyl spacers in linking pyridyltriazolyl chelating arms leads to the formation of novel polynuclear clusters with an exciting topology and structure.5
In this work, we present four novel Cu(II) complexes based on recently described5a bis((pyridin-2-yl)-1,2,4-triazol-3-yl)methane (H2L1) and 1,2-bis((pyridin-2-yl)-1,2,4-triazol-3-yl)ethane (H2L2) ligands. Both ligands have two interrelated characteristic features, a flexible backbone and the possibility to fulfill variable coordination modes, so that, depending on the coordination requirements. Varying the reaction conditions (counter-ions and pH value) allowed us to successfully isolate new complexes for the previously described ligands.
2. Results and discussion
Structure of complexes
In our previous studies,5 the products of the reaction between H2L1 and copper perchlorate were investigated. It was shown that by varying the molar ratio between the reagents we were able to prepare the tetranuclear complexes with different degrees of ligand protonation: [Cu4(H2L1)4]8+ and [Cu4(HL1)4]4+, respectively. In all cases, the perchlorate anions are non-coordinating and occupy the cavities of the crystalline lattice. In this paper, we investigated the interaction of a bis(5-(pyridin-2-yl)-1,2,4-triazol-3-yl)methane (H2L1) with two different copper salts containing potentially coordinating anions – CuCl2·2H2O and Cu(CH3COO)2·H2O.The complex 1 was formed upon reaction between H2L1 and CuCl2·2H2O in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio. In contrast to the previous studies the H2L1 ligand now acts as a bis-bidentate bridging ligand in the dinuclear double helicate [Cu2(H2L1)Cl2]Cl2, in which two ligands are wrapped helically around a pair of metal ions. The crystal structure of 1 is shown in Fig. 1.
:1 molar ratio. In contrast to the previous studies the H2L1 ligand now acts as a bis-bidentate bridging ligand in the dinuclear double helicate [Cu2(H2L1)Cl2]Cl2, in which two ligands are wrapped helically around a pair of metal ions. The crystal structure of 1 is shown in Fig. 1.
 | ||
| Fig. 1 Molecular structure of 1. Counter-anions and solvent molecules are omitted for clarity. Selected bond lengths (in Å): Cu1–N1 1.970(3), Cu1–N8 1.989(3), Cu1–N4 2.091(3), Cu1–N5 2.187(3), and Cu1–Cl1 2.2709. | ||
The Cu⋯Cu separation is 6.083(2) Å and this reflects the length and flexibility of the methylene spacer group of the H2L1 ligand. The Cu(II) ions are five coordinated by two bidentate N-donor ligand fragments and one chlorido ligand. According to Addison classification, the coordination polyhedra of the Cu(II) atoms adopt distorted square pyramidal geometry (SPY, the parameter τ is 0.36).6 The heavy distortion from the ideal SPY coordination geometry arises from the very non-equivalent Cu–N bond lengths: d(Cu–NPy) = 1.989 and 2.091 Å, d(Cu–NTz) = 1.970 and 2.187 Å (NPy stands for pyridine nitrogen atoms and NTz stands for triazole nitrogen atoms). Both near-planar pyridyltriazolyl fragments are twisted relative to each other by the angle of 69.88(4)°. The angle C(5)–C(1)–C(6), which represents the flexure of the methylene ‘belt’ within the dimer, was found to be 112.64(3)° on each strand, suggesting that minimal distortion of the ligand is required in order to conform to the helical topology for similar ligands.7
Replacement of the chloride ligand by acetate-anions led to the preparation of tetranuclear complex 2 in which the H2L1 ligand is fully deprotonated (L12−) in contrast to previous studies.5,8 The molecular structure of 2 is shown in Fig. 2 and Fig. S1†
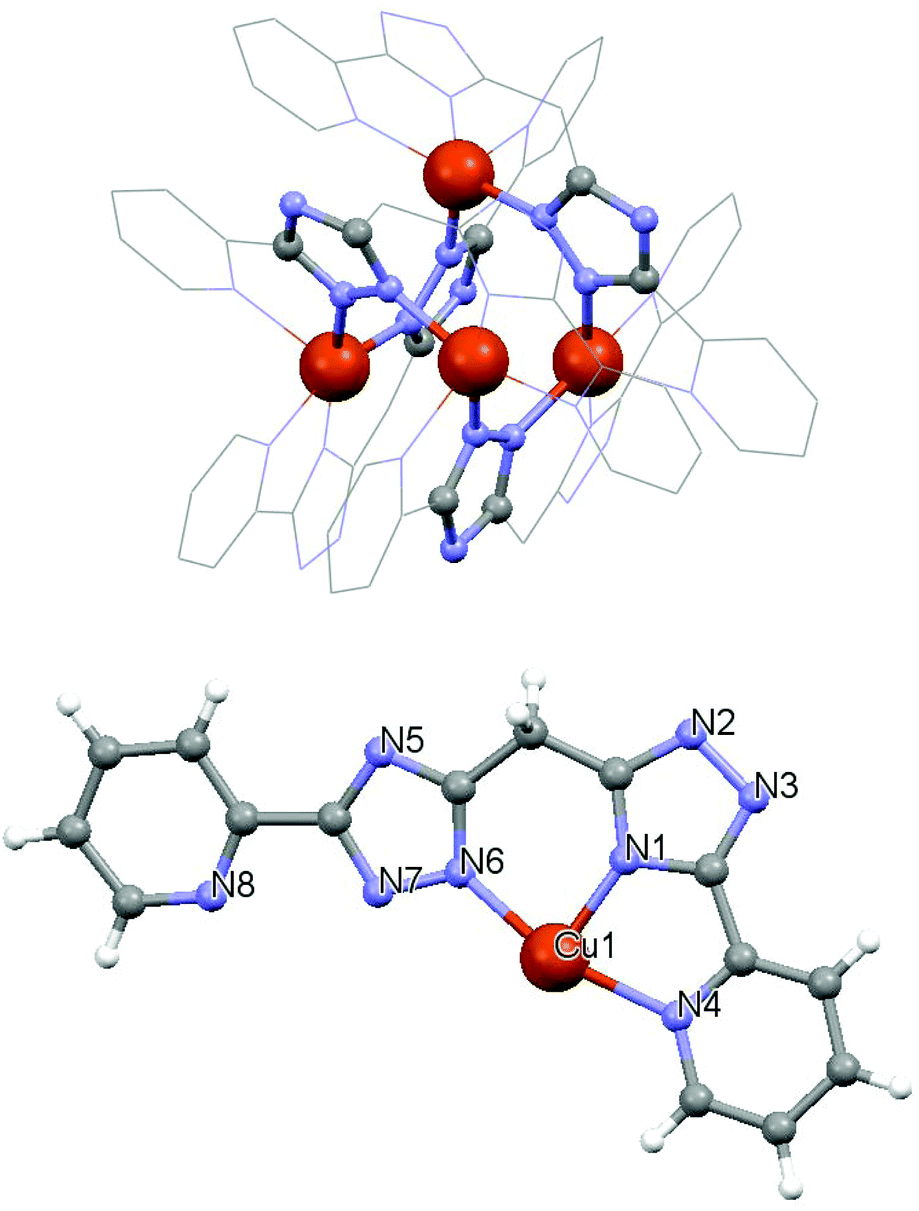 | ||
| Fig. 2 (above) Molecular structure of 2 with highlighted Cu4 core. Counter-anions, H-atoms and solvent molecules are omitted for clarity. (below) The asymmetric unit of 2. Selected bond lengths (in Å). Cu1–N1 1.912(4), Cu1–N7 1.974(3), Cu1–N6 2.040(4), Cu1–N4 2.136(3), and Cu1–N8 2.254(4). | ||
The complex [Cu4(L1)4]·6H2O·2.8MeOH (2) is a homoleptic [2 × 2] grid involving four copper(II) centres bridged by the μ-N1,N2-triazolyl units. All the copper atoms are pentacoordinated with the N5 donor set. Each copper(II) atom displays slightly distorted square-pyramidal geometry with the nitrogen atom from the pyridine ring in the axial position (τ = 0.13). Four copper(II) centres are arranged into an unusual Cu4N8 trigonal pyramidal core (Fig. S1). The [2 × 2] array of Cu(II) ions has the Cu⋯Cu separations of 4.031–4.471 Å, which are slightly longer than in the case of the complexes involving protonated forms of the ligand (3.93–3.98 Å for [Cu4(H2L1)]8+ and 3.97–4.06 Å for [Cu4(HL1)]4+)5 and these values are typical of the μ-triazolyl coordination mode.8 The ligand strands are oriented in a “head-to-tail” arrangement at the Cu(II) sites; the “head” and “tail” terms refer to the tridentate, and bidentate donor pockets, respectively. The L12− ligand has a substantially planar structure with the average deviation of the atoms from their best-fit mean plane of 0.13 Å.
The reaction of H2L2 with Cu(ClO4)2·6H2O in MeOH afforded a blue solution from which the [Cu(H2L2)(ClO4)2] (3) compound was obtained. X-ray crystallography (Fig. 3) showed that in 3 the Cu ion has the CuN4O2 coordination environment with the H2L2 ligand acting as an equatorial tetradentate chelator (d(Cu–NPyr) = 2.0631(17) Å, d(Cu–NTz) = 1.9690(17) Å) and with two perchlorate ligands in axial positions. The coordination geometry is an axially elongated octahedron due to the Jahn–Teller effect (d(Cu–O) = 2.521(2) Å). The two bidentate pyridyltriazolyl arms are not quite coplanar, which can be described by the angle 9.77° between the two mean planes involving each bidentate fragment. It appears that the elongation of the bridging aliphatic part between the two coordinating fragments makes the tetradentate coordination mode more favorable, when compared with the related ones.5
 | ||
| Fig. 3 Molecular structure of 3. Selected bond lengths (in Å): Cu1–N1 = 2.0631(17), Cu1–N2 = 1.9690(17), Cu1–O3 = 2.521(2). | ||
A very interesting result was obtained by studying the reaction of H2L2 with copper salts in an alkaline medium in an attempt to determine the coordination modes of the deprotonated form of 1,2-bis(5-(pyridin-2-yl)-1,2,4-triazol-3-yl)ethane. In order to achieve this goal we tried various ratios of reagents and different types of copper salts (CuCl2·2H2O, Cu(CH3COO)2·H2O and Cu(ClO4)2·6H2O) and bases (NaOH, MeONa, Et3N). In all cases, unidentifiable mixtures of compounds were obtained. However, prolonged reflux (6 h) of H2L2 in combination with two equivalents of Cu(ClO4)2·6H2O and in the presence of an excess of NaOH allowed us to isolate the crystals of the unusual complex, which, according to elemental analysis and electrospray ionization (ESI) data, might have an approximate formula [Cu3(OH)Na2(L′)6](ClO4) where HL′ is 3,5-bis-(pyridin-2-yl)-1,2,4-triazole. The ESI spectra of 4 contain several predominant peaks corresponding to the molecular ion and its decay products. A characteristic signal for a single charged ion at m/z 1586.9 is assigned to the species [Cu3(OH)Na2(L′)6]+, based on a reasonable agreement of the isotopic distribution pattern and this structural motif was further confirmed by the single-crystal XRD measurements. The presence of water and acetone molecules in the complex has been confirmed by thermogravimetric analysis. The TGA plot recorded for 4 shows two consecutive and distinct weight losses of 2.8% and 5.2% at about 55–60 °C and at 95–110 °C, respectively, and associated with the presence of one acetone molecule (calculated m m−1 loss = 2.94%) and five water molecules (calculated m m−1 loss = 4.91%) in the crystal packing of 4 (Fig. S2†).
By our assumption, the presence of a 3,5-bis-(pyridin-2-yl)-1,2,4-triazolate anion is associated with the oxidative degradation of H2L2 in an alkaline medium and subsequent symmetrization. Both the perchlorate anion and copper ion could act as the oxidizing agents under reaction conditions. The formation of the 3,5-bis-(pyridin-2-yl)-1,2,4-triazolate-ligand from the H2L1 can be rationalised by the mechanism depicted in Scheme 1. It should be noted that W.-Q. Lin and co-workers9 recently showed the possibility of spacer group oxidation in bis(5-(pyridin-2-yl)-1,2,4-triazol-3-yl)alkanes under hydrothermal conditions. Coordination compounds of transition metals on the 3,5-bis-(pyridin-2-yl)-1,2,4-triazolate anion basis are well known and are summarized in several reviews.8 However, the structure of complex 4 was not previously described.
 | ||
| Scheme 1 The schematic representations of the H2L1 and H2L2 ligands. | ||
Complex 4 crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) as a solvate with 11 water molecules (some solvate molecules that were disordered were removed by the SQUEEZE procedure to achieve reasonable refinements.10). Differences in the solvate composition for a polycrystalline sample and a single crystal are associated with partial desolvation of the sample upon drying. The crystalline lattice of 4 consists of trinuclear [Cu3(μ3-OH)Na2(L′)6]+ complex molecules and perchlorate counter anions. The structure of 4 is shown in Fig. 4.
as a solvate with 11 water molecules (some solvate molecules that were disordered were removed by the SQUEEZE procedure to achieve reasonable refinements.10). Differences in the solvate composition for a polycrystalline sample and a single crystal are associated with partial desolvation of the sample upon drying. The crystalline lattice of 4 consists of trinuclear [Cu3(μ3-OH)Na2(L′)6]+ complex molecules and perchlorate counter anions. The structure of 4 is shown in Fig. 4.
 | ||
| Fig. 4 (a) Molecular structure of 4 (H atoms and counter-anion are omitted for clarity). (b) Coordination mode of 3,5-bis-(pyridin-2-yl)-1,2,4-triazolate ligands in 4. | ||
Three Cu and two Na cations define a trigonal–bipyramidal polyhedron, in which the Cu⋯Na, Cu⋯Cu, and Na⋯Na average distances are 4.564, 3.510, and 8.177 Å, respectively.
Six 3,5-bis-(pyridin-2-yl)-1,2,4-triazolate mono-deprotonated ligands wrap around the complex pentaheteronuclear cluster. Two homochiral [Na(L′)3] units are placed in the apical positions of the molecule and they simultaneously coordinate the {(μ3-OH)Cu3}5+ central core. In this manner, the ligands coordinate to the Cu3Na2 unit with helical arrangement, while the whole complex cluster has no intrinsic D3 symmetry. Note that enantiomers (ΔΔ, ΛΛ) of the cation unit induced by the helical coordination arrangement of the homochiral unit coexist as a racemic form in the crystal.
The apical Na ions are in distorted octahedral environments with six nitrogen atoms from three 3,5-bis-(pyridin-2-yl)-1,2,4-triazolate ligands. The average Na–NPy and Na–Ntz bond lengths are 2.507 and 2.445 Å, respectively.
Three equatorial Cu(II) atoms possess the distorted N4O trigonal-pyramidal geometry (τ = 0.76, 0.74, 0.68), in which four nitrogen atoms come from the remaining coordination sites of two L′− ligands (Fig. 4b). The structure of the trinuclear core is noticeably asymmetric, which is reflected in various Cu–O bond lengths and Cu–Cu distances as well as Cu–O–Cu angles Fig. S3.† The oxygen atom from the hydroxido ligand is slightly raised above the plane (by 0.157 and 0.353 Å for two positions of the disordered OH-group) formed by three copper atoms. The phase purity of the bulk samples was confirmed by XRD analyses as shown in Fig. S4.†
Solvent water molecules form H-bonds with each other (O⋯O 2.712–3.299 Å), oxygen atoms of the ClO4− anion (O2S⋯O3 W 2.901 Å) and nitrogen atom of the triazole fragment (N⋯O 2.782–2.986 Å).
In summary, the geometry of a complex cation is similar to those described in previously reported studies,11 in which two terminal triple-stranded [Na(L′)3] units wrapped a planar [Cu3(μ3-OH)]5+ core to form a bis(triple-helical) complex with trigonal–bipyramidal topology. The rigid bisbidentate L′− ligand links one apical Na ion and one equatorial copper(II) ion in a cis-bridging mode.
Magnetic properties
Variable-temperature direct current magnetization data for polynuclear compounds 1, 2, and 4 were collected on powdered microcrystalline samples over a temperature range from 2 to 300 K and under an applied dc field of 5000 Oe. The magnetic data of dinuclear complex 1 are shown in Fig. 5. The significant decrease of the effective magnetic moment below 50 K and the presence of the maximum on the Mmolvs. T curve at T = 6 K confirm the weak antiferromagnetic exchange. Therefore, the following spin Hamiltonian was used for fitting the magnetic data | (1) |
 | (2) |
 | ||
| Fig. 5 Temperature dependence of the effective magnetic moment (calculated from magnetization at B = 0.5 T) of 1 with the low-temperature region expanded in the inset. Circles = experimental points, lines = calculated using the best-fit parameters: J = −6.4 cm−1, g = 2.04 and χTIP = 5.25 × 10−9 m3 mol−1. | ||
The magnetic properties of tetranuclear complex 2 are similar, the effective magnetic moment decreases on lowering the temperature practically to zero and there is also the maximum on the Mmolvs. T curve at T = 70 K; all these data suggest the presence of the strong antiferromagnetic exchange. The most probable superexchange pathway leads through the bridging μ-N1,N2-triazolyl units; thus the following spin Hamiltonian was postulated
 | (3) |
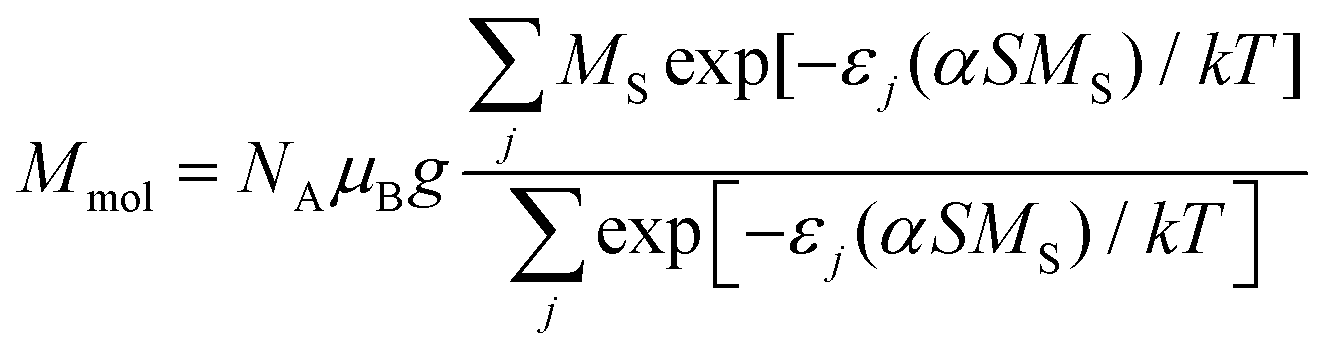 | (4) |
The best-fit to the experimental magnetic data was obtained with J = −58.7 cm−1 and g = 2.09 and the resulting energy levels are also depicted (Fig. 6).
 | ||
| Fig. 6 Top: Temperature dependence of the effective magnetic moment (calculated from magnetization at B = 0.5 T) of 2 with the low-temperature region expanded in the inset. Circles = experimental points, lines = calculated using the best-fit parameters: J = −58.7 cm−1 and g = 2.09. Bottom: The calculated energy levels according to spin S. | ||
It is interesting to compare magnetic properties of the complexes [Cu4(H2L1)(ClO4)8],5b [Cu4(HL1)(ClO4)4],5d from our previous articles and [Cu4(L1)4] from the current work. Despite the general similarity, these complex exchange parameters are different: −70, −53.7 and −58.7 cm−1, respectively. All complexes are structurally tetranuclear and the main difference between them lies in the degree of protonation of the ligand which causes distortion of the Cu–N–N–Cu′ bridge fragment. From the literature18 it is known that the absolute J value decreases with the asymmetry of the Cu–N–N–Cu′ framework. Asymmetry of the bridge could be evaluated as the Cu–N–N and N–N–Cu′ angle difference – here noted as Δ. Notable that the highest value Δ (10.83°) is observed for the complex [Cu4(HL1)(ClO4)4] for which the value of the exchange parameter is the smallest in this series and, in contrast, the lowest value of Δ (8.35°) corresponds to the maximum value of the J-parameter in [Cu4(H2L1)(ClO4)8].
The magnetic data of trinuclear complex 4 displayed in Fig. 7 exhibit very strong antiferromagnetic exchange, as it is evident from the room temperature value of the effective magnetic moment (2.3μB), which is significantly lower than the theoretical value of 3.0μB for three non-interacting spin centers (Si = 1/2 with g = 2.0). The effective magnetic moment is constantly lowering on cooling down to ≈100 K, and then the plateau with a value of 1.8 μB is reached in the temperature range from 80 to 40 K, which agrees well with the S = 1/2 ground state. However, further cooling results in a drop of μeff down to 1.1μB at T = 2 K. This drop can be explained by the antisymmetric exchange among copper atoms within the triangle arrangement. The concept of this interaction was developed by Dzyaloshinsky and Moriya,13 recently reviewed,14 and is typical of antiferromagnetically coupled trinuclear copper complexes.11,15 Then, the suitable spin Hamiltonian is of the form
 | (5) |
 | (6) |
θcosϕ,sinθsinϕ,cosθ). Then, the averaged molar magnetization of the powder sample was calculated as the integral (orientational) average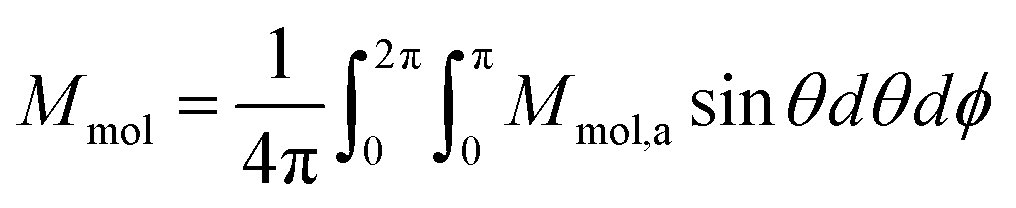 | (7) |
 | ||
| Fig. 7 Top: Temperature dependence of the effective magnetic moment (calculated from magnetization at B = 0.5 T) of 4 with the low-temperature region expanded in the inset and the isothermal magnetization data measured at T = 2, 3 and 5 K. Circles are experimental points, blue lines are calculated using the best-fit parameters: J = −213 cm−1 and g = 2.02, red lines = calculated using the best-fit parameters: J12 = −236 cm−1, J13 = J23 = −223 cm−1, g = 2.05 and |dz| = 16.8 cm−1. Bottom: The calculated energy levels in magnetic field using J = −213 cm−1 and g = 2.02 (blue color) and J12 = −236 cm−1, J13 = J23 = −223 cm−1, g = 2.05 and |dz| = 16.8 cm−1 (red color). | ||
First, the experimental data were analyzed without introducing the antisymmetric exchange (ASE), which resulted in J = −213 cm−1 and g = 2.02; however, this model cannot account for low temperature data as it is evident from Fig. 7. Therefore, the ASE was included into the fitting procedure. The best-fits were acquired with J12 = −236 cm−1, J13 = J23 = −223 cm−1, g = 2.05 and |dz| = 16.8 cm−1 (Fig. 7) or alternatively with J12 = −218 cm−1, J13 = J23 = −231 cm−1, g = 2.05 and |dz| = 16.7 cm−1 (Fig. S5†). It must be noted that the equilateral model with J12 = J23 = J13 = J was unable to concurrently describe temperature and field dependent magnetic data for 4 and therefore the isosceles model was applied. The respective energy levels are shown in Fig. 7 and Fig. S5,† where the energy pattern corresponds to two S = 1/2 and one S = 3/2 spin states split due to the antiferromagnetic coupling. Also, it is apparent that the ASE generates large magnetic anisotropy of two S = 1/2 levels, which can be quantified by calculating the g-factors for the lowest Kramers doublets using effective spin Seff = 1/2, which results in gxy = 0.83 and gz = 2.05.
EPR studies
The X-band EPR spectra of complexes 1 and 2 were recorded on powder samples at room and 50 K temperatures. The magnetic exchange properties of the polynuclear Cu(II) species resulting from the antiferromagnetic interactions very often cause no EPR signals to be observed at room temperature (apparently, because relaxation phenomena hamper the observation) and only badly resolved spectra at low temperatures. In the present case, the room temperature EPR spectra of title complexes exhibit anisotropic signals (Fig. 8 and Fig. S6†) typical of square pyramidal/trigonal–bipyramidal Cu(II) centres and show a poorly resolved rhombic feature both at room temperature and at 50 K. | ||
| Fig. 8 Experimental and simulated X-band EPR spectra of a polycrystalline sample of complex 2 at 50 K. | ||
The EPR spectra of polycrystalline samples 1 and 2 contain anisotropic resonances in the half-field region at 1600 G, assignable to a forbidden transition (ΔMS = ±2, g ≈ 4.2), as well as anisotropic signals around the typical 3200 G region (ΔMS = ± 1).
The spectra are described by a rhombically distorted spin Hamiltonian with a fine structure
| Ĥ = β(gxSxHx + gySyHy + gzHzSz) + D(Sz2 − S(S + 1)/3) + E(Sx2 − Sy2) | (8) |
| Complex | S | T | g x | g y | g z | D (cm−1) | E (cm−1) |
|---|---|---|---|---|---|---|---|
| 1 | 1 | 293 | 2.10 | 2.04 | 2.21 | 0 | 0 |
| 2 | 1 | 50 | 2.01 | 2.17 | 2.25 | 0.2067 | 0.0645 |
| 293 | 2.01 | 2.17 | 2.25 | 0.2067 | 0.0645 | ||
| 1 | 50 | 2.06 | 2.04 | 2.18 | 0.0421 | 0.0140 | |
| 293 | 2.07 | 2.04 | 2.18 | 0.0421 | 0.0140 | ||
| 2 | 50 | 2.10 | 2.10 | 2.28 | 0.0290 | 0.0043 | |
| 293 | 2.08 | 2.08 | 2.27 | 0.0309 | 0.0037 |
The EPR spectrum of the tetramer was simulated via the Belford (eigenfield) method16 as the sum of the spectra of four complexes, three of which have spin 1 (but the parameters of the complexes with E2(1) = E3(1) = 0 were assumed to be identical) and one is spin 2. The complex concentrations in different spin states were calculated from Boltzmann's populations of the corresponding levels at given temperatures (Fig. 8). The best fit parameters D and E and the components of the g-tensor are given in Table 1.
Electron paramagnetic resonance spectra of the ground crystalline sample of 4 have been recorded at room temperature and at 2 K at X-band frequencies. At room temperature, the EPR spectrum of the trimer 4 contains one weak unresolved peak (Fig. S7†). Upon cooling to 2 K, however in addition to the main signal at g = 2.04 the EPR spectrum exhibits a second band at lower fields with g = 1.01 (Fig. 9). These features are associated with the presence of the antisymmetric exchange resulting in large anisotropy of the g-tensor of the ground state with the effective spin 1/2.11 Moreover, the experimental value of gxy = 1.01 is in good agreement with the value of gxy = 0.83 obtained from simulation of the magnetic data.
 | ||
| Fig. 9 X-Band EPR spectra of a polycrystalline sample of complex 4 at 2 K. | ||
3. Conclusions
In summary we have successfully synthesized diverse copper-based architectures using symmetric bis-chelating ligands bis((pyridin-2-yl)-1,2,4-triazol-3-yl)methane and 1,2-bis((pyridin-2-yl)-1,2,4-triazol-3-yl)ethane. The crystal structures of four complexes were determined, and it was shown that both the conformational freedom and coordination flexibility of the semirigid ligands have an important influence on the resulting structures. One of the promising results features a reaction of 1,2-bis((pyridin-2-yl)-1,2,4-triazol-3-yl)ethane and Cu(ClO4)2 under alkaline conditions, which results in in situ rearrangements of the ligand to 3,5-bis(pyridine-2-yl)-1,2,4-triazole. The antiferromagnetic interactions between the Cu(II) atoms of different strengths were found in the polynuclear complexes 1, 2 and 4 including the antisymmetric exchange for trinuclear complex 4 evidenced also by X-band EPR spectroscopy.4. Experimental
General
The reagents and solvents employed were commercially available and used as received without further purification. The C, H, and N microanalyses were carried out with a PerkinElmer 240 elemental analyser. Electrospray mass spectra of complexes were recorded on a Finnagan TSQ 700 mass spectrometer in the positive ion mode. Samples were prepared at a concentration of ∼2 mg ml−1 MeOH. Spectra were acquired over an m/z range of 50–2000; several scans were averaged to provide the final spectrum. Magnetic susceptibility measurements were performed with the use of a Quantum Design magnetometer/susceptometer PPMS-9 under an external magnetic field of 0.5 T in the temperature range of 2–300 K. The diamagnetic contributions of the samples were estimated from Pascal's constants. The X-band EPR spectra were recorded on an Elexsys E-680X radiofrequency spectrometer (Bruker). The commercially available, CuCl2·2H2O, Cu(CH3COO)2·H2O and Cu(ClO4)2·6H2O were used as reactants. The synthesis of H2L1 and H2L2 was described previously.5Synthetic procedures
:1 v/v) (10 cm3) and a solution of CuCl2·2H2O (0.171 g, 1 mmol) in MeOH (10 cm3) was added and the resulting blue solution was vigorously stirred for 1 h at 50 °C. The reaction mixture was filtered, and the solution was left to stand for a few days and yielded greenish blue X-ray-quality single crystals of 1.
Data for [Cu2(H2L1)Cl2]Cl2·4CH3OH (1): yield 51%. Anal. Found: C, 40.47; H, 4.12; N, 22.16%. Required for C34H40Cl4Cu2N16O4: C, 40.60; H, 4.01; N, 22.28%.
Data for [Cu4L14]·4H2O (2): yield 63%. Anal. Found: C, 47.08; H, 3.38; N, 29.24%. Required for C60H48Cu4N32O4: C, 46.93; H, 3.15; N, 29.19%.
:1 v/v) (10 cm3) and a solution of Cu(ClO4)2·6H2O (0.378 g, 1 mmol) in MeOH (10 cm3) was added. The resulting blue solution was vigorously stirred for 3 h at room temperature. Then, the solution was left to stand for a few days at room temperature and blue single crystals of X-ray quality precipitated.
Data for [Cu(H2L2)](ClO4)2: yield 55%. Anal. Found: C, 33.22; H, 2.73; N, 19.10%. Required for C16H14Cl2CuN8O8: C, 33.08; H, 2.43; N, 19.29%.
:1 v/v) (10 cm3) and a solution of Cu(ClO4)2·6H2O (0.756 g, 2 mmol) in acetone water (1:1 v/v) (20 cm3) was added and the resulting blue solution was vigorously stirred for 1 h at room temperature. Then, 2 ml of 30% NaOH solution (20 mmol) was added and the deep green reaction mixture was refluxed for 6 h. After cooling, the mixture was filtered and the solution was left to stand at room temperature. Big green X-ray-quality single crystals were collected after three days.
Data for [Cu3OHNa2(L′)6](ClO4)·5H2O·C3H6O (HL′ – 3,5-bis-(pyridin-2-yl)-1,2,4-triazole): yield 11%. Anal. Found: C, 48.88; H, 3.71; N, 23.03%. Required for C75H65ClCu3Na2N30O11: C, 49.10; H, 3.57; N, 22.90%.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the financial support from the Russian foundation for basic research (project no. 16-03-00386) and Russian science foundation (project no. 18-13-00024). MAK, NNE, EAU, VVM and ILE acknowledge the State Assignment on Fundamental Research of the Kurnakov Institute of General and Inorganic Chemistry. IN would like to acknowledge financial support from The Ministry of Education, Youth and Sports of the Czech Republic (LO1305). RH acknowledges the financial support from institutional sources of the Department of Inorganic Chemistry, Palacky University Olomouc, Czech Republic. Single crystal X-ray diffraction (compounds 2 and 4) were performed at the User Facilitiy Centers of IGIC RAS.Notes and references
- (a) J.-M. Lehn, Science, 2002, 295, 2400 CrossRef CAS PubMed; (b) J.-M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4763 CrossRef CAS PubMed.
- (a) S. O. Scott, E. L. Gavey, S. J. Lind, K. C. Gordon and J. D. Crowley, Dalton Trans., 2011, 40, 12117 RSC; (b) U. R. Pokharel, F. R. Fronczek and A. W. Maverick, Dalton Trans., 2013, 42, 14064 RSC; (c) R. A. S. Vasdev, D. Preston and J. D. Crowley, Dalton Trans., 2017, 46, 2402 RSC; (d) A. M. Castilla, W. J. Ramsay and J. R. Nitschke, Acc. Chem. Res., 2014, 47, 2063–2073 CrossRef CAS PubMed; (e) N. J. Young and B. P. Hay, Chem. Commun., 2013, 49, 1354–1379 RSC; (f) M. M. J. Smulders, I. A. Riddell, C. Browne and J. R. Nitschke, Chem. Soc. Rev., 2013, 42, 1728–1754 RSC; (g) M. L. Saha, S. De, S. Pramanik and M. Schmittel, Chem. Soc. Rev., 2013, 42, 6860–6909 RSC.
- (a) L. N. Dawe, K. V. Shuvaev and L. K. Thompson, Chem. Soc. Rev., 2009, 38, 2334 RSC; (b) M. Fujita, M. Tominaga, A. Hori and B. Therrien, Acc. Chem. Res., 2005, 38, 369 CrossRef CAS PubMed; (c) D. Fiedler, D. H. Leung, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2005, 38, 349 CrossRef CAS PubMed; (d) T. D. Hamilton and L. R. MacGillivray, Cryst. Growth Des., 2004, 4, 419 CrossRef CAS; (e) A. L. Garay, A. Pichon and S. L. James, Chem. Soc. Rev., 2007, 36, 846 RSC; (f) S. Alvarez, Dalton Trans., 2006, 2209 Search PubMed.
- (a) M. D. Ward, Chem. Commun., 2009, 4487 RSC; (b) B. R. Hall, L. E. Manck, I. S. Tidmarsh, A. Stephenson, B. F. Taylor, E. J. Blaikie, D. A. Vander Griend and M. D. Ward, Dalton Trans., 2011, 40, 12132 RSC; (c) A. Stephenson, S. P. Argent, T. Riis-Johannessen, I. S. Tidmarsh and M. D. Ward, J. Am. Chem. Soc., 2011, 133, 858 CrossRef CAS PubMed; (d) H. Fenton, I. S. Tidmarsh and M. D. Ward, Dalton Trans., 2009, 4199 RSC; (e) S. P. Argent, H. Adams, T. Riis-Johannessen, J. C. Jeffery, L. P. Harding, W. Clegg, R. W. Harrington and M. D. Ward, Dalton Trans., 2006, 2996 Search PubMed; (f) A. M. Najar, C. Avci and M. D. Ward, Inorg. Chem. Commun., 2012, 15, 126 CrossRef CAS; (g) S. P. Argent, H. Adams, L. P. Harding, T. Riis-Johannessen, J. C. Jeffery and M. D. Ward, New J. Chem., 2005, 29, 904 RSC; (h) A. Stephenson, D. Sykes and M. D. Ward, Dalton Trans., 2013, 42, 6756 RSC.
- (a) A. N. Gusev, V. F. Shulgin, E. Beyjyyev, G. G. Alexandrov, I. L. Eremenko and W. Linert, Polyhedron, 2015, 85, 525 CrossRef CAS; (b) A. N. Gusev, I. Nemec, R. Herchel, E. Bayjyyev, G. A. Nyshchimenko, G. G. Alexandrov, I. L. Eremenko, Z. Travnicek, M. Hasegawa and W. Linert, Dalton Trans., 2014, 43, 7153 RSC; (c) A. N. Gusev, M. Hasegawa, G. A. Nishchymenko, V. F. Shulgin, S. B. Meshkova, P. Doga and W. Linert, Dalton Trans., 2013, 42, 6936 RSC; (d) A. N. Gusev, V. F. Shulgin, I. O. Ryush, M. Hasegawa, M. A. Kiskin, N. N. Efimov, K. A. Lyssenko, I. L. Eremenko and W. Linert, Eur. J. Inorg. Chem., 2017, 704 CrossRef CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349 RSC.
- C. S. Hawes, C. M. Fitchett and P. E. Kruge, Supramol. Chem., 2012, 24, 553 CrossRef CAS.
- (a) J. G. Haasnoot, Coord. Chem. Rev., 2000, 200–202, 131 CrossRef CAS; (b) G. Aromí, L. A. Barrios, O. Roubeau and P. Gamez, Coord. Chem. Rev., 2011, 255, 485 CrossRef.
- W.-Q. Lin, Y.-Y. Peng, L. Tong, J.-H. Jia, J.-L. Liu, Y.-C. Chen, W.-B. Chen and M.-L. Tong, Chem. – Asian J., 2017, 12, 2172 CrossRef CAS PubMed.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9 CrossRef CAS PubMed.
- (a) S. Ferrer, F. Lloret, E. Pardo, J. M. Clemente-Juan, M. Liu-González and S. García-Granda, Inorg. Chem., 2012, 51, 985 CrossRef CAS PubMed; (b) P. A. Angaridis, P. Baran, R. Boca, F. Cervantes-Lee, W. Haase, G. Mezei, R. G. Raptis and R. Werner, Inorg. Chem., 2002, 41, 2219 CrossRef CAS PubMed; (c) C. Di Nicola, F. Garau, M. Gazzano, M. Monari, L. Pandolfo, C. Pettinari and R. Pettinari, Cryst. Growth Des., 2010, 10, 3120 CrossRef CAS.
- R. Boča, A Handbook of Magnetochemical Formulae, Elsevier, Amsterdam, 2012 Search PubMed.
- (a) I. Dzyaloshinsky, J. Phys. Chem. Solids, 1958, 4, 241 CrossRef CAS; (b) T. Moriya, Phys. Rev., 1960, 120, 91 CrossRef CAS.
- R. Boča and R. Herchel, Coord. Chem. Rev., 2010, 254, 2973 CrossRef.
- E. M. Zueva, M. M. Petrova, R. Herchel, Z. Trávníček, R. G. Raptis, L. Mathivathanan and J. E. McGrady, Dalton Trans., 2009, 5924 RSC.
- G. Belford, R. L. Belford and J. F. Burkhaven, J. Magn. Reson., 1973, 11, 251 CAS.
- (a) G. M. Sheldrick, SADABS. Program for Scanning and Correction of Area Detector Data, Guttinngen Univ., Guttingen, 1997 Search PubMed; (b) G. M. Sheldrick, SHELX97. Program for the Solution of Crystal Structures, Guttinngen Univ., Guttingen, 1997 Search PubMed; (c) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- S. Ferrer, P. J. van Koningsbruggen, J. G. Haasnoot, J. Reedijk, H. Kooijman, A. L. Spek, L. Lezama, A. M. Arif and J. S. Miller, J. Chem. Soc., Dalton Trans., 1999, 4269 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1860806–1860809. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8dt04816a |
| This journal is © The Royal Society of Chemistry 2019 |