
Metal oxide/hydroxide-based materials for supercapacitors
Fan Shi,
Lu Li,
Xiu-li Wang,
Chang-dong Gu and
Jiang-ping Tu*
State Key Laboratory of Silicon Materials, Key Laboratory of Advanced Materials and Applications for Batteries of Zhejiang Province, Department of Materials Science and Engineering, Zhejiang University, Hangzhou 310027, China. E-mail: tujp@zju.edu.cn; tujplab@zju.edu.cn; Fax: +86-571-87952573; Tel: +86-571-87952856
First published on 18th August 2014
Abstract
Supercapacitors are promising energy storage and conversion devices with high power densities. However, their low energy densities limit their practical application. The electrode is a key component that determines the performance of supercapacitors. As electrode materials, transition metal oxides/hydroxides usually exhibit high capacitance, leading to high energy densities. This review discusses the advantages and disadvantages of different metal oxides/hydroxides, in order to synthesize high-performance electrode materials. Two main strategies to enhance the supercapacitive performance are proposed: developing composites and nanostructured materials.
1. Introduction
In the past several decades, the global economy has developed rapidly, as has the world population, which has resulted in an increasingly strong demand for energy. Since global energy consumption is increasing continuously, the exhaustion of global energy may soon threaten human society. One research report suggests that the global energy needs will quickly double by the mid-century and even triple by 2100.1 To date, fossil fuels have been used widely and largely in our society. However, fossil fuel reserves are limited, and will be depleted soon. Shafiee et al. have computed that the depletion time for oil, coal, and gas, are about 35, 107 and 37 years, respectively.2 Thus, research into developing clean, reliable, and sustainable alternative energies, such as solar, tidal and wind, have been implemented worldwide to replace fossil fuels. An important intermediate step between these energy resources and versatile energy applications is energy storage, and this has attracted significant attention in academia and industry. Clearly, it is necessary to develop low-cost, efficient, and environmentally friendly energy storage devices that can satisfy the needs of modern life.Among various energy storage systems, supercapacitors (also referred to as electrochemical capacitors) are considered to be one of the most promising future energy storage systems, due to their high power density (>10 kW kg−1), high rate capability and long cycle life (>1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000 cycles).3 One of their unique advantages is that the power densities of supercapacitors are superior to batteries. In addition, their energy densities are several orders of magnitude higher than traditional capacitors, although they are still much lower than batteries and fuel cells.4,5 Clearly, supercapacitors can play a role to bridge the power and energy gap between batteries/fuels and traditional dielectric capacitors. Since the first electrochemical capacitor patent was applied by Becker in 1957,6 considerable efforts have been put into researching efficient supercapacitors. Especially in recent years, to meet the ever-increasing demand of modern applications requiring high power density, such as energy back-up systems, portable electronic devices, and hybrid electric vehicles,7–9 there has been an explosion in supercapacitors research, and researchers have made great progress in this field. Their main target has been to enhance the energy densities of supercapacitors without sacrificing their high power densities, with the aim that supercapacitors will be able to function as a primary power source, just like batteries.10–12
000000 cycles).3 One of their unique advantages is that the power densities of supercapacitors are superior to batteries. In addition, their energy densities are several orders of magnitude higher than traditional capacitors, although they are still much lower than batteries and fuel cells.4,5 Clearly, supercapacitors can play a role to bridge the power and energy gap between batteries/fuels and traditional dielectric capacitors. Since the first electrochemical capacitor patent was applied by Becker in 1957,6 considerable efforts have been put into researching efficient supercapacitors. Especially in recent years, to meet the ever-increasing demand of modern applications requiring high power density, such as energy back-up systems, portable electronic devices, and hybrid electric vehicles,7–9 there has been an explosion in supercapacitors research, and researchers have made great progress in this field. Their main target has been to enhance the energy densities of supercapacitors without sacrificing their high power densities, with the aim that supercapacitors will be able to function as a primary power source, just like batteries.10–12
2. Fundamentals of supercapacitors
2.1. Types of supercapacitors
Supercapacitors can be categorized into two types based on the energy storage mechanism: electrochemical double layer capacitor (EDLC) or a pseudocapacitor (Fig. 1).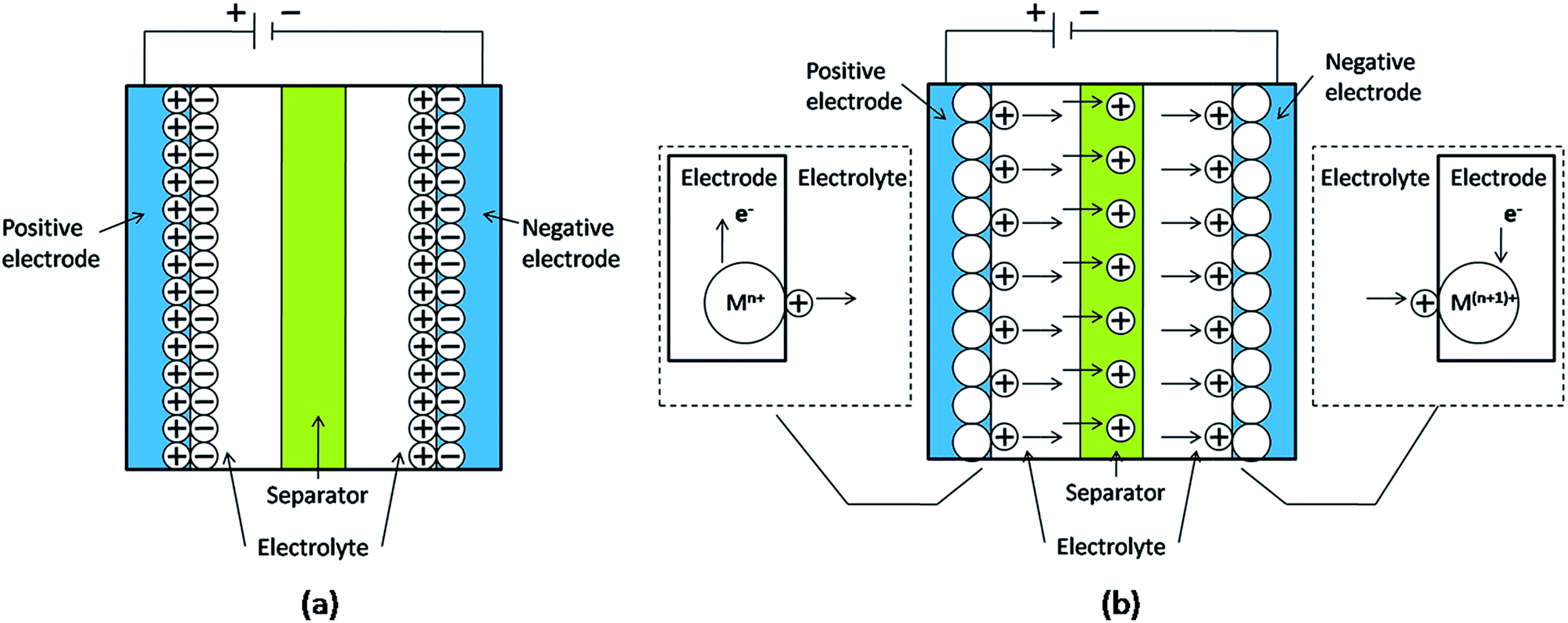 | ||
| Fig. 1 Schematic diagram of supercapacitors: (a) EDLC; (b) pseudocapacitor (M represents the metal atom. If anions in the electrolyte take part in the reversible redox reaction, they will move in the opposite direction to the cations). | ||
In EDLCs, the energy storage and transport mechanism is similar to the two-plate conventional capacitors (Fig. 1a). There is a pure physical charge accumulation at the interface between the electrode and electrolyte.6,13 During the charging process, the electrons move from the negative electrode to the positive electrode through the external loop, with anions moving towards the positive electrode, while the cations move towards the negative electrode in the electrolyte. During the discharging process, the electrons and ions move reversely.6 Because no charges transfer across the interface between the electrode and electrolyte, the energy storage mechanism is non-Faradaic and there are no redox reactions. Since only physical charge transfer occurs, there is almost no volume or morphology change of the electrode material, which results in the long cycle life of EDLC.
In contrast to EDLCs, pseudocapacitors involve faradaic redox reactions. When a potential is applied to a pseudocapacitor, fast and reversible faradaic reactions occur on the electrode materials, generating charges and resulting in the charge transfer across the double layers. This is similar to the charging and discharging processes that take place in batteries (Fig. 1b). There are three types of faradaic processes in the electrodes: reversible adsorption (e.g., the adsorption of hydrogen ions on the surface of the platinum electrode), redox reactions of transition metal oxides, and reversible electrochemical doping–dedoping process in the electrodes based on conductive polymers. Commonly, pseudocapacitors provide higher specific capacitance and energy density than EDLCs, because the faradaic processes can occur not only on the surface of pseudocapacitor electrodes but also in the interior of these electrodes.7,8,13 However, since redox reactions are involved, pseudocapacitors often suffer from a lack of stability during cycling. The pseudocapacitors also have a lower power density than EDLCs because faradaic processes are slower than non-faradaic processes.6
2.2 Parameters for supercapacitors
Supercapacitors can be evaluated based on these following criteria:3 (1) a high specific capacitance (SC); (2) a high power density with a relatively high energy density (>10 W h kg−1); (3) an excellent cyclability; (4) fast charging/discharging rates within seconds; (5) low self-discharging; (6) safe operation; (7) low cost. Like a battery, a full supercapacitor device consists of several parts: positive and negative electrodes, a separator, and an electrolyte. Some parameters of these parts affect the performance of the supercapacitor, such as the specific surface area, the pore size distribution of electroactive materials, the intrinsic properties of the electrolyte, the morphology, the electric conductivity of electroactive materials, and the interface between the electrode and electrolyte.14A large specific surface area is a key point for electrode materials, and can facilitate the contact between the electrode and electrolyte, provide more active sites, and lead to an enhanced capacitance.6 Usually, porous structure materials possess large specific areas, which can offer more channels for the electrolyte as well, resulting in less electrochemical polarization. However, the capacitance does not linearly increase with the increasing specific surface area, since the pore size of electrode materials plays an important role and the materials must have desirable pore distribution.7 Macropores (>50 nm) do not contribute much to the specific surface area. They serve as ion-buffering reservoirs and conduits to deliver electrolytes to the interior. Micropores (<2 nm) are too small for ions to move in or out. Largeot et al. report that when the pore size of electrode materials is very close to the ion size, the capacitance reaches the maximum.4
The two significant parameters to evaluate the performance of a supercapacitor are energy density and power density, which can be obtained by the following eqn (1) and (2):
| E = CV2/2m | (1) |
| P = V2/4mR | (2) |
Usually, there are two methods to calculate the SC of the electrode materials. We can calculate the SC from the cyclic voltammetry (CV) curves according to eqn (3):
| C = (∫IdV)/(vmV) | (3) |
| C = IΔt/mΔV | (4) |
In eqn (3), C (F g−1) is the specific capacitance, I(A) is the response current, V is the potential (V), v is the potential scan rate (V s−1), and m (g) is the mass of the electroactive materials in the electrode. In eqn (4), I represents the discharge current, m represents the mass of active materials, ΔV represents the potential drop during the discharge and Δt represents the total discharge time.
Because of the high SC, pseudocapacitor materials intensively arouse researchers' interest. In this review, we provide a brief summary of the recent progress on transition metal oxide/hydroxide materials for high-performance supercapacitor electrodes.
3. Electrode materials of transition metal oxides/hydroxides
Among the parts of a full supercapacitor device, the electrode is considered to be a key component affecting the electrochemical performance of a supercapacitor. Therefore, choosing and designing efficient electrode materials is of great importance to fabricate high-performance supercapacitors.Early studies in supercapacitors have focused on carbon materials due to their easy accessibility, large surface area, and relatively good electric conductivity. However, these carbon materials usually suffer from low SC. Hence, researchers have shifted their attention from pure carbon materials to pseudocapacitor materials (such as transition metal oxides/hydroxides and conducting polymers).6,15–17 SCs of the latter type are 3–7 times larger than the former. Transition metal oxides/hydroxides are considered to be the best candidates. Among them, the most commonly used electroactive materials can be divided into two types: noble (RuO2,18 IrO219) and cheap transition metal oxides/hydroxides (MnO2,20,21 Co3O4,22–24 NiO,25–27 Co(OH)228,29 and Ni(OH)230,31). However, the transition metal oxides/hydroxides usually have relatively low power density and poor cycling stability. The low power density can be ascribed to the poor electric conductivity of metal oxides/hydroxides, which limits the electron transfer rates. Moreover, the damage to the morphology caused by the swelling and shrinkage of the electrode materials during the charging and discharging processes leads to the lack of cycling stability. To solve these problems, the two main methods, involve combining metal oxides/hydroxides with conductive materials and developing porous nanostructures: the former can accelerate the reaction kinetics; the latter can buffer the stress from the swelling and shrinkage of the electrodes and provide more ion adsorption or active sites for the charge transfer reactions, as well as shorten the diffusion and transfer pathways of the electrolyte ions (Fig. 2).
 | ||
| Fig. 2 Strategies to enhance the performance of supercapacitors. | ||
3.1. Noble metal oxides
The earliest studied transition metal oxide for a supercapacitor was RuO2, and it is still recognized as one of the most promising electrode materials, because of the high theoretical SC (≈2000 F g−1),32 long cycle life, wide potential window, high electric conductivity, high rate capability and good electrochemical reversibility.15,33–36 The pseudocapacative behaviour of RuO2 in an acidic electrolyte can be described as a fast and reversible faradaic reaction, according to eqn (5):7| RuO2 + xH+ + xe− ↔ RuO2−x(OH)x | (5) |
In fact, the SCs of RuO2 electrode materials are usually smaller than the theoretical value because of the high crystalline and power constraints.32 The supercapacitive performance of RuO2 depends on the amount of combined water, the crystallinity, annealing temperature and particle size.
It is known that the reaction of RuO2 in the acidic system involves the insertion and extraction of H+ according to eqn (5), and that RuO2·nH2O is a good proton conductor.38 So, the combined water in hydrous RuO2 can accelerate the diffusion of H+ in the electrode. The hydrous Ru oxides exhibit high SCs of 861 F g−1 and 900 F g−1,39,40 which are much larger than the anhydrous form. In addition, as the content of the combined water decreases from RuO2·0.5H2O to RuO2·0.03H2O, the SC decreases from 720 F g−1 to 19.2 F g−1.41
The crystallinity also affects the supercapacitive performance of RuO2. The well-crystallized RuO2 is so compact that the insertion and extraction of ions/electrons are difficult, which leads to an increase in the electrochemical impedance and a decrease in the supercapacitive performance. In contrast, the redox reactions of the amorphous RuO2 take place not only on the surface but also in the bulk of the materials. Hence, there are many more articles reporting the superior performance of amorphous RuO2 materials compared with crystallized ones.41,42 The amorphous RuO2 thin films anodically deposited on stainless steel substrates demonstrate stable electrochemical capacitor properties, with an ultrahigh maximum SC of 1190 F g−1 in H2SO4.43
The annealing temperature also has an important influence on the supercapacitive performance. When annealed at high temperature, RuO2·nH2O possesses a good crystallinity and low water content.44 And a sharp decrease in capacitance is observed at an annealing temperature above 200 °C.45 It is reported that when the annealing temperature is close to the crystallization temperature of Ru oxide, RuO2·nH2O can still remain amorphous and hydrous,41,46 with the optimal annealing temperature being around 150 °C.35,41
The particle size of RuO2 is another important factor for the electrochemical performance. Small-sized particles have short diffusion and transport pathways of electrolyte ions, as well as high specific surface areas. As a result, the utilization of electroactive materials and the high rate charge/discharge capability of the supercapacitors will be improved. The ion diffusion time constant (τ) can be obtained by eqn (6):47
| τ = L2/2D | (6) |
Despite all the advantages, RuO2 is still not suitable for commercial application with supercapacitors, due to its high cost and scarce source. To reduce the cost, two ways have been frequently proposed: composing RuO2 with other cheap metal oxides to synthesize composites, such as MnO2, Co3O4, NiO, TiO2 and SnO,54–59 or depositing RuO2 on low-cost substrates to form composites, such as various kinds of carbons and conducting polymers.18,33,34,60–64 The RuO2·nH2O/TiO2 nanocomposite synthesized by microwave-assisted hydrothermal synthesis exhibits a maximum SC of 992 F g−1, with quite a high power density of 400 kW kg−1.65 In this composite, the TiO2 crystallites are formed before RuO2·nH2O nucleation, thus enhancing the exposure and utilization efficiency of RuO2·nH2O. The PEDOT-PSS-RuO2·nH2O electrode formed by embedding hydrous RuO2 particles into a conductive PEDOT-PSS matrix, which provides a 3D matrix for loading and stabilizing RuO2·nH2O, achieves a maximum SC of 653 F g−1.61 The SC of using RuO2·nH2O/multi-walled carbon nanotubes/Ti as electrodes in H2SO4 aqueous solution can be up to 1652 F g−1, which is attributed to the nanopore structure that allows fast and reversible faradaic processes to occur on the electrode surface.66 The conductive substrates improve the conductivity of RuO2, leading to a shorter ion transfer distance.35
3.2. Cheap metal oxides/hydroxides
In order to reduce the cost of supercapacitor electrode materials, a feasible scheme is to develop cheap metal oxides/hydroxides as alternative candidates to replace RuO2.| MnO2 + C+ + e− ↔ MnOOC | (7) |
The second one is based on the surface adsorption of electrolyte cations on the MnO2 electrode:67,68
| (MnO2)surface + C+ + e− ↔ (MnOOC)surface | (8) |
Both the mechanisms involve a redox reaction between the III and IV oxidation states of Mn.
MnO2 materials have various crystal structures (α-, β-, γ-, δ-, and λ-), which determine their electrochemical performance, especially when the size of the tunnels limit the intercalation of cations.69 Birnessite δ-MnO2, with a 2D tunnel structure doped with potassium, has the advantage of providing a relatively high SC (110 F g−1) with a moderate BET surface area (17 m2 g−1). The SC followed is by λ-MnO2 with a 3D tunnel structure. β- and γ-MnO2, with 1D tunnel structures, just provide a pseudofaradaic surface capacitance that is correlated with the BET surface area of the crystalline materials.69 Different synthesis conditions can lead to various MnO2 structures. Gradually increasing the precursor acidity can result in a progression from layered birnessite δ-MnO2, through α-MnO2 with a relatively large tunnel structure, to a more compact and dense β-MnO2 phase.70
Although the theoretical SC of MnO2 is pretty high, the practical SC of unmodified MnO2 is usually lower than 350 F g−1, which is not comparable to RuO2.20,71 The SC and rate capability of thick MnO2 electrode are limited by its poor electric conductivity and electrochemical dissolution during cycling.32,72 To improve the electric conductivity and charge storage capability, the incorporation of other metal elements into MnO2 compounds or composing MnO2 nanostructured composites with well-conductive materials are both feasible ways.73–77 The former method can introduce more defects and charge carriers to enhance the conductivity.78–80 A hybrid structure consisting of nanoporous gold and nanocrystalline MnO2 with an enhanced electric conductivity had a high SC of 1145 F g−1, which is close to the theoretical SC. The nanoporous gold acts not only as a double-layer capacitor, but also as a good electronic/ionic conductor to improve the pseudocapacitive performance of the MnO2.73 Moreover, Co doping can prevent the dissolution of MnO2, V doping can inhibit the crystal growth of MnO2, and Fe doping can improve the cycle stability due to reducing the concentration of unstable Mn3+ ions.81 Based on the latter method, various MnO2 composites, such as MnO2/carbon nanotubes,72,82 MnO2/graphene,83 MnO2/carbon nanofibers,84 MnO2/nanoporous gold,85 and MnO2/PANI,86 were also fabricated. Nanowire arrays in which a gold core nanowire is encapsulated inside a hemicylindrical shell of MnO2 show an ultrahigh SC of 1020 ± 100 F g−1, with an extremely poor cycle stability.74 However, when the nanowire core/shell arrays are transferred into an ultradry acetonitrile electrolyte, the cycle stability improved dramatically. Recently, Chen et al. fabricated a MnO2/CNT/sponge composite hybrid electrode, which achieved a remarkable SC of 1230 F g−1, with an energy density of 31 W h kg−1 and a power density of 63 kW kg−1, as well as excellent cycle stability—the SC only degraded about 4% after 10000 cycles at a current density of 5 A g−1.87 Graphene, a unique two-dimensional carbon material, has attracted a lot of attention from researchers recently due to its large surface area, high electric conductivity, outstanding mechanical properties, and good chemical stability. It is considered as one of the best substrates to deposit MnO2.83,88 Several kinds of MnO2/graphene with good capacitance have been prepared.88–91 A 3D macroporous MnO2/graphene composite, demonstrating a SC of 389 F g−1, was fabricated by using a sacrificial template of polystyrene colloidal particles.92 MnO2-nanoflowers-coated graphene with a SC of 328 F g−1 was fabricated by electrodeposition.91 The large specific surface area of graphene facilitates rapid ionic transport within the electrodes, while the excellent conductivity accelerates the redox reactions.
During cycling, the partial dissolution of MnO2 in even mildly acidic and near-neutral electrolyte, which can be described in eqn (9) and (10), is a serious problem confining the practical application of MnO2 in supercapacitors.20,93,94
| MnO2 + H+ + e− → MnOOH | (9) |
| MnOOH + 3H+ + e− → Mn2+ + 2H2O | (10) |
To solve this problem, preparing a protective polymer shell on MnO2 is usually employed by researchers. Various conducting polymer shells (PANI, PPy, PTh, and their derivatives) have been fabricated, which can not only prevent the dissolution of MnO2, but also improve the mechanical stability and flexibility.95–99 The PANI/mesoporous carbon/MnO2 ternary composite synthesized by chemical oxidative polymerization has an 88% capacitance retention after 1000 cycles, and the MnO2/CNT/PEDOT-PSS composite exhibits an extremely long cycle life (99% retention of SC after 1000 cycles).100,101 MnO2/conducting polymer composites also show high SCs. For example, the hierarchical MnO2/PPy@carbon nanofiber composite reaches a SC of 705 F g−1, and the PANI/mesoporous carbon/MnO2 ternary nanocomposite possesses a SC of 695 F g−1.99,100
| NiO + OH− ↔ NiOOH + e− | (11) |
| NiO + H2O ↔ NiOOH + H+ + e− | (12) |
| Ni(OH)2 ↔ NiOOH + H+ + e− | (13) |
| Ni(OH)2 + OH− ↔ NiOOH + H2O + e− | (14) |
The two theories reach a consensus that Ni2+ oxidizes to NiOOH through losing an electron, resulting in the supercapacitive reactions. Most researchers tend to favour the first theory, but the second one is reasonable as well, because NiO can combine with OH− in the alkaline electrolyte to generate Ni(OH)2, which will contribute to part of the capacitance.
There are two main issues for NiO electrode materials in supercapacitors: (1) NiO is a kind of p-type semiconductor with low electric conductivity; (2) the cycle stability is poor. To address these problems, fabricating nanostructured NiO and composing NiO with other materials are both feasible.
Nanostructured NiO materials can provide a large specific surface area, as well as short diffusion and transport pathways of ions and electrons, resulting in fast reaction kinetics.27,107–113 Meanwhile, nanostructures can buffer the stress from swelling of the electrodes and inhibit pulverization, to enhance the cycling performance. Porous NiO with macropores from using electrophoresis and electrodeposition have much larger SC (351 F g−1) than bare NiO film (140 F g−1).114 Various NiO nanostructures, such as nanobelts, nanowires, nanorods, nanoplatelets, and nanoflowers, have been fabricated.115–120 The hierarchically porous NiO films synthesized through different methods also show good electrochemical performance. Xia et al. developed chemical bath deposition with a colloidal crystal template to synthesize a hierarchically porous NiO film, which delivered a SC of 309 F g−1 at 1 A g−1, with a capacitance retention of 89% after 4000 cycles (Fig. 3).27 The structure of the film consists of two parts: the understructure, which is a NiO monolayer hollow-sphere array, and the superstructure, which constitutes net-like NiO nanoflakes. The degradation mechanism of NiO capacitance is mainly attributed to two factors: the self-discharge, due to the partial dissolution of NiOOH and the bubbling effect of oxygen evolution. The enhanced electrochemical capacitive performance is mainly attributed to the large surface area and high porosity of this unique hierarchically porous architecture. Zhang et al. used a facile ammonia-evaporation method to synthesize a hierarchical structure consisting of triangular prism-like NiO and randomly porous NiO nanoflakes.25 This hierarchically porous NiO film exhibits a SC of 232 F g−1 at 2 A g−1, with the SC increasing to 441 F g−1 after around 1500 cycles.
 | ||
| Fig. 3 Morphological and structural characterizations of hierarchically porous NiO film prepared by a 1 mm PS sphere: (a) SEM images of top and side views (side view presented in inset); (b) an enlarged view. Reproduced from ref. 27. | ||
Composing NiO with carbon materials or porous metals can efficiently enhance the electric conductivity and surface area, leading to a high utilization of active materials and an excellent rate capability.26,111,121–128 The NiOx/CNT (carbon nanotube) electrodes prepared by electrochemical deposition show an ultrahigh SC of 1701 F g−1 with 8.9 wt% NiOx in the composite electrode.129 However, when the weight percentage of NiOx is increased to 36.6%, the SC drops significantly to 671 F g−1. This decrease in SC can be attributed to the larger dead volume of the oxides, high equivalent series resistance for heavier deposits, and a more ineffective ionic transportation resulting from the reduced pore size and pore clogging.
Ni(OH)2 is a hexagonal-layered structure and has two polymorphs, α- and β-Ni(OH)2, corresponding to γ- and β-NiOOH after oxidizing.130–133 α-Ni(OH)2 is a hydroxyl-deficient phase with interlayered anions and water molecules. β-Ni(OH)2 possesses a brucite structure without water molecules. The α-Ni(OH)2/γ-NiOOH couple shows a higher SC than the β-Ni(OH)2/β-NiOOH couple due to the greater change in valence. The transformation between Ni(OH)2 and NiOOH is described in Fig. 4. Under the common charging condition, Ni(OH)2 transforms between β-Ni(OH)2 and β-NiOOH. When the material is overcharged, β-NiOOH will change to γ-NiOOH, and then α-Ni(OH)2 after discharging. α-Ni(OH)2 is not very stable, since it can transform to β-Ni(OH)2 easily through aging in alkaline electrolyte.134–136
 | ||
| Fig. 4 Transformation between Ni(OH)2 and NiOOH. | ||
The theoretical SC of Ni(OH)2 is about 3650 F g−1, and its practical SC is much higher than NiO.132,133,136–138 The powder materials usually show SCs of 500–600 F g−1, while the modified film can even reach an extremely high SC of 3000 F g−1, which is very close to the theoretical SC (Fig. 5).139 Porous α-Ni(OH)2 electrodeposited on Ni foam exhibits a very high SC of 3152 F g−1 at a current density of 4 A g−1, but the SC drops quickly as the current density increases. The amazing SC can be attributed to its loosely crystal structure, which contributes to the easy insertion/extraction of ions and the hydratability of Ni(OH)2 in favor of electrochemical reactions.140 The terrible rate capability, due to the poor electric conductivity of Ni(OH)2, and the poor cycle stability limit the application of Ni(OH)2.140 The Ni(OH)2/carbon composites exhibit good rate capabilities and cycling performance. A Ni(OH)2/graphene sheet electrode has demonstrated a high SC of 1335 F g−1 at a current density of 2.8 A g−1 and 953 F g−1 at 45.7 A g−1 with good rate capability, and there is no obvious capacitance drop at a current density of 28.6 A g−1.141 α-Ni(OH)2 nanosheets composited with low defect density CNTs have been fabricated by a one-step hydrothermal method.142 They delivered a higher SC of 1302.5 F g−1 than their individual components (372.1 F g−1 for Ni(OH)2 and 101.4 F g−1 for carbon nanotubes), demonstrating the synergistic effects of the hybrid materials to enhance electrochemical performance.
 | ||
| Fig. 5 (a) FESEM photographs of Ni(OH)2 deposited on the nickel foam branch; (b) an enlarged view. Reproduced from ref. 132. | ||
| Co3O4 + OH− + H2O ↔ 3CoOOH + e− | (15) |
| CoOOH + OH− ↔ CoO2 + H2O + e− | (16) |
Co3O4 is a p-type semiconductor with low electronic and ionic conductivity, leading to poor rate capability. It has a large volume change, even with pulverization during the cycle process, resulting in a short cycle life.22,144,152 The nanoscale Co3O4 shows a larger SC than bulk Co3O4. The Co3O4 nanosheet arrays on Ni foam exhibit a SC of 2735 F g−1 by electrodepositing Co(OH)2 and then undergoing a thermal transformation to Co3O4.153 The binder-free Co3O4 nanowire arrays synthesized by a facial hydrothermal method deliver a SC of 1160 F g−1 at 2 A g−1, and the SC degradation is only 9.6% after 5000 cycles.154 Xia et al. also synthesized a single-crystalline Co3O4 nanowire array through a hydrothermal method (Fig. 6). It shows a SC of 754 F g−1 at 2 A g−1 and a long cycle life, due to the unique 1D nanostructure, which can provide fast ionic diffusion paths, alleviate the expansion stress, and restrain the pulverization of Co3O4.22 Gao et al. prepared Co3O4 nanowire arrays, displaying a SC of 746 F g−1 at 5 mA cm−2 by a hydrothermal method.155 However, the capacitance loss is about 14% after 500 cycles. The Co3O4 nanotubes were prepared by chemically depositing Co(OH)2 into anodic aluminum oxide templates with heat treatment at 500 °C.156 This unique architecture displays a good supercapacitive performance, with a SC of 574 F g−1 at 0.1 A g−1 and 95% capacitance retention after 1000 cycles. Composites can also significantly enhance supercapacitive performance.157–162 Co3O4 nanowires fabricated on 3D graphene foam grown by the CVD method show a SC of ≈1100 F g−1 at 10 A g−1, and with good cycle stability.163 A facile one-step hydrothermal treatment is employed to synthesize reduced graphene oxide (rGO)/Co3O4 composites. The specific surface area increases from 42.65 to 124.89 m2 g−1, and the SC also increases from 22.6 to 263.0 F g−1 at 0.2 A g−1 with the increment of the mass ratio of rGO/Co3O4 from 0 to 1:2.157 Liang et al. incorporated Co3O4 into 1D nanoporous carbons through a controlled thermolysis of organometallic precursors.164 The SC of this composite can reach as high as 1066 F g−1.137
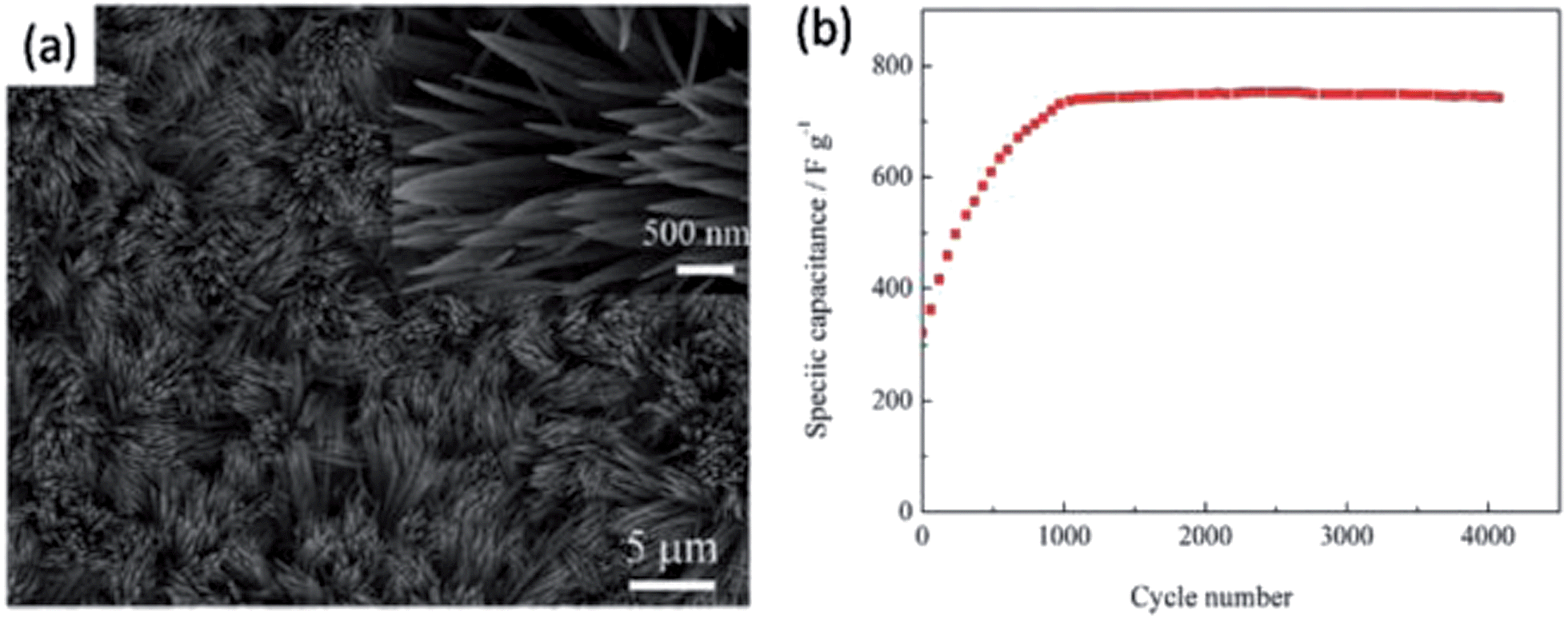 | ||
| Fig. 6 (a) SEM images of Co3O4 nanowire array grown on nickel foam (magnified top view presented in insets); (b) cycling performance of the Co3O4 nanowire array at a current density of 2 A g−1. Reproduced from ref. 22. | ||
Co(OH)2 has similar properties and structure to Ni(OH)2, which is also a hexagonal-layered structure. It can also be divided into α- and β-Co(OH)2, where α-Co(OH)2 has a better supercapacitive performance than β-Co(OH)2. The reactions can be expressed by eqn (17) and then (16):164–170
| Co(OH)2 + OH− ↔ CoOOH + H2O + e− | (17) |
The theoretical SC of Co(OH)2 is about 3600–3700 F g−1, and also the practical SC can reach a high value.171–173 Its shortcomings are still poor rate capability and cycle stability. β-Co(OH)2 nanoflakes potentiodynamically deposited on stainless steel provide a SC of 890 F g−1 and a capacitance retention of 84% after 10000 cycles.174 Material possessing one of the highest SC was obtained by Zhou et al.175 They electrodeposited mesoporous Co(OH)2 on Ni foam, which then exhibited a maximum SC of 2646 F g−1. However, when they used Ti plate as the substrate to replace Ni foam, the film only demonstrated a SC of 1018 F g−1 because of the smaller specific surface area than Ni foam. The Co(OH)2 nanoflakes electrodeposited on 3D porous nano-Ni also exhibited a high SC of 2028 F g−1 at 2 A g−1.30 It is important that the film also demonstrates an ultrahigh SC of 1920 F g−1 at 40 A g−1 and maintains 94.7% capacitance after 2000 cycles.
However, Co3O4/Co(OH)2 and NiO/Ni(OH)2 have a similar drawback, in that their potential window is low, which restricts their practical application.
However, iron oxides are not ideal materials for supercapacitors, until researchers can solve their poor capacitance and cycling stability.
 | ||
| Fig. 7 Spinel structure (M represents the metal atom). | ||
4. Conclusions
As a kind of electrochemical energy storage device, supercapacitors play an important role in our society. The behaviour of supercapacitors mainly depends on the electrode materials. We have reviewed various transition metal oxides/hydroxides as electrode materials of supercapacitors, due to their high capacitance, as well as high energy density with faradaic reactions. Properties, such as a large surface area, excellent electric conductivity and short diffusion paths for ions and electrons, lead to high specific capacitance and good rate capability. Thus, developing nanostructured materials and composing metal oxides/hydroxides can efficiently enhance supercapacitive performance. Moreover, these two methods are able to alleviate the stress from the swelling and shrinkage of the electrodes during charge/discharge processes and reduce the partial dissolution of some metal oxides/hydroxides in the electrolyte, which together can improve the cycling stability. In addition to the excellent electrochemical performance, many other factors of electrode materials should be taken into consideration, such as price, resource accessibility, and impact on the environment. If we combine all the strategies above, it should be much easier to fabricate high-performance electrode materials for supercapacitors in practical applications.Acknowledgements
This work was supported by the Program for Innovative Research Team in University of Ministry of Education of China (IRT13037) and Key Science and Technology Innovation Team of Zhejiang Province (2010R50013).Notes and references
- D. G. Nocera, Chem. Soc. Rev., 2009, 38, 13–15 RSC.
- S. Shafiee and E. Topal, Energy Policy, 2009, 37, 181–189 CrossRef PubMed.
- L. L. Zhang and X. S. Zhao, Chem. Soc. Rev., 2009, 38, 2520–2531 RSC.
- C. Largeot, C. Portet, J. Chmiola, P.-L. Taberna, Y. Gogotsi and P. Simon, J. Am. Chem. Soc., 2008, 130, 2730–2731 CrossRef CAS PubMed.
- S. G. Kandalkar, D. S. Dhawale, C.-K. Kim and C. D. Lokhande, Synth. Met., 2010, 160, 1299–1302 CrossRef CAS PubMed.
- G. P. Wang, L. Zhang and J. J. Zhang, Chem. Soc. Rev., 2012, 41, 797–828 RSC.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- J. R. Miller and P. Simon, Science, 2008, 321, 651–652 CrossRef CAS PubMed.
- D. Pech, M. Brunet, H. Durou, P. Huang, V. Mochalin, Y. Gogotsi, P.-L. Taberna and P. Simon, Nat. Nanotechnol., 2010, 5, 651–654 CrossRef CAS PubMed.
- P.-C. Chen, G. Z. Shen, Y. Shi, H. T. Chen and C. W. Zhou, ACS Nano, 2010, 4, 4403–4411 CrossRef CAS PubMed.
- J. Yan, Z. J. Fan, W. Sun, G. Q. Ning, T. Wei, Q. Zhang, R. F. Zhang, L. J. Zhi and F. Wei, Adv. Funct. Mater., 2012, 22, 2632–2641 CrossRef CAS PubMed.
- X. H. Xia, D. L. Chao, Z. X. Fan, C. Guan, X. H. Cao, H. Zhang and H. J. Fan, Nano Lett., 2014, 14, 1651–1658 CrossRef CAS PubMed.
- B. E. Conway, Electrochemical supercapacitors: Scientific fundamentals and technological applications, Kluwer Academic/Plenum, New York, 1999 Search PubMed.
- A. G. Pandolfo and A. F. Hollenkamp, J. Power Sources, 2006, 157, 11–27 CrossRef CAS PubMed.
- Y. Zhang, H. Feng, X. B. Wu, L. Z. Wang, A. Q. Zhang, T. C. Xia, H. C. Dong, X. F. Li and L. S. Zhang, Int. J. Hydrogen Energy, 2009, 34, 4889–4899 CrossRef CAS PubMed.
- K. Wang, H. P. Wu, Y. N. Meng and Z. X. Wei, Small, 2014, 10, 14–31 CrossRef CAS PubMed.
- S. Liu, S. H. Sun and X. Z. You, Nanoscale, 2014, 6, 2037–2045 RSC.
- N. Soin, S. S. Roy, S. K. Mitra, T. Thundat and J. A. McLaughlin, J. Mater. Chem., 2012, 22, 14944–14950 RSC.
- Y. M. Chen, J. H. Cai, Y. S. Huang, K. Y. Lee and D. S. Tsai, Nanotechnology, 2011, 22, 115706 CrossRef CAS PubMed.
- W. F. Wei, X. W. Cui, W. X. Chen and D. G. Ivey, Chem. Soc. Rev., 2011, 40, 1697–1721 RSC.
- Y. W. Cheng, S. T. Lu, H. B. Zhang, C. V. Varanasi and J. Liu, Nano Lett., 2012, 12, 4206–4211 CrossRef CAS PubMed.
- X. H. Xia, J. P. Tu, Y. Q. Zhang, Y. J. Mai, X. L. Wang, C. D. Gu and X. B. Zhao, RSC Adv., 2012, 2, 1835–1841 RSC.
- X. H. Xia, J. P. Tu, X. L. Wang, C. D. Gu and X. B. Zhao, Chem. Commun., 2011, 47, 5786–5788 RSC.
- X. H. Xia, J. P. Tu, Y. J. Mai, X. L. Wang, C. D. Gu and X. B. Zhao, J. Mater. Chem., 2011, 21, 9319–9325 RSC.
- Y. Q. Zhang, X. H. Xia, J. P. Tu, Y. J. Mai, S. J. Shi, X. L. Wang and C. D. Gu, J. Power Sources, 2012, 199, 413–417 CrossRef CAS PubMed.
- X. H. Xia, J. P. Tu, Y. J. Mai, R. Chen, X. L. Wang, C. D. Gu and X. B. Zhao, Chem.–Eur. J., 2011, 17, 10898–10905 CrossRef CAS PubMed.
- X. H. Xia, J. P. Tu, X. L. Wang, C. D. Gu and X. B. Zhao, J. Mater. Chem., 2011, 21, 671–679 RSC.
- T. Xue, X. Wang and J.-M. Lee, J. Power Sources, 2012, 201, 382–386 CrossRef CAS PubMed.
- J. Jiang, J. P. Liu, R. M. Ding, J. H. Zhu, Y. Y. Li, A. Z. Hu, X. Li and X. T. Huang, ACS Appl. Mater. Interfaces, 2011, 3, 99–103 CAS.
- X. H. Xia, J. P. Tu, Y. Q. Zhang, Y. J. Mai, X. L. Wang, C. D. Gu and X. B. Zhao, J. Phys. Chem. C, 2011, 115, 22662–22668 CAS.
- J. Yan, W. Sun, T. Wei, Q. Zhang, Z. J. Fan and F. Wei, J. Mater. Chem., 2012, 22, 11494–11502 RSC.
- J. Yan, Q. Wang, T. Wei and Z. J. Fan, Adv. Energy Mater., 2014, 4, 1300816 Search PubMed.
- J. T. Zhang, J. W. Jiang, H. L. Li and X. S. Zhao, Energy Environ. Sci., 2011, 4, 4009–4015 CAS.
- H. L. Wang, Y. Y. Liang, T. Mirfakhrai, Z. Chen, H. S. Casalongue and H. J. Dai, Nano Res., 2011, 4, 729–736 CrossRef CAS PubMed.
- Z.-S. Wu, D.-W. Wang, W. C. Ren, J. P. Zhao, G. M. Zhou, F. Li and H.-M. Cheng, Adv. Funct. Mater., 2010, 20, 3595–3602 CrossRef CAS PubMed.
- I. H. Kim and K. B. Kim, J. Electrochem. Soc., 2006, 153, A383–A389 CrossRef CAS PubMed.
- Y. F. Su, F. Wu, L. Y. Bao and Z. H. Yang, New Carbon Mater., 2007, 22, 53–58 CrossRef CAS.
- K. E. Swider, C. I. Merzbacher, P. L. Hagans and D. R. Rolison, Chem. Mater., 1997, 9, 1248–1255 CrossRef CAS.
- J. T. Zhang, J. Z. Ma, L. L. Zhang, P. Z. Guo, J. W. Jiang and X. S. Zhao, J. Phys. Chem. C, 2010, 114, 13608–13613 CAS.
- J. W. Long, K. E. Swider, C. I. Merzbacher and D. R. Rolison, Langmuir, 1999, 15, 780–785 CrossRef CAS.
- J. P. Zheng, P. J. Cygan and T. R. Jow, J. Electrochem. Soc., 1995, 142, 2699–2703 CrossRef CAS PubMed.
- K. Kuratani, T. Kiyobayashi and N. Kuriyama, J. Power Sources, 2009, 189, 1284–1291 CrossRef CAS PubMed.
- V. D. Patake, S. M. Pawar, V. R. Shinde, T. P. Gujar and C. D. Lokhande, Curr. Appl. Phys., 2010, 10, 99–103 CrossRef PubMed.
- Z. R. Cormier, H. A. Andrea and P. Zhang, J. Phys. Chem. C, 2011, 115, 19117–19128 CAS.
- H. Kim and B. N. Popov, J. Power Sources, 2002, 104, 52–61 CrossRef CAS.
- M. K. Seo, A. Saouab and S. J. Park, Mater. Sci. Eng., B, 2010, 167, 65–69 CrossRef CAS PubMed.
- C. Liu, F. Li, L.-P. Ma and H.-M. Cheng, Adv. Mater., 2010, 22, E28–E62 CrossRef CAS PubMed.
- W. Sugimoto, K. Yokoshima, Y. Murakami and Y. Takasu, Electrochim. Acta, 2006, 52, 1742–1748 CrossRef CAS PubMed.
- A. Ananth, M. S. Gandhi and Y. S. Mok, J. Phys. D: Appl. Phys., 2013, 46, 155202 CrossRef.
- Y. F. Ke, D. S. Tsai and Y. S. Huang, J. Mater. Chem., 2005, 15, 2122–2127 RSC.
- J. Y. Kim, K. H. Kim, H. K. Kim, S. H. Park, K. Y. Chung and K. B. Kim, RSC Adv., 2014, 4, 16115–16120 RSC.
- W. Sugimoto, H. Iwata, Y. Yasunaga, Y. Murakami and Y. Takasu, Angew. Chem., Int. Ed., 2003, 42, 4092–4096 CrossRef CAS PubMed.
- C.-C. Hu, K.-H. Chang, M.-C. Lin and Y.-T. Wu, Nano Lett., 2006, 6, 2690–2695 CrossRef CAS PubMed.
- T. S. Hyun, J. E. Kang, H. G. Kim, J. M. Hong and I. D. Kim, Electrochem. Solid State Lett., 2009, 12, A225–A228 CrossRef CAS PubMed.
- Y. H. Li, K. L. Huang, D. M. Zeng, S. Q. Liu and Z. F. Yao, J. Solid State Electrochem., 2010, 14, 1205–1211 CrossRef CAS.
- X. M. Liu and X. G. Zhang, Electrochim. Acta, 2004, 49, 229–232 CrossRef CAS PubMed.
- F. Pico, J. Ibanez, T. A. Centeno, C. Pecharroman, R. M. Rojas, J. M. Amarilla and J. M. Rojo, Electrochim. Acta, 2006, 51, 4693–4700 CrossRef CAS PubMed.
- Y. G. Wang and X. G. Zhang, Electrochim. Acta, 2004, 49, 1957–1962 CrossRef CAS PubMed.
- C. C. Hu, K. H. Chang and C. C. Wang, Electrochim. Acta, 2007, 52, 4411–4418 CrossRef CAS PubMed.
- C. C. Hu and W. C. Chen, Electrochim. Acta, 2004, 49, 3469–3477 CrossRef CAS PubMed.
- L.-M. Huang, H.-Z. Lin, T.-C. Wen and A. Gopalan, Electrochim. Acta, 2006, 52, 1058–1063 CrossRef CAS PubMed.
- C. J. Zhang, H. H. Zhou, X. Q. Yu, D. Shan, T. T. Ye, Z. Y. Huang and Y. F. Kuang, RSC Adv., 2014, 4, 11197–11205 RSC.
- Z. Zhou, Y. R. Zhu, Z. B. Wu, F. Lu, M. J. Jing and X. B. Ji, RSC Adv., 2014, 4, 6927–6932 RSC.
- B. K. Balan, H. D. Chaudhari, U. K. Kharul and S. Kurungot, RSC Adv., 2013, 3, 2428–2436 RSC.
- C. C. Hu, Y. L. Yang and T. C. Lee, Electrochem. Solid State Lett., 2010, 13, A173–A176 CrossRef CAS PubMed.
- T.-F. Hsieh, C.-C. Chuang, W.-J. Chen, J.-H. Huang, W.-T. Chen and C.-M. Shu, Carbon, 2012, 50, 1740–1747 CrossRef CAS PubMed.
- M. Toupin, T. Brousse and D. Belanger, Chem. Mater., 2004, 16, 3184–3190 CrossRef CAS.
- J. X. Zhu, W. H. Shi, N. Xiao, X. H. Rui, H. T. Tan, X. H. Lu, H. H. Hng, J. Ma and Q. Y. Yan, ACS Appl. Mater. Interfaces, 2012, 4, 2769–2774 CAS.
- T. Brousse, M. Toupin, R. Dugas, L. Athouel, O. Crosnier and D. Belanger, J. Electrochem. Soc., 2006, 153, A2171–A2180 CrossRef CAS PubMed.
- S. W. Donne, A. F. Hollenkamp and B. C. Jones, J. Power Sources, 2010, 195, 367–373 CrossRef CAS PubMed.
- J. F. Zang and X. D. Li, J. Mater. Chem., 2011, 21, 10965–10969 RSC.
- J. M. Ko and K. M. Kim, Mater. Chem. Phys., 2009, 114, 837–841 CrossRef CAS PubMed.
- X. Lang, A. Hirata, T. Fujita and M. W. Chen, Nat. Nanotechnol., 2011, 6, 232–236 CrossRef CAS PubMed.
- W. B. Yan, J. Y. Kim, W. D. Xing, K. C. Donavan, T. Ayvazian and R. M. Penner, Chem. Mater., 2012, 24, 2382–2390 CrossRef CAS.
- S. Hassan, M. Suzuki, S. Mori and A. Abd El-Moneim, RSC Adv., 2014, 4, 20479–20488 RSC.
- G. Q. Han, Y. Liu, E. J. Kan, J. Tang, L. L. Zhang, H. H. Wang and W. H. Tang, RSC Adv., 2014, 4, 9898–9904 RSC.
- Y. Gao, Y. S. Zhou, M. Qian, H. M. Li, J. Redepenning, L. S. Fan, X. N. He, W. Xiong, X. Huang, M. Majhouri-Samani, L. Jiang and Y. F. Lu, RSC Adv., 2013, 3, 20613–20618 RSC.
- J. Yan, E. Khoo, A. Sumboja and P. S. Lee, ACS Nano, 2010, 4, 4247–4255 CrossRef CAS PubMed.
- J. P. Liu, J. Jiang, C. W. Cheng, H. X. Li, J. X. Zhang, H. Gong and H. J. Fan, Adv. Mater., 2011, 23, 2076–2081 CrossRef CAS PubMed.
- D. F. Yang, J. Power Sources, 2012, 198, 416–422 CrossRef CAS PubMed.
- H. N. Yoo, D. H. Park and S.-J. Hwang, J. Power Sources, 2008, 185, 1374–1379 CrossRef CAS PubMed.
- A. L. M. Reddy, M. M. Shaijumon, S. R. Gowda and P. M. Ajayan, J. Phys. Chem. C, 2010, 114, 658–663 CAS.
- L. J. Deng, G. Zhu, J. F. Wang, L. P. Kang, Z. H. Liu, Z. P. Yang and Z. L. Wang, J. Power Sources, 2011, 196, 10782–10787 CrossRef CAS PubMed.
- J. G. Wang, Y. Yang, Z. H. Huang and F. Y. Kang, Electrochim. Acta, 2011, 56, 9240–9247 CrossRef CAS PubMed.
- X. Y. Lang, H. T. Yuan, Y. Iwasa and M. W. Chen, Scr. Mater., 2011, 64, 923–926 CrossRef CAS PubMed.
- Jaidev, R. I. Jafri, A. K. Mishra and S. Ramaprabhu, J. Mater. Chem., 2011, 21, 17601–17605 RSC.
- W. Chen, R. B. Rakhi, L. B. Hu, X. Xie, Y. Cui and H. N. Alshareef, Nano Lett., 2011, 11, 5165–5172 CrossRef CAS PubMed.
- J. Yan, Z. J. Fan, T. Wei, W. Z. Qian, M. L. Zhang and F. Wei, Carbon, 2010, 48, 3825–3833 CrossRef CAS PubMed.
- Z. P. Li, J. Q. Wang, S. Liu, X. H. Liu and S. R. Yang, J. Power Sources, 2011, 196, 8160–8165 CrossRef CAS PubMed.
- Z. S. Wu, W. C. Ren, D. W. Wang, F. Li, B. L. Liu and H. M. Cheng, ACS Nano, 2010, 4, 5835–5842 CrossRef CAS PubMed.
- Q. Cheng, J. Tang, J. Ma, H. Zhang, N. Shinya and L. C. Qin, Carbon, 2011, 49, 2917–2925 CrossRef CAS PubMed.
- B. G. Choi, M. Yang, W. H. Hong, J. W. Choi and Y. S. Huh, ACS Nano, 2012, 6, 4020–4028 CrossRef CAS PubMed.
- S. Bakardjieva, P. Bezdicka, T. Grygar and P. Vorm, J. Solid State Electrochem., 2000, 4, 306–313 CrossRef CAS.
- S. Nijjer, J. Thonstad and G. M. Haarberg, Electrochim. Acta, 2000, 46, 395–399 CrossRef CAS.
- Q. Lu and Y. K. Zhou, Funct. Mater. Lett., 2011, 4, 31–36 CrossRef CAS.
- Q. Lu and Y. K. Zhou, J. Power Sources, 2011, 196, 4088–4094 CrossRef CAS PubMed.
- G. H. Yu, L. B. Hu, N. Liu, H. L. Wang, M. Vosgueritchian, Y. Yang, Y. Cui and Z. N. Bao, Nano Lett., 2011, 11, 4438–4442 CrossRef CAS PubMed.
- H. Jiang, J. Ma and C. Z. Li, J. Mater. Chem., 2012, 22, 16939–16942 RSC.
- J.-G. Wang, Y. Yang, Z.-H. Huang and F. Kang, J. Mater. Chem., 2012, 22, 16943–16949 RSC.
- Y. F. Yan, Q. L. Cheng, V. Pavlinek, P. Saha and C. Z. Li, Electrochim. Acta, 2012, 71, 27–32 CrossRef CAS PubMed.
- Y. Hou, Y. W. Cheng, T. Hobson and J. Liu, Nano Lett., 2010, 10, 2727–2733 CrossRef CAS PubMed.
- F. B. Zhang, Y. K. Zhou and H. L. Li, Mater. Chem. Phys., 2004, 83, 260–264 CrossRef CAS PubMed.
- M. S. Wu, Y. A. Huang, J. J. Jow, W. D. Yang, C. Y. Hsieh and H. M. Tsai, Int. J. Hydrogen Energy, 2008, 33, 2921–2926 CrossRef CAS PubMed.
- V. Srinivasan and J. W. Weidner, J. Electrochem. Soc., 1997, 144, L210–L213 CrossRef CAS PubMed.
- V. Srinivasan and J. W. Weidner, J. Electrochem. Soc., 2000, 147, 880–885 CrossRef CAS PubMed.
- X. H. Xia, J. P. Tu, J. Zhang, X. L. Wang, W. K. Zhang and H. Huang, Sol. Energy Mater. Sol. Cells, 2008, 92, 628–633 CrossRef CAS PubMed.
- Y. Zhang, Y. G. Gui, X. B. Wu, H. Feng, A. Q. Zhang, L. Z. Wang and T. C. Xia, Int. J. Hydrogen Energy, 2009, 34, 2467–2470 CrossRef CAS PubMed.
- X. M. Liu, X. G. Zhang and S. Y. Fu, Mater. Res. Bull., 2006, 41, 620–627 CrossRef CAS PubMed.
- K. R. Prasad and N. Miura, Appl. Phys. Lett., 2004, 85, 4199–4201 CrossRef CAS PubMed.
- B. R. Tao, J. A. Zhang, F. J. Miao, S. C. Hui and L. J. Wan, Electrochim. Acta, 2010, 55, 5258–5262 CrossRef CAS PubMed.
- J. B. Wu, Z. G. Li and Y. Lin, Electrochim. Acta, 2011, 56, 2116–2121 CrossRef CAS PubMed.
- X. Ge, C. D. Gu, Y. Lu, X. L. Wang and J. P. Tu, J. Mater. Chem. A, 2013, 1, 13454–13461 CAS.
- M. Khairy and S. A. El-Safty, RSC Adv., 2013, 3, 23801–23809 RSC.
- M. S. Wu, M. J. Wang and J. J. Jow, J. Power Sources, 2010, 195, 3950–3955 CrossRef CAS PubMed.
- B. Wang, J. S. Chen, Z. Wang, S. Madhavi and X. W. Lou, Adv. Energy Mater., 2012, 2, 1188–1192 CrossRef CAS PubMed.
- H. Pang, Q. Y. Lu, Y. Z. Zhang, Y. C. Li and F. Gao, Nanoscale, 2010, 2, 920–922 RSC.
- Z. Y. Lu, Z. Chang, J. F. Liu and X. M. Sun, Nano Res., 2011, 4, 658–665 CrossRef CAS PubMed.
- C. Z. Yuan, J. Y. Li, L. R. Hou, L. Yang, L. F. Shen and X. G. Zhang, Electrochim. Acta, 2012, 78, 532–538 CrossRef CAS PubMed.
- C.-Y. Cao, W. Guo, Z.-M. Cui, W.-G. Song and W. Cai, J. Mater. Chem., 2011, 21, 3204–3209 RSC.
- X. H. Xia, J. P. Tu, J. Zhang, X. L. Wang, W. K. Zhang and H. Huang, Electrochim. Acta, 2008, 53, 5721–5724 CrossRef CAS PubMed.
- Q. Lu, M. W. Lattanzi, Y. Chen, X. Kou, W. Li, X. Fan, K. M. Unruh, J. G. Chen and J. Q. Xiao, Angew. Chem., Int. Ed., 2011, 50, 6847–6850 CrossRef CAS PubMed.
- W. Lv, F. Sun, D.-M. Tang, H.-T. Fang, C. Liu, Q.-H. Yang and H.-M. Cheng, J. Mater. Chem., 2011, 21, 9014–9019 RSC.
- X. H. Cao, Y. M. Shi, W. H. Shi, G. Lu, X. Huang, Q. Y. Yan, Q. C. Zhang and H. Zhang, Small, 2011, 7, 3163–3168 CrossRef CAS PubMed.
- B. Gao, C. Z. Yuan, L. H. Su, S. Y. Chen and X. G. Zhang, Electrochim. Acta, 2009, 54, 3561–3567 CrossRef CAS PubMed.
- M. L. Huang, C. D. Gu, X. Ge, X. L. Wang and J. P. Tu, J. Power Sources, 2014, 259, 98–105 CrossRef CAS PubMed.
- Z. Y. Ji, X. P. Shen, Y. L. Xu, H. Zhou, S. Bai and G. X. Zhu, RSC Adv., 2014, 4, 13601–13609 RSC.
- M. E. Plonska-Brzezinska, D. M. Brus, A. Molina-Ontoria and L. Echegoyen, RSC Adv., 2013, 3, 25891–25901 RSC.
- Y. G. Zhu, G. S. Cao, C. Y. Sun, J. Xie, S. Y. Liu, T. J. Zhu, X. B. Zhao and H. Y. Yang, RSC Adv., 2013, 3, 19409–19415 RSC.
- K. W. Nam, K. H. Kim, E. S. Lee, W. S. Yoon, X. Q. Yang and K. B. Ki, J. Power Sources, 2008, 182, 642–652 CrossRef CAS PubMed.
- R. S. Jayashree and P. V. Kamath, J. Appl. Electrochem., 2001, 31, 1315–1320 CrossRef CAS.
- J. T. Li, W. Zhao, F. Q. Huang, A. Manivannan and N. Q. Wu, Nanoscale, 2011, 3, 5103–5109 RSC.
- Q. L. Xian and J. A. Li, J. Inorg. Mater., 2010, 25, 1268–1272 CrossRef CAS.
- U. M. Patil, K. V. Gurav, V. J. Fulari, C. D. Lokhande and O. S. Joo, J. Power Sources, 2009, 188, 338–342 CrossRef CAS PubMed.
- Y. Ren and L. A. Gao, J. Am. Ceram. Soc., 2010, 93, 3560–3564 CrossRef CAS PubMed.
- M. C. Bernard, P. Bernard, M. Keddam, S. Senyarich and H. Takenouti, Electrochim. Acta, 1996, 41, 91–93 CrossRef CAS.
- H. M. Du, L. F. Jiao, K. Z. Cao, Y. J. Wang and H. T. Yuan, ACS Appl. Mater. Interfaces, 2013, 5, 6643–6648 CAS.
- J. W. Lang, L. B. Kong, M. Liu, Y. C. Luo and L. Kang, J. Solid State Electrochem., 2010, 14, 1533–1539 CrossRef CAS.
- A. K. Mondal, D. W. Su, S. Q. Chen, B. Sun, K. F. Li and G. X. Wang, RSC Adv., 2014, 4, 19476–19481 RSC.
- G. W. Yang, C. L. Xu and H. L. Li, Chem. Commun., 2008, 6537–6539 RSC.
- H. Jiang, C. Z. Li, T. Sun and J. Ma, Chem. Commun., 2012, 48, 2606–2608 RSC.
- H. L. Wang, H. S. Casalongue, Y. Y. Liang and H. J. Dai, J. Am. Chem. Soc., 2010, 132, 7472–7477 CrossRef CAS PubMed.
- S. Chen, J. W. Zhu, H. Zhou and X. Wang, RSC Adv., 2011, 1, 484–489 RSC.
- H. Cheng, Z. G. Lu, J. Q. Deng, C. Y. Chung, K. L. Zhang and Y. Y. Li, Nano Res., 2010, 3, 895–901 CrossRef CAS.
- Q. Yang, Z. Y. Lu, Z. Chang, W. Zhu, J. Q. Sun, J. F. Liu, X. M. Sun and X. Duan, RSC Adv., 2012, 2, 1663–1668 RSC.
- X. X. Qing, S. Q. Liu, K. L. Huang, K. Z. Lv, Y. P. Yang, Z. G. Lu, D. Fang and X. X. Liang, Electrochim. Acta, 2011, 56, 4985–4991 CrossRef CAS PubMed.
- H.-K. Kim, T.-Y. Seong, J.-H. Lim, W. I. Cho and Y. S. Yoon, J. Power Sources, 2001, 102, 167–171 CrossRef CAS.
- V. Srinivasan and J. W. Weidner, J. Power Sources, 2002, 108, 15–20 CrossRef CAS.
- P. J. Kulesza, S. Zamponi, M. A. Malik, M. Berrettoni, A. Wolkiewicz and R. Marassi, Electrochim. Acta, 1997, 43, 919–923 CrossRef.
- Z. Y. Xun, C. X. Cai, W. Xing and T. H. Lu, J. Electroanal. Chem., 2003, 545, 19–27 CrossRef CAS.
- Y. Q. Zhang, L. Li, S. J. Shi, Q. Q. Xiong, X. Y. Zhao, X. L. Wang, C. D. Gu and J. P. Tu, J. Power Sources, 2014, 256, 200–205 CrossRef CAS PubMed.
- Y. Shan and L. Gao, Mater. Chem. Phys., 2007, 103, 206–210 CrossRef CAS PubMed.
- H. Wang, C. M. Holt, Z. Li, X. Tan, B. S. Amirkhiz, Z. Xu, B. C. Olsen, T. Stephenson and D. Mitlin, Nano Res., 2012, 5, 605–617 CrossRef CAS.
- C. Z. Yuan, L. Yang, L. R. Hou, L. F. Shen, X. G. Zhang and X. W. Lou, Energy Environ. Sci., 2012, 5, 7883–7887 CAS.
- F. Zhang, C. Z. Yuan, X. J. Lu, L. J. Zhang, Q. Che and X. G. Zhang, J. Power Sources, 2012, 203, 250–256 CrossRef CAS PubMed.
- Y. Y. Gao, S. L. Chen, D. X. Cao, G. L. Wang and J. L. Yin, J. Power Sources, 2010, 195, 1757–1760 CrossRef CAS PubMed.
- J. Xu, L. Gao, J. Y. Cao, W. C. Wang and Z. D. Chen, Electrochim. Acta, 2010, 56, 732–736 CrossRef CAS PubMed.
- G. J. Liu, L. Q. Fan, F. D. Yu, J. H. Wu, L. Liu, Z. Y. Qiu and Q. Liu, J. Mater. Sci., 2013, 48, 8463–8470 CrossRef CAS.
- R. T. Wang, X. B. Yan, J. W. Lang, Z. M. Zheng and P. Zhang, J. Mater. Chem. A, 2014, 2, 12724–12732 CAS.
- L. Yang, S. Cheng, Y. Ding, X. B. Zhu, Z. L. Wang and M. L. Liu, Nano Lett., 2012, 12, 321–325 CrossRef CAS PubMed.
- C. Z. Yuan, L. Yang, L. R. Hou, J. Y. Li, Y. X. Sun, X. G. Zhang, L. F. Shen, X. J. Lu, S. L. Xiong and X. W. Lou, Adv. Funct. Mater., 2012, 22, 2560–2566 CrossRef CAS PubMed.
- J. W. Lang, X. B. Yan and Q. J. Xue, J. Power Sources, 2011, 196, 7841–7846 CrossRef CAS PubMed.
- J. Yan, T. Wei, W. M. Qiao, B. Shao, Q. K. Zhao, L. J. Zhang and Z. J. Fan, Electrochim. Acta, 2010, 55, 6973–6978 CrossRef CAS PubMed.
- X.-C. Dong, H. Xu, X.-W. Wang, Y.-X. Huang, M. B. Chan-Park, H. Zhang, L.-H. Wang, W. Huang and P. Chen, ACS Nano, 2012, 6, 3206–3213 CrossRef CAS PubMed.
- Y. Liang, M. G. Schwab, L. Zhi, E. Mugnaioli, U. Kolb, X. Feng and K. Muellen, J. Am. Chem. Soc., 2010, 132, 15030–15037 CrossRef CAS PubMed.
- D. T. Dam, X. Wang and J.-M. Lee, RSC Adv., 2012, 2, 10512–10518 RSC.
- Z. J. Yu, Y. Dai and W. Chen, J. Chin. Chem. Soc., 2010, 57, 423–428 CAS.
- V. Gupta, T. Kusahara, H. Toyama, S. Gupta and N. Miura, Electrochem. Commun., 2007, 9, 2315–2319 CrossRef CAS PubMed.
- X. Ge, C. D. Gu, X. L. Wang and J. P. Tu, J. Phys. Chem. C, 2014, 118, 911–923 CAS.
- X. H. Xia, J. P. Tu, Y. Q. Zhang, J. Chen, X. L. Wang, C. D. Gu, C. Guan, J. S. Luo and H. J. Fan, Chem. Mater., 2012, 24, 3793–3799 CrossRef CAS.
- Y. Q. Zhang, X. H. Xia, J. Kang and J. P. Tu, Chinese Sci. Bullet., 2012, 57, 4215–4219 CrossRef CAS.
- J. A. Jiang, J. P. Liu, R. M. Ding, J. H. Zhu, Y. Y. Li, A. Z. Hu, X. Li and X. T. Huang, ACS Appl. Mater. Interfaces, 2011, 3, 99–103 CAS.
- Y. L. Chen, Z. A. Hu, Y. Q. Chang, H. W. Wang, G. R. Fu, X. Q. Jin and L. J. Xie, Chin. J. Chem., 2011, 2011(29), 2257–2262 CrossRef PubMed.
- L. J. Xie, X. Q. Jin, G. R. Fu, Y. L. Xie, Y. X. Wang and Z. A. Hu, Chem. J. Chin. Univ., 2010, 31, 353–356 CAS.
- A. D. Jagadale, V. S. Jamadade, S. N. Pusawale and C. D. Lokhande, Electrochim. Acta, 2012, 78, 92–97 CrossRef CAS PubMed.
- W.-J. Zhou, M.-W. Xu, D.-D. Zhao, C.-L. Xu and H.-L. Li, Microporous Mesoporous Mater., 2009, 117, 55–60 CrossRef CAS PubMed.
- X. H. Xia, Y. Q. Zhang, D. L. Chao, G. Cao, Y. J. Zhang, L. Li, X. Ge, I. M. Bacho, J. P. Tu and H. J. Fan, Nanoscale, 2014, 6, 5008–5048 RSC.
- Q. Q. Xiong, J. P. Tu, Y. Lu, J. Chen, Y. X. Yu, X. L. Wang and C. D. Gu, J. Mater. Chem., 2012, 22, 18639–18645 RSC.
- K. Y. Xie, J. Li, Y. Q. Lai, W. Lu, Z. A. Zhang, Y. X. Liu, L. M. Zhou and H. T. Huang, Electrochem. Commun., 2011, 13, 657–660 CrossRef CAS PubMed.
- J. Chen, K. L. Huang and S. Q. Liu, Electrochim. Acta, 2009, 55, 1–5 CrossRef CAS PubMed.
- S.-Y. Wang, K.-C. Ho, S.-L. Kuo and N.-L. Wu, J. Electrochem. Soc., 2006, 153, A75–A80 CrossRef CAS PubMed.
- W. H. Shi, J. X. Zhu, D. H. Sim, Y. Y. Tay, Z. Y. Lu, X. Zhang, Y. Sharma, M. Srinivasan, H. Zhang, H. H. Hng and Q. Y. Yan, J. Mater. Chem., 2011, 21, 3422–3427 RSC.
- X. F. Xia, Q. L. Hao, W. Lei, W. J. Wang, D. P. Sun and X. Wang, J. Mater. Chem., 2012, 22, 16844–16850 RSC.
- Y.-H. Kim and S.-J. Park, Curr. Appl. Phys., 2011, 11, 462–466 CrossRef PubMed.
- X. Y. Liu, Y. Q. Zhang, X. H. Xia, S. J. Shi, Y. Lu, X. L. Wang, C. D. Gu and J. P. Tu, J. Power Sources, 2013, 239, 157–163 CrossRef CAS PubMed.
- Z. Wang, X. Zhang, Y. Li, Z. T. Liu and Z. P. Hao, J. Mater. Chem. A, 2013, 1, 6393–6399 CAS.
- Q. Q. Xiong, J. P. Tu, S. J. Shi, X. Y. Liu, X. L. Wang and C. D. Gu, J. Power Sources, 2014, 256, 153–159 CrossRef CAS PubMed.
- S. W. Kim, H. W. Lee, P. Muralidharan, D. H. Seo, W. S. Yoon, D. K. Kim and K. Kang, Nano Res., 2011, 4, 505–510 CrossRef CAS.
- G. Q. Zhang, L. Yu, H. B. Wu, H. E. Hoster and X. W. Lou, Adv. Mater., 2012, 24, 4609–4613 CrossRef CAS PubMed.
- K. Karthikeyan, D. Kalpana and N. Renganathan, Ionics, 2009, 15, 107–110 CrossRef CAS.
- B. Liu, B. Y. Liu, Q. F. Wang, X. F. Wang, Q. Y. Xiang, D. Chen and G. Z. Shen, ACS Appl. Mater. Interfaces, 2013, 5, 10011–10017 CAS.
- L. Yu, L. Zhang, H. B. Wu, G. Q. Zhang and X. W. Lou, Energy Environ. Sci., 2013, 6, 2664–2671 CAS.
- L. Huang, D. C. Chen, Y. Ding, S. Feng, Z. L. Wang and M. L. Liu, Nano Lett., 2013, 13, 3135–3139 CrossRef CAS PubMed.
- W. L. Yang, Z. Gao, J. Ma, X. M. Zhang, J. Wang and J. Y. Liu, J. Mater. Chem. A, 2014, 2, 1448–1457 CAS.
- X. Y. Liu, S. J. Shi, Q. Q. Xiong, L. Li, Y. J. Zhang, H. Tang, C. D. Gu, X. L. Wang and J. P. Tu, ACS Appl. Mater. Interfaces, 2013, 5, 8790–8795 CAS.
- C. H. An, Y. J. Wang, Y. A. Huang, Y. A. Xu, C. C. Xu, L. F. Jiao and H. T. Yuan, CrystEngComm, 2014, 16, 385–392 RSC.
- L. Li, Y. Q. Zhang, X. Y. Liu, S. J. Shi, X. Y. Zhao, H. Zhang, X. Ge, G. F. Cai, C. D. Gu, X. L. Wang and J. P. Tu, Electrochim. Acta, 2014, 116, 467–474 CrossRef CAS PubMed.
- S. L. Kuo and N. L. Wu, Electrochem. Solid-State Lett., 2005, 8, A495–A499 CrossRef CAS PubMed.
- S. N. Pusawale, P. R. Deshmukh and C. D. Lokhande, Bull. Mater. Sci., 2011, 34, 1179–1183 CrossRef CAS PubMed.
- N.-L. Wu, Mater. Chem. Phys., 2002, 75, 6–11 CrossRef CAS.
- M. Sathiya, A. S. Prakash, K. Ramesha, J. M. Tarascon and A. K. Shukla, J. Am. Chem. Soc., 2011, 133, 16291–16299 CrossRef CAS PubMed.
- Q. H. Do, C. C. Zeng, C. Zhang, B. Wang and J. Zheng, Nanotechnology, 2011, 22, 365402 CrossRef PubMed.
- T. Gujar, V. Shinde, C. Lokhande and S.-H. Han, J. Power Sources, 2006, 161, 1479–1485 CrossRef CAS PubMed.
- F.-L. Zheng, G.-R. Li, Y.-N. Ou, Z.-L. Wang, C.-Y. Su and Y.-X. Tong, Chem. Commun., 2010, 46, 5021–5023 RSC.
- J. Rajeswari, P. S. Kishore, B. Viswanathan and T. K. Varadarajan, Electrochem. Commun., 2009, 11, 572–575 CrossRef CAS PubMed.
- L. Zheng, Y. Xu, D. Jin and Y. Xie, J. Mater. Chem., 2010, 20, 7135–7143 RSC.
- X. H. Xia, Z. Y. Zeng, X. L. Li, Y. Q. Zhang, J. P. Tu, N. C. Fan, H. Zhang and H. J. Fan, Nanoscale, 2013, 5, 6040–6047 RSC.
- A. Ramadoss, G.-S. Kim and S. J. Kim, CrystEngComm, 2013, 15, 10222–10229 RSC.
| This journal is © The Royal Society of Chemistry 2014 |