OF-DFT SCF calculations for an H2 molecule using a nonlocal kinetic energy functional defined on energy coordinate
Received
28th October 2025
, Accepted 15th January 2026
First published on 15th January 2026
Abstract
A nonlocal kinetic energy density functional in the orbital-free density functional theory (OF-DFT) is utilized to perform the SCF calculations for an H2 molecule. The functional is formulated within the framework of the novel DFT [Takahashi, J. Phys. B: At., Mol. Opt. Phys., 2018, 51, 055102] that uses the electron distribution ne(ε) on the energy coordinate ε as a fundamental variable of DFT. The response function χe0 in the integral kernel of the nonlocal functional is also represented on the coordinate ε. As a notable feature in our approach, χe0 of the whole molecule is given by the composite of the response functions of the isolated fragments constituting the molecule. The electron density and the potential energy curve of the H2 molecule are computed by the OF-DFT SCF calculations, showing reasonable agreements with those obtained by the Kohn–Sham DFT calculations.
1 Introduction
Density-functional theory (DFT) for electrons1–3 is an indispensable theoretical tool to study the electronic properties of molecules and materials. A number of studies are published annually for the applications of DFT and for the theoretical developments. The theoretical framework of DFT was first provided by the Hohenberg–Kohn (HK) theorem4 which proves that the external potential υext applied to an N-electron system uniquely corresponds to the resultant ground state electron density n of the system. The HK theorem offers a theoretical basis to develop the density functional to describe the energy components of the total electronic energy Etot of the system of interest. Actually, Etot[n] as a functional of the density n can be given by the sum of the energy contributions, thus,| | 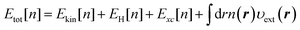 | (1) |
where r represents the coordinate of electrons. The term Ekin[n] on the right-hand side of eqn (1) is the kinetic energy functional, and EH[n] stands for the Hartree potential of the density n(r). The functional Exc[n], referred to as the exchange–correlation energy, represents the non-classical interaction between the electrons. The last term provides the Coulomb interaction between the electron density and the external potential. The functional Etot[n] provides the ground state energy E0 when the υ-representable density1n coincides with the density n[υext] that corresponds to the υext of the system of interest.
We note that the existence of the functionals in eqn (1) is guaranteed solely by the HK theorem. Therefore, the range of definition of the functionals must be the set of υ-representative electron densities.1 However, it can be extended to the set of N-rep. electron densities by virtue of the DFT formalism based on Levy's constraint search.5 Thus, the ground state energy E0 for a given external potential υext can be obtained at least formally through minimization of the energy functional of eqn (1) with respect to N-rep. electron densities on the basis of the variation principle. These discussions guarantee the existence of the route to find the energy E0 and the corresponding density n using only the electron density as a fundamental variable. Such an approach is, of course, quite advantageous as compared with the wave-function theory because the number of variables to be optimized during the variational calculation can be drastically reduced. However, unfortunately, the accurate kinetic energy functional Ekin in eqn (1) is not currently available, although substantial efforts have been devoted to developing the functional.6,7 To resolve this problem, a compromise was made by Kohn and Sham,8 who introduced a ‘non-interacting reference system’ represented with a set of wave functions {φi} for N electrons. Then, the kinetic energy of the non-interacting system with the density n is defined as the sum of the kinetic energies of N electrons, thus,
| | 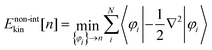 | (2) |
Accordingly, the energy functional of
eqn (1) is rewritten as
| | 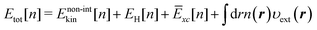 | (3) |
where
Ēxc[
n] is supposed to include the difference
Ekin–
Enon-intkin in the kinetic energy. In the approach of Kohn–Sham DFT (KS-DFT), the minimization of
Etot[
n] with respect to
n is replaced by the search in the orbital space {
φi} as shown in
eqn (2). The minimization under the orthonormalization conditions 〈
φi|
φj〉 =
δij between the orbital pairs (
i,
j) leads to the set of KS equations,
| | 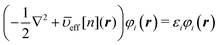 | (4) |
for
N electrons. The effective potential
![[small upsilon, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0d5.gif) eff
eff in
eqn (4) is given by the functional differentiation of the last three terms in
eqn (3), thus
| | 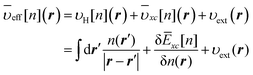 | (5) |
The exchange–correlation potential
xc in
eqn (5) plays a decisive role in determining the accuracy of the KS-DFT calculation.
The reliability and accuracy of the KS-DFT have been well established through a lot of applications. However, as noted above, the introduction of the orbitals necessitates calculations to ensure the orthonormalization conditions between all the orbital pairs. This causes a drastic increase in the computational cost with the increase in the number of electrons in the system of interest, which prevents the application of KS-DFT to larger systems. It is, thus, desirable to develop a kinetic energy functional Ekin[n] which enables one to perform DFT calculations without orbitals, that is, orbital-free DFT (OF-DFT) calculations.6,7,9 In the following paragraphs, we provide a brief review of the development of OF-DFT.
Thomas10 and Fermi11 provided a formula for the exact kinetic energy for a homogeneous electron gas (HEG) with density n. By using the formula, the Thomas–Fermi (TF) functional ETFkin[n] is explicitly written as
| | 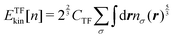 | (6) |
with
CTF being
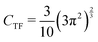
.
σ in
eqn (6) represents the index for spin (
σ =
α,
β). The first OF-DFT functional, referred to as the TF model, is then given by
| | 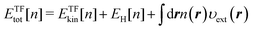 | (7) |
The TF model can be improved by adding the LDA (local-density approximation) exchange functional
ELDAx[
n] to
eqn (7), which gives the Thomas–Fermi–Dirac (TFD) model
1 written as
| | 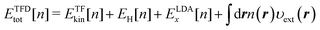 | (8) |
The LDA functional
ELDAx[
n] is explicitly given by
| | 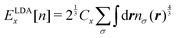 | (9) |
with the coefficient
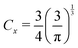
. Unfortunately, it was proved that neither the TF nor the TFD model realizes the formation of chemical bonds in principle.
12,13 The first primitive approach to resolve this issue was to add the von Weizsäcker (vW) correction
14 to
eqn (6). The vW correction term
EvW[
n] is given by
| | 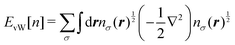 | (10) |
It can be readily recognized that
EvW[
n] itself gives the exact kinetic energy of a one-electron system or a two-electron system with a closed shell. The TF kinetic energy with the vW correction is simply given by
| | | ETFWkin[n] = ETFkin[n] + λEvW[n] | (11) |
where
λ is a weight parameter to mix the vW correction. The analysis of the response function of
eqn (11) in momentum space
9 shows that
ETFWkin[
n] with
λ = 1 reproduces the correct behavior at large
k limit of the response function of the HEG with density
n. On the other hand,
λ = 1/9 realizes the correct behavior of the response function at small
k. It was shown in ref.
15 that an intermediate value
λ = 1/5 gives the best result for atoms with large nuclear charges. It is naturally anticipated that the inclusion of higher order gradient terms leads to better results. It is known, however, that it gives only a slight improvement on the kinetic energy.
16
An effort has also been devoted to develop a series of GGA (generalized gradient approximation) kinetic energy functionals,17–19 where the inhomogeneity parameter sσ of the electron density plays a role. The definition of sσ is common to those in the GGA corrections to Exc functionals and is given by
| | 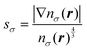 | (12) |
It was demonstrated in ref.
19 that several GGA kinetic functionals applied to a set of 77 molecules show rather good agreements with the kinetic energies given by Hartree–Fock (HF) wave functions. Note, however, that the densities used as the inputs to the kinetic functionals were those obtained through HF-SCF calculations at their equilibrium structures. We note that optimizing the density self-consistently using a kinetic energy functional has its own difficulty. The gradient term in
eqn (12) tends to flatten the density when the GGA correction term is applied to the density during the SCF procedure. As a consequence, the shell structures inherent in the electron density will be destroyed.
So far, we have reviewed the local or semi-local kinetic energy functionals, which refer only to the density n(r) or gradients of the density. It seems that inclusion of the nonlocal term into the kinetic functional is needed to realize the detailed structure in the electron density of the system of interest. To our knowledge, the nonlocal kinetic energy functional was first developed by Chacón et al. who utilized the weighted average of the density to construct the functional.20 The weight function was determined so that the resultant kinetic functional realizes the response function of the HEG. In a development by Wang and Teter (WT),21 they also referred the response function of the HEG to construct the nonlocal term of the kinetic energy functional. The nonlocal term EnlocWT[n] in the WT functional is explicitly written as
| | 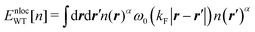 | (13) |
where
ω0 is the kernel of the integral and only depends on the distance |
r −
r′|.
kF is the Fermi wave number for the HEG with the density
n0. Explicitly,
kF is determined as
kF = (3π
2n0)
1/3. For the bulk system, the average electron density of the unit cell can be taken as the density
n0. The parameter
α in
eqn (13) was set at 5/6 in the original study. The kernel
ω0 is directly related to the response function of the HEG with density
n0. Importantly, it was demonstrated in their study
21 that the shell structure in the density profile of the Ne atom was found to be maintained after the SCF calculation employing the nonlocal term of
eqn (13). A lot of developments have been conducted to improve the WT functional.
22–27 In the work by Huang and Carter,
24 they introduced a integration kernel
ω0 into
eqn (13) that depends on the parameter
sσ in
eqn (12) for applications with semiconductors. Interestingly, the functional was also applied to covalent bonds in homonuclear diatomic molecules.
28 It is quite impressive that their method is capable of describing the chemical bonds adequately. However, the method for determining the average electron density
n0 in a covalent bond seems somewhat ambiguous. We refer the reader to the literature
6,7 for a more detailed review of the recent advances in the OF-DFT.
In recent works, H. T. developed a series of kinetic energy functionals.29–32 Importantly, some of the functionals29,32 are based on the novel framework of the density-functional theory (DFT) for electrons,33,34 where the electron distribution ne(ε) defined on the energy coordinate ε plays a fundamental role in the DFT. Hereafter, we refer to the distribution ne(ε) as the energy electron density. The explicit definitions of ne(ε) and of the energy coordinate ε will be given in the next section. The external potential υext can also be defined on the energy coordinate ε, that is, υeext(ε). As proved in the literature,33 there exists one-to-one correspondence between a subset {ne(ε)} of the energy electron densities and a subset {υeext(ε)} of the external potentials completely in parallel to the HK theorem.4 Thus, it is possible to formulate a kinetic energy functional Eekin[ne] specific to each subset. The author developed a nonlocal kinetic energy functional29 using the response function χe0(ε, ε′) defined on the energy coordinate for describing covalent bonds in molecules. An important feature in the construction of the response function of a molecule is that it is given as a ‘composite’ of the response functions of its constituent atoms. The functional was applied to the calculation of the potential energy curve of the H2 molecule. However, we note that the energy values were those obtained solely using a post SCF density by the KS-DFT.
The main purpose of the present work is to devise a method to optimize the electron density self-consistently using the nonlocal kinetic energy functional given in the literature.29 A detailed analyses of the property of the composite response function χe,cmp0(ε, ε′) is also an important issue. It will be shown that the composite response function χe,cmp0(ε, ε′) of H2 strictly reproduces the full response function χe,full0(ε, ε′) obtained for the whole molecular system. The mechanism underlying the agreement of χe,cmp0(ε, ε′) with χe,full0(ε, ε′) will be discussed. The nonlocal kinetic functional using χe,cmp0(ε, ε′) is applied in the OF-DFT SCF calculation to calculate the potential energy curve of the H2 molecule. The result will be compared with that obtained by the corresponding KS-DFT calculation. We consider in the future the present method to be applied mainly to the condensed molecular systems such as proteins embedded in water.
The organization of the remainder of this article is as follows. In the next section, we first provide a brief review of the DFT that uses the energy electron density as the fundamental variable. Then, it will be followed by an outline of the construction of the nonlocal kinetic energy functional. The method for updating the electron density during the SCF of the OF-DFT is also described. The subsequent section will be devoted to providing the numerical details to implement the OF-DFT using the real-space grid formalism.35–40 In the section ‘Results and discussion’ we first compare the composite response function χe,cmp0(ε, ε′) with χe,full0(ε, ε′) for H2 molecules to show their equivalence. The potential energy curve of H2 obtained by the SCF calculation of the OF-DFT will be compared with that given by the KS-DFT. We also discuss the advantages and deficiencies of our development in OF-DFT. In the last section, we summarize the present work, and provide a perspective for future developments.
2 Theory and method
This section is devoted to describing the theory and the method for the construction of the nonlocal kinetic energy functional. In the first subsection, the energy electron density ne(ε), which serves as the fundamental variable in the novel DFT,33 is introduced. Then, in the next subsection, the nonlocal kinetic energy functional (KEF) is formulated in terms of the functional of ne(ε). In the third subsection, details of the methodology used to construct the composite response function are addressed. A method to update the electron density in the SCF of the OF-DFT is described in the last subsection.
2.1 Energy electron density
In this subsection, we briefly review the DFT for electrons33,34 based on the energy electron density ne(ε). To do this, we first introduce the definition of the function ne(ε). Suppose that nσ(r) is the electron density with spin σ of a molecule under consideration. Then, neσ(ε) is given by the projection of nσ(r) onto the energy coordinate ε,| | 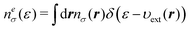 | (14) |
where υext(r) is the external potential felt by the N electrons included in the system of interest. Eqn (14) indicates that the external potential υext is used (though not necessarily) as a function to define the energy coordinate ε for a given spatial coordinate r. Hereafter, in this subsection, let us denote this special potential υext as υdef to avoid confusion. Provided that contour surfaces of an external potential 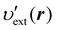 are completely parallel to those of the potential υdef(r), then it is possible to identify the potential
are completely parallel to those of the potential υdef(r), then it is possible to identify the potential 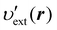 using the potential
using the potential 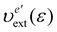 defined on the one-dimensional coordinate ε, thus,
defined on the one-dimensional coordinate ε, thus,| | 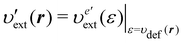 | (15) |
Specifically, for υdef(r) itself, the corresponding potential υedef(ε) satisfies the relationship υedef(ε) = ε by definition. Thus, a defining potential υdef identifies a subset I of external potentials {υext(r)}I. Using the potential υdef(r) as the defining potential for ε, all potentials in the subset can be represented by the potential on the coordinate ε without loss of information, that is, {υeext(ε)}I. For each subset I of the external potentials, we obtain the corresponding set of ‘ground state’ electron densities {n(r)}I. Projecting the set of the densities {n(r)}I using eqn (14) leads to the corresponding set of energy electron densities {ne(ε)}I. Quite importantly, it is possible to prove that there exists one-to-one correspondence between the set of potentials {υeext(ε)}I and the set of energy electron densities {ne(ε)}I.33 The method of proof is completely parallel to the HK theorem.4 Thus, it is possible to formulate an energy functional such as Eexc[ne] with ne being an argument for each subset I.33,34 In the literature,29,32 the author developed kinetic energy functionals Eekin[ne] including nonlocal terms.29 In the following subsection, the outline of the method to construct the nonlocal kinetic energy functional for the energy electron density will be provided.
2.2 Nonlocal kinetic energy functional
An important feature of our development of the nonlocal kinetic energy functional (KEF) is that it refers to the response function of the constituent atoms in the molecule of interest, rather than that of the homogeneous electron gas (HEG). In the following, we first provide the general formulation for the KEF described with the inverse of the response function of the system of interest. To do this, we minimize the energy Etot[n] in eqn (1) with respect to the density n under constraint 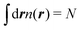 , leading to the condition at the stationary state with the ground state density n0,
, leading to the condition at the stationary state with the ground state density n0,| |  | (16) |
where μ is a chemical potential, and υeff[n](r) is defined as| |  | (17) |
The differentiation of eqn (16) with respect to n(r) at n0 gives
| | 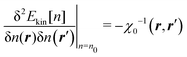 | (18) |
where
χ0(
r,
r′) is the response function of the system with the density
n0 and defined as
| | 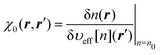 | (19) |
Apart from the system of interest, we now introduce a reference system of which the ground state density is given as n0. Then, for an arbitrary density n close to n0, its kinetic energy Ekin[n] can be approximated by the second-order Taylor expansion with respect to the deviation δn = n − n0, thus,
| | 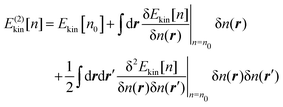 | (20) |
Using the relationship of
eqn (18), we get
| | 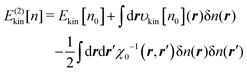 | (21) |
This equation constitutes the basis for the developments in the following.
The integration kernel of the nonlocal term of eqn (21) is a 6-dimensional function. Therefore, direct evaluation of eqn (21) requires a huge computational cost. However, when referring to the HEG as the reference system, the corresponding response function χ0 can be reduced to a function of |r − r′| as in the kernel ω0 in eqn (13). In our approach, we take a different approach based on the DFT that uses ne(ε) as a fundamental variable. We consider the response function between the energy coordinates ε and ε′, not between the spatial coordinates r and r′. To this end, χ0(r, r′) is projected onto the energy coordinate to yield χe0(ε, ε′),
| | 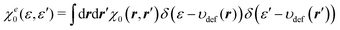 | (22) |
It is supposed that the external potential
υext(
r) of the system of interest is used as the defining potential
υdef(
r) in
eqn (22). Due to the projection of
eqn (22), large amounts of the information contained by the response function
χ0(
r,
r′) will be lost. However, note that when the contour surfaces of the polarization density δ
n(
r) =
n(
r) −
n0(
r) are parallel to those of the external potential
υext(
r) used as
υdef(
r), the projection does not degrade the energy of the nonlocal term in
eqn (21). This can be readily verified as follows.
| | 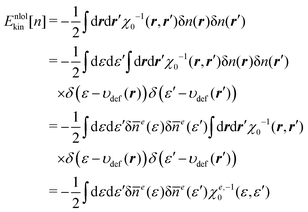 | (23) |
To derive the third equality, we used the assumption that δ
n(
r) is constant over the iso-energy surface of the potential
υdef. The quantity
![[n with combining macron]](https://www.rsc.org/images/entities/i_char_006e_0304.gif) e
e(
ε) is the value of δ
n(
r) at
r specified by
ε =
υdef(
r).
e(
ε) can also be obtained by dividing
ne(
ε) by the spatial volume
Ω(
ε) with the coordinate
ε, that is,
| | | e(ε) = ne(ε)·Ω(ε)−1 | (24) |
where
Ω(
ε) is defined as
| | 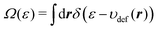 | (25) |
Eqn (23) is strictly valid, for example, when a spherically symmetric electron density of an atom is to be polarized due to a change in its nuclear charge.
29 Of course, in general,
eqn (23) holds only approximately, and its validity also depends on the choice of the reference density
n0. This issue will be examined in the ‘Results and discussion’ section for H
2 molecules. Anyway, we now reduce the 6-dimensional integration kernel in
eqn (21) to the 2-dimensional one; thus,
| | 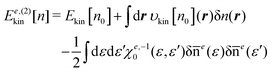 | (26) |
With this kinetic energy, the total energy
EOF-DFTtot[
n] of the OF-DFT is given by
| | 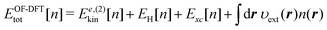 | (27) |
For later references, we also provide the potential
υtot[
n](
r) given as the functional derivative of the total energy, thus,
| | 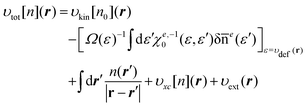 | (28) |
2.3 Composite response function
The next issue is how to construct the response function χe0 in eqn (26) for a reference system. A naive and time-consuming method to obtain χ0 or χe0 is to perform the inverse KS-DFT method41–47 for a given reference density n0. χ0 is then constructed with the KS wave functions by exploiting the second-order perturbation theory. This approach is robust, but useless because the computational cost is much higher than that for the KS-DFT calculation itself for the whole system. Our strategy to solve this problem is to construct χ0 as a ‘composition’ of the response functions of the subsystems comprising the entire system.29 In what follows, we illustrate how to construct the composite response function. Suppose that a molecule of interest consists of two subsystems A and B. Then we solve the KS equation separately for each isolated subsystem. The reference electron density n0(r) of the entire system is then given by the sum of the ground state densities nA(r) and nB(r) for the subsystems A and B, respectively, that is,| | | n0(r) = nA(r) + nB(r) | (29) |
This method is completely the same as the Harris approach,48 where the density of the coupled fragments is approximated by the sum of the isolated fragment densities. There might be another elaborate method to construct the reference density n0. This issue will be discussed later. For the density n0 of eqn (29), the corresponding reference system is defined as the system that has n0 as its ground state density. Assuming that n0 is non-interacting υ-representable, the KS equation for the reference system is explicitly written as| | 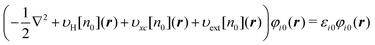 | (30) |
It is quite important to note in the equation that the external potential is given as a functional υext[n0] of the density n0. Hence, υext[n0] is not equal to the external potential υext of interest in general. For later discussion, we introduce the full response function 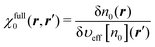 for the system identified by eqn (30). χfull0(r, r′) is then projected onto the energy coordinate to yield χe,full0(ε, ε′) given by
for the system identified by eqn (30). χfull0(r, r′) is then projected onto the energy coordinate to yield χe,full0(ε, ε′) given by| | 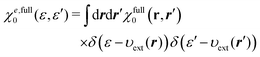 | (31) |
Note that the potential υext in eqn (31) is the external potential for the system of interest and not for the reference system. On the other hand, the composite response function χe,cmp0(ε, ε′) defined on the energy coordinate is given by| | | χe,cmp0(ε, ε′) = χeA(ε, ε′) + χeB(ε, ε′) | (32) |
In parallel to eqn (31), the individual response function χeX(ε, ε′) for subsystem X = A or B is also given by| | 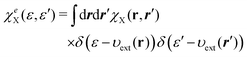 | (33) |
where the response function χX(r, r′) is defined as 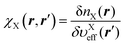 with υXeff being the KS effective potential of the isolated subsystem X.
with υXeff being the KS effective potential of the isolated subsystem X.
Our primary concern to the composite response function χe,cmp0(ε, ε′) is whether it works in eqn (26) as an approximation to χe,full0. Note that a response property is nonlocal and, therefore, the correlation between the two subsystems is being incorporated into the ‘real’ response function. From this consideration, seemingly, the construction of the response function shown in eqn (32) does not work correctly. However, surprisingly, as will be demonstrated in the Results and discussion section, the full response function χe,full0 can be safely replaced by χe,cmp0 in describing the chemical bond in H2 molecules. The mechanism underlying the equivalence of χe,cmp0 to χe,full0 for H2 will also be elucidated. As shown in eqn (26), the calculation of the energy Ee,(2)kin[n] requires the inversion of the response function χe,cmp0 or χe,full0. Since the response function has an eigenvector with a null eigenvalue, its inversion requires some devises.29 Details of the numerical procedure for the inversion will be provided in the Computational details section.
2.4 OF-DFT SCF
For a post-SCF electron density given by e.g. KS-DFT calculations, the TFD model (eqn (8)) or a kinetic GGA functional provides rather appropriate kinetic energies. However, the density determined self-consistently with the TFD or a kinetic GGA functional tends to deviate from the KS density. It is well known that the shell structures in the density profiles of atoms disappear in the SCF calculations using the TFD model. The GGA correction to the model does not improve the situation because the gradient term in the functional tends to flatten the shell structures inherent in the density profile. In our preliminary SCF calculations using eqn (26), we find that the full optimization of the electron density n(r) in the real space gives rise to erroneous behaviors in the electron density. More precisely, a sharp peak was found to arise in a confined spatial region during the SCF procedure. We note that the nonlocal term in eqn (26) is always positive with respect to the deviation δne. Therefore, the nonlocal term effectively suppresses the deviation of the density ne from the reference density ne0. However, the integration kernel χe,−10 applies to the projected density difference δne(ε). It is thus possible that n(r) deviates seriously from n0(r) within a confined region of r without changing the amount of δne(ε). This constitutes the major drawback of the method using ne(ε) in the SCF calculations.
To avoid such an unfavorable situation, we impose a constraint on the polarization density δn(r). To be explicit, δn(r) is subject to the constraint
| | | δn(r) = n(r) − n0(r) = δne(ε)|ε=υdef(r) | (34) |
Thus, the amount of the density polarization at
r is solely specified by its energy coordinate
ε =
υdef(
r). The update of the density is then performed through the equation as follows
| |  | (35) |
where
η is supposed to a small positive real number, and the initial guess of the density is supposed to be the reference density
n0 in
eqn (28).
υetot[
n](
ε) in
eqn (35) is given by the projection of the potential
υtot[
n](
r) shown in
eqn (28). Explicitly,
υetot[
n](
ε) is given by
| |  | (36) |
It is easy to see that
υetot[
n](
ε) = const. is achieved when the density converges,
i.e.,
ni+1(
r) ≃
ni(
r). Remember that the initial guess of the density for the SCF is supposed to be
n0 in
eqn (29). Hence, the density at the SCF convergence varies depending on the choice of the reference density
n0 taken as the initial guess.
It is helpful to see in more detail the integrand in eqn (35). First, we note that the following equation can be obtained directly from eqn (18), that is,
| | 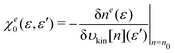 | (37) |
We also note that
υetot can be expressed as
υetot =
υekin +
υeeff. Then the integrand can be formulated as
| | 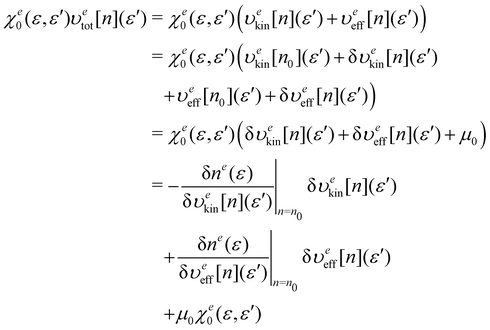 | (38) |
The notation δ
υekin[
n] in the above equation means the potential change due to the variation of density from
n0 to
n. In deriving the third equality in
eqn (38), we used the fact that
υekin[
n0](
ε) +
υeeff[
n0](
ε) =
μ0. The last term in the right hand side of the last equality becomes zero after integrating over
ε′. The last equality in
eqn (38) shows that the density change δ
n due to the variation δ
υekin of the kinetic potential compensates that given by the variation δ
υeeff of the effective potential when the SCF convergence is achieved.
3 Computational details
This section provides the numerical details for the implementation of the OF-DFT calculations. The first subsection describes the numerical techniques used in the SCF of the OF-DFT. In our implementation of the OF-DFT, the real-space grid approach is employed to represent the potentials as well as density, where nonlocal pseudopotentials are usually used to describe the electron-nuclei potential. To perform the OF-DFT calculations for the H2 molecule, a local potential is introduced for the hydrogen atom in the second subsection. In the third subsection, the numerical details for the implementation of the real-space grid method are provided. The fourth and fifth subsections describe the construction of the response function and the method of its inversion, respectively. The last subsection provides the method to construct the energy electron densities and the response functions.
3.1 OF-DFT SCF
In the OF-DFT SCF calculation, the electron density n(r) will be updated using eqn (35), where the non-negativity of the density must be guaranteed. To this end, we introduce a variable ϕ(r), satisfying n(r) = ϕ(r)2. Then, eqn (35) is revised to update ϕ(r), thus29,| |  | (39) |
where ![[small chi, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e11b.gif) e0 is defined as29
e0 is defined as29| |  | (40) |
The acceleration factor η in eqn (39) is set in the range 0.03 ≤ η ≤ 0.08 depending on the convergence behavior of the SCF. The criterion for the SCF convergence is specified by the condition −5.0 × 10−6 ≤ ΔE = Ei+1 − Ei ≤ 0.0 for the energy EOF-DFTtot[n] in eqn (27) given in the atomic units.
3.2 Local potential for H2
As will be described in the following subsection, the present OF-DFT calculations are performed using the real-space grids.35–39 Usually, in the KS-DFT calculations, the Coulomb potential formed by the nuclei is represented with nonlocal pseudopotentials to avoid steep behaviors in the resultant wave functions. However, to perform the OF-DFT calculations, the external potential υext must be local. Thus, we introduce a local potential for H atoms to represent a chemical bond of the H2 molecule. As a consequence, the resultant chemical bond is not for a real hydrogen molecule, but for a ‘pseudo’ H2 molecule. Note, however, that it makes sense because the result given by the OF-DFT can be fully compared with that by the corresponding KS-DFT calculation using the same potential.
The local potential for the pseudo H atom is constructed by replacing the charge of the proton with a Gaussian charge distribution. Explicitly, the delta function δ(r − R) for the charge of a proton placed at R is replaced by a normalized Gaussian, thus,
| | 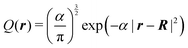 | (41) |
The exponent
α of the charge distribution was determined using the exponent
α′ presented in Table I in the literature
49 which conducted the non-Born–Oppenheimer calculation for the H
2 molecule. The exponent
α′ = 21.95 used in the literature for the initial guess of the proton wave function was translated to the exponent
α of the density through
α = 2 × 21.95. Then, the potential that corresponds to the distribution is given by
| | 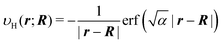 | (42) |
This local potential is used to describe the interaction between the electrons and the pseudo hydrogen atom H
A placed at
RA. The same is true for the potential of pseudo hydrogen atom H
B at
RB. The local potential of a pseudo hydrogen molecule H
2 is thus constructed.
3.3 Real-space grid method
The OF-DFT calculations are performed by conducting a Fortran module29 combined with the ‘Vmol’ package33,37–40,50–52 based on the real-space grid formalism.35,36 The grid width h of the real-space cell is set at h = 0.2867869 a.u., which corresponds to the cutoff energy Ec = 60 a.u. of the calculation using the plane waves. Then, h is multiplied by the number Ng = 64 of grids along each axis of the cell, leading to the cell size L = 18.35436 a.u. The H2 molecule is enclosed in the cubic cell of the size L, where the mass center of the molecule is set at the center of the box and the molecular axis is aligned parallel to the x axis of the real-space cell. Each axis of the cell is uniformly discretized by 64 grids, leading to the grid width h = 0.2867869 a.u. The Hartree energy EH[n] in eqn (27) is computed using the method in the literature.53 The exchange–correlation energy Exc[n] in eqn (27) is evaluated using the BLYP functional.54,55 The KS-DFT calculations are also performed to yield the wave functions which are used to construct the response functions of the reference systems. The Laplacian in the KS equation shown in eqn (4) is represented with the 4th-order finite difference method.35,36
3.4 Response function
As shown in eqn (31), the full response function χe,full0(ε, ε′) is obtained by projecting χfull0(r, r′) onto the energy coordinate. χfull0(r, r′) can be built from the wave functions {φi0} given as the eigenvectors of the KS equation in eqn (30). Using the second-order perturbation theory (PT2), χfull0(r, r′) is given by| | 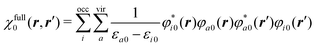 | (43) |
The effective potential υeff[n0](r) in eqn (30) can be obtained by the inverse KS-DFT method44–47 for example. However, in the case of the H2 molecule, υeff[n0](r) is directly obtained from the relationship;| | 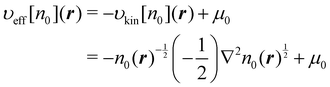 | (44) |
In applying eqn (44) to eqn (30), μ0 is simply set to 0 since the value of μ0 does not affect the construction of the response function. Eqn (44) is also used to determine the kinetic potential υkin[n0](r). And the kinetic energy Ekin[n0] in eqn (26) can be evaluated using n0(r)1/2 as the wave function of the H2 molecule with the density n0. Note that these methods can only be used for the one-electron system or two-electron system with the closed shell structure. However, for a ‘frozen’ electron density, its kinetic energy and the potential might be obtained adequately utilizing a series of kinetic GGA functionals.19,32
The composite response function χe,cmp0(ε, ε′) in eqn (32) can also be obtained from the solutions of the individual KS equations for the isolated subsystems HA and HB. The number of orbitals employed to construct the response function of H2 is set to 10 for both χe,full0 and χe,cmp0.
3.5 Inversion of response function
When computing the energy Ee,(2)kin[n] in eqn (26), it is necessary to invert the response function χe0(ε, ε′). As proved in Appendix A of the literature,29 the projected response function χe0(ε, ε′) is positive semi-definite. Thus, it can be inverted by excluding the eigenvector with a null eigenvalue from the matrix. Furthermore, the eigenvectors with effectively null eigenvalues are also excluded to avoid unphysical oscillations that arise in the resultant kinetic potential. This approach is also known as the truncated singular value decomposition (TSVD)56 in the context of the optimized effective potential (OEP) method.2,57,58 In our present application, it is found that only a single eigenvector is dominant in the spectral decomposition of the response function. Indeed, for the hydrogen molecule with a bond length of 1.4 a.u., the ratio of the second largest eigenvalue to the largest eigenvalue in χe,full0(ε, ε′), defined in eqn (31), is found to be only 3 × 10−4. Thus, when constructing the inversion of χe0(ε, ε′), only the eigenvector with the largest eigenvalue is used as in the case of the literature.29 The effect of this treatment was fully discussed in Appendix A of the literature.29
3.6 Energy electron density
The energy electron density neσ(ε) defined in eqn (14) is constructed on a natural logarithmic grid. The coordinate εk of the kth grid for the energy in the range from εmin to εmax is specified by| | 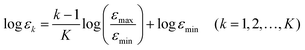 | (45) |
where K is the number of grids on the energy axis. For the pseudo H2 molecule described by the potential shown in eqn (42), we employ the following settings; εmin = 0.12 a.u., εmax = 8.3 a.u., and K = 20. The energy coordinate ε of the real-space grid specified with the index M = (l, m, n) is evaluated using the external potential with its opposite sign. Suppose that the coordinate of a grid is given by rM = (xl, ym, zn), then its energy coordinate ε(rM) is given by using eqn (42), thus,| | 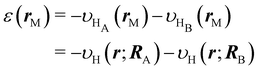 | (46) |
where RA and RB are the coordinates of the hydrogen atoms. To increase the number of sampling points for constructing the distribution neσ(ε), we introduce a double grid. Then, the electron densities on the double grids are evaluated using 4th-order Lagrange interpolations of the coarse grids. The width of the double grid is set at h/5 with h being the width of the coarse grid. However, for the projection of the response function χ0(r, r′) onto ε and ε′, the double grid is not employed.
4 Results and discussion
In the first subsection of this section, we elucidate the mechanism underlying the equivalence between the full response function χe,full0 and the composite response function χe,full0 for the H2 molecule. The density profiles of the H2 molecule obtained by the OF-DFT SCF calculation are shown in the second subsection, where the densities are compared with those given by the corresponding KS-DFT calculations. The potential energy curves of the OF-DFT for the hydrogen molecule are presented in the third subsection and compared with the KS-DFT curve.
4.1 Response function
Before discussing the potential energy curve of H2 given by the OF-DFT SCF calculation, we examine the composite response function χe,cmp0(ε, ε′) of eqn (32) by comparing it with the full response function χe,full0(ε, ε′) of eqn (31). Seemingly, the correlation between the two subsystems HA and HB is not incorporated in the composite response function χe,cmp0(ε, ε′), since it is merely given by the sum of the individual response functions of the isolated subsystems as shown in eqn (32). In Fig. 1(a), the full response function χe,full0(ε, ε′) is presented, and it is compared with the composite response function χe,cmp0(ε, ε′) in Fig. 1(b). Surprisingly enough, the two response functions are shown to be hardly discernible. This means that χe,full0 is essentially the same as χe,cmp0 at least for the H2 molecule.
 |
| | Fig. 1 Response functions of the H2 molecule with bond distance R(H–H) = 1.4 a.u. (a) The full response function χe,full0(ε, ε′) defined in eqn (31). (b) The composite response function χe,cmp0(ε, ε′) in eqn (32). | |
In fact, the projection of the response function onto the energy coordinate plays an essential role in the fact that χe,cmp0 coincides with χe,cmp0. For an intuitive understanding, we consider a density polarization due to the HOMO–LUMO transition in the full response function χfull0(r, r′) of eqn (43). Fig. 2 shows a schematic illustration of the profile of the product φHOMO(r) × φLUMO(r) in eqn (43) of the H2 molecule. It can be seen in the figure that the transition from φHOMO to φLUMO induces a density polarization along the molecular axis. Thus, the correlation between the subsystems HA and HB is realized in the response function given by eqn (43). Such a correlation cannot be described with the composite response function. However, interestingly, the product φHOMO × φLUMO vanishes completely when projected onto the energy coordinate ε, because the contributions from HA and HB with opposite signs cancel with each other as illustrated in Fig. 2. One might think that the cancellation caused by the projection is nothing but a sort of degradation. However, it should be noted that the polarization from n0(r) of eqn (29) to the ground state density n(r) does not include polarization that induces a dipole moment along the molecular axis. Thus, the cancellation of φHOMO × φLUMO due to the projection does not degrade the response function as long as the polarization from n0 to n is concerned. On the other hand, polarization due to the transition from φHOMO to the orbital given by the linear combination of the two 2s orbitals on the hydrogen atoms may contribute to the variation from n0 to n. Importantly, such a polarization in the response function does not vanish when it is projected onto the energy coordinate. Furthermore, the polarization can also be described with the composite response function.
 |
| | Fig. 2 Profile of the product of the HOMO (highest occupied molecular orbital) and the LUMO (lowest unoccupied molecular orbital) of the H2 molecule. The bold horizontal lines indicate the values of the product on the same energy coordinate ε defined by υdef(r). | |
Now we provide mathematical proof of the fact that χe,cmp0(ε, ε′) coincides with χe,full0(ε, ε′). It is important to notice that n0 is not independent from nA. n0 is solely determined from nA due to the symmetry of the H2 molecule. Then, χe,full0 can be reformulated as
| | 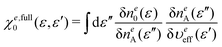 | (47) |
By projecting
eqn (29) onto the energy coordinate, we directly obtain the following relationship,
| | | ne0(ε) = neA(ε) + neB(ε) | (48) |
Since the potential
υeff(
r) specified by
υeeff(
ε) has the same symmetry as the molecular symmetry of H
2,
neA(
ε) in
eqn (48) is completely identical to
neB(
ε). Thus, the relationship
holds. It should also be noted that
| |  | (50) |
holds for the subsystem response functions in H
2 molecules. Then,
eqn (47) can be rewritten as
| | 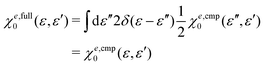 | (51) |
where we use the relationship of
eqn (32). Thus, it is proved that the full response function
χe,full0 built from the wave functions of the whole molecule can be replaced by
χe,cmp0 given as the composite of the individual response functions
χeA and
χeB of the isolated subsystems. Of course,
eqn (51) holds only when the system under consideration has high symmetry. However, it can be expected that the major part of the correlation between the subsystems can be incorporated into the composite response function even for systems with lower symmetry. Since the present study serves only as a preliminary test calculation using the simplest model system, more detailed analyses especially for hetero-nuclear diatomic molecules must be conducted in the future work. It should also be noted that the computational cost of constructing
χe,cmp0 scales only linearly with system size in contrast to the cost of
χe,full0.
Fig. 3(a) shows the contour plots of the local potential of the pseudo H2 molecule introduced in Subsection 3.2. It is compared with the polarization density δn(r) = δnKS(r) − δn0(r) shown in Fig. 3(b). It should be noted that the contour lines of δn(r) are almost parallel to those of the external potential of the pseudo H2 molecule, although the profile of δn along the molecular axis is rather different from that of the potential. Indeed, Fig. 3(b) shows that the polarization can be characterized by an increase in the electron population in the region of large energy coordinate ε and a decrease in the region of small ε. Therefore, it is reasonable to utilize a response function represented with the energy coordinate instead of the real-space coordinate. In fact, the response function χe,full0, or equivalently, χe,cmp0 presented in Fig. 1 shows bipolar behavior along ε in agreement with the profile of δn.
 |
| | Fig. 3 (a) Contour plot of the electron-nuclei potential 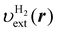 for the pseudo H2 molecule. The potential of each hydrogen atom is given in eqn (42). The value of for the pseudo H2 molecule. The potential of each hydrogen atom is given in eqn (42). The value of  is plotted. (b) Polarization density δn(r) defined by the KS-DFT density nKS(r) subtracted by the reference density n0(r) of eqn (29). Unit of the density is a.u.−3. The white dots represent the positions of the hydrogen atoms. is plotted. (b) Polarization density δn(r) defined by the KS-DFT density nKS(r) subtracted by the reference density n0(r) of eqn (29). Unit of the density is a.u.−3. The white dots represent the positions of the hydrogen atoms. | |
4.2 OF-DFT SCF density
Here we present the electron density nOF optimized through the OF-DFT SCF procedure. Fig. 4 shows the profile of nOF along the molecular axis of the pseudo H2 molecule with a bond length of 1.4 a.u. The reference electron density n0(r) defined in eqn (29) as well as the density optimized by KS-DFT are also provided to make comparisons. As shown in the figure, after the SCF calculation of OF-DFT starting from the reference density n0, the electron population on the two hydrogen atoms increases significantly as compared with n0. The density profile can be directly compared with the density nKS obtained by the KS-DFT calculation that uses the same exchange–correlation functional Exc as in the OF-DFT calculation. It is seen in the figure that nKS also increases on the hydrogen atoms as compared to n0 in agreement with nOF, although the peak height of nKS is lower than that of the density nOF. However, as a major difference between nOF and nKS, the density nOF exhibits a depression at the center of the hydrogen atoms, which shows a clear contrast to nKS. The source of the deficiency of the density nOF can be attributed to the fact that the density polarization is constrained in the direction of the energy coordinate ε. As illustrated in eqn (35) or in eqn (39), in our approach, the amount of the density polarization δn is constrained to be constant on the surface with the same energy coordinate. However, in the spatial region between the two hydrogen atoms, δn remains almost constant as shown in Fig. 3(b), while the energy coordinate 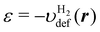 varies significantly within this region as in Fig. 3(a). A possible way to avoid the undesirable behavior in the polarization δn(r) is to construct n0, for example, by means of the frozen density embedding (FDE) method.6,59,60 In the FDE method, the electron population in the region between the hydrogen atoms will be increased, because the density of each subsystem is to be optimized under the influence of the surrounding environment. Thus, the polarization density δ = δnKS − δnFDE will decrease in the region between the hydrogen atoms in contrast to δn = δnKS − δn0 plotted in Fig. 3(b). As a result, the description of the polarization δ using the response function χe0(ε, ε′) might be justified.
varies significantly within this region as in Fig. 3(a). A possible way to avoid the undesirable behavior in the polarization δn(r) is to construct n0, for example, by means of the frozen density embedding (FDE) method.6,59,60 In the FDE method, the electron population in the region between the hydrogen atoms will be increased, because the density of each subsystem is to be optimized under the influence of the surrounding environment. Thus, the polarization density δ = δnKS − δnFDE will decrease in the region between the hydrogen atoms in contrast to δn = δnKS − δn0 plotted in Fig. 3(b). As a result, the description of the polarization δ using the response function χe0(ε, ε′) might be justified.
 |
| | Fig. 4 Profile of the spin densities nσ(r) along the molecular axis of the pseudo H2 molecule with its bond distance being 1.4 a.u. The positions of the two hydrogen atoms are indicated with the arrows. The OF-DFT density is determined self-consistently by using the full response function χe,full0 in eqn (39). The KS-DFT density is also shown for comparisons. The density n0 is given by eqn (29). The BLYP functional54,55 is used for the construction of these densities. | |
We also provide in Fig. 5 the density profile of the pseudo H2 molecule along the y axis perpendicular to the molecular axis. It is seen in the figure that the density nOF shows rather good agreement with the density nKS, although the densities of nOF at y = ∼±0.6 a.u. are observed to be smaller than those of nKS by 0.02 a.u.−3.
 |
| | Fig. 5 Profile of the spin densities nσ(r) on the line parallel to the y axis, provided for the same H2 molecule as in Fig. 4. The line intersects the x axis at the grid of x = −0.574 a.u. where the density exhibits its maximum value along the x axis. The other notations are the same as in Fig. 4. | |
4.3 Potential energy curve
The potential energy curves (PECs) of the pseudo H2 molecule are presented in Fig. 6. The OF-DFT SCF calculations using the full response function χe,full0 of eqn (31) and the composite response function χe,cmp0 of eqn (32) are performed for the production of these PECs. To make comparisons, the PEC by the KS-DFT calculation using the same Exc functional is also provided. Note that the von Weizsäcker kinetic energy functional EvW[n] of eqn (10) gives the same potential energy curve as the KS-DFT, since EvW[n] is equivalent to the KS-DFT in the case of the H2 molecule. To see the effect of the SCF procedure on the energies of the OF-DFT, the PEC is also computed using eqn (27) with the reference density n0 as an argument.
 |
| | Fig. 6 Potential energy curves (PECs) of the pseudo H2 molecule. ‘OF-DFT(full)’ stands for the OF-DFT SCF calculation with the full response function χe,full0 defined in eqn (31). ‘OF-DFT(cmp)’ means the calculation with the composite response function χe,cmp0 defined in eqn (32). The KS-DFT calculation is performed using the same Exc functional (BLYP)54,55 as in the OF-DFT calculations. For the PEC of ‘n0’, the density n0 of eqn (29) is used as the argument of the functional Etot[n] defined in eqn (27). All the PECs are shifted so that their minimums become zero. | |
First, it should be noted that the PEC given by the OF-DFT with χe,full0 shows an excellent agreement with that of the OF-DFT with χe,cmp0. This is a direct consequence of the fact that χe,full0(ε, ε′) is equivalent to χe,cmp0(ε, ε′) as shown in Fig. 1. As shown in the PEC by the KS-DFT, the pseudo H2 molecule has the potential minimum at R(H–H) = ∼1.5 a.u., which differs from the fact that the real H2 has the energy minimum at the bond length of ∼1.4 a.u. It is found that the equilibrium bond length is elongated by ∼0.1 a.u. in the OF-DFT SCF calculations. Thus, it is revealed that the OF-DFT calculation using the response function represented on the energy coordinate ε degrades the shape of the PEC. However, the overall behavior of the PEC is reasonably described with the OF-DFT method. It is possible that some correction to the kinetic energy functional will improve the behavior of the PEC. A possible method for the correction is to improve the reference density n0 given in the form of eqn (29). Constructing the density of each subsystem under the influence of its surroundings will deliver a better reference density for the initial guess of the interacting system. As noted above, the frozen-density embedding (FDE) approach59,60 is a promising candidate to build the reference density. Another approach to improve the OF-DFT might be to make a correction to the kinetic energy functional itself. For example, it is reasonable to tune the kinetic energy density at a coordinate r according to the gradient of the external potential υext(r) used as the defining potential for the energy coordinate. These issues will be addressed in future works. In the comparisons with the PEC of the KS-DFT, the behavior of the PEC given by eqn (27) using the non SCF density n0 as an argument is found to be apparently worse than the PECs given by the self-consistent density. Actually, the equilibrium bond length is found to be further elongated by ∼0.1 a.u. as compared to the result of the OF-DFT SCF calculation. It is thus found that the present OF-DFT SCF procedure works successfully in describing the σ bond in the hydrogen molecule.
It will be useful to analyze the kinetic energy density dkin(r) to see which spatial region in the molecule gives the larger kinetic energy error. For the H2 molecule, we define dvWkin(r) of the von Weizsäcker functional or equivalently of the KS-DFT, thus,
| | 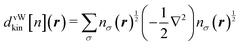 | (52) |
Note that the integration of the kinetic energy density
dvWkin(
r) leads to the accurate total kinetic energy
EvWkin[
n] of the hydrogen molecule with the density
n. For the kinetic energy functional
Ee,(2)kin[
n] of
eqn (26), given as the present work, the corresponding kinetic energy density
dPWkin(
r) is defined as
| | 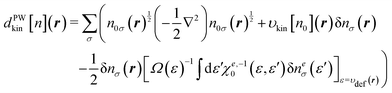 | (53) |
where δ
n is defined as δ
n =
n −
n0. Then, we define the difference Δ
dkin(
r) as
| | | Δdkin(r) = dPWkin[nPW](r) − dvWkin[nvW](r) | (54) |
where
nPW is the density optimized through the OF-DFT SCF calculation shown in subsection 2.4 and
nvW is that given by the KS-DFT calculation. Δ
dkin(
r) on the molecular plane is plotted in
Fig. 7. In accordance with the trend that the present approach overestimates the electron densities at the atomic core regions as shown in
Fig. 4, Δ
dkin(
r) exhibits sharp positive peaks at the core regions. Note that the larger curvatures of the density
nPW than
nvW at the atomic cores may also contribute to the growth of the peaks. Meanwhile, Δ
dkin(
r) shows a significant decrease in the region between the atomic cores. This is the direct consequence of the fact that the density
nPW becomes smaller than
nvM in this region. However, interestingly, it is shown that the depression of Δ
dkin(
r) is alleviated near the center of the chemical bond. This can be attributed to the increase in curvature of the density
nPW at the center of the bond as compared to that of
nvW. Anyway, it is revealed that the cancellation of Δ
dkin(
r) takes place between the overestimations of Δ
dkin at the atomic cores and the underestimation in the region of the chemical bond.
 |
| | Fig. 7 The difference Δdkin(r) of the kinetic energy difference defined in eqn (54) for the H2 molecule placed on the xy plane with R(H–H) = 1.5 a.u. The white dots on the contour plot represent the positions of the hydrogen atoms. | |
In closing this subsection, we make a remark on the accuracy of the present approach for the atomic kinetic energy. In Fig. 3 of ref. 29, kinetic energies of a pseudo hydrogen atom with respect to the variation of its nuclear charge Zv were compared with those given by the KS-DFT. It was shown in the figure that the graph given by the present approach agrees well with that by the KS-DFT over the range of the core charge Zv investigated, though the deviation becomes larger in the small value of Zv. It is of substantial interest to compare the OF-DFT kinetic energies of atoms with the accurate reference values obtained e.g. by sophisticated molecular orbitals theories. However, unfortunately, the present calculations utilized the real-space grids to represent the density and the potentials, which necessitates the use of the pseudopotentials to ensure the smooth behaviors of the resultant densities on the potentials. This prevents the direct comparisons of the present method with some reference benchmark data. It should also be noted that the kinetic functional of eqn (26) will violate some physical constraint to be satisfied by the rigorous functionals. As an example of the constraint, it is known that the von Weizsäcker kinetic energy functional EvW[n] serves as the lower bound of the non-interacting kinetic energy functional Ekin[n],7 that is, EvW[n] ≤ Ekin[n]. Actually, for the hydrogen molecule with R(H–H) = 1.4 a.u., the kinetic energy of eqn (26) is evaluated as 0.9181 a.u., while EvW[n] is computed as 1.0856 a.u. for the density n optimized by the present OF-DFT SCF calculation. Thus, it was revealed that the present kinetic functional violates the lower bound constraint. It is also possible in general that the kinetic potential becomes negative since the functional was not designed so that it fulfills these desired physical constraints. These unfavorable properties should be eliminated from the functional in the future developments.
5 Conclusions
In this article, a nonlocal kinetic energy functional Enlockin was applied to the OF-DFT SCF calculation to describe the covalent bond in the H2 molecule. An important feature of the functional Enlockin is that it is based on a novel framework of DFT where the electron distribution on the energy coordinate serves as a fundamental variable of the DFT.29,33,34 Thus, the response function χe0(ε, ε′) as well as the polarization density δne(ε) in the nonlocal functional are described on the energy coordinate ε.
Another notable feature of the present approach is that the response function χe0(ε, ε′) of the H2 molecule is given as the sum of the response functions of the isolated subsystems. Surprisingly, it was demonstrated for the H2 molecule that the response function χe,full0 of the fully correlated system with a reference density n0 can be faithfully reproduced by the composite response function χe,cmp0 built from the response functions of the subsystems. The underlying mechanism for the equivalence between χe,full0 and χe,cmp0 was discussed, and a mathematical proof for the fact was provided with respect to the H2 molecule.
A method to update the electron density for the SCF of the OF-DFT was also provided. The key of the method is to impose a constraint on the polarization density δn(r). That is, δn(r) is constrained to be constant on the surface with the same energy coordinate ε. In other words, the density polarization is considered only along the energy coordinate ε. The SCF density of the OF-DFT showed a reasonable agreement with the density given by the KS-DFT for the H2 molecule although a significant density depression was observed in the region between the two hydrogen atoms. The potential energy curves (PECs) of the hydrogen molecule were also computed using the SCF densities of the OF-DFT. Although the equilibrium bond length was found to be elongated by ∼0.1 a.u. compared with the PEC by the KS-DFT, it was demonstrated that the SCF density can adequately describe the covalent bond. It was also found that the PEC can be significantly improved by the optimization of the density through the SCF that employed the reference density n0 as an initial guess.
It is possible that the results given by the present approach can be improved e.g. by modifying the method to construct the reference density n0. It might also be effective to modulate the kinetic energy density at a coordinate r according to the gradient of the external potential υext(r). These issues will be examined in future works.
Finally, we also remark on the computational cost of the OF-DFT. Unfortunately, at the present stage of development, the computational time for the OF-DFT is found to be much larger than that for the KS-DFT. The major computational cost in our approach of the OF-DFT arises from the construction of the energy electron density and the 2-dimensional response function χe0(ε, ε′). Note, however, that the cost associated with these calculations increases only linearly with the size of the system of interest. And we also expect that these calculations are suitable for the application of GPGPU (General-Purpose computing on Graphics Processing Units) and thus, they can be accelerated effectively.
Author contributions
Hideaki Takahashi: conceptualization; formal analysis; funding acquisition; investigation; methodology; software; visualization; writing – original draft; writing – review and editing.
Conflicts of interest
There are no conflicts to declare.
Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information provides the raw data used to produce the graphs in Fig. 1(a) and (b). See DOI: https://doi.org/10.1039/d5cp04133c.
Acknowledgements
This paper was supported by the Grant-in-Aid for Scientific Research(C) (no. 17K05138, no. 22K12055, and no. 25K08570) from the Japan Society for the Promotion of Science (JSPS) and the Grant-in-Aid for Challenging Exploratory Research (no. 25620004) from the Japan Society for the Promotion of Science (JSPS).
Notes and references
-
R. G. Parr and W. Yang, Density-functional theory of atoms and molecules, Oxford university press, New York, 1989 Search PubMed.
-
R. M. Martin, Electronic Structure, Basic Theory and Practical Methods, Cambridge University Press, Cambridge, 2004 Search PubMed.
-
F. Jensen, Introduction to Computational Chemistry, Wiley, 2009 Search PubMed.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864–B871 Search PubMed.
- M. Levy, Proc. Natl. Acad. Sci. U. S. A., 1979, 76, 6062–6065 CrossRef CAS PubMed.
- in Recent Progress in Orbital Free Density Functional Theory (Recent Advances in Computational Chemistry), ed. T. A. Wesolowski and Y. A. Wang, World Scientific, Singapore, 2013 Search PubMed.
- W. Mi, K. Luo, S. B. Trickey and M. Pavanello, Chem. Rev., 2023, 123, 12039–12104 CrossRef CAS PubMed.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
-
Y. A. Wang and E. A. Carter, Theoretical Methods in Condensed Phase Chemistry (Progress in Theoretical Chemistry and Physics), Kluwer, Dordrecht, 2000, ch. 5, vol. 5 Search PubMed.
- L. H. Thomas, Proc. Cambridge Philos. Soc., 1927, 23, 541–548 Search PubMed.
- E. Fermi, Z. Phys., 1928, 48, 73–79 CrossRef CAS.
- E. Teller, Rev. Mod. Phys., 1962, 34, 627–631 CrossRef CAS.
- N. L. Balàzs, Phys. Rev., 1967, 156, 42–47 CrossRef.
- C. F. von Weizsacker, Z. Phys., 1935, 96, 431–458 CrossRef.
- K. Yonei and Y. Tomishima, J. Phys. Soc. Jpn., 1965, 20, 1051–1057 CrossRef CAS.
- J. P. Perdew, M. Levy, G. S. Painter, S. Wei and J. B. Lagowski, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 838–843 CrossRef CAS PubMed.
- H. Lee, C. Lee and R. G. Parr, Phys. Rev. A: At., Mol., Opt. Phys., 1991, 44, 768–771 Search PubMed.
- F. Tran and T. A. Wesołowski, Int. J. Quantum Chem., 2002, 89, 441–446 CrossRef CAS.
- A. J. Thakkar, Phys. Rev. A: At., Mol., Opt. Phys., 1992, 46, 6920–6924 Search PubMed.
- E. Chacón, J. E. Alvarellos and P. Tarazona, Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 32, 7868–7877 Search PubMed.
- L.-W. Wang and M. P. Teter, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13196–13220 CrossRef CAS PubMed.
- Y. A. Wang, N. Govind and E. A. Carter, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 13465–13471 CrossRef CAS.
- Y. A. Wang, N. Govind and E. A. Carter, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60, 16350–16358 CrossRef CAS.
- C. Huang and E. A. Carter, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 1–15 Search PubMed.
- L. A. Constantin, E. Fabiano and F. Della Sala, Phys. Rev. B, 2018, 97, 1–10 Search PubMed.
- W. Mi, A. Genova and M. Pavanello, J. Chem. Phys., 2018, 148, 184107 CrossRef PubMed.
- W. Mi and M. Pavanello, Phys. Rev. B, 2019, 100, 1–6 Search PubMed.
- J. Xia, C. Huang, I. Shin and E. A. Carter, J. Chem. Phys., 2012, 136, 084102 Search PubMed.
- H. Takahashi, Int. J. Quantum Chem., 2022, 122, e26969 CrossRef CAS.
- H. Takahashi, J. Chem. Phys., 2023, 158, 014102 CrossRef CAS PubMed.
- H. Takahashi, J. Chem. Phys., 2023, 159, 124118 Search PubMed.
- H. Takahashi, Electron. Struct., 2025, 7, 025003 Search PubMed.
- H. Takahashi, J. Phys. B: At., Mol. Opt. Phys., 2018, 51, 055102 CrossRef.
- H. Takahashi, J. Phys. B: At., Mol. Opt. Phys., 2020, 53, 245101 CrossRef CAS.
- J. R. Chelikowsky, N. Troullier and Y. Saad, Phys. Rev. Lett., 1994, 72, 1240–1243 CrossRef CAS PubMed.
- J. R. Chelikowsky, N. Troullier, K. Wu and Y. Saad, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 11355–11364 Search PubMed.
- H. Takahashi, T. Hori, T. Wakabayashi and T. Nitta, Chem. Lett., 2000, 222–223 Search PubMed.
- H. Takahashi, T. Hori, T. Wakabayashi and T. Nitta, J. Phys. Chem. A, 2001, 105, 4351 Search PubMed.
- H. Takahashi, T. Hori, H. Hashimoto and T. Nitta, J. Comput. Chem., 2001, 22, 1252–1261 CrossRef CAS.
- H. Takahashi, N. Matubayasi, M. Nakahara and T. Nitta, J. Chem. Phys., 2004, 121, 3989–3999 Search PubMed.
- W. M. C. Foulkes and R. Haydock, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 39, 12520–12536 CrossRef PubMed.
- R. van Leeuwen and E. J. Baerends, Phys. Rev. A: At., Mol., Opt. Phys., 1994, 49, 2421–2431 Search PubMed.
- Q. Wu and W. Yang, J. Chem. Phys., 2003, 118, 2498–2509 CrossRef CAS.
- E. S. Kadantsev and M. J. Stott, Phys. Rev. A: At., Mol., Opt. Phys., 2004, 69, 012502 Search PubMed.
- B. Kanungo, P. M. Zimmerman and V. Gavini, Nat. Commun., 2019, 10, 4497 Search PubMed.
- Y. Shi and A. Wasserman, J. Phys. Chem. Lett., 2021, 12, 5308–5318 Search PubMed.
- H. Takahashi, J. Chem. Phys., 2024, 161, 104108 CrossRef CAS PubMed.
- J. Harris, Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 31, 1770–1779 CrossRef CAS PubMed.
- Y. Shigeta, H. Takahashi, S. Yamanaka, M. Mitani, H. Nagao and K. Yamaguchi, Int. J. Quantum Chem., 1998, 70, 659–669 Search PubMed.
- H. Takahashi, H. Hashimoto and T. Nitta, J. Chem. Phys., 2003, 119, 7964–7971 Search PubMed.
- T. Hori, H. Takahashi and T. Nitta, J. Chem. Phys., 2003, 119, 8492–8499 Search PubMed.
- H. Takahashi, S. Sakuraba and A. Morita, J. Chem. Inf. Model., 2020, 60, 1376–1389 Search PubMed.
- R. N. Barnett and U. Landman, J. Chem. Phys., 1993, 48, 2081–2097 Search PubMed.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 Search PubMed.
- A. J. C. Felipe, A. Bulat, T. Heaton-Burgess and W. Yang, J. Chem. Phys., 2007, 127, 174101 CrossRef PubMed.
- R. T. Sharp and G. K. Horton, Phys. Rev., 1953, 90, 317 Search PubMed.
- J. D. Talman and W. F. Shadwick, Phys. Rev. A: At., Mol., Opt. Phys., 1976, 14, 36–40 CrossRef CAS.
- T. A. Wesolowsk and A. Warshel, J. Phys. Chem., 1993, 97, 8050–8053 CrossRef.
- T. A. Wesolowski, S. Shedge and X. Zhou, Chem. Rev., 2015, 115, 5891–5928 CrossRef CAS PubMed.
|
| This journal is © the Owner Societies 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence






 . σ in eqn (6) represents the index for spin (σ = α, β). The first OF-DFT functional, referred to as the TF model, is then given by
. σ in eqn (6) represents the index for spin (σ = α, β). The first OF-DFT functional, referred to as the TF model, is then given by


 . Unfortunately, it was proved that neither the TF nor the TFD model realizes the formation of chemical bonds in principle.12,13 The first primitive approach to resolve this issue was to add the von Weizsäcker (vW) correction14 to eqn (6). The vW correction term EvW[n] is given by
. Unfortunately, it was proved that neither the TF nor the TFD model realizes the formation of chemical bonds in principle.12,13 The first primitive approach to resolve this issue was to add the von Weizsäcker (vW) correction14 to eqn (6). The vW correction term EvW[n] is given by



 are completely parallel to those of the potential υdef(r), then it is possible to identify the potential
are completely parallel to those of the potential υdef(r), then it is possible to identify the potential  using the potential
using the potential  defined on the one-dimensional coordinate ε, thus,
defined on the one-dimensional coordinate ε, thus,
 , leading to the condition at the stationary state with the ground state density n0,
, leading to the condition at the stationary state with the ground state density n0,












 for the system identified by eqn (30). χfull0(r, r′) is then projected onto the energy coordinate to yield χe,full0(ε, ε′) given by
for the system identified by eqn (30). χfull0(r, r′) is then projected onto the energy coordinate to yield χe,full0(ε, ε′) given by

 with υXeff being the KS effective potential of the isolated subsystem X.
with υXeff being the KS effective potential of the isolated subsystem X.


















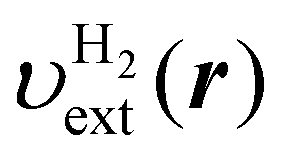 for the pseudo H2 molecule. The potential of each hydrogen atom is given in eqn (42). The value of
for the pseudo H2 molecule. The potential of each hydrogen atom is given in eqn (42). The value of  is plotted. (b) Polarization density δn(r) defined by the KS-DFT density nKS(r) subtracted by the reference density n0(r) of eqn (29). Unit of the density is a.u.−3. The white dots represent the positions of the hydrogen atoms.
is plotted. (b) Polarization density δn(r) defined by the KS-DFT density nKS(r) subtracted by the reference density n0(r) of eqn (29). Unit of the density is a.u.−3. The white dots represent the positions of the hydrogen atoms. varies significantly within this region as in Fig. 3(a). A possible way to avoid the undesirable behavior in the polarization δn(r) is to construct n0, for example, by means of the frozen density embedding (FDE) method.6,59,60 In the FDE method, the electron population in the region between the hydrogen atoms will be increased, because the density of each subsystem is to be optimized under the influence of the surrounding environment. Thus, the polarization density δ
varies significantly within this region as in Fig. 3(a). A possible way to avoid the undesirable behavior in the polarization δn(r) is to construct n0, for example, by means of the frozen density embedding (FDE) method.6,59,60 In the FDE method, the electron population in the region between the hydrogen atoms will be increased, because the density of each subsystem is to be optimized under the influence of the surrounding environment. Thus, the polarization density δ




