
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotocatalytic synthesis of imidazo[1,2-a]pyridines via C(sp)3–H functionalization using ethylarene as a sustainable surrogate of acetophenone and luminescence studies
Priya
Mahaur
,
Harshita
Pandey
,
Vandana
Srivastava
 * and
Sundaram
Singh
*
* and
Sundaram
Singh
*
Indian Institute of Technology BHU, Department of Chemistry, Varanasi, 221005, Uttar Pradesh, India. E-mail: sundaram.apc@itbhu.ac.in
First published on 6th October 2025
Abstract
A metal-free method for synthesizing imidazopyridines utilizing ethylarene, 2-aminopyridine, and NBS under visible light using Eosin-Y as a photocatalyst has been developed. Eosin-Y activates and oxidizes the C–H bonds of ethylarene, followed by bromination and coupling with aminopyridine. This technique effectively transforms ethylbenzene and aminopyridine into desirable products, and it also employs catalytic amounts of the halogenating agent, which is continuously regenerated. This approach is suitable for biologically active compounds with luminescence properties due to its high atom efficiency, metal-free nature, environmental friendliness, and use of visible light as a renewable energy source.
Introduction
Imidazo[1,2-a]pyridines have been very popular building blocks in synthetic organic chemistry over the last 20 years and have attracted much attention. This is because of their numerous biological effects, which include antituberculosis,1 antipyretic,2 antiepileptic,3 antiulcer,4 anticonvulsant,2 antifungal,5 antiviral,6 anticancer,7 antiprotozoal,8 antibacterial,9 anti-inflammatory,2 anthelmintic,10 and antitumor properties.11 A wide range of medications, including zolpidem (used to treat insomnia), zolimidine (for peptic ulcer treatment), olprinone (for acute heart failure), alpidem, necopidem and saripidem (anxiolytic agents), and miroprofen (a prostanoid signaling modulator, analgesic, and NSAID), share the core structure of imidazo[1,2-a]pyridines (Fig. 1).12,13 | ||
| Fig. 1 Some biologically active compounds containing the imidazo[1,2-a]pyridine framework. | ||
Due to its involvement in the synthesis of numerous natural products, imidazo[1,2-a]pyridine is recognized as a drug prejudice scaffold.14 A significant amount of work has been invested in the creation of imidazo[1,2-a]pyridine and its derivatives. Since the imidazole moiety is fused with the pyridine ring and increases biological activity because of a nitrogen-containing heterocycle, these molecules are regarded as keystone heterocycles.15 They are incredibly useful in the fields of pharmaceuticals and natural products.16 Because of its wide spectrum of pharmacological characteristics, scientists have focused a lot of effort on developing mild yet effective synthesis methods for the imidazo[1,2-a]pyridine scaffold. Several imidazo[1,2-a]pyridine synthesis methods have thus emerged in the last several years.16
Various techniques have been established to date for the synthesis of imidazole rings. The conventional method often entails cyclizing and directly condensing α-haloacetophenone with 2-aminopyridine (Scheme 1a).3 Acetophenone can also be used as the starting material. The carbonyl group is first ortho-halogenated, and then it is condensed and cyclized with 2-aminopyridine (Scheme 1b).17 New synthetic techniques have been developed in recent years. An oxidant and a halogenating reagent, for instance, can be added to ethylbenzene18 and styrene19 to generate α-haloacetophenone (Schemes 1c and d). Furthermore, techniques for activating C–H bonds in phenylacetylene, catalyzed by transition metals, have been documented (Scheme 1e).20–22 In the previous literature, α-bromoacetophenone has been utilized more frequently, and its reactions are more developed.3 However, as an eye irritant that causes tears, α-bromoacetophenone can seriously affect both the environment and the scientific community. α-Bromoacetophenone has also been documented by Shimokawa et al. as a significant intermediate in the reaction involving ethylarenes with N-bromosuccinimide (NBS).23 Motivated by this research, we thought about possibilities to synthesize a range of imidazoles from ethylarenes. Various metal catalysts have been used previously to synthesize imidazo[1,2-a]pyridines under conventional conditions, which also affect the environment.
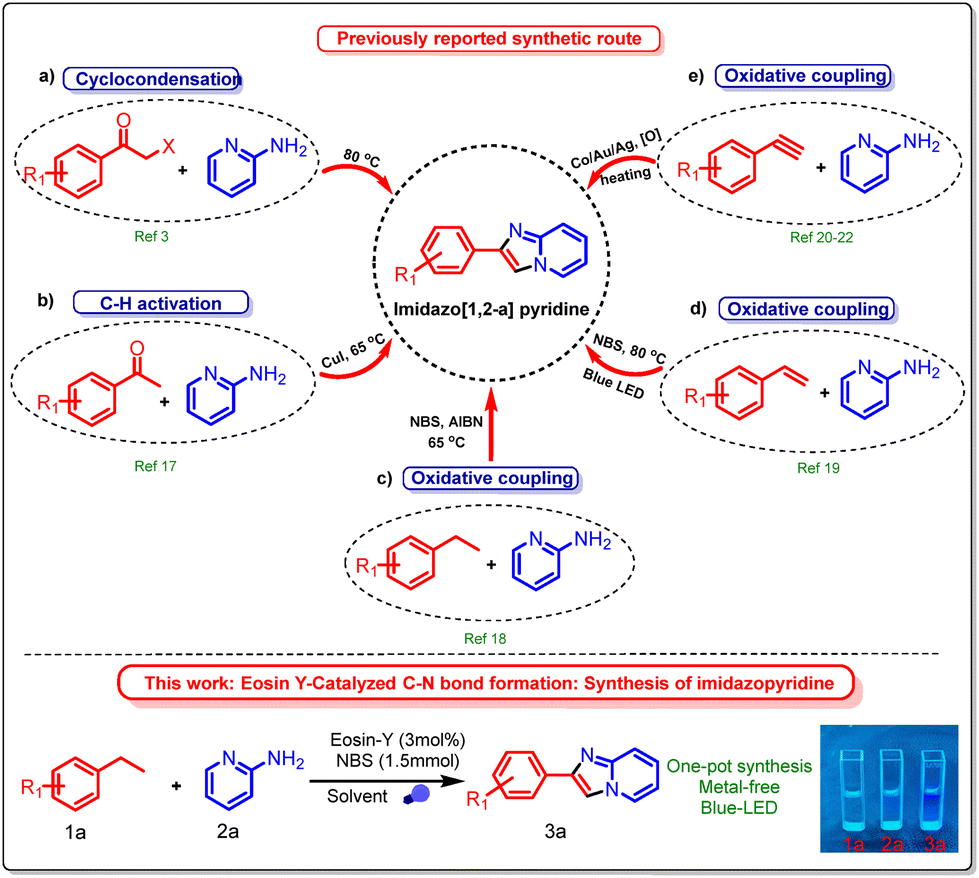 | ||
| Scheme 1 Approach for the synthesis of imidazo[1,2-a]pyridines. | ||
C(sp3)–H functionalization is a unique and efficient approach for converting simple substrates into useful compounds. Catalytic C–H functionalization has significantly expanded in scope and transformed substrates into useful compounds in recent decades. Various catalytic approaches, such as transition metal, enzymatic, photochemical, electrochemical, and photoredox systems, have been utilized to selectively functionalize C–H bonds and create useful compounds.24,25 The field of photoredox catalysis is rapidly expanding, with applications in new reaction development, renewable energy, chemical feedstocks, natural product synthesis, and biology.26,27 Visible light and photoredox catalysis are increasingly being used for organic synthesis due to their environmental friendliness and potency.28 Eosin-Y is an organic dye that promotes photoredox catalysis in organic reactions. It has gained popularity due to its ease of use, environmental friendliness, and potential for use in visible-light-mediated organic transformations.25
There is a need to develop effective methods for preparing imidazo[1,2-a]pyridines using visible light and Eosin-Y as a photocatalyst. Photocatalytic synthesis of imidazo[1,2-a]pyridines from ethylbenzene and 2-aminopyridine has not been previously described. N-Bromosuccinimide (NBS) is widely used in organic synthesis, particularly in green chemistry, due to its efficiency and environmental friendliness. NBS functions as a selective brominating agent, enabling controlled and moderate halogenation reactions with minimal byproducts, in accordance with the waste reduction principles of green chemistry. However, the direct functionalization of ethylbenzene as a raw material is uncommon. Here, we present the synthesis of imidazopyridines from ethylarenes and 2-aminopyridine, highlighting their potential applicability in pharmaceuticals and natural products. Additionally, the synthesized compounds exhibit promising photophysical properties, making them useful in optoelectronic materials, fluorescent probes, biological imaging, and excellent quantum yields.29,30 The quantum yields of the synthesized products 3a, 3e, 3k, and 3s were 82.8%, 79.3%, 105.4%, and 121.8%, respectively, using quinine sulfate in 0.5 M H2SO4 solution (Table S4 in the SI).
Results and discussion
In the preliminary investigation, ethylbenzene (1a) was utilized as a model substrate and treated with NBS in the presence of photoredox catalysts in the solvent. Subsequently, 2-aminopyridine (2a) was introduced to produce the desired product. The SI (see page S4) provides a detailed account of the optimization studies. We were pleased to achieve a 45% product yield at the start of our optimization efforts, employing ethylbenzene (1 equiv.), NBS, and 2-aminopyridine (1 equiv.), Eosin-Y as the photocatalyst, and ethanol as the solvent. The reaction was performed in ambient air at room temperature for 12 hours, which served as our model conditions (Table 1, entry 1). The expected product yield was significantly reduced when Eosin-Y was substituted with other photocatalysts, such as rhodamine B, Rose Bengal, and Acridine Red, indicating that Eosin-Y is more efficient under optimal conditions (Table 1, entry 2).|
|
||
|---|---|---|
| Entry | Deviation from the standard reaction | Yieldb (%) |
| a Reaction conditions: ethylbenzene (1 mmol), NBS (1.5 mmol), 2-aminopyridine (1 mmol), Eosin-Y (2 mol%), and EtOH (10 mL) under a blue LED (12 h) in open air at room temperature. b Isolated yield. c Different reaction times (2–12 h). | ||
| 1 | Nonea | 45 |
| 2 | With different photocatalysts | 12–18 |
| 3 | Use of MeOH rather than EtOH | 35 |
| 4 | Use of H2O rather than EtOH | 25 |
| 5 | Use of DMSO rather than EtOH | 47 |
| 6 | Use of DMF rather than EtOH | 42 |
| 7 | Use of DCM rather than EtOH | 40 |
| 8 | Use of MeCN rather than EtOH | 58 |
| 9 | Use of nonpolar solvents rather than EtOH | 15 |
| 10 | 1, 2, 3, and 5 mol% of Eosin-Yc | 45–85 |
| 11 | In the dark | n.r. |
| 12 | Different LEDs instead of a blue LED | 48–60 |
| 13 | Without the PC | n.r. |

Furthermore, various solvents were screened in the place of ethanol (Table 1, entries 3–9) with Eosin-Y (2 mol%) as the optimal catalyst. The results indicated that acetonitrile was the most effective solvent, providing the highest yield (Table 1, entry 8). The Eosin-Y loading and reaction time were varied in order to conduct the optimization study (Table 1, entry 10). When 2 mol% Eosin-Y was utilized, the yield was 58% after 12 hours (see Table S2, entry 1). Under the same conditions, decreasing the catalyst loading to 1 mol% reduced the yield to 45% (see Table S2, entry 2). Increasing the catalyst loading to 3 mol% increased the yield to 85% (see Table S2, entry 3), while increasing it to 5 mol% lowered the yield to 75% (see Table S2, entry 4). The influence of reaction time was then investigated at the optimum catalyst loading (3 mol%). The yield remained uniform at 85% even when the time was shortened from 12 hours to 8, 6, 4, and 2 hours (see Table S2, entries 5–8). This shows that the reaction occurs effectively with 3 mol% Eosin-Y and that just 2 hours are required to produce the maximal yield of 85% (see Table S2, entry 8). Without light, no reaction (n.r.) was observed, which verifies that the reaction is dependent on light and is driven by photochemistry (Table 1, entry 11). The yield of the desired product was reduced when the reaction was carried out under LEDs of different colours other than blue (Table 1, entry 12). Next, we explored our reaction in the absence of a photocatalyst but did not obtain our desired product (Table 1, entry 13).
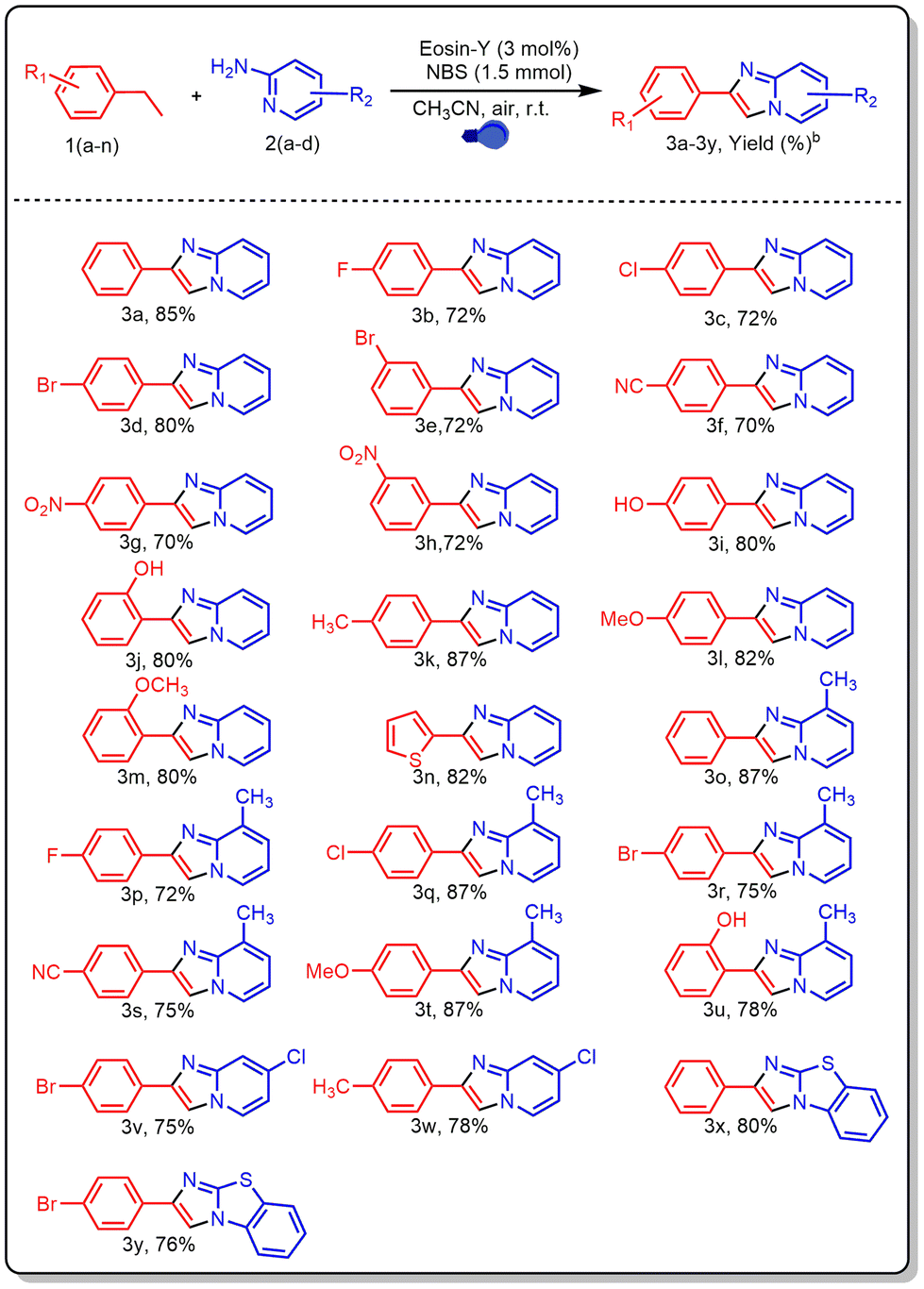
After optimizing the reaction conditions, a variety of ethylarenes and 2-aminopyridines were tested to determine the method's versatility and suitability for synthesising various imidazo[1,2-a]pyridines (Table 2). Ethylarenes containing electron-withdrawing substituents on the aromatic ring, such as halogens, nitro, and cyano groups, reacted efficiently with 2-aminopyridine (2a), affording products (3b–3h) in moderate to good yields (70–80%). Similarly, ethylarenes with electron-donating groups like hydroxy, methyl, and methoxy provided high yields of imidazo[1,2-a]pyridines (3i–3m), ranging from 80% to 87%. A heteroarene subjected to the same reaction with 2-aminopyridine also produced product 3n in a high yield of 82%. The method showed good tolerance for both electron-rich and electron-deficient substituents on 2-aminopyridine. Electron-rich groups such as methyl led to excellent yields (72–87%, 3o–3u), whereas electron-withdrawing groups like halogens gave slightly lower yields (75% and 78%, 3v and 3w). Additionally, using ethylarenes with 2-aminobenzothiazole under the same conditions afforded products 3x and 3y in 80% and 76% yields, respectively.
We further investigated the feasibility of this methodology on a multigram scale (Scheme 2). Under the optimized reaction conditions, a 10 mmol scale reaction was performed in a 50 mL round-bottom flask equipped with a magnetic stir bar. Ethylbenzene (10 mmol, 1220 μL, 1.0 equiv.), N-bromosuccinimide (15 mmol, 2.67 g, 1.5 equiv.), 2-aminopyridine (10 mmol, 0.94 g, 1.0 equiv.), and Eosin-Y (3 mol%) were dissolved in 25 mL of acetonitrile and irradiated with blue LED light under open-air conditions for 2 h, with progress monitored by TLC. Upon completion, the reaction mixture was extracted with ethyl acetate (3 × 25 mL), and the combined organic layers were dried over anhydrous Na2SO4. Removal of the solvent under reduced pressure and purification by silica gel column chromatography (10% ethyl acetate in n-hexane) furnished the desired product in 80% yield (1.55 g).
 | ||
| Scheme 2 Gram scale synthesis of 2-phenylimidazo[1,2-a]pyridine (3a). | ||
Next, we shifted our focus to a mechanistic study for proposing a reaction mechanism and carried out several control experiments (Scheme 3a–e). To determine the reaction pathway, an experiment was conducted using the radical scavenger 2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO) (Scheme 3a). The results showed a significant suppression in the yield of product 3a (12%) and the formation of new adducts 4a and 5a, which was confirmed by high-resolution mass spectrometry (HRMS) data, indicating the generation of a free radical and supporting the involvement of a free radical mechanism in the reaction. Additionally, to evaluate the role of O2 (oxygen), the reaction was conducted under both oxygen and nitrogen atmospheres (Scheme 3b). When the reaction was performed under nitrogen (Scheme 3b), the obtained yield of the product was 0%, whereas a significantly higher yield (85%) of the desired product was achieved in the presence of oxygen. These results suggest that oxygen is essential for this transformation. The absence of a catalyst resulted in no product formation, highlighting the crucial role of the photocatalyst in the reaction (Scheme 3b). To further understand the reaction pathway, analysis under standard conditions was performed, resulting in the detection of intermediates H (α-bromoacetophenone, 6a) and E (acetophenone, 7a) (Scheme 3c and d). The structures of these intermediates were confirmed through 1H and 13C NMR spectroscopy. The presence of H2O2 was qualitatively verified using the potassium iodide (KI) test. In this assay, H2O2 oxidizes iodide ions (I−) to molecular iodine (I2), which imparts a characteristic brown colouration when a few drops of KI solution are added to the reaction mixture (Scheme 3e).
 | ||
| Scheme 3 Control experiments. | ||
Next, the UV-Visible spectra were recorded for the (0.8 mM) concentration of individual reactants (1a, 2a, and EY) as well as for the reaction mixtures containing 1a, 2a, NBS, and EY in ethanol solvent (Fig. 2a). The UV-Vis analysis indicated that both the reactants, Eosin-Y and the reaction mixture, exhibited absorption in the blue region of the spectrum (detailed information in the SI, page no. S6–S8). Furthermore, Stern–Volmer quenching experiments revealed efficient quenching of the photoexcited state of Eosin-Y by compounds 1a and 2a (Fig. 2b and c). The extent of quenching increased proportionally with the concentrations of 1a and 2a, as shown in Fig. 2(d and e) (detailed information in the SI, page no. S8–S10). The Stern–Volmer graph is linear at high concentrations, as shown in Fig. 2(d and e), indicating a dynamic quenching mechanism. Additionally, the light–dark cycling experiment revealed that continuous light irradiation is essential for the reaction to proceed (Fig. 2f).
 | ||
| Fig. 2 (a) UV-vis absorption spectra of the reactants and the reaction mixture. (b and c) Fluorescence emission spectra of EY with different concentrations of quenchers 1a and 2a. (d and e) Stern–Volmer fluorescence quenching plot of EY with different concentrations of quenchers 1a and 2a. (f) Light–dark cycle experiment. | ||
In light of prior reports18,25,31,32 and control experiment findings, a potential pathway for the overall reaction mechanism was proposed (Scheme 4). Upon irradiation with blue light, Eosin-Y (EY) is promoted to its excited state (EY*). This potent photooxidant initiates a single electron transfer (SET) with ethylbenzene (1a), yielding the radical cation intermediate A. The resulting reduced form of EY transfers an electron to molecular oxygen, regenerating the ground-state catalyst and producing a superoxide anion. The radical cationic intermediate A undergoes deprotonation to generate the benzylic radical B. This reactive species rapidly reacts with molecular oxygen to form the peroxyl intermediate C. A subsequent hydrogen atom abstraction yields benzyl hydroperoxide D, which undergoes dehydration to furnish the key intermediate E. Next, intermediate E reacts with NBS, resulting in α-bromoacetophenone (intermediate H) and succinimide by selective bromination. Furthermore, reactant 2a under blue light irradiation gives a radical cation intermediate (2a′) (formed by the SET of EY), which then couples with intermediate I to form intermediate J, in which a new C–N bond is formed. After deprotonation, intermediate J is converted into K, which is then dehydrated to produce intermediate L. From intermediate L, the adjacent nitrogen undergoes intramolecular nucleophilic attack on the benzylic carbon, resulting in the displacement of the bromide ion and the formation of the cyclized intermediate. The last deprotonation of intermediate M restores aromaticity, resulting in the imidazo[1,2-a]pyridine derivative (3a) as the end product.
 | ||
| Scheme 4 Proposed mechanism. | ||
The UV-Vis absorbance spectra of various imidazo[1,2-a] pyridine derivatives were recorded in ethanol (EtOH) as the solvent. For example, the UV-vis spectra of compounds 3a, 3e, 3k, and 3s were recorded at a concentration of ∼0.6 μM and are shown in Fig. 3, displaying maximum absorption bands with peak absorbance at 300, 312.5, 322.5, and 263 nm, respectively. These wavelength (red/blue) shifts may be attributed to enhanced electronic delocalization facilitated by the presence of functionalized phenacyl substituents attached to the imidazo[1,2-a]pyridine core.
 | ||
| Fig. 3 UV-Vis absorbance spectra of 3a, 3e, 3k, and 3s in EtOH. | ||
The photoluminescence properties of the imidazo[1,2-a] pyridine derivatives were examined under UV light excitation in the 254–365 nm range at the same concentrations used in UV-vis experiments, as shown in the inset images of the samples in Fig. 4(a–d). As illustrated in Fig. 4(a–d), all the synthesized derivatives exhibited pronounced emission in the blue to violet spectral regions when dispersed in EtOH solvent. Maximum emission wavelengths were observed at 346, 370, 372, and 412 nm for compounds 3a, 3e, 3k, and 3s, respectively, as shown in Fig. 4(a–d).
 | ||
| Fig. 4 Fluorescence emission spectra of different imidazo[1,2-a]pyridine derivatives 3a, 3e, 3k, and 3s. The inset shows photographs of these derivatives in EtOH solvent under visible and 365 nm UV irradiation. | ||
The quantum yields of the samples were determined using a single-point method by comparison with quinine sulfate, which possesses a quantum yield of approximately 54% at 350 nm. The following equation was employed for the calculation:

Quinine sulfate in a 0.5 M H2SO4 solution was used as the reference standard, and the quantum yields of the synthesized imidazo[1,2-a] pyridine derivatives 3a, 3e, 3k, and 3s were determined to be 82.8%, 79.3%, 105.4%, and 121.8%, respectively (as shown in Table S4 of the SI, page no. S10). The quantum yield can sometimes exceed 100%, possibly due to multiple exciton generation, where a single absorbed photon generates more than one electron–hole pair, resulting in an effective quantum yield greater than 100%.33
Conclusions
In conclusion, we have synthesized imidazopyridines from ethylbenzene, Eosin-Y, NBS, and 2-aminopyridine under blue LED irradiation in acetonitrile (CH3CN), demonstrating a successful and efficient photoredox approach for constructing nitrogen-containing heterocycles. The reaction relies critically on visible-light activation, as shown by light-on/off experiments, where product formation occurs only during illuminated periods. Eosin-Y acts as a metal-free photoredox catalyst, initiating benzylic radical formation via NBS-mediated bromination of ethylbenzene, followed by nucleophilic cyclization with 2-aminopyridine to afford the imidazopyridine scaffold.Notably, several imidazopyridine derivatives obtained through this protocol exhibit promising photophysical properties, such as fluorescence, making them attractive candidates for applications in optoelectronic materials, fluorescent probes, and biological imaging. The mild conditions, metal-free catalysis, and potential for functional material development highlight the synthetic and applicative value of this light-driven methodology.
Author contributions
All authors have approved the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Data availability
Data supporting this study with 1H, 13C, HRMS and 1H-1H COSY of (3h), etc, are included in the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5ob01406a.Acknowledgements
Priya Mahaur acknowledges the University Grants Commission (UGC) for a Senior Research Fellowship (SRF). The authors are thankful to the Central Instrumental Facility Center (CIFC), IIT (BHU), for NMR.References
- M. W. Majewski, R. Tiwari, P. A. Miller, S. Cho, S. G. Franzblau and M. J. Miller, Bioorg. Med. Chem. Lett., 2016, 26, 2068–2071 CrossRef CAS PubMed.
- L. Almirante, L. Polo, A. Mugnaini, E. Provinciali, P. Rugarli, A. Biancotti, A. Gamba and W. Murmann, Derivatives of Imidazole. I. Synthesis and Reactions of Imidazo[1,2-α]pyridines with Analgesic, Antiinflammatory, Antipyretic, and Anticonvulsant Activity, J. Med. Chem., 1965, 8(3), 305–312 CrossRef CAS PubMed.
- S. Ulloora, A. V. Adhikari and R. Shabaraya, Chin. Chem. Lett., 2013, 24, 853–856 CrossRef CAS.
- J. E. Starrett, T. A. Montzka, A. R. Crosswell and R. L. Cavanagh, J. Med. Chem., 1989, 32, 2204–2210 CrossRef CAS PubMed.
- Z. A. Kaplancikli, G. Turan-Zitouni, A. Özdemr and G. Revial, J. Enzyme Inhib. Med. Chem., 2008, 23, 866–870 CrossRef CAS PubMed.
- K. S. Gudmundsson and B. A. Johns, Bioorg. Med. Chem. Lett., 2007, 17, 2735–2739 CrossRef CAS PubMed.
- J. Wang, W. Liu, G. Luo, Z. Li, C. Zhao, H. Zhang, M. Zhu, Q. Xu, X. Wang, C. Zhao, Y. Qu, Z. Yang, T. Yao, Y. Li, Y. Lin, Y. Wu and Y. Li, Energy Environ. Sci., 2018, 11, 3375–3379 RSC.
- T. Biftu, D. Feng, M. Fisher, G.-B. Liang, X. Qian, A. Scribner, R. Dennis, S. Lee, P. A. Liberator, C. Brown, A. Gurnett, P. S. Leavitt, D. Thompson, J. Mathew, A. Misura, S. Samaras, T. Tamas, J. F. Sina, K. A. McNulty, C. G. McKnight, D. M. Schmatz and M. Wyvratt, Bioorg. Med. Chem. Lett., 2006, 16, 2479–2483 CrossRef CAS PubMed.
- N. S. Rao, C. Kistareddy, B. Balram and B. Ram, Pharma Chem., 2012, 4, 2408–2415 CAS.
- S. Zhou, G. Chen and G. Huang, Chem. Biol. Drug Des., 2019, 93, 503–510 CrossRef CAS PubMed.
- M. Hranjec, M. Kralj, I. Piantanida, M. Sedić, L. Šuman, K. Pavelić and G. Karminski-Zamola, J. Med. Chem., 2007, 50, 5696–5711 CrossRef CAS PubMed.
- Y. Wang, S. Li, X. Wang, Y. Yao, L. Feng and C. Ma, RSC Adv., 2022, 12, 5919–5927 RSC.
- H. Sanaeishoar, R. Nazarpour and F. Mohave, RSC Adv., 2015, 5, 68571–68578 RSC.
- R. Rawat and S. M. Verma, Synth. Commun., 2020, 50, 3507–3534 CrossRef CAS.
- S. M. Roopan, S. M. Patil and J. Palaniraja, Res. Chem. Intermed., 2016, 42, 2749–2790 CrossRef.
- A. Altaher, M. Adris and S. Aliwaini, Syst. Rev. Pharm., 2021, 12, 79–86 CAS.
- D. C. Mohan, S. N. Rao, C. Ravi and S. Adimurthy, Asian J. Org. Chem., 2014, 3, 609–613 CrossRef CAS.
- L. Chen, H. Zhu, J. Wang and H. Liu, Molecules, 2019, 24, 893 CrossRef PubMed.
- M. H. Shinde and U. A. Kshirsagar, Green Chem., 2016, 18, 1455–1458 RSC.
- Sk. Rasheed, D. N. Rao and P. Das, Asian J. Org. Chem., 2016, 5, 1213–1218 CrossRef.
- E. P. A. Talbot, M. Richardson, J. M. McKenna and F. D. Toste, Adv. Synth. Catal., 2014, 356, 687–691 CrossRef CAS PubMed.
- C. He, J. Hao, H. Xu, Y. Mo, H. Liu, J. Han and A. Lei, Chem. Commun., 2012, 48, 11073–11075 RSC.
- S. Shimokawa, Y. Kawagoe, K. Moriyama and H. Togo, Org. Lett., 2016, 18, 784–787 CrossRef CAS PubMed.
- J.-Y. Chen, W. Wu, Q. Li and W.-T. Wei, Adv. Synth. Catal., 2020, 362, 2770–2777 CrossRef CAS.
- A. K. Kushwaha, S. K. Maury, A. Kamal, H. K. Singh, S. Pandey and S. Singh, Chem. Commun., 2023, 59, 4075–4078 RSC.
- S. D. Tambe, R. S. Rohokale and U. A. Kshirsagar, Eur. J. Org. Chem., 2018, 2018, 2117–2121 CrossRef CAS.
- M. Bu, G. Lu, J. Jiang and C. Cai, Catal. Sci. Technol., 2018, 8, 3728–3732 RSC.
- X. Zhao, B. Li and W. Xia, Org. Lett., 2020, 22, 1056–1061 CrossRef CAS PubMed.
- J. C. Rodríguez, R. A. Maldonado, G. Ramírez-García, E. D. Cervantes and F. N. De La Cruz, J. Heterocycl. Chem., 2020, 57, 2279–2287 CrossRef.
- Y. Rani, M. P. Km and P. Tripathi, ACS Appl. Mater. Interfaces, 2024, 16, 68936–68949 CrossRef CAS PubMed.
- A. K. Kushwaha, A. Kamal, H. K. Singh, S. K. Maury, T. Mondal and S. Singh, Org. Lett., 2024, 26, 1416–1420 CrossRef CAS PubMed.
- S. Pandey, A. Singh, A. K. Kushwaha and S. Singh, J. Org. Chem., 2024, 89, 12576–12582 CrossRef CAS PubMed.
- O. E. Semonin, J. M. Luther, S. Choi, H.-Y. Chen, J. Gao, A. J. Nozik and M. C. Beard, Science, 2011, 334, 1530–1533 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |