
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Computational exploration of M-sites with chemical order and disorder in M′2M′′B2 and M′4M′′B3 compounds†
Adam
Carlsson
,
Rodrigo Mantovani
Ronchi
,
Johanna
Rosen
and
Martin
Dahlqvist
 *
*
Materials Design, Department of Physics, Chemistry and Biology (IFM), Linköping University, SE-581 83 Linköping, Sweden. E-mail: martin.dahlqvist@liu.se
First published on 9th December 2024
Abstract
Boron-based materials are highly desirable for their promising mechanical properties, rendering them ideal for various industrial applications. In this study, we take advantage of the two unique metal Wyckoff sites in the prototype structures V3B2 (4h and 2a) and Cr5B3 (16h and 4c). These two sites were populated by two different metals, M′ and M′′, forming novel B-based ternary M′2M′′B2 and M′4M′′B3 compounds. A high-throughput phase stability search encompassing a total of 2738 compositions with M′ and M′′ being occupied systematically with Al, Si, Sc, Ti, V, Cr, Mn, Fe, Co, Ni, Cu, Zn, Ga, Ge, Y, Zr, Nb, Mo, Tc, Ru, Rh, Pd, Ag, Cd, In, Sn, Hf, Ta, W, Re, Os, Ir, Pt, Au, Hf, Tl, and Pb was performed. The thermodynamic phase stability of the simulated materials was assessed by evaluating the formation enthalpy, ΔHcp for all compostions with chemical order and disorder for selective compositions. In total, 56 ternary phases were identified as stable when considering a configurational entropy contribution at 2000 K. Out of the 56 predicted stable phases, 32 displayed a preference for chemical order whereas 24 favored disorder of M′ and M′′. Initial attempts at synthesis of the predicted stable Ta2MoB2 compound were made, showing indications for the presence of ordered Ta2MoB2 or disordered (Ta0.667Mo0.333)3B2.
Introduction
High-throughput (HT) computational searches of materials are centered around the concepts of constructing extensive databases of materials with calculated electronic properties and then extrapolating the results based on certain criteria, i.e., predicted synthesizability,1 or the hardness of the materials.2,3 Notable examples of publicly accessible databases of materials, such as the Materials Project,4 OQMD,5,6 and AFLOWLIB,7 all encompass a diverse range of both thermodynamic and electronic properties.8,9 In practice, the construction of a materials database typically follows one of two distinct approaches. The database is formed either by focusing on a single materials system, through methodologies such as crystal structure prediction frameworks,10–13 or by screening already known low-energy structures with the hope of discovering additional novel compounds of similar structure.14–17We herein consider the latter approach by focusing on two identified boron-based binary structures with two unique metal Wyckoff sites, namely V3B2 (P4/mbm),18 originating from U3Si2 discovered by Zachariasen in 1949,19 and Cr5B3 (I4/mcm).20 The V3B2 and Cr5B3 structures were selected because of the experimentally demonstrated order of Cr and W in W2CrB2 (V3B2) and W4CrB3 (Cr5B3),21 and serves as the foundation for discovering additional ternary boron-based materials in this work. In addition, a multitude of materials with similar structures have previously been studied in which discussions regarding ordered and disordered lattice configurations vary.22–28
Both V3B2 and Cr5B3 prototype structures consist of two Wyckoff sites occupied by transition metals, herein denoted as M, with a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 4:1 ratio, respectively. The remaining sites are occupied by boron, denoted as B. To distinguish between the metal sites, we use the M′2M′′B2 and M′4M′′B3 notation to describe their compositions and the M′:M′′ ratio. The implicated metal order in M′2M′′B2 and M′4M′′B3 herein serve as potential alloying structures in the quest for novel synthesizable boron-based materials. Multi-component boron-based materials have recently gained an extensive amount of interest due to the possibility of tuning the materials’ properties by combining atomic species of desired nature to achieve materials with exotic properties suitable for industrial applications.29–32 The increased number of components does, however, increase the complexity of the material, which often leads to more challenging synthesis procedures. This is where theoretical studies may be used to pave the way toward the identification of stable and synthesizable materials to guide experimental procedures.
:1 and 4:1 ratio, respectively. The remaining sites are occupied by boron, denoted as B. To distinguish between the metal sites, we use the M′2M′′B2 and M′4M′′B3 notation to describe their compositions and the M′:M′′ ratio. The implicated metal order in M′2M′′B2 and M′4M′′B3 herein serve as potential alloying structures in the quest for novel synthesizable boron-based materials. Multi-component boron-based materials have recently gained an extensive amount of interest due to the possibility of tuning the materials’ properties by combining atomic species of desired nature to achieve materials with exotic properties suitable for industrial applications.29–32 The increased number of components does, however, increase the complexity of the material, which often leads to more challenging synthesis procedures. This is where theoretical studies may be used to pave the way toward the identification of stable and synthesizable materials to guide experimental procedures.
The HT phase stability screening consisted of evaluating the formation enthalpy, ΔHcp, for 2664 ternary and 74 binary phases for the two prototype structures, M′2M′′B2 and M′4M′′B3, by systematically alloying the M′ and M′′ sites with Al, Si, Sc, Ti, V, Cr, Mn, Fe, Co, Ni, Cu, Zn, Ga, Ge, Y, Zr, Nb, Mo, Tc, Ru, Rh, Pd, Ag, Cd, In, Sn, Hf, Ta, W, Re, Os, Ir, Pt, Au, Hf, Tl and Pb. The screening considered both ordered and disordered arrangement of M′ and M′′ in which simulated solid solutions were modeled on the M-sites when ΔHcp was in a region chosen to represent a metastable compound based on the configurational entropic contribution.
Out of 41 M′2M′′B2 compositions predicted to be stable, 23 favored a chemically ordered arrangement on the M-sites and 18 a disordered counterpart. The M′4M′′B3 compounds, with an extra MB layer, yielded 9 stable materials favoring ordered M-sites and 6 with a disordered distribution of M′ and M′′ on the M-sites. For one of the predicted stable materials, Ta2MoB2, initial synthesis attempts were made. Analysis of the XRD diffractogram indicated synthesis of Ta2MoB2. These results are further corroborated by a bonding analysis.
Theoretical and experimental methods
Density functional theory
The structural optimization procedure was performed using the density functional theory (DFT) framework. The Vienna Ab Initio Simulation Package (VASP), version 5.4.1,33–35 was used where the exchange–correlation was modeled using the generalized gradient approximation (GGA)36 with the all-electron projector augmented wave (PAW)37,38 method. The cutoff energy was set to 520 eV, a Γ-centered 2π × 0.027 Å−1k-point density was used, and with an energy and force convergence criteria of 10−7 eV and −10−2 eV Å−2, respectively. These settings to ensure compatability with the Materials Project database.4 Additional competing phases considered from ref. 14 and 39 were all reoptimized using the above VASP settings. A ferromagnetic spin configuration was considered for every material where the initial magnetic moment was set to 3 for materials with M sites populated by Cr, Mn, Fe, Co, or Ni while remaining materials had an initial spin of 0.5.Phonon dispersion spectra were calculated for M′2M′′B2 and M′4M′′B3 through 2 × 2 × 3 (120 atoms) and 2 × 2 × 2 (128 atoms) supercells, respectively, by using the finite displacement method as implemented in Phonopy.40 Furthermore, the LOBSTER code was used to retrieve the density of states (DOS) and crystal orbital Hamiltonian population (COHP) where the calculated band-structure energy is reconstructed into orbital interactions.41–44 Positive COHP values indicate antibonding interactions while negative COHP values indicate bonding interactions. All crystal structures illustrated herein were generated using VESTA.45
Phase stability predictions
The phase stability predictions was evaluated using the formation enthalpy, which compares the energy of an arbitrary M′-M′′-B phase, denoted M′xM′′yBz, with all possible linear combinations of competing phases within the M′-M′′-B material system. The set of most competing phases is retrieved by solving | (1) |
| ΔHcp(M′xM′′yBz) = E(M′xM′′yBz) − min Ecp(bM′, bM′′, bB) | (2) |
A phase is considered stable if ΔHcp < 0, indicating a phase being energetically favored as compared to decomposing into competing phases. ΔHcp > 0 indicates that decomposing into identified competing phases is more energetically favorable. The complete set of competing phases considered herein is composed of data from the Materials Project database4 and ternary MAB phases reported in ref. 39. In addition, the considered M′2M′′B2 phases are included within the set of competing phases when evaluating M′4M′′B3 and M′4M′′B3 when evaluating the stability for M′2M′′B2. Tables of all competing phases considered in this work, with corresponding total energy values, can be found in the ESI.†
Furthermore, to model configurational disorder where M′ and M′′ are randomly mixed on the two M-sites, we use the special quasi-random structures (SQS) method.46 The integer notations in M′2M′′B2 and M′4M′′B3 are herein used to represent chemical order of M′ and M′′ sites whereas parenthesis representation, (M′0.67M′′0.33)3B2 and (M′0.8M′′0.2)5B3, correspond to chemical disorder mixture on both M′ and M′′ sites. The corresponding mixing free energy for structures with disordered M′ and M′′ is approximated as:
| ΔGdiscp [T] = ΔHdiscp − TΔS, | (3) |
| ΔS = −kB[nln(n) + (1 − n) ln(1 − n)], | (4) |
Synthesis of Ta2MoB2
The boride powder was synthesized by mixing TaB2 (99.5%, Sigma Aldrich) and MoB (99.5%, <325 mesh, Aldrich) with elemental powders of Ta (99.97%, <325 mesh, Alfa Aesar), Mo (99.5%, APS 3–7 μm, Thermo Scientific) and amorphous B (95%, Aldrich) in molar ratios 0.66 TaB2 + 0.66 MoB + 1.33 Ta + 0.33 Mo. The powders were mixed in an agate mortar and cold-pressed using a load corresponding to a stress of 50 MPa. The discs were placed in an alumina crucible and kept at 1600 °C for 20 h under 5 sccm of argon flow (heating and cooling rates of 5 °C min−1). After cooling down to room temperature, the sintered disc was crushed and sieved through a 36 μm sieve.Characterization methods
Structural analysis and phase identification were performed through X-ray diffraction (XRD), using a PANalytical diffractometer, with a Cu Kα radiation source and 1/2° and 5 mm divergence and receiving slits. The diffraction pattern was recorded using a step size and time per step of 0.0084° and 15.24 s, respectively. Scanning electron microscopy (SEM), using a LEO 1550 instrument, combined with an energy dispersive X-ray spectrometer (EDX), from Oxford Instruments, was used for the analysis of morphology and chemical composition, primarily the metal content. A minimum of 10 individual particles were examined.Results & discussion
Binary boron-based precursors
Materials like V3B2, Cr5B3, and Mo5SiB2 all exhibit tetragonal crystal structures with two M-sites. V3B2 features V at Wyckoff sites 4h and 2a,18 Cr5B3 with Cr at Wyckoff sites 16h and 4c,20 and Mo5SiB2 with Mo at Wyckoff sites 16h and 4c.47 Having two different M-sites can allow for site-specific population of two carefully chosen metals that lead to formation of chemical order. Examples of such materials are W2CrB2 (prototype V3B2 with M sites 4h and 2a), W4CrB3 (prototype Cr5B3 with M sites 16h and 4c),21 and for Ti4MoSiB2 and Ta4VSiB2 (prototype Mo5SiB2 with M sites 16h and 4c).47,48In this work, we focus on two prototype binary structures V3B2 (P4/mbm) and Cr5B3 (I4/mcm) and take advantage of the two independent M sublattices. To distinguish the two different metal sites, we use the notation M′ and M′′, which yields the general compositions M′2M′′B2 and M′4M′′B3, respectively.
The V3B2 prototype structure has been identified in a total of 10 material systems forming the P4/mbm symmetry.18,23–27,49 However, structural information regarding the ordered or disordered arrangement on the M-sublattice is typically scarce or neglected, except in ref. 23 and 24. These two references suggest an ordered arrangement of Mo and Fe in Mo2FeB2 in addition to W and Cr in W2CrB2. This is in contrast to the Cr5B3 prototype structure, which has been reported in binary Cr5B3,20 ternary W4CrB3,21,50 and quaternary M′4M′′SiB2 systems.47,48,51,52 However, the two identified prototype structures display a strong similarity to each other, with the Cr5B3 structure closely resembling the V3B2 structure, albeit with an additional metal and boron layer. A schematic illustration comparing the two ternary structures is shown in Fig. 1 in which the additional metal boron layer may be observed when viewed along the [100] zone axis.
 | ||
| Fig. 1 Schematic illustration of (a) M′2M′′B2 (P4/mbm) and (b) M′4M′′B3 (I4/mcm) along the [001] and [100] zone axes, with atoms of the two metal sites represented by blue and red while boron atoms are in green. | ||
The M′2M′′B2 structure (space group P4/mbm and prototype V3B2) is displayed in Fig. 1a and is composed of Wyckoff sites 4h (M′), 2a (M′′), and 4g (B). A layered stacking composed of alternating layers of M′′-B and M′ is apparent along the [100] zone axis. The structure viewed from the [001] zone axis illustrates the B-layer to be composed of alternating B-pairs rotated 90 degrees around the [001] axis.
The structure of M′4M′′B3 (space group I4/mcm and prototype Cr5B3) is illustrated in Fig. 1b. This structure has additional layers of M′ and B, as compared to M′2M′′B2, which is apparent when viewed along the [100] zone axis. The additional B-layer contributes to a more complicated structure, where the alternating B-pairs have an additional B-layer located within the neighboring layer but rotated 90 degrees around the [001] zone axis. This structure is composed of four different sites, M′ at 16h, M′′ at 4c, and B at 8h and 4a, respectively. Note that the M′4M′′B3 structure bears a close resemblance to the T2 phase, in which the 4a site is occupied by Si instead of B, resulting in M′4M′′SiB2. The latter formula, with a different combination of M′ and M′′, has previously been predicted to include at least 11 thermodynamically stable phases energetically favoring ordered M′ and M′′ on the M sites and 40 with a disordered distribution of M′ and M′′.47 Note that several M′4M′′SiB2, with ordered M′ and M′′, have been subsequently synthesized.47,48,51,52 The close resemblance of the selected prototype structures with respect to the T2 phase, which resulted in 51 predicted stable and several synthesized materials, further strengthens the motivation for finding novel ternary compounds with chemical order of M′ and M′′.
Thermodynamical phase stability predictions
The high-throughput stability screening was performed by considering the two selected crystal structures (V3B2 and Cr5B3 prototypes) where the two M-sites were selectively populated by Al, Si, Sc, Ti, V, Cr, Mn, Fe, Co, Ni, Cu, Zn, Ga, Ge, Y, Zr, Nb, Mo, Tc, Ru, Rh, Pd, Ag, Cd, In, Sn, Hf, Ta, W, Re, Os, Ir, Pt, Au, Hf, Tl and Pb. Note the use of M′ and M′′ as general notations for elements where M′ ≠ M′′ represents ternary phases and M′ = M′′ represents binary phases. Solid solution structures (SQS) were modeled on the M′ and M′′ sites for ordered ternary phases identified as stable (ΔHcp < 0 meV per atom) or close to stable (0 ≤ ΔHcp < 70 meV per atom). The chosen close-to-stable energy limit of 70 meV per atom was motivated by the configurational entropy term, TΔS, which in eqn (3) for an arbitrary M′2M′′B2 phase and M′4M′′B3 phase is equal to −70 and −54 meV per atom, respectively, at 2000 K. The reason for choosing 2000 K is motivated by the typical temperature used for powder synthesis of similar boron-based materials. Since both ordered and disordered distributions of M′ and M′′ are considered herein, we choose to account for the contribution from configurational entropy to the Gibbs free energy for structures modeled with a disordered distribution of M′ and M′′ (SQS) at a temperature expected to be used for synthesis attempts. This is an approach used to identify whether an ordered or disordered distribution of M′ and M′′ are to be expected upon synthesis and has previously been demonstrated to confirm experimental observations of boron-based materials14,47,53 and for other related layered materials.54,55Phase stability predictions for M′2M′′B2 phases
The phase stability predictions for the M′2M′′B2 phases are displayed in Fig. 2a for both binary (M′ = M′′) and ternary (M′ ≠ M′′) phases arranged by the group number of the considered elements, ranging from high to low. Note that a limited region of the considered M′ and M′′ combinations are shown as most of the phases located outside this region did not fulfill ΔHcp < 150 meV per atom. The complete heatmap is displayed in Fig. S2.† Disordered phases were modeled as a solid solution of M′ and M′′ for ordered phases evaluated with ΔHcp < 70 meV per atom. Hence, disordered configurations were evaluated as ΔGcp according to eqn (3) in contrast to structures with ordered M sites, which were evaluated using ΔHcp in eqn (2). The background color represents the calculated thermodynamic stability for the chemical order/disorder of lowest energy with stable phases (ΔHcp < 0 or ΔGcp < 0 meV per atom) in blue, and not stable or at best metastable (ΔHcp > 0 or ΔGcp > 0 meV per atom) phases in red. Grey represents M′2M′′B2 phases predicted far from stable (ΔHcp > 150 meV per atom). The symbols used in Fig. 2a represent the M site configuration of the lowest energy evaluated at 2000 K, with filled circles illustrating ordered M′2M′′B2 phases and open circles disordered (M′0.667M′′0.333)3B2 phases. Experimentally known phases in which ordered and disordered configurations have been verified are marked by black squares. | ||
| Fig. 2 (a) Calculated formation enthalpy ΔHcp (chemical order) or Gibbs free energy of formation ΔGcp (solid solution) at 2000 K for M′2M′′B2 where the stability is represented by the color in blue (stable) and red (not stable). Phases with ΔHcp > 150 meV per atom are colored grey and classified far from stable. Symbols represent the chemical order/disorder of lowest energy for a given combination of M′ and M′′ with filled circles for ordered M′2M′′B2 and open circles for solid solution (M′0.667M′′0.333)3B2. Black squares represent synthesized compounds. Schematic illustrations of (b) ordered and (c) disordered arrangements of M′ and M′′ when viewed along the [001] zone axis, with M′, M′′, and B in blue, red, and green, respectively. | ||
Fig. 2a shows 44 phases predicted to be stable at 2000 K, including 3 binary and 41 ternary phases. Among the ternaries, 23 phases were predicted to energetically favor chemical order of M′ and M′′ whereas 18 were found with a disordered arrangement of M′ and M′′. The experimentally known phases, marked with black squares, display formation enthalpy values that correlate well with values found in the Materials Project database. This includes Mo3B2, which has been experimentally reported but is predicted to be unstable with +29 meV per atom. The majority of materials denoted as experimentally known were discovered during the mid-20th century with, in some cases, limited information regarding the chemical composition, structural information, and arrangement of their atomic species.18,23–27,49 The systems modelled herein therefore represent the ideal compositions and structures, while defects and potential divergence from the ideal stoichiometry have not been considered. A complete list of predicted stable phases along with their sets of most competing phases and preferred M-arrangement at 2000 K is found in Table S1.† In addition, the phonon dispersion spectrum has been calculated for all stable M′2M′′B2 with order of M′ and M′′, i.e., ΔHcp < 0. Note that Tc-based phases have not been considered due to the unstabale and radioactive Tc. These are shown in Fig. S3 to S27.† The absence of imaginary frequencies indicates that 24 out of 25 M′2M′′B2 are dynamically stable. The only dynamically unstable phase is Ta2MnB2 as indicated by the presence of imaginary frequencies.
Hu et al., previously predicted the degree of the chemical configuration of forming ordered or disordered M sites within M′2M′′B2 and M′4M′′B3 compounds to correlate with the atomic number of the alloying M′ and M′′ elements. According to Hu et al., compounds with M′ of a greater atomic number than M′′ typically displayed an ordered arrangement when considering the elements Ni, Co, Cr, W, and Mo.21 We herein constructed a stability heatmap with the elements arranged by their atomic number, as illustrated in Fig. S2,† to investigate this hypothesis. The results illustrate that groups of both stable and metastable phases are formed when M′ is occupied by transition metals of Period 4 (Ti, V, Cr, Mn, Fe), 5 (Zr, Nb, Mo, Tc), and Period 6 (Hf, Ta, W), whereas M′′ is occupied by elements of Period 3 (Al, Si), Period 4 (Sc, Ti, C, Cr, Mn, Fe, Co, Nu Cu), Period 5 (Nb, Mo, Tc, Ru, Rh) and Period 6 (Ta, W, Re, Os, Ir Pt). Every formed group of predicted stable compounds includes phases with both chemically ordered and disordered arrangements on the M-sites, thus illustrating no distinct connection between the M-site arrangement and the atomic number of M. This is exemplified by Ti2ReB2, predicted stable with ΔHcp = −41 meV per atom, which is chemically ordered even though the atomic number of Re (Z = 75), occupying the 2a sites, is larger than Ti (Z = 22), occupying the 4h site. Furthermore, the neighboring phase (Ti0.667W0.333)3B2 is also predicted stable, with ΔGcp = −26 meV per atom, but with a disordered distribution of M-elements when occupied by Ti and W (Z = 74).
Phase stability predictions for M′4M′′B3 phases
We continue the HT phase stability screening by considering the M′4M′′B3 structure. Fig. 3 illustrates stability for a limited elemental space whereas a complete heatmap may be found in Fig. S28.† The background color represents the calculated thermodynamic stability for the chemical order/disorder of lowest energy with blue regions representing stable phases (ΔHcp < 0 or ΔGcp < 0 meV per atom). The symbols, again, represent the metal arrangement of lowest energy evaluated at 2000 K with filled circles illustrating ordered M′4M′′B3 phases and open circles disordered (M′0.8M′′0.2)5B3 phases. Again, experimentally known phases with verified ordered or disordered configurations are marked by a black edge.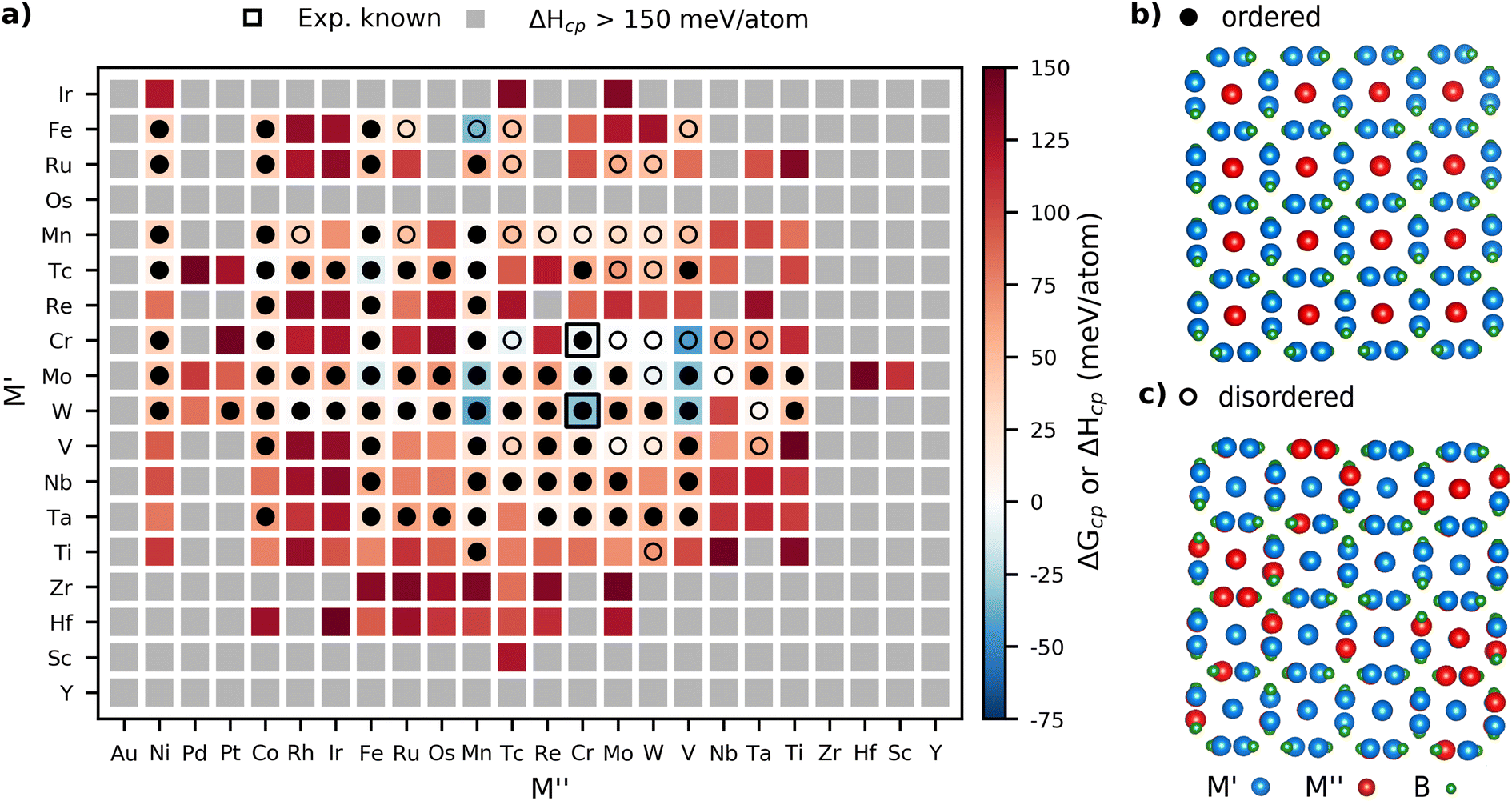 | ||
| Fig. 3 (a) Calculated formation enthalpy ΔHcp (chemical order) or Gibbs free energy of formation ΔGcp (solid solution) at 2000 K for M′4M′′B3 where the stability is represented by the color ranging from blue (stable) to red (not stable). Phases with ΔHcp > 150 meV per atom are colored grey and classified far from stable. Symbols represent the chemical order of the lowest energy for a given combination of M′ and M′′ with filled circles for ordered M′4M′′B2 and open circles for solid solution (M′0.8M′′0.2)5B3. Black squares mark synthesized compounds. Schematic illustrations of (b) ordered and (c) disordered arrangements of M′ and M′′ when viewed along the [001] zone axis, with M′, M′′, and B in blue, red, and green, respectively. | ||
A total of 15 ternary phases within Fig. 3a were predicted stable out of which nine favor an ordered M-site arrangement of M′ and M′′ whereas six prefer the M-sites to be disorderly arranged at 2000 K. The only experimentally verified M′4M′′B3 phase, W4CrB3, was predicted stable with ΔHcp = −31 meV per atom, further corroborating the formation enthalpy approach for phase stability predictions. A complete list of predicted stable phases along with their set of most competing phases and the preferred M-arrangement at 2000 K is found in Table S2.† In addition, the phonon dispersion spectrum has been calculated for all stable M′4M′′B3 structures with order of M′ and M′′, i.e., ΔHcp < 0. These are shown in Fig. S29–S35.† Out of seven M′4M′′B3 phases, three are found to be dynamically stable, specifically Mo4VB3, Mo4FeB3, and W4VB3. Note that all phases identified as dynamically unstable contain magnetic elements such as Cr, Mn, or Fe. Since only non-magnetic and ferromagnetic spin configurations have been considered, a more in-depth analysis with phonon dispersion spectra calculated for various anti-ferromagnetic spin configurations may lead to dynamically stable structures.
The complete stability heatmap for the M′4M′′B3 compounds is shown in Fig. S28,† with metals arranged based on their atomic number, which illustrates a strikingly similar appearance to that for the M′2M′′B2 compounds shown in Fig. S2.† Once again, groups of stable compositions are formed when M′ is occupied by metals from Period 4 (Ti, V, Cr, Mn, Fe), Period 5 (Nb, Mo, Tc, Ru), and Period 6 (Hf, Ta, W, Re) whereas M′′ is occupied by elements from Period 3 (Al, Si), Period 4 (Ti, C, Cr, Mn, Fe, Co, Nu Cu, Zn), Period 5 (Nb, Mo, Tc, Ru, Rh, Pd) and Period 6 (Ta, W, Re, Os, Ir Pt). Similarly to the M′2M′′B2 composition, the complete heatmap illustrates no distinct correlation between the atomic numbers of the metal and the arrangement of the M-sites. This may, however, be traced to the few numbers of predicted disordered phases with the M′4M′′B3 composition. Further correlations may have been observed if chemical disorder on the M-sites was considered for the complete chemical phase space for both prototype structures, which is herein left for future studies.
Experimental realization
An attempt to verify the predicted stable compounds shown in Fig. 2 was made by selecting Ta2MoB2 for synthesis, with a predicted formation enthalpy ΔHcp = −53 meV per atom with a chemically ordered M-element configuration of Ta and Mo. The selection of Ta2MoB2, in contrast to Ta2VB2 and Ta2CrB2, which displayed similar ΔHcp values, is motivated by Ta and Mo being of different group numbers in addition to avoiding magnetic elements, such as Cr. The XRD pattern shown by the blue line in Fig. 4 is obtained from a sample with initial elemental powder ratios corresponding to stoichiometric Ta2MoB2. The data reveal the presence of TaB and Ta2MoB2 (peaks consistent with either chemical order or disorder on the M-sites) with a few peaks that could not be assigned to a specific compound. A comparison of the measured XRD pattern with simulated patterns of Ta2MoB2, (Ta0.667Mo0.333)3B2, and TaB is shown in Fig. S37.† It is worth noting that simulated XRD peaks, the positions of which are indicated by vertical bars at the bottom of Fig. 4 or in full in Fig. S37,† are slightly shifted towards lower angles, indicating larger calculated lattice parameters, which can be attributed to the underestimation of lattice parameters by GGA functionals. EDX measurements on individual particles revealed a distribution of metal concentrations, ranging from 0.8 to 2.9 in terms of Ta/Mo ratios. Importantly, certain particles were identified with Ta/Mo ratios that aligned with the correct stoichiometry, i.e., Ta/Mo = 2. To assess whether Ta2MoB2 is chemically ordered, we attempted to compare the theoretical XRD pattern of both chemically ordered (Ta2MoB2) and disordered ((Ta0.667Mo0.333)3B2) structures. However, both of these structures display very similar characteristics and could therefore not be distinguished. Thus, further insights concerning chemical order or not may be based on the performed phase stability predictions, which showed a lower formation enthalpy for the ordered Ta2MoB2 compared to the disordered (Ta0.667Mo0.333)3B2. Representative SEM images and the measured chemical concentrations are shown in Fig. S38 and Table S3.† | ||
| Fig. 4 XRD pattern of the sample from the attempted synthesis of Ta2MoB2 with assigned peaks for Ta2MoB2, (Ta0.667Mo0.333)3B2, and TaB, given by green, red, and yellow vertical bars respectively. | ||
Bonding analysis
In addition, the bonding properties of the chemically ordered symmetries for both Ta2MoB2 and Mo2TaB2 were studied through the LOBSTER code, to identify possible preference for either of the metals to occupy one of the metal sites from an interaction perspective. These two phases were selected as the former compound was predicted stable (ΔHcp = −53 meV per atom) and the latter not (ΔHcp = +95 meV per atom). Fig. 5 displays the DOS and COHP plots for Ta2MoB2 and Mo2TaB2, respectively. The bonding characteristics for Ta2MoB2, shown in Fig. 5a, indicate the presence of minor to negligible bonding/antibonding states at or below the Fermi level, Ef. Notably, the total DOS exhibits a local minimum at the Fermi energy Ef that further emphasizes the preference for bonding within Ta2MoB2. This is in contrast to Mo2TaB2, in Fig. 5b, where the Fermi level is shifted away from the local minima in the total DOS, due to the presence of additional valence electrons. An examination of the DOS and COHP curves reveals a modest increase in non-bonding states for Mo2TaB2 compared to Ta2MoB2, albeit small in contrast to the difference in DOS at Ef. These observations indicate that Ta has a slight preference for occupying the majority of the 4h Wykoff sites compared to Mo. This, in combination with the XRD data and the phase stability predictions, suggests that the M-site configuration for Ta2MoB2 may be chemically ordered.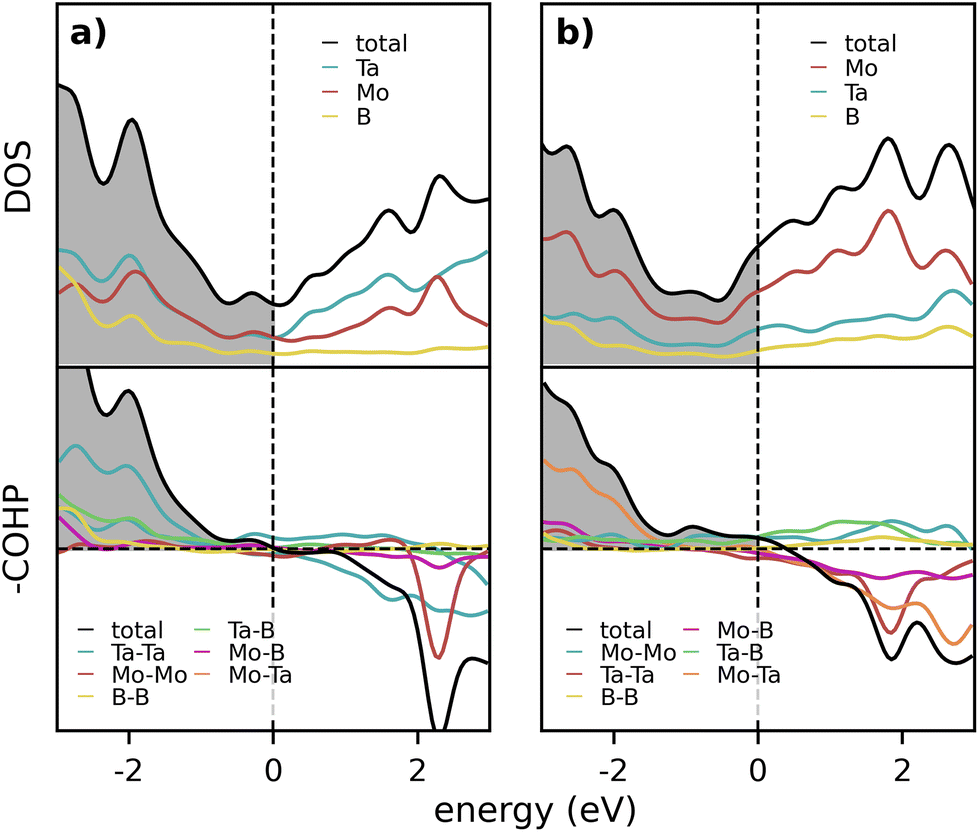 | ||
| Fig. 5 DOS and COHP plots for (a) Ta2MoB2 and (b) Mo2TaB2, respectively, with the Fermi energy Ef denoted by a vertical dashed line. | ||
Furthermore, a comparative analysis of bond strength for selected M′ and M′′ has been carried out for M′2M′′B2 in terms of the integrated projected crystal orbital Hamiltonian population (IpCOHP). A more negative value of IpCOHP indicates stronger bonding. This is shown in Fig. 6 for different interactions in M′2M′′B2. We find that the B–B interactions are the strongest, indicating that B plays a crucial role in stabilizing the bonding structure. B–B is also the shortest bond, around 1.7 to 1.9 Å depending on M′ and M′′. The weakest interactions are found for M′–M′ and M′′–M′′, consistent with their longer bond distance (∼<3 Å). M′–B and M′′–B, and M′–M′′ bonds are found in-between, where metals from Period 5 (Mo) or Period 6 (Hf, Ta, W) generally form stronger bonds with B than metals from Period 4 (Ti, V, Cr). Metals from Group 6 (Cr, Mo, W) show stronger bonding with B compared to metals from Groups 4 and 5. The latter (V, Ta) form moderately strong bonds with B, with Ta being stronger than V. Increased valence for M′′ generally leads to stronger M′–B and M′′–B bonds but weaker M′′–M′′ and B–B bonds. This indicates that finding the optimal bonding in M′2M′′B2 is challenging.
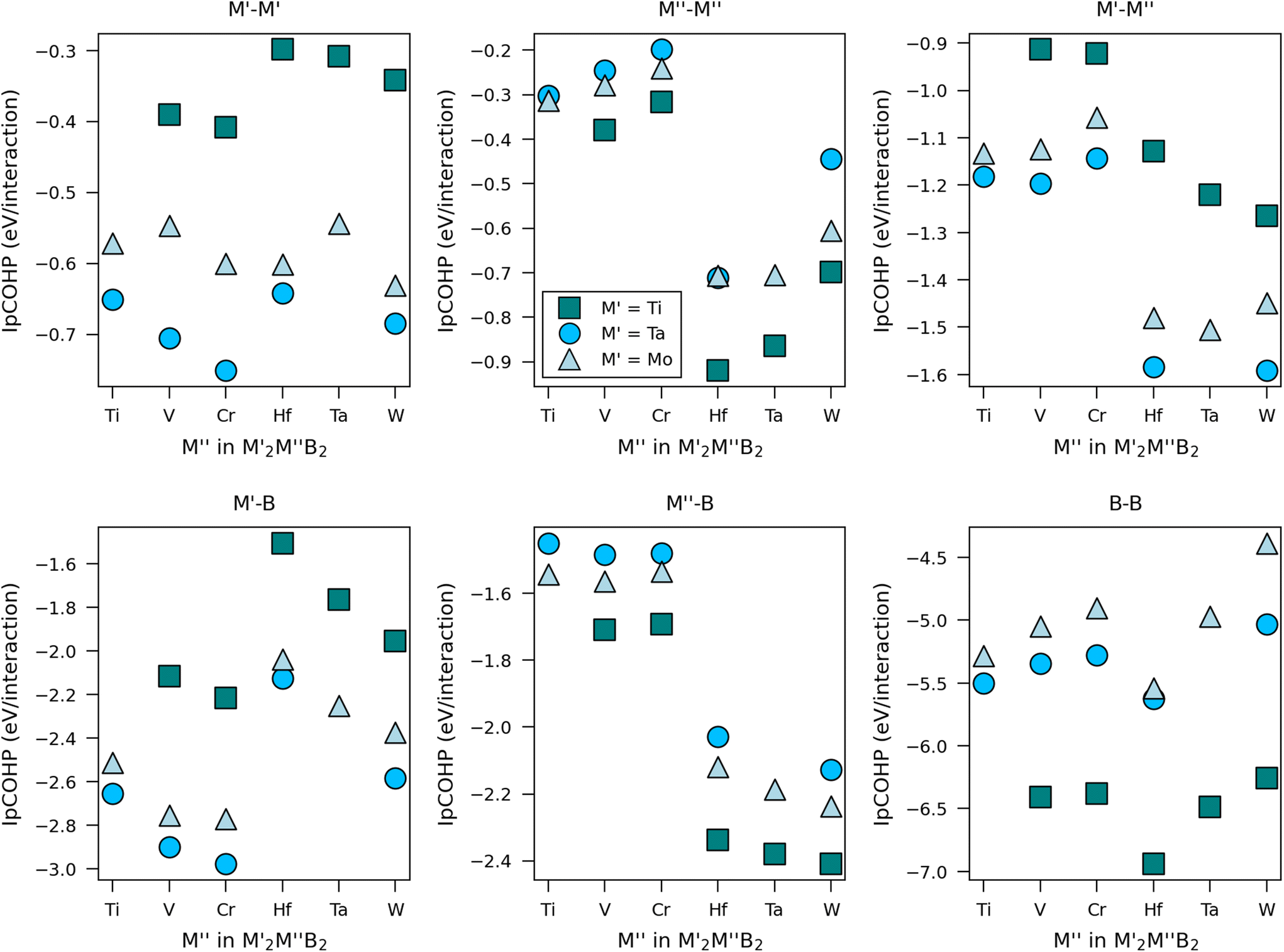 | ||
| Fig. 6 Bond strengths in terms of integrated partial COHP (IpCOHP) for six different interactions in M′2M′′B2 where M′ = Ti, Ta, Mo and M′′ = Ti, V, Cr, Hf, Ta, Mo. | ||
Conclusions
We have performed a high-throughput search to identify theoretically stable ternary boron-based materials that are feasible for synthesis. The thermodynamical phase stability was evaluated for a total of 2378 phases, where M′ and M′′ were systematically occupied by elements ranging from Period 3 to Period 6 and by taking advantage of the two different M sublattices in M3B2 (prototype V3B2) and M5B3 (prototype Cr5B3) structures. We identified 56 ternary materials to be stable at 2000 K, out of which 41 belong to the M′2M′′B2 composition and 15 to the M′4M′′B3 composition. Initial attempts at synthesizing the predicted stable Ta2MoB2 were made through powder synthesis, with XRD indicating the presence of ordered Ta2MoB2 or disordered (Ta0.667Mo0.333)3B2. However, distinguishing ordered (Ta2MoB2) from disordered ((Ta0.66Mo0.33)3B2) requires in-depth TEM analysis. The bonding characteristics of Ta2MoB2 and Mo2TaB2 were further analyzed by designing COHP and DOS plots. Ta2MoB2 illustrated better characteristics thus suggesting Ta occupies the 4h Wyckoff site of the ordered symmetries.Author contributions
M.D and J.R. supervised the project, which was conducted by A.C, R.R., and M.D. The data analysis, computations, and writing of the main parts of the manuscript drafts were conducted by A.C. whereas R.R. governed the experimental procedures. All authors reviewed and edited the manuscript.Data availability
The data supporting the results presented herein are available upon reasonable request from the corresponding author.Conflicts of interest
The authors declare no competing interests.Acknowledgements
M. D. and J. R. acknowledge support from the Swedish Research Council through projects 2019-05047 and 2022-06099. J. R. also acknowledges funding from the Knut and Alice Wallenberg (KAW) Foundation for a Fellowship/Scholar Grant and Project funding (KAW 2020.0033). The calculations were conducted using supercomputer resources provided by the National Academic Infrastructure for Supercomputing in Sweden (NAISS) and the Swedish National Infrastructure for Computing (SNIC) at the National Supercomputer Centre (NSC) and the High-Performance Computing Center North (HPC2N), partially funded by the Swedish Research Council through grant agreements no. 2022-06725 and no. 2018-05973.References
- C. J. Bartel, A. Trewartha, Q. Wang, A. Dunn, A. Jain and G. Ceder, npj Comput. Mater., 2020, 6, 97 CrossRef.
- A. Mansouri Tehrani, A. O. Oliynyk, M. Parry, Z. Rizvi, S. Couper, F. Lin, L. Miyagi, T. D. Sparks and J. Brgoch, J. Am. Chem. Soc., 2018, 140, 9844–9853 CrossRef CAS PubMed.
- C. Nyshadham, C. Oses, J. E. Hansen, I. Takeuchi, S. Curtarolo and G. L. W. Hart, Acta Mater., 2017, 122, 438–447 CrossRef CAS.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder and K. A. Persson, APL Mater., 2013, 1, 011002 CrossRef.
- J. E. Saal, S. Kirklin, M. Aykol, B. Meredig and C. Wolverton, JOM, 2013, 65, 1501–1509 CrossRef CAS.
- S. Kirklin, J. E. Saal, B. Meredig, A. Thompson, J. W. Doak, M. Aykol, S. Rühl and C. Wolverton, npj Comput. Mater., 2015, 1, 15010 CrossRef CAS.
- S. Curtarolo, W. Setyawan, S. Wang, J. Xue, K. Yang, R. H. Taylor, L. J. Nelson, G. L. W. Hart, S. Sanvito, M. Buongiorno-Nardelli, N. Mingo and O. Levy, Comput. Mater. Sci., 2012, 58, 227–235 CrossRef CAS.
- S. Curtarolo, G. L. W. Hart, M. B. Nardelli, N. Mingo, S. Sanvito and O. Levy, Nat. Mater., 2013, 12, 191–201 CrossRef CAS PubMed.
- C. J. Bartel, J. Mater. Sci., 2022, 57, 10475–10498 CrossRef CAS.
- C. W. Glass, A. R. Oganov and N. Hansen, Comput. Phys. Commun., 2006, 175, 713–720 CrossRef CAS.
- A. O. Lyakhov, A. R. Oganov, H. T. Stokes and Q. Zhu, Comput. Phys. Commun., 2013, 184, 1172–1182 CrossRef CAS.
- D. C. Lonie and E. Zurek, Comput. Phys. Commun., 2011, 182, 372–387 CrossRef CAS.
- S. Hajinazar, A. Thorn, E. D. Sandoval, S. Kharabadze and A. N. Kolmogorov, Comput. Phys. Commun., 2021, 259, 107679 CrossRef CAS.
- M. Dahlqvist, Q. Tao, J. Zhou, J. Palisaitis, P.OÅ. Persson and J. Rosen, J. Am. Chem. Soc., 2020, 142, 18583–18591 CrossRef CAS PubMed.
- V. I. Hegde, M. Aykol, S. Kirklin and C. Wolverton, Sci. Adv., 2020, 6, eaay5606 CrossRef CAS PubMed.
- G. H. Jóhannesson, T. Bligaard, A. V. Ruban, H. L. Skriver, K. W. Jacobsen and J. K. Nørskov, Phys. Rev. Lett., 2002, 88, 255506 CrossRef.
- A. G. Van Der Geest and A. N. Kolmogorov, Calphad, 2014, 46, 184–204 CrossRef CAS.
- H. Nowotny and A. Wittmann, Monatsh Chem., 1958, 89, 220–224 CrossRef CAS.
- W. H. Zachariasen, Acta Cryst., 1949, 2, 94–99 CrossRef CAS.
- C. N. Guy and A. A. Uraz, J. Less-Common Met., 1976, 48, 199–203 CrossRef CAS.
- X. B. Hu, H. Y. Niu, X. L. Ma, A. R. Oganov, C. A. J. Fisher, N. C. Sheng, J. D. Liu, T. Jin, X. F. Sun, J. F. Liu and Y. Ikuhara, Acta Mater., 2018, 149, 274–284 CrossRef CAS.
- A. M. Filippi, J. Less-Common Met., 1973, 30, 153–158 CrossRef CAS.
- E. I. Gladyshevskii, T. F. Fedorov, Y. B. Kuz'ma and R. V. Skolozdra, Sov. Powder Metall. Met. Ceram., 1966, 5, 305–309 CrossRef.
- V. S. Telegus and Y. B. Kuz'ma, Sov. Powder Metall. Met. Ceram., 1968, 7, 133–138 CrossRef.
- A. S. Sobolev, Y. B. Kuz'ma, T. E. Soboleva and T. F. Fedorov, Sov. Powder Metall. Met. Ceram., 1968, 7, 48–51 CrossRef.
- V. S. Telegus and Y. B. Kuz'ma, Sov. Powder Metall. Met. Ceram., 1971, 10, 52–56 CrossRef.
- Y. B. Kuz'ma, Sov. Powder Metall. Met. Ceram., 1971, 10, 298–300 CrossRef.
- Y. B. Kuz'ma, T. I. Ts'olkovskii and O. P. Baburova, Neorg. Mater., 1968, 4, 1081–1085 Search PubMed.
- V. Moraes, H. Riedl, C. Fuger, P. Polcik, H. Bolvardi, D. Holec and P. H. Mayrhofer, Sci. Rep., 2018, 8, 9288 CrossRef.
- H. Wu, H. Fan and Y. Hu, Phys. Rev. B, 2021, 103, L041203 CrossRef CAS.
- B. D. Sahoo and K. D. Joshi, J. Appl. Phys., 2023, 134 CAS.
- M. A. Ali, M. M. Hossain, A. K. M. A. Islam and S. H. Naqib, J. Alloys Compd., 2021, 857, 158264 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B:Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B:Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B:Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B:Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- A. Carlsson, J. Rosen and M. Dahlqvist, Phys. Chem. Chem. Phys., 2022, 24, 11249–11258 RSC.
- A. Togo, F. Oba and I. Tanaka, Phys. Rev. B:Condens. Matter Mater. Phys., 2008, 78, 134106 CrossRef.
- R. Dronskowski and P. E. Bloechl, J. Phys. Chem., 1993, 97, 8617–8624 CrossRef CAS.
- V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, J. Phys. Chem. A, 2011, 115, 5461–5466 CrossRef CAS PubMed.
- S. Maintz, V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, J. Comput. Chem., 2016, 37, 1030–1035 CrossRef CAS PubMed.
- R. Nelson, C. Ertural, J. George, V. L. Deringer, G. Hautier and R. Dronskowski, J. Comput. Chem., 2020, 41, 1931–1940 CrossRef CAS PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- A. Zunger, S. H. Wei, L. G. Ferreira and J. E. Bernard, Phys. Rev. Lett., 1990, 65, 353–356 CrossRef CAS.
- M. Dahlqvist, J. Zhou, I. Persson, B. Ahmed, J. Lu, J. Halim, Q. Tao, J. Palisaitis, J. Thörnberg, P. Helmer, L. Hultman, P.OÅ. Persson and J. Rosen, Adv. Mater., 2021, 33, 2008361 CrossRef CAS PubMed.
- A. S. Etman, J. Halim, H. Lind, M. Dorri, J. Palisaitis, J. Lu, L. Hultman, P.OÅ. Persson and J. Rosen, Cryst. Growth Des., 2023, 23, 4442–4447 CrossRef CAS.
- A. Wittmann, H. Nowotny and H. Boller, Monatsh Chem., 1960, 91, 608–615 CrossRef CAS.
- X. B. Hu, Y. L. Zhu, N. C. Sheng and X. L. Ma, Sci. Rep., 2014, 4, 7367 CrossRef CAS PubMed.
- J. Halim, P. Helmer, J. Palisaitis, M. Dahlqvist, J. Thörnberg, P.OÅ. Persson and J. Rosen, Inorg. Chem., 2023, 62, 5341–5347 CrossRef CAS PubMed.
- Q. Tao, J. Halim, J. Palisaitis, A. Carlsson, M. Dahlqvist, U. Wiedwald, M. Farle, P. Persson and J. Rosen, Cryst. Growth Des., 2023, 23, 3258–3263 CrossRef CAS.
- M. Dahlqvist and J. Rosen, Nanoscale, 2021, 13, 18311 RSC.
- M. Dahlqvist and J. Rosen, Nanoscale, 2020, 12, 785–794 RSC.
- M. Dahlqvist and J. Rosen, Nanoscale, 2022, 14, 10958–10971 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: The SQS convergence, and complete heatmaps of the studied materials systems arranged based on the atomic number and tabulated formation enthalpy values, structural configuration, set of most competing phases, and phonon dispersion spectrum can be found in the attached supplementary material. See DOI: https://doi.org/10.1039/d4nr02948h |
| This journal is © The Royal Society of Chemistry 2025 |