
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sodium compensation: a critical technology for transforming batteries from sodium-starved to sodium-rich systems
Bin
Zhu
 a,
Wei
Zhang
*bg,
Zhenjing
Jiang
b,
Jie
Chen
b,
Zheng
Li
a,
Jingqiang
Zheng
a,
Naifeng
Wen
a,
Ruwei
Chen
b,
Hang
Yang
b,
Wei
Zong
c,
Yuhang
Dai
c,
Chumei
Ye
d,
Qi
Zhang
e,
Tianyun
Qiu
f,
Yanqing
Lai
a,
Jie
Li
a and
Zhian
Zhang
*a
a,
Wei
Zhang
*bg,
Zhenjing
Jiang
b,
Jie
Chen
b,
Zheng
Li
a,
Jingqiang
Zheng
a,
Naifeng
Wen
a,
Ruwei
Chen
b,
Hang
Yang
b,
Wei
Zong
c,
Yuhang
Dai
c,
Chumei
Ye
d,
Qi
Zhang
e,
Tianyun
Qiu
f,
Yanqing
Lai
a,
Jie
Li
a and
Zhian
Zhang
*a
aSchool of Metallurgy and Environment, Engineering Research Center of the Ministry of Education for Advanced Battery Materials, Hunan Provincial Key Laboratory of Nonferrous Value-Added, Metallurgy, Central South University, Changsha 410083, P. R. China. E-mail: zhangzhian@csu.edu.cn
bDepartment of Chemistry, University College London, London WC1H 0AJ, UK. E-mail: wei.zhang.21@ucl.ac.uk
cDepartment of Engineering Science, University of Oxford, Oxford OX1 3PJ, UK
dDepartment of Materials Science and Metallurgy, University of Cambridge, Cambridge CB3 0FS, UK
eSchool of Engineering, University of Warwick, Coventry CV4 7AL, UK
fSchool of Photovoltaic and Renewable Energy Engineering, University of New South Wales, Sydney, NSW 2052, Australia
gDepartment of Chemical System Engineering, School of Engineering, The University of Tokyo, Hongo 7-3-1, Bunkyo-ku, Tokyo 113-8656, Japan
First published on 9th August 2024
Abstract
Sodium-ion batteries (SIBs) have attracted wide attention from academia and industry due to the low cost and abundant sodium resources. Despite the rapid industrialization development of SIBs, it still faces problems such as a low initial coulombic efficiency (ICE) leading to a significant decrease in battery energy density (e.g., 20%). Sodium compensation technology (SCT) has emerged as a promising strategy to effectively increase the ICE to 100% and drastically boost battery cycling performance. In this review, we emphasize the importance of SCT in high-performance SIBs and introduce its working principle. The up-to-date advances in different SCTs are underlined in this review. In addition, we elaborate the current merits and demerits of different SCTs. This review also provides insights into possible future research directions in SCT for high-energy SIBs.
 Bin Zhu | Bin Zhu received his Master's degree from Central South University in 2022. He is now a PhD student at Central South University under the supervision of Prof. Zhian Zhang. His research interests focus on cathode Li/Na compensation materials for Li/Na-ion batteries. |
 Wei Zhang | Wei Zhang is currently a JSPS Fellow at the University of Tokyo. He did his postdoctoral research at University College London (UCL). He received his PhD degree in Chemistry from UCL in 2024. His research focuses on the development of electrodes and electrolytes and their interfacial behaviors for next-generation batteries and advanced characterizations, with the goal of building sustainable energy storage systems from environmentally friendly and available materials and processes. |
 Zhian Zhang | Zhian Zhang is currently a Professor of the School of Metallurgy and Environment at Central South University. He received his PhD degree from the University of Electronic Science and Technology of China in 2005. His research interest focuses on advanced energy storage materials for battery applications. |
1 Introduction
Lithium-ion batteries (LIBs) play a dominant role in electrochemical energy storage,1–4 whereas energy storage systems require larger installed capacity, faster frequency regulation, and lower-cost batteries to support the rapid development of renewable energy sources.5–8 Moreover, the relatively low content of lithium in the Earth's crust (0.0017%), the high cost of extraction and refining, and its uneven distribution are the main factors limiting the application of lithium batteries for energy storage at scale. Hence, it is necessary to find lower cost alternatives to lithium.9–13 Sodium-ion batteries (SIBs) are a promising alternative due to the abundant raw materials and cost advantages.14,15 Besides, SIBs can use aluminum foil as the current collector for both electrodes due to the absence of the alloying reaction between Na and Al, which further reduces the overall costs.16 Moreover, SIBs exhibit greater stability at high temperatures and are less sensitive to temperature changes compared to their lithium-ion counterparts (LIBs). Research also shows that Na+ has a lower solvation energy, a faster interface ion diffusion capability, a higher ionic conductivity, and good safety.16,17 These attributes enable SIBs to achieve long-life performance across a wide temperature range, making them suitable for large-scale energy storage systems such as power grids and renewable energy storage.18–20 However, several technical challenges must still be addressed for widespread commercialization. The relatively poor electrochemical performance of SIBs compared to LIBs is not only attributed to the physical and chemical properties of electrode materials but also to the formation of a solid-electrolyte interphase (SEI) by the irreversible decomposition of the electrolyte as well as to a number of side reactions at the anode side, inducing a low initial coulombic efficiency (ICE).21,22 The cathode acts as the sole source of active Na+ ions for transport between the cathode and anode. The SEI formation process is also accompanied by the depletion of active sodium and irreversible capacity loss. Up to 15% of the active sodium ions will be inevitably consumed during the initial cycle, which may decrease the battery energy density by 20%.23 Therefore, it is of great significance to improve the ICE and maximize the performance of SIBs.In order to effectively improve the ICE, researchers have proposed some methods to this end, mainly including: (i) reducing the consumption of sodium ions by decreasing the defects in the material;24 (ii) reducing the specific surface area of the material to decrease the consumption of the electrolyte;24 (iii) adding film-forming additives22,24 to the electrolyte that are beneficial for stabilizing the SEI film; and (iv) using the sodium compensation technique (SCT) to donate additional Na+ to compensate for the irreversible loss of Na+ during the cycling process.25,26 However, the effect of material optimization is relatively limited for reaching a high ICE of >90%.27 It is also difficult to accurately form a favorable SEI layer by suitable film-forming electrolyte additives. Meanwhile, the method is not widely used due to the lack of suitable additive species. In contrast, SCT is an effective means to boost the ICE by introducing additional sodium ions, which has the prospect of large-scale industrialization to realize a 100% ICE. As displayed in Fig. 1, in comparison to the practical conditions, using the sodium compensation technique (SCT) to donate additional Na+ to compensate for the irreversible loss of Na+ during the cycling process realizes a 100% ICE. Upon battery operation, the SCT-treated anodes showed a flattened and decreased potential profile, indicating a higher energy density and wider voltage range for the full cell.28 The SCT treatment on cathodes is able to supplement the Na+ loss especially for the sodium-deficient cathode (e.g., P2-phase oxide, Na2/3Ni1/3Mn2/3O2),29 which helps to stabilize the electrolyte concentration and cathode material structure. This method of supplementing the loss of Na+ plays a crucial role in stabilizing the material structure and extending the battery life.18,22 SCT for the initial sodium loss by introducing additional sodium ions into the system can ensure a sufficient concentration of Na+ ions in the electrolyte right from the beginning. This process not only helps maintain the overall electrochemical balance but also effectively mitigates irreversible sodium loss. As a result, it preserves the structural integrity of the electrode, enhances the capacity and stability of the active material, and promotes more stable battery operation. Moreover, the uniform distribution of Na+ ions significantly reduces the likelihood of dendrite formation, thus minimizing the risk of battery failure.
 | ||
| Fig. 1 Schematic representation of the (de)-intercalation of sodium ions under (a) practical conditions; (b) the conditions with cathode sodium compensation; (c) the conditions with anode sodium compensation. | ||
SCT has emerged as the most straightforward and efficient way to address the cathode and anode material state matching issue by boosting energy density and offsetting active sodium-ion loss, two factors that are essential for the practical use of SIBs. By adding extra active sodium ions to the electrode material beforehand, the goal is to completely eradicate the permanent capacity loss upon the initial cycle. This work provides a timely extensive overview of the most recent advancements in the field of SCTs, including a chemical method using a Na complex, electrochemical approach, short circuit with sodium metal, cathode sacrificial additives, and sodium-rich cathode materials (Scheme 1). The merits and demerits of current approaches are carefully examined and deliberated in terms of the reaction mechanism, safety, compatibility, effectiveness, and scalability. After the SCT treatment, the ICE of the battery can become ∼100%, which indicates that the energy density can be effectively improved. The two most promising approaches for commercial applications—chemical method and cathode sacrificial additives—are highlighted as state-of-the-art developments. We also explore the unsolved scientific issues and technical challenges from a practical perspective. This timely review may encourage continued development of high-energy SIBs and offer recommendations for the study of cutting-edge sodium compensation technology.
 | ||
| Scheme 1 Outline of the most studied sodium compensation technologies. | ||
2 Sodium compensation technology
2.1. Direct contact method
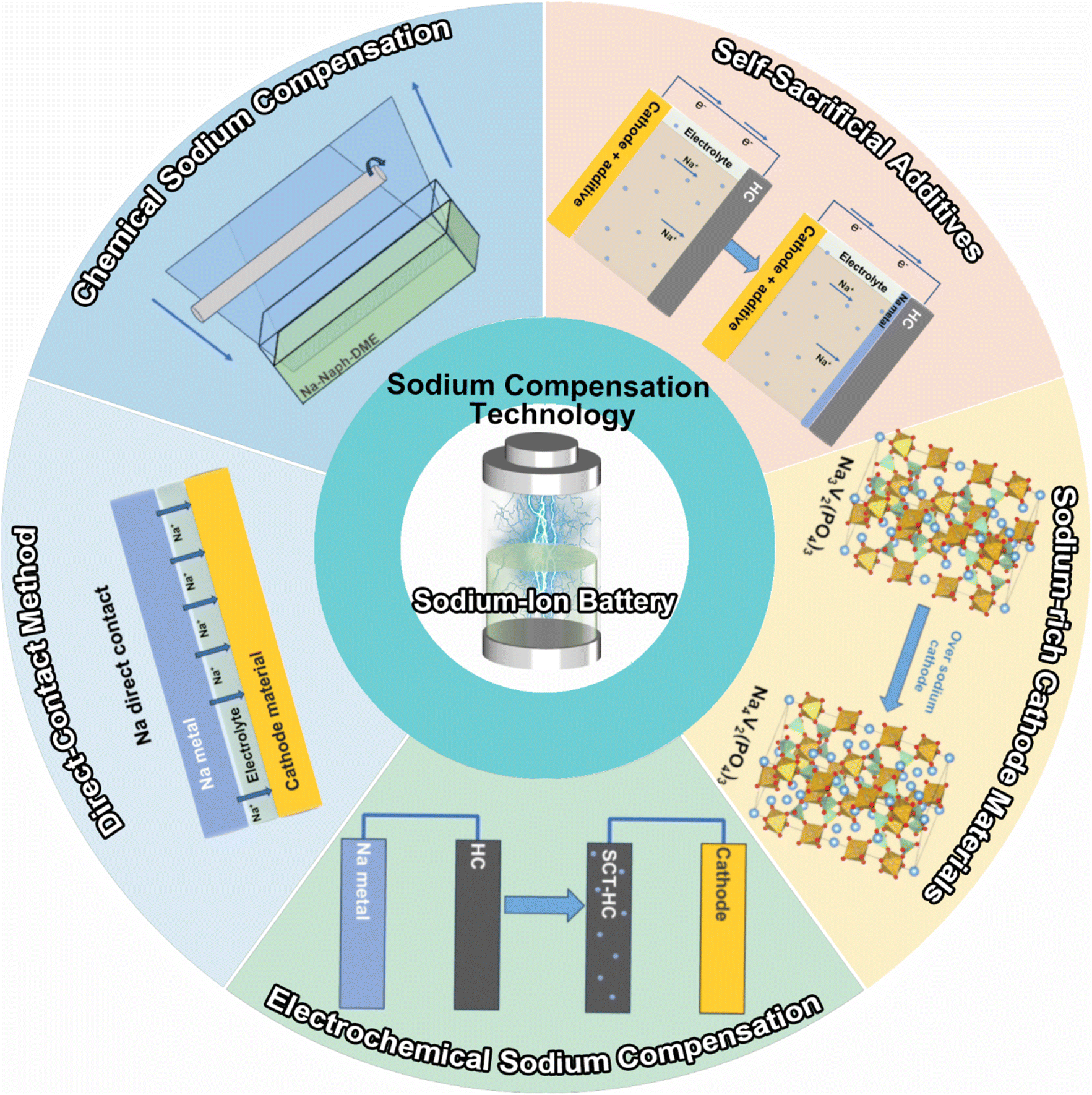
The direct contact sodium compensation method is performed by directly contacting sodium metal with an electrode (e.g., hard carbon) using a small amount of electrolyte. To illustrate, sodium metal is placed in direct contact with the anode after adding a small amount of electrolyte (e.g., 0.5 ml) and applying external pressure for a certain period (e.g., 30 min). Using the large potential difference (e.g., 2.7 V) between the anode (hard carbon) and the sodium metal, sodium metal is converted into sodium ions by losing electrons. These sodium ions are subsequently driven into the anode by a current that compensates for the depletion of active sodium within it as displayed in Fig. 2a. This process can occur at room temperature or elevated temperatures, depending on the materials and the desired rate of reaction. The sodium ions migrate into the electrode materials, occupying sites within the structure that will later accommodate sodium during battery operation. And they can react with the electrode materials forming sodium compounds (e.g., Na2Ti3O7 from Ti3O7) that are stable within the electrode matrix. These compounds act as sodium reservoirs, providing sodium ions upon battery operation. This reaction is hinged on the electrochemical self-discharge principle, and the extent of sodium compensation can be roughly controlled by adjusting the length of the contact time.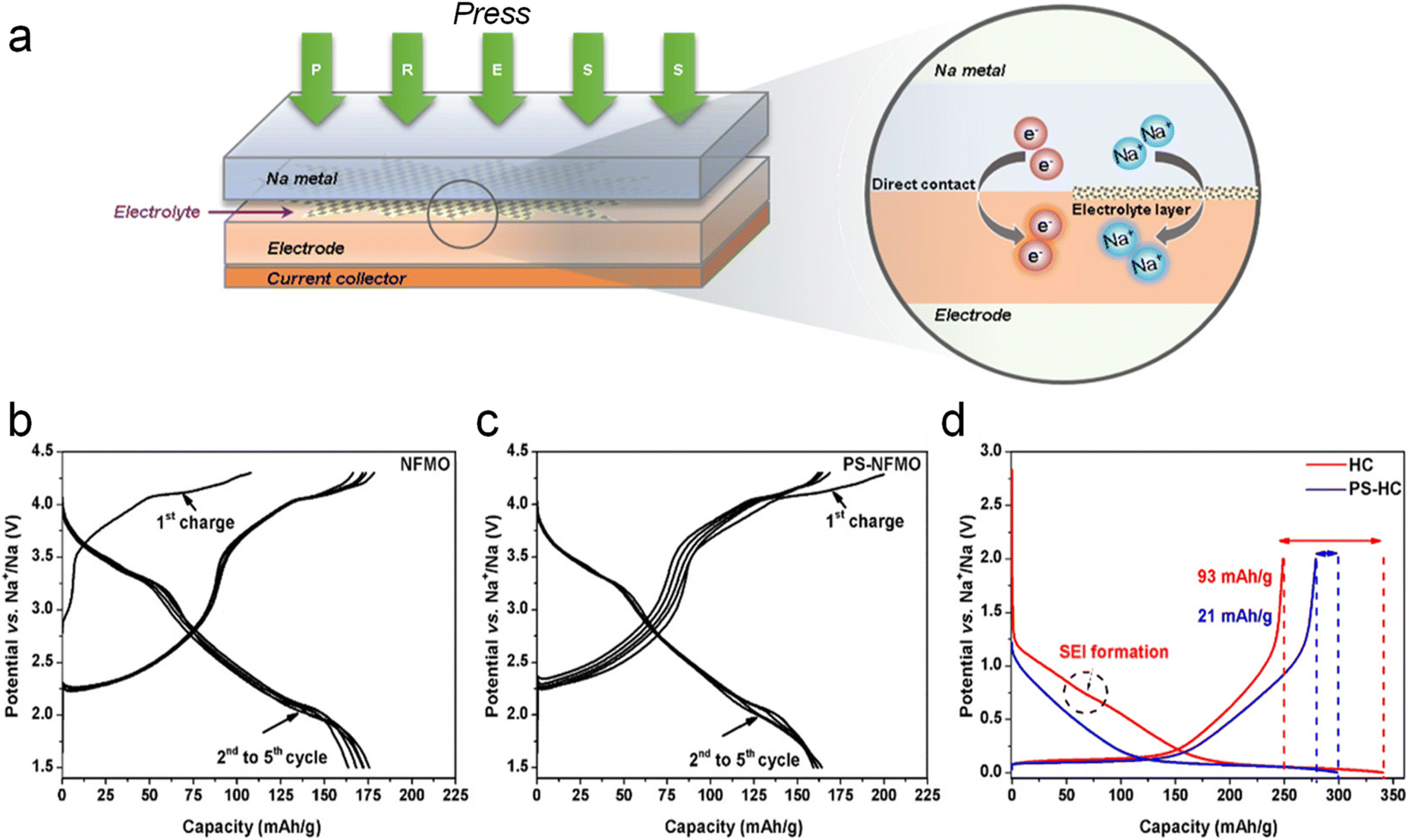 | ||
| Fig. 2 (a) Schematic illustration of direct contact SCT. Charge–discharge profiles of Na0.67Fe0.5Mn0.5O2 (b) before and (c) after sodium compensation. (d) Galvanostatic charge–discharge curves of HC electrodes with/without SCT. Reproduced with permission.30 Copyright 2019, American Chemical Society. | ||
Moeez et al.30 proposed the use of Na metal in direct contact with a hard carbon (HC) anode and a sodium-deficient P2-type Na0.67Fe0.5Mn0.5O2 cathode, respectively, which not only succeeded in compensating for the sodium loss on the anode side, but also provided the cathode with more active sodium ions. It is found that a favorable passivating layer can be formed on the cathode surface to suppress material dissolution. As such, the initial charging capacity of the Na0.67Fe0.5Mn0.5O2 cathode (Fig. 2b) was increased from 109 mA h g−1 to 200 mA h g−1 (SCT-treated Na0.79Fe0.5Mn0.5O2; Fig. 2c); the ICE of the HC electrode was improved from 73% to 94% with a significant reduction of the irreversible capacity loss by 70 mA h g−1 (Fig. 2d).
It shows that the direct contact method can pre-introduce active sodium ions into the electrode material by a facile and effective process. However, it is difficult to accurately control the SCT extent of the electrode material. This is due to the fact that for electrode materials, when the SCT extent is high, sodium can be easily precipitated at the anode side, resulting in safety hazards; a low SCT extent, nevertheless, is insufficient to make up for the loss of active sodium.
2.2. Electrochemical sodium compensation
Electrochemical sodium compensation is reported to accurately adjust the extent of SCT by controlling the current density and cut-off voltage of the half cell. The prepared electrode (e.g., HC) is pre-cycled against a metallic sodium counter electrode to drive sodium ions into the electrode material by an external voltage, followed by removing the SCT-treated electrode for further full-cell assembly.Dating back to 2015, Wang et al.31 pioneeringly reported this SCT on the hard carbon anode in a three-electrode configuration (HC serves as the working electrode, and Na foil is the reference/counter electrode) under 10 mA g−1 as shown in Fig. 3a. Compared with the pristine HC‖NaNi0.5Ti0.5O2 full cell, the ICE of the SCT treated one increased from 64.5% to 73.0% (Fig. 3b). It also presented better cycling stability with 72.0% capacity retention after 100 cycles, higher than the 69.5% of the pristine cell. Inspired by this work, loads of reports adopted this SCT to pre-introduce enough active sodium ions into HC anodes and to form a favorable SEI layer with Na metal before assembling full cells in order to accurately evaluate the real application potential of the as-prepared cathodes.19,33–38
 | ||
| Fig. 3 Electrochemical sodium compensation method. (a) Schematic diagram of a three-electrode configuration toward the sodium compensation of a hard carbon anode and (b) the initial charge–discharge curve of the full cell with the NaNi0.5Ti0.5O2 cathode and HC anodes with/without SCT. Reproduced with permission.31 Copyright 2015, the Royal Society of Chemistry. (c) Schematic diagram of phase transformation of P2-Na0.67Li0.1Fe0.37Mn0.53O2 to P′2-type Na0.99Li0.1Fe0.37Mn0.53O2 after SCT treatment and corresponding cycling performance comparison. Reproduced with permission.26 Copyright 2024, American Chemical Society. (d) Seven HC electrodes with different SOCs and corresponding potentials. Reproduced with permission.32 Copyright 2021, Elsevier. | ||
Other than HC anodes, this SCT can be employed in cathodes as well. Notably, P2-type layered oxides hold great promise as cathode materials for SIBs due to the outstanding rate capability and cycling stability. The main obstacle for them lies in the intrinsic Na deficiency (e.g., Na2/3Ni1/3Mn2/3O2), greatly limiting the capacity performance of the full cell since it is the only source of active sodium in the full-cell configuration. In this regard, Fang et al.26 very recently precisely injected 0.32 excess active sodium per chemical formula unit into P2-phase Na0.67Li0.1Fe0.37Mn0.53O2 to realize P′2-type Na0.99Li0.1Fe0.37Mn0.53O2 by using the electrochemical SCT (discharging the Na‖Na0.67Li0.1Fe0.37Mn0.53O2 half battery to 1.5 V). It is reported that this process significantly improved the specific capacity from 61 mA h g−1 to 125 mA h g−1 for full cells coupled with HC anodes. It also presented noticeably better cycling performance compared to not only the pristine full cell but also the full cell with an SCT-treated HC anode (Fig. 3c).
With the purpose of disclosing the relationship between the sodium compensation degree of electrodes and full-cell performance, Pi et al.32 performed this SCT on the HC electrode with Na metal in half cells and subjected them to a certain state of charge (SOC) at a constant current rate of ∼0.05C (Fig. 3d), followed by removing the SCT treated HC and coupling it with an Na3V2(PO4)2F3 cathode to build a full cell. It is found that the electrochemical behavior of the HC‖Na3V2(PO4)2F3 full cell is highly dependent on different SOCs (i.e., sodium compensation degree). The 0% SOC (fully discharged) HC could enable an optimal electrochemical performance: an initial discharge capacity of 114.1 mA h g−1 and a capacity retention of 71.8% throughout 600 cycles at 10C. This HC is able to offer sufficient active sodium for compensating for the loss of active sodium from SEI generation and irreversible intercalated sodium sites. This work sufficiently proves the advantage of electrochemical SCT in controlling the extent of sodium compensation.
Thus, electrochemical sodium compensation is an effective approach to improve the ICE of the electrode by modulating the external circuitry to precisely adjust the SCT degree and regulate the rate of SEI film formation, thus improving the performance of the subsequent full cell. In comparison to other SCTs, it may also maximally preserve the actual state of sodiation in the first cycle without causing additional undesirable reactions, which has the least detrimental impact on battery performance. However, half-cell disassembly and reassembly of the full cell greatly increase the process complexity and difficulty. In addition, the residual sodium metal in the process has high reactivity and is difficult to handle, increasing the overall cost and limiting its practical application.
2.3. Physical sodium compensation of the sodium-based metal
In comparison to the above two SCTs, the potential for large-scale application of sodium metal physical SCT is higher. This is attributed to the high theoretical capacity of sodium (1166 mA h g−1) and low redox potential (−2.741 V versus standard hydrogen electrode), which can be directly physically compounded with the electrode active material by using a small amount of sodium metallic powder or sodium foil to quantitatively and controllably pretreat the electrode with active sodium. Several sodium metal physical SCTs have been developed.In 2016, Zhang et al.39 for the first time reported a facile and scalable ball milling method on Sb and P powders to prepare various Na-based alloys such as Na3Sb and Na3P as the sacrificial sodium source with high initial charging capacities (Fig. 4a). Specifically, Na3P and Na3Sb powders were obtained through a solid-state reaction by ball milling stoichiometric amounts of bulk metallic sodium red phosphorus (P) or antimony (Sb) powders respectively in an argon atmosphere to prevent oxidation of sodium for 2 hours based on a ball-to-material mass ratio of 35. The underlying mechanism can be the solid-state alloying reaction, which allows sodium atoms to diffuse into the lattice of P or Sb, forming Na3P and Na3Sb alloys respectively under high-energy ball-milling conditions. These compounds are formed through direct combination reactions: 3Na + P → Na3P; 3Na + Sb → Na3Sb. In addition, this approach is also suitable for sodium compensation of a sodium-deficient insertion layered oxide (Na0.67Fe0.5Mn0.5O2) and polyanion compound (Na3V2(PO4)2F3) to build pure Na1Fe0.5Mn0.5O2 and Na4V2(PO4)2F3 phases with a noticeable capacity improvement. The as-prepared Na-rich cathode materials effectively make up for sodium loss from the SEI formation process and thus enable a substantial increase in energy density of full cells when coupled with HC anodes. The HC‖Na1Fe0.5Mn0.5O2 full cell presented an increased discharge capacity from 71 mA h g−1 to 128 mA h g−1, and the energy density was boosted by 30% compared to the pristine one.
 | ||
| Fig. 4 (a) Charge–discharge profiles of Na0.67[Fe0.5Mn0.5]O2 and Na1[Fe0.5Mn0.5]O2 by ball milling with Na metal. Reproduced with permission.39 Copyright 2016, Springer Nature. (b) Pulsed ultrasonic SCT dispersion method. Reproduced with permission.40 Copyright 2018, Elsevier. (c) Diagrammatic illustration of the metallurgical pre-alloying process of Sn–Na. Reproduced with permission.41 Copyright 2019, American Chemical Society. (d) Schematic illustration of solid-state electrochemical SCT on the HC electrode. Reproduced with permission.42 Copyright 2024, Wiley-VCH. | ||
Additionally, Tang et al.40 proposed an ultrasonic dispersion method for SCT on HC anodes (Fig. 4b). Specifically, Na chunks in mineral oil were melted at 98 °C and ultrasonically treated to form a hexane-based Na powder suspension (2–16 μm in size) in an inert atmosphere, followed by a spin coating process on the HC anode surface. It decreases irreversible ICE to 8% from the initial 19.3%. ∼5% increment in energy density of the HC‖NaCrO2 full cell is thus realized.
Furthermore, Liu et al.43 performed SCT on a Sn anode using a roll-to-roll process to metallurgically generate Na–Sn alloys in situ from sodium metal foil and Sn anodes (Fig. 4c). To perform the metallurgical pre-alloying process of sodium, Na foil was overlayed on a Sn foil and the two foils were rolled to form a Na–Sn alloy. This is based on the solid-state alloying reaction Na + Sn → Na–Sn. It has been found that when pure Sn foil was used as the anode, Sn easily catalyzed the rapid decomposition of the electrolyte to produce large amounts of CO2 and H2 gases at a specific voltage. An excessively thick SEI film on the electrode surface deactivated the Sn electrode accordingly. In contrast, the as-formed Sn–Na alloy lowers the open-circuit potential, thus inhibiting the gas-related side reactions. Thus, the ICE of the Sn–Na‖Na3V2(PO4)2F3 full cell increased significantly from 24.68% to 75%. In addition, the Na–Sn alloy presents a good air stability after exposure to air for 2 h. It has good potential for large-scale application considering the operability of the roll-to-roll process.
Wang et al.42 very recently reported a new vacuum thermal evaporation SCT method on HC anodes by controlling the thickness of deposited sodium metal to 1 and 2.3 μm from a quartz crystal microbalance, followed by being aged for 3–5 days under an Ar atmosphere (Fig. 4d). The chemical potential difference between Na metal and HC drives the sodium compensation process and brings about a thin Na2O SEI layer for fast Na+ transport. The full cell with the Na3V2(PO4)3 cathode presents an improved capacity (from 66.1 mA h g−1 to 102.2 mA h g−1), a boosted ICE (from 61% to 94%), and enhanced capacity retention. Besides, the as-prepared HC displays good air stability. This technology can accurately adjust the SCT degree and avoids direct usage of metallic sodium, which should be paid more attention in practical application.
Although sodium-based metal physical SCT is simple, the residual sodium metal will easily turn into “dead sodium”, leading to an increase in the polarization of the battery and sodium precipitation phenomenon. And sodium powder or sodium foil has technical difficulties (soft texture) and safety risks (high reactivity; high sensitivity to water and air) during production, transportation and use. How to improve their air stability is the key to realize their large-scale application.
2.4. Chemical sodium compensation by a Na complex
The high reactivity and risky properties of metallic sodium should be kept in mind. Instead, chemical sodium compensation by a Na complex is considered one of the most promising SCTs by virtue of its safety, practicality, and ease of industrialization, and thus has received increasing attention in recent years. This is achieved by employing the large potential difference between the chemical sodiation reagent and electrode material. Chemical sodiation reagents are usually generated in situ from sodium metal and aromatic compounds in organic solvents. Commonly used aromatic compounds include biphenyl (Biph), naphthalene (Naph), etc., and the organic solvents usually are ethylene glycol dimethyl ether (DME), tetrahydrofuran (THF), etc. In the process, the electrons and sodium ions of the chemical sodiation reagent will be automatically transferred to the electrode material (e.g., HC) for sodium compensation within a short period due to the strong reducing capability of the chemical sodiation reagent. Accurate control of the SCT depth (ICE can reach ∼100%) can be attained by controlling the reaction time and temperature as well as the variety and concentration of the sodiation reagents.In 2019, Cao et al.44 performed chemical SCT on a Na2Ti6O13 anode by immersing it into sodium naphthalene solutions (Na-Naph-DME) at different concentrations. The Na-Naph@DME solution was obtained by adding naphthalene to ethylene glycol dimethyl ether (DME) with the same number of moles of metallic sodium. Na2Ti6O13 was then put into the above solution for sodium compensation. The ICE of the pretreated Na2Ti6O13‖Na3V2(PO4)3 full cell was increased from 40% to 80%.
Afterwards, Wang et al.45 reported a roll-to-roll chemical SCT by simultaneously immersing a P2-type Na0.67Fe0.1Al0.1Mn0.8O2 cathode and HC anode in a sodium (Na)–naphthalene (NP)@tetrahydropyran (THP) solution for a certain period (Fig. 5a). This strategy builds favorable interphases around both electrodes, which not only facilitate interfacial charge transfer but also mitigate notorious side reactions (e.g., transition metal dissolution, electrolyte decomposition, and destruction of the CEI/SEI). As such, the HC‖Na0.67Fe0.1Al0.1Mn0.8O2 pouch cell presented excellent capacity retention of 93.4% throughout 100 cycles. However, it is still faced with the problems that the immersion process requires a large amount of SCT reagent and the residual reagent on the electrode surface is difficult to be completely removed.
 | ||
| Fig. 5 (a) Schematic of the Na–NP@THP solution-based chemical sodium compensation method for a commercial roll-to-roll process. Reproduced with permission.45 Copyright 2023, Wiley-VCH. (b) Illustration of spraying the Na–Naph@THF solution manufacturing process. Reproduced with permission.46 Copyright 2019, Wiley-VCH. (c) Electrostatic potential mapping and (d) boiling and flashing points of various solvents. Reproduced with permission.47 Copyright 2024, Wiley-VCH. (e) HOMO energy levels of anionic radicals of PAHs and aromatic ketone. Reproduced with permission.48 Copyright 2024, Wiley-VCH. | ||
In this regard, Sun et al.46 reported a simple but efficient spraying approach. Sodium metal and naphthalene (Naph) with an equal molar amount were added into tetrahydrofuran (THF) to form Na–Naph@THF solution. And then a certain amount of this strongly reductive solution was directly sprayed onto the HC surface for sodium compensation. During this treatment, the residual reagent (i.e., naphthalene and tetrahydrofuran) was evaporated for recycling to reduce the impact on the battery (Fig. 5b). The entire process not only achieves sodium compensation, but also recycles organic matter during the drying process, which greatly reduces the process cost. The ICE of the SCT-treated HC was increased from 67% to 87%. The energy density of the full cell with a Na0.9[Cu0.22Fe0.30Mn0.48]O2 cathode was increased from 141 W h kg−1 to 240 W h kg−1.
Nevertheless, the redox potential of Na-Naph@THF is high (∼0.2 V versus Na+/Na) and can only partially SCT-treat the electrode, impossibly achieving a 100% ICE. Alternatively, a sodium compensation reagent with a lower desodiation potential of 0.12 V (Na–Bp@DME; Bp = biphenyl) was developed by Liu et al.49 The ICE of HC thus achieved 100%, and a uniform and dense SEI film was formed on the surface, which is conducive to the improvement of its air stability. The energy density of the HC‖Na3V2(PO4)2 full cell was increased from 120 W h kg−1 to 218 W h kg−1. Afterwards, Li et al.50 reported a Na–Bp@THF solution presenting a lower desodiation potential of 0.095 V to effectively improve the ICE of Sn and P anodes. Qin et al.51 further developed an in situ electrochemical SCT via introducing Na–Bp@DME reagent as an electrolyte additive into a DME-based ether electrolyte. It realizes sodium compensation on HC anodes with a ∼100% ICE due to the theoretical calculation result that the lowest unoccupied molecular orbital (LUMO) energy level of HC resembles the highest occupied molecular orbital (HOMO) energy level of Na–Bp reagent and their symmetry matches well.
However, an extremely low redox potential of polycyclic aromatic hydrocarbons (PAHs) will cause the electrode structure to deteriorate and incur sodium accumulation with safety hazards. Zhang et al.48 very recently designed mild chemical SCT reagents' molecular structure via introducing a carbonyl group in PAHs to balance the conjugated and inductive effect. Benzophenone (BP) and 9-fluorenoneb (9-FN) stand out as the optimal candidates with favorable redox potentials of 1.07 V and 1.55 V (versus Na+/Na), respectively (Fig. 5e). 9-FN manifests versatile sodium compensating abilities for various cathode materials including layered Na0.67Ni0.33Mn0.67O2, tunneled Na0.44MnO2, polyanionic Na3V2(PO4)2F3, Na3V2(PO4)3, and Na4Fe2,91(PO4)2P2O7 with greatly boosted initial charging capacity to compensate for sodium loss in the anode side. Fang et al.28 also reported a sodium diphenyl ketone in dimethyl ether (Na-DK@DME) reagent featuring a redox potential of 0.55 V for sodium compensation of HC anodes with a greatly boosted ICE of 99.2%.
On the other hand, practical application is hindered by the high reactivity of presodiated electrodes. Also, there are safety issues for widespread use due to the volatility of the toxic aromatic reagents, low flash point and boiling point of the commonly used ether solvents, and the use of sodium metal in the production of sodium-containing aromatic radicals, which deserves more attention. Thus, Gao et al.53 applied sodium bis(2-methoxyethoxy) aluminum hydride (i.e., NaAlH2(OCH2CH2OCH3)2) for sodium compensation on HC anodes with a high ICE of 94.6%, and they found that a passivated Al2O3 nanolayer was in situ built to endow SCT-treated HC anodes with good air stability. Man et al.47 molecularly designed linear ether solvent and screened out tetraethylene glycol dimethyl ether (TEGDME) as an optimal candidate due to its high boiling point of 276 °C and flash point of 141 °C (versus 84 and −2 °C for DME) to intrinsically enhance the safety (Fig. 5c and d). The designed sodium 4-methylbiphenyl (4-MBP) in TEGDME (i.e., Na-4-MBP@TEGDME) SCT reagent greatly improved the ICE of HC from 65.28% to 99.1%.
In general, chemical sodium compensation can be controlled by adjusting the concentration of the Na complex solution and the reaction time. The danger associated with the strong activity of Na metal can be minimized. The spraying of the Na-complex solution on the electrode surface facilitates efficient and homogeneous sodium compensation compared to the other methods mentioned above, and the roll-to-roll process helps in the mass production of sodiated electrodes. In addition, sodium compensation reagents can be used for various types of electrode materials (Table 1), and the recycling of organic reagents through process optimization can significantly reduce costs. However, the air instability of treated electrodes remains to be solved.
| Electrode | Sodium compensation reagent | ICE of the half-cell before/after SCT | ICE of the full cell after sodium compensation | Ref. |
|---|---|---|---|---|
| a Naph (sodium naphthaline); DME (methoxymethane); THF (tetrahydrofuran); NaBiph (sodium biphenyl). | ||||
| Na2Ti6O13 | Na–Naph@DME | 65%/100% | 80.0% (Na2Ti6O13‖Na3V2(PO4)3) | 44 |
| Na2Ti3O7 | ∼43%/∼100% | None | 44 | |
| TiO2 | ∼75%/∼100% | None | 44 | |
| Hard carbon | Na–Naph@THF | 67%/87% | 92.9% (HC‖Na0.9[Cu0.22Fe0.30Mn0.48]O2) | 46 |
| Sb | Na–Biph@DME | 75%/∼100% | 95.4% (Sb‖Na3V2(PO4)3) | 52 |
| Sn | Na–Biph/THF | 70%/94% | None | 50 |
| P/C | 64%/94% | None | 50 | |
Different methods discussed above possess unique strengths and weaknesses. The direct contact method and the sodium-metal physical mixing approach offer advantages such as easy control, a high sodium compensation capacity, and a sufficiently low redox potential. However, their reliance on sodium metal in the preparation process presents significant safety hazards. The high reactivity of sodium metal complicates precise control of sodium compensation in the direct contact method, leading to non-uniform SEI film formation and potential material degradation. To achieve a uniform SEI film, the electrochemical sodium compensation method controls sodium compensation precisely by assembling a half-cell to regulate current density and charging time. Nevertheless, the method's complexity, involving the assembly and disassembly of half-cells, hinders its scalability. Moreover, residual metallic sodium after disassembly remains a safety concern.
In contrast, the chemical sodium compensation by a Na complex method offers simplicity and easy control of the SCT degree by adjusting reaction time. By utilizing sodium compounds instead of sodium metal, it avoids the safety hazards associated with metallic sodium. However, challenges persist such as sensitivity of sodium compounds to air and insufficient redox potential, which require resolution for optimal performance.
In summary, each sodium compensation method for SEI film formation presents trade-offs in terms of control, safety, and technical feasibility. The choice of method depends on specific application requirements, including safety considerations, scalability, and desired SEI film characteristics. Continued research and development are essential to address these challenges and advance battery technology effectively.
The above sodium compensation methods use sodium metal as a sodium source to provide Na+ ions or to form a strongly reducing sodium compensation reagent, which incurs safety hazards. In contrast, sodium compensation additives applying cathode materials are a promising and relatively safer strategy for compensating for the initial active sodium loss. It achieves sodium compensation by an extra amount of active sodium ions during the initial charging process and it can be categorized as self-sacrificial additives and sodium-rich cathode materials.
2.5. Self-sacrificial additives
The ideal sodium compensation additives should feature the following characteristics: (i) high sodium compensation capacity (>350 mA h g−1) to provide sufficient active sodium ions; (ii) appropriate decomposition potential for irreversible desodiation within the operating voltage range of the cathode; (iii) the additive itself and oxidative by-product have no impact on the subsequent cycling of the battery; (iv) technological compatibility with current processes. The performance evaluation of sodium compensation additives mainly focuses on these four perspectives. There are many types of additives for the cathode, which can be roughly divided into two categories: inorganic and organic. | ||
| Fig. 6 (a) Schematic illustration of the self-sacrificial additive strategy. (b) Schematic diagram of electrodes prepared by spraying a slurry containing Na2O2. Reproduced with permission.56 Copyright 2021, American Chemical Society. (c) Crystal structure of Na2NiO2. Reproduced with permission.57 Copyright 2015, American Chemical Society. (d) Schematic illustration of a separator coated by Na3P@rGO sodium compensation additives. Reproduced with permission.58 Copyright 2023, Wiley-VCH. | ||
Many other SCT additives similar to NaN3 were subsequently reported such as NaBH4,59 NaNH2,60 Na2O,61 Na2O2,56 Na2CO3,62 and NaNO2.63 Gas evolution, however, remains as a main obstacle and will lead to the formation of pores on the surface of the electrode, which is not conducive to the structural stability of the electrode and battery safety (H2 gas from NaBH4 or NaNH2). In this regard, Guo et al.56 developed a spraying process by spraying a slurry of acetonitrile solution containing Na2O2 additives to form a Na2O2 sodium compensation layer onto the surface of the dry Na2/3Ni1/3Mn1/3Ti1/3O2 cathode (Fig. 6b). Upon charging, this SCT layer will decompose at 4 V (versus Na+/Na; Na2O2 → 2Na+ + O2↑ + 2e−) to provide active sodium ions (charging capacity: 421.5 mA h g−1) and more importantly, avoids the porous structure of the underlying electrode by O2 gas evolution. Otherwise, O2 gas was reported to react with the SCT-treated electrode vigorously to deplete the active sodium ions of the battery, which accounts for the low utilization extent of this type of additive. The initial discharge capacity of the HC‖Na2/3Ni1/3Mn1/3Ti1/3O2 with 20% Na2O2 was 81.7 mA h g−1, and the capacity retention was 84.1% after 80 cycles at 0.5C, which were significantly higher than those of the pristine full cell (60.3 mA h g−1 and 61.3%).
To avoid the negative effect of the gas byproduct on the electrode, some other inorganic sodium compensation additives including Na3P, Na2S, Na2NiO2, etc. without generating gas entered the researchers' field of vision. Park et al.57 in 2015 applied orthorhombic Na2NiO2 (Fig. 6c) as a cathode SCT additive (Na2NiO2 → NaNiO2 + Na+ + e−, at 2.1 V versus Na+/Na) with a charging capacity of 285 mA h g−1. Only 10 wt% Na2NiO2 additive into the NaCrO2 cathode is able to increase the reversible capacity of the Sb/C‖NaCrO2 full cell from 74.6 mA h g−1 to 92.1 mA h g−1. However, the resultant crystalline NaNiO2 can serve as a cathode active species showing a low reversible capacity of 88.7 mA h g−1 in the subsequent cycles, which greatly reduced the sodium replenishment effect of the additive.
Considering the high capacity of 804 mA h g−1 of Na3P, Mao et al.58 reported a separator capacity-compensation strategy by combining Na3P with a conductive rGO coating layer, which was located on the side of the separator facing the cathode (Fig. 6d). It is found that Na3P starts to desodiate at 4.05 V versus Na+/Na with an actual capacity of 478 mA h g−1. The initial specific capacity of the HC‖NaNi1/3Fe1/3Mn1/3O2 full cell with the Na3P@rGO-coated separator was increased to 128.2 mA h g−1 from 108.5 mA h g−1 (i.e., 18.2%). This strategy can be also applied in the HC‖Na3V2(PO4)3 full cell with a capacity increase of 36.2%. Furthermore, they identified that the remaining phosphorus after the decomposition of Na3P could prevent oxygen evolution from the cathode at high voltages due to its high reactivity with active oxygen species, thus boosting battery safety. However, Na3P has high reactivity to air, and is easily converted into highly toxic PH3, which impede its use.
Comparatively, Na2S seems to be better than Na3P as an alternative sodium compensating additive, effectively avoiding the generation of toxic gases. The sodium compensation capacity of Na2S is quite close to the theoretical capacity of 687 mA h g−1.64 Another candidate is Na2S. However, the decomposition of Na2S produces electronically and ionically insulated sulfur and polysulfides, which will increase battery resistance. Besides, polysulfide dissolution in the electrolyte is unfavorable for subsequent cycling stability and self-discharge of the battery. To address the above problems, Hu et al.65 proposed to transform Na2SO4 into Na2S in situ encapsulated in a carbon matrix (Na2S-4) with a boosted electronic conductivity and smaller particle size than the commercial counterpart. As a sodium compensation additive, the Na2S/C composite was able to achieve an initial charging capacity of 407 mA h g−1 at 3.8 V versus Na+/Na. The addition of 10 wt% Na2S/C into the Na3V2(PO4)3 cathode endows the HC‖Na3V2(PO4)3 pouch cell with a capacity increase of 24.3% and with an increment in the energy density from 117.2 to 138.6 W h kg−1. Moreover, the Na2S additive doesn't produce gas upon charging and its desodiated product has no negative influence on the subsequent cycling stability and rate capability of SIBs. Although the inorganic sodium compensation additive can effectively improve the energy density of the battery, there are still problems such as residual and gas formation after SCT treatment and high price, which restrict its large-scale production. Table 2 compares various types of inorganic SCT additives from different aspects.
| Additive | Potential (V versus Na+/Na) | Theoretical/actual capacity (mA h g−1) | Main product | Problem | Ref. | |
|---|---|---|---|---|---|---|
| Inorganic additive | NaN3 | 3.55 | 412/315 | N2 | Explosive | 66 |
| Na3P | 0.5 | 804/600 | P | Toxic and flammable | 39 and 65 | |
| Na2O | 2.5–4.2 | 864/500 | O2 | Production of H2 | 67 | |
| Na2O2 | 4 | 687/421 | O2 | Production of O2 | ||
| NaNO2 | 3.3–3.8 | 427/350 | NO2 | Production of NO2 | 63 | |
| Na2CO3 | 4 | 506/253 | CO2, O2 | Production of O2 | 62 | |
| Na2NiO2 | 2.0–3.6 | 392/277 | NaNiO2 | Limited sodium compensation | 57 | |
| NaCrO2 | 3.0–4.2 | 251/229 | Na0.06CrO2 | Limited sodium compensation | 68 | |
| NaBH4 | 2.4 | 708/750 | B, H2 | Production of H2 | 59 | |
| NaNH2 | 3.8 | 686/680 | N2H4, N2, H2 | Production of H2 and N2H4 with strong reduction and instability | 60 | |
| Na2S | 3.8 | 687/553 | S | Low electronic conductivity | 64 |
Organic carbonyl compounds are usually electrochemically unstable at high voltages and undergo irreversible oxidative desodiation reactions and thus are widely used as sodium compensation additives. In 2017, Lee et al.69 for the first time discovered that the practical reversible capacity of disodium rhodizonate (Na2C6O6) is substantially lower than the theoretical specific value (501 mA h g−1). The irreversible phase transition of Na2C6O6 from the γ- to α-phase during the initial cycle accounts for the deterioration of its redox activity as displayed in Fig. 7a. Inspired by this, Zou et al.70 applied Na2C6O6 as a sodium compensation additive as shown in Fig. 7b. In typical carbonate-based battery electrolytes, Na2C6O6 with its stable sodium–oxygen bond is weakly soluble, but its desodiation product (cyclohexanehexone) is neutral and easily soluble, which may ensure the forward desodiation reaction. The actual charging capacity of Na2C6O6 was 309.8 mA h g−1 at 3.98 V versus Na+/Na, which is higher than the two-electron theoretical value of 250 mA h g−1 and indicates that there are some unknown side reactions. Thus, Marelli et al.74 employed online electrochemical mass spectroscopy, vibrational spectroscopy, and in situ XRD to further disclose that the products (rhodizonate) of Na2C6O6 desodiation catalyze the decomposition of the electrolyte or rhodizonate is further oxidized.
 | ||
| Fig. 7 (a) A schematic of the sodium storage mechanism of Na2C6O6. Reproduced with permission.69 Copyright 2017, Springer Nature. (b) Scheme of the desodiation of Na2C6O6. Reproduced with permission.70 Copyright 2020, Wiley-VCH. (c) Schematic illustration of the Na compensation mechanism in P2-NLNMO@AC by acetic acid. Reproduced with permission.71 Copyright 2023, American Chemical Society. (d) DTPA-5Na sodium compensation schematic. Reproduced with permission.72 Copyright 2020, Elsevier. (e) Schematic diagram of sodium compensation concept by Na2C4O4. Reproduced with permission.73 Copyright 2018, Wiley-VCH. | ||
Recently, Zhang et al.71 neutralized surficial residual alkalis and thus formed sodium acetate (CH3COONa, AC-Na, Fig. 7c) by introducing acetic acid (AC) into layered cathode materials, which realized the goal of “turning the waste into wealth”. AC-Na, as a Na-compensated additive, has a high specific capacity of 300 mA h g−1 and an oxidation potential of 4.1 V versus Na+/Na. Based on sodium compensation, the 2 Ah-level HC‖Na0.85Li0.12Ni0.22Mn0.66O2@AC pouch cell presented a capacity retention of 95.1% over 120 cycles and achieved an energy density increase from 112 to 130 W h kg−1.
In response to possible electrolyte decomposition at such a relatively high oxidation voltage of 4.1 V, Zou et al.75 afterwards effectively lowered the oxidation voltage plateau of sodium carboxylate to 3.45 V versus Na+/Na by molecularly optimizing the conjugated aromatic architecture and introducing a stronger electron-donating substituent and thus sodium para-aminobenzoate (PABZ-Na) was proposed. Ethylenediaminetetraacetic acid tetrasodium salt (EDTA-4Na)76 and penta-sodium diethylenetriamine pentaacetate acid salt (DTPA-5Na)72 (Fig. 7d) have also been reported by Jo et al.72 as SCT additives to show charging capacities of 420 mA h g−1 at 3.79 V and 363 mA h g−1 at 3.6 V versus Na+/Na, respectively. But both of them produce H2O during the desodiation process and H2O will be further decomposed into H2 and O2, which will threaten battery safety and cycling stability.
Shanmukaraj et al.73 proposed Na2C4O4 as an SCT additive with a charging capacity of 256 mA h g−1 at 3.6 V versus Na+/Na (Na2C4O4 → 2CO2↑ + 2C + 2e− + 2Na+) (Fig. 7e). The HC‖Na3(VOPO4)2F full cell with 30 wt% Na2C4O4 additives achieved an initial discharge capacity of 120 mA h g−1 at 0.05C, which is 50% higher than that of the unpretreated full cell (80 mA h g−1). A better battery performance was achieved compared to the NaN3-added cell, which was ascribed to the cathode porosity increase by the as-formed CO2. Thus, Pan et al.77 further investigated the decomposition process of Na2C4O4 by performing nitrogen adsorption, Raman spectroscopy and electrochemical mass spectrometry. A new decomposition mechanism of Na2C4O4 was identified: C4O42− ↔ 1.47CO2 + 1.06CO + 1.47C + 2e−. Upon charging, the generated CO was further partially proportionated into C and CO2 catalyzed by activated carbon in the electrode, which significantly improved the conductivity. Besides, the formed CO2 tends to react with the sodiation electrode to build the Na2CO3 passivating layer to effectively prolong battery lifespan.
For organic sodium compensation additives, a greater molecular weight means a lower theoretical sodium compensation capacity. Reducing the molecular weight from functional groups and increasing the proportion of sodium content is one of the effective ways to increase the sodium compensation capacity of additives. Niu et al.78 further designed and synthesized sodium oxalate (Na2C2O4) as a lightweight SCT additive with a low oxidation potential of 3.97 V versus Na+/Na and a high charging capacity of 394.6 mA h g−1. They also found that the incorporation of conductive carbon or noncarbon-based catalysts can effectively reduce the oxidation potential of sodium-compensated additives (from 4.41 to 3.97 V in this case). Similarly, Shanmukaraj et al.79 combined a sodium pyruvate (Na2C3O4) additive with KJ600 conductive carbon and then added it into a P2-Na2/3Mn0.8Fe0.1Ti0.1O2 cathode. The half-cell showed a discharge capacity of 164 mA h g−1 at 0.1C, which is 27% higher than that of the pristine one.
Table 3 compares various types of organic SCT additives. Therefore, it is foreseeable that molecular design of organic sodium compensation additives and more in-depth study of reaction mechanisms will be the future research direction.
| Additive | Potential (V versus Na+/Na) | Theoretical/actual capacity (mA h g−1) | Main product | Problem | Ref. | |
|---|---|---|---|---|---|---|
| Organic additive | Na2C6O6 | 3.6 | 250/310 | C6O6 | Copious side effect | 70 and 74 |
| Na2C6H2O6 | 3.98 | 250/265 | Unknown | Limited sodium compensation and copious side effect | 70 | |
| CH3COONa | 4.1 | 326/300 | C2H6, CO2 | High decomposition potential | 71 | |
| EDTA-4Na | 3.79 | 282/420 | CO, C3N, H2O, H2, O2 | Produce flammable gas and H2O | 76 | |
| DTPA-5Na | 3.61 | 266/363 | CO, C3N, H2O, H2, O2 | Produce flammable gas and H2O | 72 | |
| Na2C4O4 | 3.6 | 339/256 | CO2, CO, C | Low electronic conductivity | 73 | |
| Na2C2O4 | 3.97 | 400/394 | CO2 | Low electronic conductivity | 78 | |
| Na2C3O5 | 4 | 331/310 | C, CO2 | Low electronic conductivity | 79 |
2.6. Sodium-rich cathode materials
Although the sacrificial additive process is simple and convenient, it will inevitably generate some by-products (“dead mass”) and gases. They are prone to bring about safety problems and affect the subsequent cycles of the battery, limiting the practical application. Therefore, it has been proposed by some researchers that the direct use of some sodium-rich cathode materials can better realize sodium compensation, avoid adding other components in the cathode and decrease the negative effect of by-products.Sodium Superionic Conductor (NASICON)-type phosphates have emerged as a promising cathode material candidate by virtue of the sodium-rich composition (2–5 Na species per chemical formula unit), fast ionic conductivity, and robust structure.14,80 NaxV2(PO4)3 stands out due to the rich redox couples of V (V2+/V3+, V3+/V4+, and V4+/V5+). As presented in Fig. 8a, the most representative V-based NASICON (i.e., Na3V2(PO4)3, denoted as Na3VP) can be transformed to Na4VP and finally to Na5VP at 1.6 V and 0.3 V versus Na+/Na during the sodiation process.82 Each extra sodium ion in the formula can theoretically provide a capacity and 58.8 mA h g−1. Upon charging, Na3VP experiences a phase transition to Na1VP at 3.4 V versus Na+/Na and is the main phase during battery operation. Therefore, Na-rich phases including Na4VP and Na5VP are good choices as sodium compensation species for the sodium loss by HC anodes; after desodiation, the remaining Na3VP phase can reversibly function as a cathode (Na3VP ↔ Na1VP) in the subsequent cycles.
 | ||
| Fig. 8 (a) Schematic illustration for phase transformations of NaxV2(PO4)3 and corresponding electrochemical potentials. (b) Illustration of sodium compensation on NaxV2(PO4)3, battery assembly, and battery operation within two different voltage windows. Reproduced with permission.81 Copyright 2022, Wiley-VCH. | ||
Jian et al.83 and Mirza et al.84 adopted an electrochemical SCT against a sodium metal electrode as mentioned in the above section to discharge Na3V2(PO4)3 into a sodium-rich Na4V2(PO4)3 cathode. The intermediate Na4VP phase serves as an extra sodium source to compensate for HC and as a cathode. The energy density of the HC‖Na4V2(PO4)3 full cell is 265 W h kg−1, which is 76% higher than that of the Na3VP counterpart. However, as discussed above, this electrochemical SCT is faced with complex disassembling, rinsing, and battery assembly processes, which is quite time-consuming and not appropriate for mass production.
Thus, Wu et al.81 employed a direct-contact SCT to achieve Na4VP and Na5VP Na-rich phases as shown in Fig. 8b. They assembled a full cell based on a sodium-free anode (Al/C collector) and Na5VP cathode, achieving a high energy density of 400 W h kg−1 and a high-capacity retention of 93% after 130 cycles. Although this method simplifies the technological process, it is still hard to be applied in practical application since the sodium compensation extent is difficult to control and sodium metal may give rise to safety hazards.
Alternatively, the chemical method using a Na complex holds great promise in this regard due to its straightforward operation, high efficiency, high safety, and easy scalability. Liu et al.85 subjected Na3VP to Na–Bp@DME for 30 s to realize a Na-rich Na4VP phase hinged on the large potential difference between Bp−/Bp (0.12 V versus Na+/Na) and sodiation of Na3VP (1.65 V versus Na+/Na). This approach endowed an anode-free full cell with much improved capacity and prolonged cycling performance. Nonetheless, the Na–Bp@DME reagent may not be an ideal reagent for generating Na4VP owing to the potential mismatch to incur an over-extent of sodium compensation to form Na5VP (0.3 V versus 0.12 V for the Bp−/Bp redox). To this end, Xu et al.86 further proposed phenazine-sodium (PNZ-Na) to achieve the sodiation of the Na3VP precursor to build sodium-rich Na4VP based on a redox-potential matching principle. PNZ-Na stands out because of its suitable reducing potential of 1.56 V versus Na+/Na, which is significantly higher than the sodium-rich potential of Na5VP (0.28 V versus Na+/Na) but is slightly lower than that of Na4VP (1.67 V versus Na+/Na). The spontaneous sodium reaction is able to rapidly insert one Na+ ion into the Na3VP framework, obtaining a phase-pure Na4VP within only 90 s. The obtained HC‖Na4VP full cell exhibited a high energy density of 251 W h kg−1, which was 58% higher than the 159 W h kg−1 of the pristine one.
The above evidence shows that sodium-rich cathode materials are promising for sodium compensation. But this type of SCT is quite limited since there are some V-based NASICON materials that have been proposed, and the sodium compensation is significantly based on the materials themselves, which is hard to be applied in other systems with different cathodes and anodes. More sodium-rich materials deserve to be explored and synthesized.
3 Summary and outlook
Sodium-ion batteries, one of the most promising alternative technologies to lithium-ion batteries, have been constrained by a low initial coulombic efficiency and a low energy density. Sodium compensation technology has received widespread attention for its ability to effectively improve the ICE by releasing active sodium ions. This review systematically summarizes the up-to-date progress of sodium compensation technologies in recent years and provides an overview of the current most advanced sodium compensation strategies, aiming to provide researchers with a primary understanding of the significance and methods of sodium compensation. As shown in Table 4 and Fig. 9 and 10, different strategies have merits and demerits, and based on different material systems, the best means of sodium compensation remains to be selected for commercial production. Nonetheless, some challenges still require further research and optimization. Due to the high reactivity of sodium metal, which is prone to have chemical reactions with water and oxygen, sodium compensation strategies utilizing Na metal as a source of Na (electrochemical sodium compensation method, physical mixing method, and direct contact method) need to be carried out under strict anhydrous and oxygen-free conditions. Such a strict production environment leads to increased process costs. Furthermore, higher reactivity of Na metal means faster reaction times, which leads to difficulty in controlling the degree of sodium compensation and the formation of sodium dendrites, etc. And the larger ionic radius and different electrochemical behavior of sodium may affect the efficiency and longevity, while the control of the extent of sodium compensation as well as the process of disassembly and reassembly of the batteries restricts the practical application of the above technologies. Sodium-rich materials are efficient for sodium compensation and there is no additional component and any byproduct remaining in the battery. Nevertheless, this type of strategy is quite limited since there are nearly V-based NASICON materials reported and the sodium compensation is highly dependent on the materials themselves, which is hard to be applied in other systems with different cathodes and anodes. More sodium-rich materials deserve to be explored and synthesized.| Method | Agent | Merits | Demerits |
|---|---|---|---|
| Direct contact method | Metallic Na | Facile control; high sodium compensation capacity; low enough redox potential | Safety hazards with Na metal; sodium compensation extent difficulty |
| Electrochemical method | Metallic Na | Controllable SCT degree; comparable to the actual battery operation | Safety hazards with Na metal; complex process |
| Physical sodium compensation | Metallic Na | Facile control; high sodium compensation capacity; low enough redox potential | Safety hazards with Na metal |
| Chemical method | Na complex | Relatively safe; controllable SCT degree; simple process | Air-sensitive; insufficient redox potential |
| Self-sacrificial additives | Na salts | Safe; good stability; simple process | Gas formation; oxidative residues |
| Sodium-rich cathode materials | Parts of V-based NASICONs | Safe; no by-product | Less selective materials; low SCT capacity |
 | ||
| Fig. 9 Schematic illustration of the merits, demerits, and operation of different sodium compensation technologies. | ||
 | ||
| Fig. 10 Key parameter radar chart of different SCTs. | ||
In contrast, chemical sodium compensation and self-sacrificial additives are the most promising sodium compensation technologies by virtue of their simple and safe processes and controllable degree of sodium compensation. Overall, the strategies remain at the academic level for further exploration from the laboratory to industrialization.
The chemical sodium compensation method should further consider: (i) the electrode of industrial production is characterized by high loading and low porosity, which is not suitable for the wetting of the material by the sodium compensation solution, leading to inconsistency in the extent of sodium compensation at different positions of the electrode. Therefore, it is necessary to further systematically investigate how to realize the accurate control of sodium compensation. (ii) The SCT-treated electrode material has a low open-circuit potential and a strong “electronegativity”, resulting in inferior air stability. How to achieve a long-time storage of electrodes in air needs to be solved urgently. (iii) The impact of residual sodium compensation reagent on the subsequent battery performance remains to be clearly analyzed. (iv) Recovering and reusing the sodium compensation reagent and organic reagent can effectively reduce the production cost and protect the environment. (v) The safety issue should be paid more attention. The usually employed ether solvents possess a quite low flash point and boiling point. Ideal SCT solvents need to be further explored and designed at a molecular level with a high enough boiling point and flash point as well as an excellent compatibility with the Na complex. (vi) The currently used sodium reagents are hinged on polycyclic aromatic hydrocarbons with a low reducing potential (∼0.1 V versus Na+/Na), easily incurring sodium accumulation and causing the electrode's structure to deteriorate. How to modulate the reducing potential of the Na-complex to a suitable range remains to be further studied by in situ experimental characterization and theoretical simulations.
The large-scale utilization of sodium cathode compensation additives remains to solve the following problems: (i) the additive should have good air stability to avoid rapid reaction with water and oxygen. (ii) To reduce the gas generated by the decomposition of sodium compensation additives and the impact of changes in the electrode interface caused by the gas in the follow-up cycle of the battery, to prevent the decomposition products from continuing to participate in the subsequent reaction. (iii) To improve the irreversible capacity of the sodium compensation additive, to reduce its voltage polarization, and to reduce the amounts of additives in the electrodes. (iv) The operating mechanism of sodium compensation additives lacks in-depth study. There are many side reactions, and the effects on the composition and structure of SEI and CEI layers are not clear. (v) The oxidative potential of these additives should be elaborately lowered from the perspective of catalysis within the safe operating voltage range of electrolytes to prevent the over-decomposition of the electrolyte. (vi) The residual will become the “dead mass” to lower the overall energy density. Some in situ sodium compensation methods can be taken into account. For instance, the residual alkali (e.g., NaOH, Na2O, Na2CO3, etc.) around the cathode surface can be neutralized by some organic acids (e.g., acetic acid) to form a sodium organic salt as an efficient sodium compensation additive for subsequent battery operation.
In the section that follows, we also offer ideas and prospects for the advancement of SCTs in the future, addressing issues with high-throughput screening, characterization, manufacturing, equipment, and recycling.
(1) High-throughput screening. In recent years, high-throughput screening has become more crucial in the investigation of novel materials due to the advancement of high-throughput computing based on DFT. By determining the LUMO–HOMO energy level of solvents, additives, and electrolyte systems, new solvents for chemical sodium compensation and self-sacrificial sodium additives utilized in cathodes can be produced for sodium compensation. It will select a large number of chemical reagents that satisfy the fundamental parameter requirements. Some of these reagents will then be sent for higher-order parameter refinement and characterization, and machine learning will be used to analyze the correlation between the calculated data and the characterization results, thereby offering a trustworthy framework for the development of new sodium compensation reagents.
(2) New characterization. As of right now, no characterization technology is able to more accurately represent the sodium compensation process. The advancement of sodium compensation research is substantially slowed down by the fact that studies on the subject exclusively rely on electron microscope images and electrochemical performance data. Therefore, based on current electrochemical theory, it is imperative to develop new characterization approaches that can directly or indirectly indicate the sodium compensation effect.
(3) Manufacturing and equipment. Sodium compensation process control equipment design is especially crucial for large-scale production of SCTs. It can keep an eye on the condition of the electrode materials during the sodium compensation process and provide real-time feedback to the terminal equipment to make adjustments. This allows for automated and precise management of the sodium compensation degree while also improving safety.
(4) Recycling. The large-scale implementation of sodium compensation materials/reagent recycling in SIBs holds significant strategic importance for the future. Recycling sodium compensation materials and/or chemicals promotes a safer and more robust raw material supply chain in addition to easing the restrictions brought on by a shortage of raw materials and improving environmental sustainability. In the meantime, they have a higher added value for recycling and are fundamentally distinct from other conventional electrode materials.
In summary, we believe that sodium compensation is a critical approach that needs to be included in the design of the materials and electrolytes. Doing so will undoubtedly help to improve SIBs in the future.
Data availability
The type of this manuscript is a review and related data are cited from other sources. Therefore, there are no raw experimental or computational data associated with this article.Author contributions
W. Zhang, B. Zhu, and Z. Zhang designed the structure of the review. W. Zhang, B. Zhu, Z. Jiang, J. Chen, Z. Li, J. Zheng, and N. Wen contributed to the illustration. W. Zhang and B. Zhu collected the papers related to this review topic and co-wrote the paper. W. Zhang and Z. Zhang supervised this project. All authors discussed and revised the manuscript.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant No. 52274309) and Hunan province key R&D technology project (Grant No. 2023GK2070).Notes and references
- J. B. Goodenough, Nat. Electron., 2018, 1, 204 CrossRef.
- B. Zhu, W. Zhang, Q. Wang, Y. Lai, J. Zheng, N. Wen and Z. Zhang, Adv. Funct. Mater., 2024, 34, 2315010 CrossRef CAS.
- J. Li, B. Zhu, S. Li, D. Wang, W. Zhang, Y. Xie, J. Fang, B. Hong, Y. Lai and Z. Zhang, J. Electrochem. Soc., 2021, 168, 080510 CrossRef CAS.
- Z. Wang, Z. Du, Y. Liu, C. E. Knapp, Y. Dai, J. Li, W. Zhang, R. Chen, F. Guo, W. Zong, X. Gao, J. Zhu, C. Wei and G. He, eScience, 2024, 4, 100189 CrossRef.
- W. Zhang, Y. Dai, R. Chen, Z. Xu, J. Li, W. Zong, H. Li, Z. Li, Z. Zhang, J. Zhu, F. Guo, X. Gao, Z. Du, J. Chen, T. Wang, G. He and I. Parkin, Angew. Chem., Int. Ed., 2023, 62, e202212695 CrossRef CAS PubMed.
- Y. Dai, C. Zhang, W. Zhang, L. Cui, C. Ye, X. Hong, J. Li, R. Chen, W. Zong, X. Gao, J. Zhu, P. Jiang, Q. An, D. J. L. Brett, I. P. Parkin, G. He and L. Mai, Angew. Chem., Int. Ed., 2023, 62, e202301192 CrossRef CAS.
- R. Chen, C. Zhang, J. Li, Z. Du, F. Guo, W. Zhang, Y. Dai, W. Zong, X. Gao, J. Zhu, Y. Zhao, X. Wang and G. He, Energy Environ. Sci., 2023, 16, 2540–2549 RSC.
- Y. Dai, C. Zhang, J. Li, X. Gao, P. Hu, C. Ye, H. He, J. Zhu, W. Zhang, R. Chen, W. Zong, F. Guo, I. P. Parkin, D. J. L. Brett, P. R. Shearing, L. Mai and G. He, Adv. Mater., 2024, 36, 2310645 CrossRef CAS PubMed.
- C. Vaalma, D. Buchholz, M. Weil and S. Passerini, Nat. Rev. Mater., 2018, 3, 18013 CrossRef.
- W. Zhang, R. Chen, Y. Dai, X. Wu, J. Chen, W. Zong, M. Zhang, Z. Du, H. Dong, F. Zhao, H. Yang, J. Borowiec, Z. Xu, Z. Li, M. Liu, G. He and I. P. Parkin, Mater. Today, 2024 DOI:10.1016/j.mattod.2024.06.016.
- R. Chen, W. Zhang, C. Guan, Y. Zhou, I. Gilmore, H. Tang, Z. Zhang, H. Dong, Y. Dai, Z. Du, X. Gao, W. Zong, Y. Xu, P. Jiang, J. Liu, F. Zhao, J. Li, X. Wang and G. He, Angew. Chem., Int. Ed., 2024, 63, e202401987 CrossRef CAS PubMed.
- W. Zong, J. Li, C. Zhang, Y. Dai, Y. Ouyang, L. Zhang, J. Li, W. Zhang, R. Chen, H. Dong, X. Gao, J. Zhu, I. P. Parkin, P. R. Shearing, F. Lai, K. Amine, T. Liu and G. He, J. Am. Chem. Soc., 2024, 146, 21377–21388 CrossRef.
- H. Dong, X. Hu, R. Liu, M. Ouyang, H. He, T. Wang, X. Gao, Y. Dai, W. Zhang, Y. Liu, Y. Zhou, D. J. L. Brett, I. Parkin, P. Shearing and G. He, Angew. Chem., Int. Ed., 2023, 62, e202311268 CrossRef CAS PubMed.
- H. Li, W. Zhang, K. Sun, J. Guo, K. Yuan, J. Fu, T. Zhang, X. Zhang, H. Long, Z. Zhang, Y. Lai and H. Sun, Adv. Energy Mater., 2021, 11, 2100867 CrossRef CAS.
- W. Zhang, Z. Xu, H. Li, M. Xu, S. Wang, Z. Li, A. Wang, L. Zhang, L. He, S. Li, B. Zhu, Z. Zhang and Y. Lai, Chem. Eng. J., 2022, 433, 133542 CrossRef CAS.
- R. Usiskin, Y. Lu, J. Popovic, M. Law, P. Balaya, Y.-S. Hu and J. Maier, Nat. Rev. Mater., 2021, 6, 1020–1035 CrossRef CAS.
- W. Zhang, Development and Investigation of NASICON-Type Cathode Materials for High-Power and Long-Lasting Sodium-Ion Batteries, UCL (University College London), 2024 Search PubMed.
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636–11682 CrossRef CAS.
- W. Zhang, Y. Wu, Y. Dai, Z. Xu, L. He, Z. Li, S. Li, R. Chen, X. Gao, W. Zong, F. Guo, J. Zhu, H. Dong, J. Li, C. Ye, S. Li, F. Wu, Z. Zhang, G. He, Y. Lai and I. P. Parkin, Chem. Sci., 2023, 14, 8662–8671 RSC.
- W. Zhang, Y. Wu, Z. Xu, H. Li, M. Xu, J. Li, Y. Dai, W. Zong, R. Chen, L. He, Z. Zhang, D. J. L. Brett, G. He, Y. Lai and I. P. Parkin, Adv. Energy Mater., 2022, 12, 2201065 CrossRef CAS.
- Y. Li, Y. Lu, Q. Meng, A. C. S. Jensen, Q. Zhang, Q. Zhang, Y. Tong, Y. Qi, L. Gu, M. M. Titirici and Y. S. Hu, Adv. Energy Mater., 2019, 9, 1902852 CrossRef CAS.
- C. Li, H. Xu, L. Ni, B. Qin, Y. Ma, H. Jiang, G. Xu, J. Zhao and G. Cui, Adv. Energy Mater., 2023, 13, 2301758 CrossRef CAS.
- B. Liu, T. Lv, A. Zhou, X. Zhu, Z. Lin, T. Lin and L. Suo, Nat. Commun., 2024, 15, 2922 CrossRef CAS PubMed.
- X. Dou, I. Hasa, D. Saurel, C. Vaalma, L. Wu, D. Buchholz, D. Bresser, S. Komaba and S. Passerini, Mater. Today, 2019, 23, 87–104 CrossRef CAS.
- B. Zhang, R. Dugas, G. Rousse, P. Rozier, A. M. Abakumov and J. M. Tarascon, Nat. Commun., 2016, 7, 10308 CrossRef CAS PubMed.
- K. Fang, Y. Tang, J. Liu, Z. Sun, X. Wang, L. Chen, X. Wu, Q. Zhang, L. Zhang, Y. Qiao and S. G. Sun, Nano Lett., 2023, 23, 6681–6688 CrossRef CAS.
- M. Zhang, Y. Li, F. Wu, Y. Bai and C. Wu, Nano Energy, 2021, 82, 105738 CrossRef CAS.
- H. Fang, S. Gao, M. Ren, Y. Huang, F. Cheng, J. Chen and F. Li, Angew. Chem., Int. Ed., 2023, 62, e202214717 CrossRef CAS PubMed.
- Y. B. Niu, Y. J. Guo, Y. X. Yin, S. Y. Zhang, T. Wang, P. Wang, S. Xin and Y. G. Guo, Adv. Mater., 2020, 32, 2001419 CrossRef CAS.
- I. Moeez, H. G. Jung, H. D. Lim and K. Y. Chung, ACS Appl. Mater. Interfaces, 2019, 11, 41394–41401 CrossRef CAS.
- H. Wang, Y. Xiao, C. Sun, C. Lai and X. Ai, RSC Adv., 2015, 5, 106519–106522 RSC.
- Y. Pi, Z. Gan, M. Yan, C. Pei, H. Yu, Y. Ge, Q. An and L. Mai, Chem. Eng. J., 2021, 413, 127565 CrossRef CAS.
- E. de la Llave, V. Borgel, K. J. Park, J. Y. Hwang, Y. K. Sun, P. Hartmann, F. F. Chesneau and D. Aurbach, ACS Appl. Mater. Interfaces, 2016, 8, 1867–1875 CrossRef CAS.
- H. Li, M. Xu, C. Gao, W. Zhang, Z. Zhang, Y. Lai and L. Jiao, Energy Storage Mater., 2020, 26, 325–333 CrossRef.
- W. Zhang, H. Li, Z. Zhang, M. Xu, Y. Lai and S.-L. Chou, Small, 2020, 16, 2001524 CrossRef CAS PubMed.
- Y. Zhou, G. Xu, J. Lin, Y. Zhang, G. Fang, J. Zhou, X. Cao and S. Liang, Adv. Mater., 2023, 35, 2304428 CrossRef CAS.
- Q. Shi, R. Qi, X. Feng, J. Wang, Y. Li, Z. Yao, X. Wang, Q. Li, X. Lu, J. Zhang and Y. Zhao, Nat. Commun., 2022, 13, 3205 CrossRef PubMed.
- Y. Xiao, Y. F. Zhu, W. Xiang, Z. G. Wu, Y. C. Li, J. Lai, S. Li, E. Wang, Z. G. Yang, C. L. Xu, B. H. Zhong and X. D. Guo, Angew. Chem., Int. Ed., 2020, 59, 1491–1495 CrossRef CAS.
- B. Zhang, R. Dugas, G. Rousse, P. Rozier, A. M. Abakumov and J. M. Tarascon, Nat. Commun., 2016, 7, 10308 CrossRef CAS.
- J. Tang, D. K. Kye and V. G. Pol, J. Power Sources, 2018, 396, 476–482 CrossRef CAS.
- W. Liu, X. Chen, C. Zhang, H. Xu, X. Sun, Y. Zheng, Y. Yu, S. Li, Y. Huang and J. Li, ACS Appl. Mater. Interfaces, 2019, 11, 23207–23212 CrossRef CAS.
- Y. Wang, J. Lu, W. Dai, X. Cheng, J. Zuo, H. Lei, W. Liu and Z. Fu, Adv. Funct. Mater., 2024, 2403841, DOI:10.1002/adfm.202403841.
- W. Liu, X. Chen, C. Zhang, H. Xu, X. Sun, Y. Zheng, Y. Yu, S. Li, Y. Huang and J. Li, ACS Appl. Mater. Interfaces, 2019, 11, 23207–23212 CrossRef CAS.
- Y. Cao, T. Zhang, X. Zhong, T. Zhai and H. Li, Chem. Commun., 2019, 55, 14761–14764 RSC.
- Z. Wang, S. Chen, J. Qiu, J. Li, W. Zhao, Y. Ji, X. Yao, Z. Chen, K. Li, M. Dong, F. Pan and L. Yang, Adv. Energy Mater., 2023, 13, 2302514 CrossRef CAS.
- X. Liu, Y. Tan, T. Liu, W. Wang, C. Li, J. Lu and Y. Sun, Adv. Funct. Mater., 2019, 29, 1903795 CrossRef CAS.
- Q. Man, C. Wei, K. Tian, H. Shen, X. Zhang, X. Bai, B. Xi, S. Xiong and J. Feng, Adv. Energy Mater., 2024, 14, 2401016 CrossRef CAS.
- S. Zhang, J. Wang, K. Chen, X. Pu, H. Zhu, Y. Zhao, A. Zhao, X. Chen, Y. Fang, Z. Chen and Y. Cao, Angew. Chem., Int. Ed., 2024, 63, e202317439 CrossRef CAS PubMed.
- M. Liu, J. Zhang, S. Guo, B. Wang, Y. Shen, X. Ai, H. Yang and J. Qian, ACS Appl. Mater. Interfaces, 2020, 12, 17620–17627 CrossRef CAS.
- F. Li, X. Yu, K. Tang, X. Peng, Q. Zhao and B. Li, J. Appl. Electrochem., 2022, 53, 9–18 CrossRef.
- N. Qin, Y. Sun, C. Hu, S. Liu, Z. Luo, X. Cao, S. Liang and G. Fang, J. Energy Chem., 2023, 77, 310–316 CrossRef CAS.
- M. Liu, Z. Yang, Y. Shen, S. Guo, J. Zhang, X. Ai, H. Yang and J. Qian, J. Mater. Chem., 2021, 9, 5639–5647 RSC.
- X. Gao, Y. Sun, B. He, Y. Nuli, J. Wang and J. Yang, ACS Energy Lett., 2024, 9, 1141–1147 CrossRef CAS.
- G. Singh, B. Acebedo, M. C. Cabanas, D. Shanmukaraj, M. Armand and T. Rojo, Electrochem. Commun., 2013, 37, 61–63 CrossRef CAS.
- J. Martinez De Ilarduya, L. Otaegui, J. M. López del Amo, M. Armand and G. Singh, J. Power Sources, 2017, 337, 197–203 CrossRef CAS.
- Y. J. Guo, Y. B. Niu, Z. Wei, S. Y. Zhang, Q. Meng, H. Li, Y. X. Yin and Y. G. Guo, ACS Appl. Mater. Interfaces, 2021, 13, 2772–2778 CrossRef CAS.
- K. Park, B.-C. Yu and J. B. Goodenough, Chem. Mater., 2015, 27, 6682–6688 CrossRef CAS.
- Y. Mao, C. Zhou, H. Gong, S. Zhang, X. Wang, X. Liu, Q. Xiang and J. Sun, Small, 2023, 19, e2303259 CrossRef.
- P. Jeżowski, O. Crosnier and T. Brousse, Open Chem., 2021, 19, 432–441 CrossRef.
- P. Jeżowski, A. Chojnacka, X. Pan and F. Béguin, Electrochim. Acta, 2021, 375, 137980 CrossRef.
- Q. Zhang, X.-W. Gao, Y. Shi, W.-B. Luo, Y. Li, Q.-F. Gu, H.-N. Fan, F. Li and H.-K. Liu, Energy Storage Mater., 2021, 39, 54–59 CrossRef.
- M. Sathiya, J. Thomas, D. Batuk, V. Pimenta, R. Gopalan and J.-M. Tarascon, Chem. Mater., 2017, 29, 5948–5956 CrossRef CAS.
- C.-H. Jo, J. U. Choi, H. Yashiro and S.-T. Myung, J. Mater. Chem. A, 2019, 7, 3903–3909 RSC.
- X. Pan, A. Chojnacka, P. Jeżowski and F. Béguin, Electrochim. Acta, 2019, 318, 471–478 CrossRef CAS.
- L. Hu, J. Li, Y. Zhang, H. Zhang, M. Liao, Y. Han, Y. Huang and Z. Li, Small, 2023, 19, 2304793 CrossRef CAS.
- J. Martinez De Ilarduya, L. Otaegui, J. M. López del Amo, M. Armand and G. Singh, J. Power Sources, 2017, 337, 197–203 CrossRef CAS.
- Q. Zhang, X.-W. Gao, Y. Shi, W.-B. Luo, Y. Li, Q.-F. Gu, H.-N. Fan, F. Li and H.-K. Liu, Energy Storage Mater., 2021, 39, 54–59 CrossRef.
- B. Shen, R. Zhan, C. Dai, Y. Li, L. Hu, Y. Niu, J. Jiang, Q. Wang and M. Xu, J. Colloid Interface Sci., 2019, 553, 524–529 CrossRef CAS PubMed.
- M. Lee, J. Hong, J. Lopez, Y. Sun, D. Feng, K. Lim, W. C. Chueh, M. F. Toney, Y. Cui and Z. Bao, Nat. Energy, 2017, 2, 861–868 CrossRef CAS.
- K. Zou, P. Cai, Y. Tian, J. Li, C. Liu, G. Zou, H. Hou and X. Ji, Small Methods, 2020, 4, 1900763 CrossRef CAS.
- T. Zhang, J. Kong, C. Shen, S. Cui, Z. Lin, Y. Deng, M. Song, L. Jiao, H. Huang, T. Jin and K. Xie, ACS Energy Lett., 2023, 8, 4753–4761 CrossRef CAS.
- J. H. Jo, J. U. Choi, Y. J. Park, J. K. Ko, H. Yashiro and S.-T. Myung, Energy Storage Mater., 2020, 32, 281–289 CrossRef.
- D. Shanmukaraj, K. Kretschmer, T. Sahu, W. Bao, T. Rojo, G. Wang and M. Armand, ChemSusChem, 2018, 11, 3286–3291 CrossRef CAS.
- E. Marelli, C. Marino, C. Bolli and C. Villevieille, J. Power Sources, 2020, 450, 227617 CrossRef CAS.
- K. Zou, Z. Song, H. Liu, Y. Wang, A. Massoudi, W. Deng, H. Hou, G. Zou and X. Ji, J. Phys. Chem. Lett., 2021, 12, 11968–11979 CrossRef CAS PubMed.
- J. H. Jo, J. U. Choi, Y. J. Park, J. Zhu, H. Yashiro and S.-T. Myung, ACS Appl. Mater. Interfaces, 2019, 11, 5957–5965 CrossRef CAS.
- X. Pan, A. Chojnacka and F. Béguin, Energy Storage Mater., 2021, 40, 22–30 CrossRef.
- Y. B. Niu, Y. J. Guo, Y. X. Yin, S. Y. Zhang, T. Wang, P. Wang, S. Xin and Y. G. Guo, Adv. Mater., 2020, 32, e2001419 CrossRef.
- A. J. Fernández-Ropero, M. Zarrabeitia, G. Baraldi, M. Echeverria, T. Rojo, M. Armand and D. Shanmukaraj, ACS Appl. Mater. Interfaces, 2021, 13, 11814–11821 CrossRef.
- R. Rajagopalan, Z. Zhang, Y. Tang, C. Jia, X. Ji and H. Wang, Energy Storage Mater., 2021, 34, 171–193 CrossRef.
- J. Wu, C. Lin, Q. Liang, G. Zhou, J. Liu, G. Liang, M. Wang, B. Li, L. Hu, F. Ciucci, Q. Liu, G. Chen and X. Yu, InfoMat, 2022, 4, e12288 CrossRef CAS.
- J. Wu, C. Lin, Q. Liang, G. Zhou, J. Liu, G. Liang, M. Wang, B. Li, L. Hu, F. Ciucci, Q. Liu, G. Chen and X. Yu, InfoMat, 2022, 4, e12288 CrossRef CAS.
- Z. Jian, Y. Sun and X. Ji, Chem. Commun., 2015, 51, 6381–6383 RSC.
- S. Mirza, Z. Song, H. Zhang, A. Hussain, H. Zhang and X. Li, J. Mater. Chem. A, 2020, 8, 23368–23375 RSC.
- Y. Liu, X. Wu, A. Moeez, Z. Peng, Y. Xia, D. Zhao, J. Liu and W. Li, Adv. Energy Mater., 2023, 13, 2203283 CrossRef CAS.
- M. Xu, F. Zhang, Y. Zhang, C. Wu, X. Zhou, X. Ai and J. Qian, Chem. Sci., 2023, 14, 12570–12581 RSC.
| This journal is © The Royal Society of Chemistry 2024 |