
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvestigation of the spin crossover behaviour of a sublimable Fe(II)-qsal complex: from the bulk to a submonolayer on graphene/SiO2†
Adelais
Trapali
 a,
Mukil
Muppal
b,
Satakshi
Pandey
c,
Marie-Laure
Boillot
a,
Vincent
Repain
b,
Yannick J.
Dappe
d,
Jean-François
Dayen
c,
Eric
Rivière
a,
Régis
Guillot
a,
Marie-Anne
Arrio
e,
Edwige
Otero
f,
Amandine
Bellec
*b and
Talal
Mallah
*a
a,
Mukil
Muppal
b,
Satakshi
Pandey
c,
Marie-Laure
Boillot
a,
Vincent
Repain
b,
Yannick J.
Dappe
d,
Jean-François
Dayen
c,
Eric
Rivière
a,
Régis
Guillot
a,
Marie-Anne
Arrio
e,
Edwige
Otero
f,
Amandine
Bellec
*b and
Talal
Mallah
*a
aInstitut de Chimie Moléculaire et des Matériaux d'Orsay, Université Paris-Saclay, CNRS, UMR 8182, 91405 Orsay 12 Cedex, France. E-mail: talal.mallah@universite-paris-saclay.fr
bUniversité Paris Cit é, CNRS, Laboratoire Matériaux et Phénomènes Quantiques, F-75013, Paris, France
cUniversité de Strasbourg, CNRS, Institut de Physique et Chimie des Matériaux de Strasbourg (IPCMS), UMR 7504, 23 rue du Loess, Strasbourg, 67034, France
dSPEC, CEA, CNRS, Université Paris-Saclay, CEA Saclay, 91191 Gif-sur-Yvette, Cedex, France
eInstitutde Mineŕalogie, dePhysiquedes Mateŕiauxetde Cosmochimie, CNRS, Université Pierreet Marie Curie, UMR 7590, Paris, France
fSynchrotron SOLEIL, L'Orme des Merisiers, Saint-Aubin, 91192 Gif sur Yvette, France
First published on 9th July 2024
Abstract
We synthesized a sublimable molecular spin crossover Fe(II) complex based on the Schiff base tridentate ligand qsal-NEt2 (5-diethylamino-2-((quinolin-8-ylimino)methyl)phenol). The compound undergoes a transition in temperature with thermally induced excited spin state-trapping (TIESST) for high-temperature sweep rates, which can be suppressed by reducing the sweep rate. The X-ray absorption spectroscopy (XAS) studies on the microcrystalline powder confirm the TIESST effect. The molecules are deposited under ultra-high vacuum on a graphene/SiO2 substrate as a submonolayer. Investigation of the submonolayer by XAS reveals the molecular integrity and shows a spin crossover for the whole temperature range from 350 to 4 K, with residual HS species at low temperature and no TIESST effect. DFT calculations suggest a distribution of energetically similar adsorption configurations on graphene, i.e., with smooth crossover behaviour and the absence of TIESST, consistent with very weak intermolecular interactions and the absence of large molecular islands within the submonolayer.
Introduction
Since the early proposals for using spin crossover (SCO) molecular materials as components in memory devices, an intense research effort has been made to explore their potential for applications in different areas.1–9 Aiming to gain insights into the switching mechanism of a reduced number of molecules, research groups investigated their properties when assembled as submonolayers or thin films on different substrates of interest.10–21 Two main issues arose: (1) the integrity of the molecules on the different substrates and (2) the switching behaviour at the substrate/molecules interface. On metallic substrates, partial fragmentation was observed for some SCO molecules while others kept their integrity, even though their switching behaviour was different from that of the bulk, highlighting the importance of the chemical nature of the studied SCO molecules and the molecule/substrate interface.15,16,22 On Highly Oriented Pyrolytic Graphite (HOPG), the SCO behaviour was, generally, found to be similar to that observed in the bulk.10,14,23–26 Gruber et al. investigated the switching properties of [Fe(pypyr(CF3)2)2(phen)] (pypyr = 2-(2′-pyridyl)pyrrolide, phen = 1,10-phenanthroline) on different substrates and concluded, considering also other reports, that fragmentation is prone to occur on metallic substrates, while on substrates with reduced density of states at the Fermi level (semi-metallic, semi-conductors), the molecules tend to keep their integrity.18 Interestingly, for the graphene/Ni(111) substrate, X-ray Absorption Spectroscopy (XAS) studies of a sub-monolayer are consistent with a partial fragmentation of the molecules similar to metallic substrates, which may be due to the poor decoupling role of graphene from metallic Ni(111).The present investigation of both the integrity and the switching behaviour of a submonolayer of SCO molecules on graphene is motivated by the possibility of an electric read-out of the spin state, as already carried out on thick films and on SCO nanoparticles.27–29 We report the preparation and characterization of a sublimable SCO complex noted as Fe(qsal-NEt2)2 (qsal-NEt2 = 5-diethylamino-2-((quinolin-8-ylimino)methyl)phenol, see Scheme 1) in the bulk form and as a submonolayer grown on graphene/SiO2.
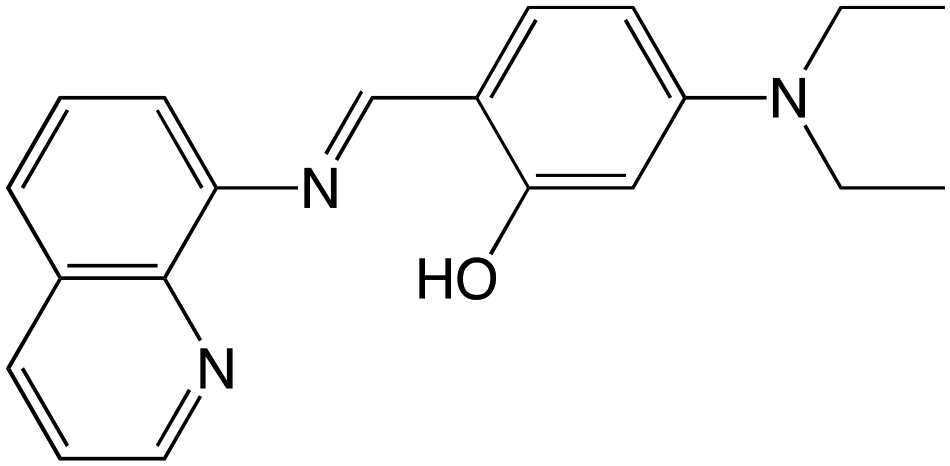 | ||
| Scheme 1 Formula of Hqsal-NEt2. | ||
To the best of our knowledge, no studies of the switching behaviour of SCO molecules assembled as a submonolayer on graphene/SiO2 have been reported to date.
The choice of the molecule was dictated mainly by its potential to keep its integrity on different substrates because of the presence of two negatively charged tridentate ligands (qsal-NEt2−).30 In this respect, it is similar to Fe(MPz)2 (MPz− = hydrotris-(3,5-dimethyl-pyrazol-1-yl) borate, a negatively charged tridentate ligand), which proved to be chemically robust when deposited on metallic substrates, with no sign of fragmentation.15,17,19,20 This probably results from the additional thermodynamic stability of this type of complex with respect to substrate–ligand interaction due to both the stabilizing electrostatic energy (enthalpic origin, positively charged metal ion and negatively charged ligands) and the tridentate character of the ligand (entropic origin) in comparison with neutral bidentate ligands such as bipyridine and phenanthroline, or related ones. The main drawback of Fe(qsal-NEt2)2 is its high air sensitivity, which requires appropriate manipulation and where a small amount of the oxidized species is difficult to avoid. It, however, constitutes an interesting case study, as we discuss below.
Experimental
Materials and methods
All reagents and solvents were purchased from commercial suppliers and used as received without any further purification. The organic ligand Hqsal-NEt2 (5-diethylamino-2-((quinolin-8-ylimino)methyl)phenol) was synthesized according to a slightly modified previously described procedure.31 The preparation method and full characterization are provided in the ESI (Fig. S1 and S2†). For the synthesis of the iron complex Fe(qsal-NEt2)2, all manipulations were carried out in an Ar-filled glovebox.Preparation of Fe(qsal-NEt2)2
FeBr2 (96 mg, 0.44 mmol, 1 eq.) was dissolved in 1.5 mL of MeOH, and the resulting solution was added to a methanolic solution (1.5 mL) of Hqsal-NEt2 (283 mg, 0.89 mmol, 2 eq.) containing 2 eq. of Et3N (90 mg, 124 μL, 0.89 mmol). The reaction mixture was stirred for 72 h at room temperature. After 72 h, a brown precipitate was formed, filtered, and successively washed with MeOH (4 × 1 mL). Fe(qsal-NEt2)2 was obtained as a light brown solid that was further dried under vacuum (262 mg, 86%). ESI-MS+ (m/z): 693.2562 [M + 1], found 692.2548 [M]+. The [M]+ corresponds to the oxidized Fe(qsal-NEt2)2 species due to its high sensitivity to O2 (ESI-MS+ studies were performed under ambient conditions, Fig. S3†). Dark green/brown plate-like crystals were obtained after a month by layering 3 mL of MeOH on top of a 1.44 mM Fe(qsal-NEt2)2 dichloromethane solution. The Fe(III) derivative ([Fe(qsal-NEt2)2]Cl) was also prepared (see the ESI and Fig. S4†).Substrate and molecular submonolayer preparation
The CVD graphene layer pre-transferred over SiO2/Si substrate (bought from Graphenea) was treated carefully to remove any contaminations resulting from the transfer process. It was first immersed in hot acetone (50 °C) for 1 hour, followed by a hot isopropyl alcohol bath (50 °C) for an additional hour, and finally baked for 4 hours at 250 °C in a glovebox.The graphene substrate was then introduced under ultra-high vacuum (UHV, base pressure around 5 × 10−10 mbar) to grow the molecular submonolayer. The graphene substrate was first annealed at 450 °C for 25 minutes. The molecules were then sublimated using a home-made Knudsen cell at a temperature of around 230 °C. The sample was then kept and measured under UHV.
Single crystal X-ray diffraction
The X-ray diffraction data for [Fe(qsal-NEt2)2] were collected using a VENTURE PHOTONIII c7 Bruker diffractometer with Micro-focus IuS source Mo Kα radiation. The crystal of [Fe(qsal-NEt2)2] was selected under an optical microscope and glued in paratone oil. The crystal was mounted on a CryoLoop (Hampton Research) with Paratone-N (Hampton Research) as the cryoprotectant and then placed in a nitrogen-gas stream at 200 K or 90 K. The temperature of the crystal was maintained at the selected value by means of a N-Helix cooling device to within an accuracy of few degrees K. Data reduction was accomplished using SAINT V7.53a. The substantial redundancy in data allowed a semi-empirical absorption correction (SADABS V2.10) to be applied on the basis of multiple measurements of equivalent reflections. CCDC 2350748 and 2350749 contain the supplementary crystallographic data for this paper (see the ESI, Fig. S5 and Tables S1, S2†).Magnetic measurements
The magnetic data were acquired using an MPMS SQUID magnetometer, on a microcrystalline powder prepared in a capsule within the glovebox, in the 150–10 K region with different sweep rates of 2.0, 0.4, and 0.1 K min−1 at an applied magnetic field of 0.5 T.X-Ray absorption spectroscopy (XAS)
The XAS measurements at the L2,3 Fe-edge were acquired on the DEIMOS beamline (synchrotron SOLEIL) under ultra-high vacuum. To prevent, as much as possible, any oxidation of the Fe ion, the bulk sample, consisting of a microcrystalline powder, was prepared in a glovebox connected to the ultra-high vacuum chamber at DEIMOS. For its part, the thin molecular film on graphene on SiO2/Si was transferred from the UHV chamber where it was prepared to the beamline using a vacuum suitcase (base pressure 3 × 10−10 mbar).Computational methods
Geometrical optimizations of different molecular configurations in crystal, on the graphene surface or isolated, as well as the corresponding Density of States (DOS), were performed using the very efficient localized-orbital Density Functional Theory (DFT) package Fireball.32 This code uses a self-consistent version of the Harris functional,33 and an LDA-like exchange–correlation scheme (McWEDA).34 An optimized numerical basis set was used for the calculations, following previous work on similar systems.35–37 For each configuration, an optimization process was performed until the forces went below 0.1 eV Å−1.Results and discussion
Single crystal X-ray structural analysis
The crystal structure was first solved at T = 200 K. [Fe(qsal-NEt2)2] crystallizes in the monoclinic P21/n space group. The asymmetric unit cell contains one iron complex and one molecule of MeOH with static disorder. The coordination sphere of Fe has two qsal-NEt2− ligands in meridional configuration (Fig. 1).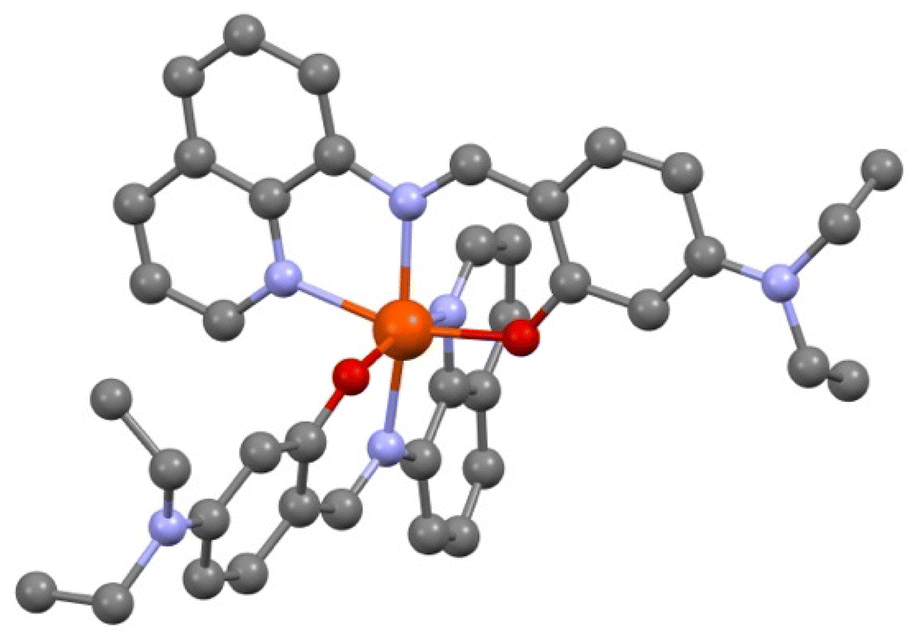 | ||
| Fig. 1 View of the molecular structure of [Fe(qsal-NEt2)2], O (red), N (blue), and C (dark grey). H atoms and solvent molecules are omitted for clarity. | ||
The crystallographic data, structure refinement details, and selected bond distances and angles in the coordination sphere of Fe are given in the ESI (Table S1†). The average bond distances 〈Fe–Nim〉, 〈Fe–Nq〉, and 〈Fe–O〉 are 2.128, 2.231, and 2.019 Å,‡ respectively, and they are in good agreement with the corresponding mean Fe–L distances of similar HS-Fe(II) Schiff base complexes reported in the literature (Tables S2 and S3†). Further support of the HS state of [Fe(qsal-NEt2)2] at 200 K comes from the high Σ and Θ octahedral distortion parameters (i.e., 80.70° and 273.07°) as calculated using the OctaDist software (Table S3†).38 Attempts to perform an X-ray diffraction study on the same crystal at 90 K by a rapid cooling from 200 K were unsuccessful due to crystal shuttering. To obtain the crystal structure at 90 K, we initially cooled down a fresh single crystal (obtained from a different preparation) from 298 K to 120 K with a temperature sweep rate of 2.0 K min−1 followed by a further decrease in temperature at a rate of 0.1 K min−1 until 90 K. The crystallographic data show the same space group as the one studied at 200 K with one molecule of [Fe(qsal-NEt2)2] in the asymmetric unit but with an additional half molecule of MeOH. At 90 K, the complex adopts a less distorted octahedral geometry (Σ = 66.30° and Θ = 235.60°), with a 〈Fe–Nim〉 bond length of 2.074 Å, which is relatively shorter than the one expected for related HS-Fe(II) compounds. The Σ parameter and 〈Fe–Nim〉 for [Fe(qsal-NEt2)2] at T = 90 K lie in between those reported for the two spin states (Table S2†). The average structural parameters indicate that at T = 90 K, the compound has a mixture spin state with around 63% residual high spin molecules, knowing that there is always an uncertainty of a few degrees on the actual temperature at which the data were collected.
Magnetic measurements
Magnetic studies were performed on a microcrystalline powder whose preparation is described in the Experimental section. The X-ray powder diffraction diagram of the material (recorded at T = 200 K, Fig. S6†) is similar to the calculated diagram of the crystal structure determined at T = 90 K and contains 1.5 MeOH molecules per Fe. It will be noted as [Fe(qsal-NEt2)2]·1.5MeOH in the following.We start the measurements at T = 150 K in order to avoid an eventual loss of solvents, with a temperature sweep rate of 2.0 K min−1. The χMT value (3.19 cm3 mol−1 K) remains constant down to T = 120 K and then decreases relatively smoothly and reaches a value of 2.36 cm3 mol−1 K at T = 78 K. Between T = 78 K and 30 K, χMT does not change, and then it decreases to 2.05 cm3 mol−1 K at T = 10 K, due to the zero-field splitting (ZFS) of anisotropic residual HS Fe(II) species. Upon heating, χMT remains constant until T = 78 K; then, it decreases to a minimum value of 1.13 cm3 mol−1 K at T = 108 K, followed by an abrupt increase. It then reaches its initial value at T = 120 K and remains constant up to T = 150 K (Fig. 2, blue curve). Such dependence of the magnetic response on the temperature sweep rate is typical of a thermally induced excited spin state-trapping (TIESST) effect, indicating that for a given sweep rate (2.0 K min−1), the molecules “freeze” in a metastable HS state and their conversion to the LS state is a kinetically slow process. Even though this is not a common phenomenon, it has been observed for several SCO complexes, and in most cases, it was found to be accompanied by structural phase transitions, among other possibilities.39–43
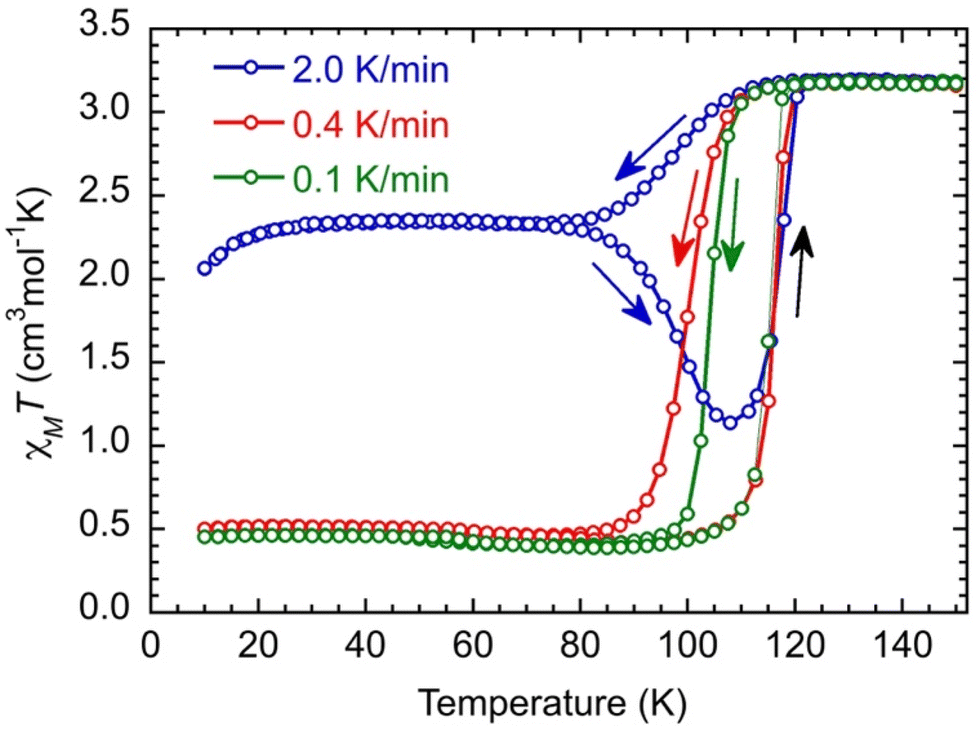 | ||
| Fig. 2 Thermal dependence of the χMT product for [Fe(qsal-NEt2)2]·1.5MeOH at different temperature sweep rates in cooling down and heating up modes between T = 150 K and 10 K. The lines are guides for the eyes. | ||
Performing the same experiment with lower temperature sweep rates (0.4 and 0.1 K min−1) prevents the trapping of the metastable Fe(II) HS species and evidences the kinetic process because the apparent widths of the hysteresis loops decrease upon diminishing the sweep rate (Fig. 2, red and green curves).
At low temperatures, χMT (0.46 cm3 mol−1 K) is constant down to T = 10 K, which may be due to some Fe(II) species still being trapped in the high spin state. However, the characteristic feature of a decrease of χMT below 30 K of HS Fe(II) species is absent. Therefore, the residual paramagnetic species is assigned to oxidized HS Fe(III) (d5, s = 5/2, χMT = 4.38 cm3 mol−1 K) that are magnetically rather isotropic and where χMT remains constant down to T = 10 K.44 Its proportion can be evaluated to be around 11% in the studied sample. The χMT value above T = 120 K (3.19 cm3 mol−1 K) allows, then, the realization that there should be some Fe(II) in the low spin state at high temperatures that can be evaluated to have a proportion around 6% assuming the χMT of HS Fe(II) to be equal to 3.25 cm3 mol−1 K.§ In summary, the relative proportion of the different species extracted from the magnetic data is found to be (FeIIHS)0.83(FeIILS)0.06(FeIIIHS)0.11 at a temperature above 120 K,§ while below 80 K when performing a sweep rate lower than 0.4 K min−1, it is found to be equal to (FeIILS)0.89(FeIIIHS)0.11. The investigation of the kinetic trapping effect is beyond the scope of this work, and it will be discussed in detail in a future publication.
X-Ray absorption spectroscopy study
Complementary XAS spectra were recorded on a microcrystalline powder of [Fe(qsal-NEt2)2]·1.5MeOH at T = 350, 100, and 4 K (Fig. 3). As shown in Fig. 3, the spectra show the presence of Fe at the two oxidation states II and III, as can be determined by linear combination (see the ESI† for reference spectra and the fitting procedure, Fig. S7†). At 350 K, adjusting the experimental curve while fixing the proportion of molecules in the LS state in agreement with the SQUID measurement, we found a proportion of 87 ± 2% of Fe(II) molecules – decomposed as 81 ± 2% in the HS state and 6% in the LS state – and 13 ± 2% of Fe(III) molecules.¶ At low temperatures, only some of the Fe(II) molecules are converted to the LS state, while the proportion of Fe(III) molecules is constant. We determined the proportion of HS Fe(II) molecules to be 70 ± 2% at 100 K, and 78 ± 2% at 4 K (see the fitting procedure in ESI Fig. S8†). With respect to the SQUID data, it is important to note that the sample was first cooled down from 300 K to 4 K at a sweep rate of around 6 K min−1 before acquiring the XA spectra at 4 K, 100 K, and finally at 350 K, so the XAS data correspond to the heating up mode. We observe at T = 4 K, the effect of X-ray exposure that converts about 7% of the LS species to HS (Fig. S9†).||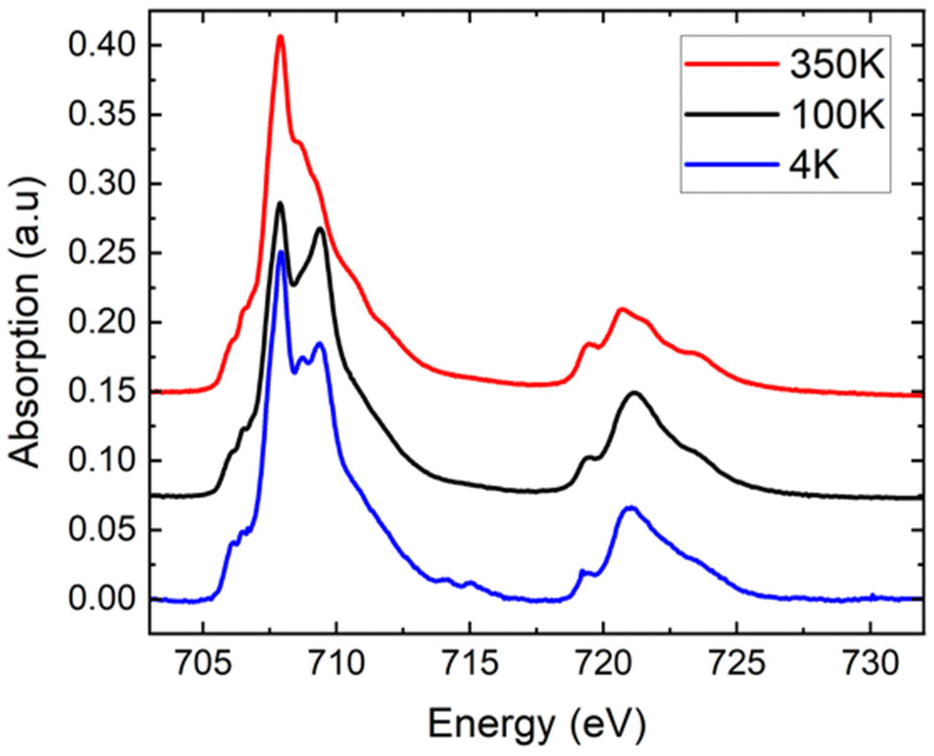 | ||
| Fig. 3 XAS spectra recorded at the Fe L2,3 edges at different temperatures for [Fe(qsal-NEt2)2]·1.5MeOH microcrystalline powder. The curves are normalized to their integral value for comparison. 350 K: average over 5 spectra, 100 K: average over 4 spectra, 4 K: single curve (first one of the series, to minimize X-ray effects). For clarity, the black and red curves are shifted upward by 0.075 and 0.150, respectively. | ||
Even though we did not collect spectra over the full temperature range to enable a quantitative comparison with the SQUID data, we can clearly see that the proportion of HS species decreases between 4 and 100 K and then increases to a value above that of 4 K when heating to 350 K. These XAS experiments on the microcrystalline powder sample are valuable because they confirm qualitatively the kinetic trapping observed from the SQUID data. In summary, the XAS data on the powder are consistent with the SQUID data due to (i) the presence of a small amount of oxidized species and (ii) the presence of the kinetic trapping.
For the molecular film on graphene, we recorded the Fe L3-edge spectra at 300 K, 140 K, and 4 K (Fig. 4a). Interestingly, the sample does not contain any Fe(III) species, as no signal is measured around 709.5 eV, irrespective of the temperature. The sublimation selects, therefore, only the neutral Fe(II) species (even though oxidized species are present in the microcrystalline powder, they may have a different sublimation temperature). More importantly, the graphene substrate does not alter the oxidation state of the molecules. Finally, decomposition of the molecules can be ruled out because no extra XAS signal constant at all temperature is observed.
 | ||
| Fig. 4 Conversion of the submonolayer with temperature. (a) Fe L3-edge spectra at 300 K (red curve), 140 K (black curve), and 4 K (blue curve) for the molecules on graphene. The spectra are normalized so that the L3-edge jump can be read directly as a percentage of the background signal. The black and red curves are shifted for clarity by 0.01 and 0.02, respectively, (b) evolution of the proportion of Fe(II) molecules in the high spin state as a function of the temperature. For the submonolayer, the evolution has been measured while warming up (red up triangles) and cooling down (red down triangles) the sample with a ramp of 1 K min−1. The proportions of HS Fe(II) molecules for stabilized temperatures for the submonolayer (red stars) and the bulk powder (black rectangles) are displayed. | ||
At room temperature, all the molecules are in the Fe(II) HS state. The XAS spectrum is also used to estimate the density of molecules on the graphene substrate, as unfortunately the system Fe(qsal-NEt2)2/graphene/SiO2 is difficult to image using STM and no calibration could be realized to estimate the direct coverage. As detailed in the ESI,† the L3-edge jump is measured and compared to the calibration of a similar molecular system.45 We thus determined that the molecular density on graphene/SiO2 is 0.5 molecule per nm2, which would correspond to a submonolayer coverage of 0.6 ML, if the molecules were arranged in a dense plane. However, as the structure of the film is unknown, the density of the molecules cannot directly be transposed into a coverage in monolayer (see the ESI†). The molecules may be assembled in packed islands or randomly adsorbed on graphene/SiO2.
For the submonolayer coverage, decreasing the temperature leads to a decrease of the signal at 707.9 eV (HS state) and to an increase of the one at 709.2 eV (LS state) (Fig. 4). The first interesting point is that the energy of the peak characteristic of the LS state is shifted toward lower energy compared to the position measured for the bulk (at 709.5 eV). This shift can originate from an alteration of the molecular orbital energy diagram of the isolated molecules adsorbed on graphene (see the calculations below). Upon cooling, the proportion of HS Fe(II) molecules decreases from 100% at 300 K to 73 ± 2% at 4 K (red stars in Fig. 4b), which can be compared to the proportion of HS Fe(II) molecules at the three temperatures measured in the bulk (black squares in Fig. 4b). We also followed the thermal conversion by measuring the absorption signal while ramping the temperature up and down at a rate of 1 K min−1. Fig. 4b presents the evolution of the HS proportion as a function of temperature (Fig. S10,† the fitted data between 33 and 145 K are shown as a movie in the ESI†). The conversion with temperature is partial and smooth, and no sign of trapping is observed in contrast to the bulk.
DFT calculations
In order to get insights into the adsorption mode of the molecules on graphene/SiO2, we performed DFT calculations for two specific cases, namely (1) the molecules in the unit cell, and (2) the molecule on graphene for three different configurations as depicted in Scheme 2. The calculations were performed for the LS state (s = 0) by optimizing the structure experimentally determined at T = 90 K. | ||
| Scheme 2 Different configurations (cf1, cf2, and cf3 from left to right) of the molecules with respect to the graphene monolayer that is horizontal and perpendicular to the plane (not shown). | ||
The calculations show the occurrence of a weak electron transfer (0.16, 0.12, and 0.17 for cf1, cf2, and cf3, respectively) that is similar to the three configurations, suggesting that there is no energetically privileged adsorption mode (Fig. S11†). A comparison of the molecules’ optimized structures in the crystal and on the substrate reveals slight distortion for all configurations due to the interaction of both the quinoline and the ethyl groups with graphene. A comparison of the total DOS of the Fe atoms projected on the d orbitals (molecules/graphene and bulk) shows a reduction of the energy difference between the fully occupied (close to the Fermi level) and the empty d orbitals (close to 2.8 eV) of 0.17 to 0.27 eV depending on the configuration of the molecule on the substrate (Fig. 5, see ESI Fig. S12†), which is qualitatively consistent with the assumed shift towards low energy in the XAS signal of the LS spin species.**
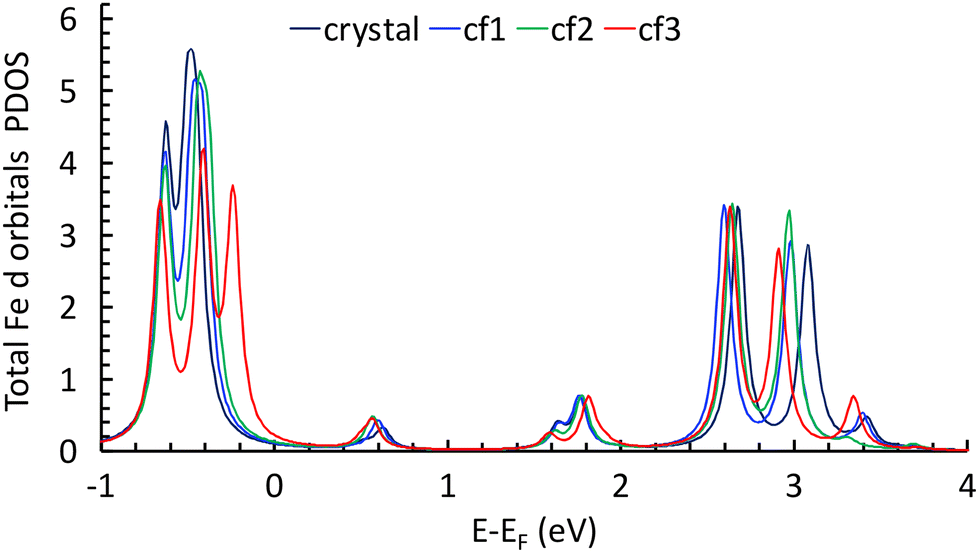 | ||
| Fig. 5 Total DOS of Fe atoms projected on the d orbitals for the molecule in the crystal and for the three configurations (see text). | ||
Conclusions
We prepared a sublimable Fe(II) complex bearing a tridentate negatively charged ligand that brings a large thermodynamic stability with respect to ligand/substrate interaction. Based on the magnetic data, the bulk exhibits a thermally induced spin state trapping at high temperature sweep rates, which is suppressed when cooling down slowly, leading to an abrupt transition with an apparent thermal hysteresis of ca. 10 K. The XAS data of the bulk confirm the magnetic measurements.For the submonolayer obtained by sublimation of the molecules, no oxidized species were detected, which is a strong indication of the integrity of the molecules on graphene/SiO2. The shift of −0.3 eV for the LS XAS signal of the submonolayer cannot be reasonably attributed to a fragmentation because no additional signals are observed. No sign of spin state trapping is observed for the submonolayer. The crossover of the submonolayer is very smooth over the whole temperature range, with a large residual HS species at low temperatures. Finally, the calculations are qualitatively consistent with the shift towards low energy of the LS XAS signal because they show a reduction of the crystal field parameter when the molecules are on graphene/SiO2, with no privileged adsorption mode.
All these characteristics suggest an arrangement of the molecules on the surface different from that of the bulk, with very weak (or no) intermolecular interactions. Only a local technique such as scanning tunnelling microscopy would give a definitive answer, but such an experiment is difficult to perform on the poorly conductive graphene/SiO2 substrate.
Author contributions
M. L. B. proposed the complex to be studied. A. B., M. L. B., T. M., and V. R. conceptualized the experiments. A. T. made the complex and performed preliminary characterization studies. R. G. solved the crystal structure. A. T., R. G., and M. L. B. analyzed the crystallographic data. E. R. and A. T. performed the magnetic studies on the bulk compound. A. T., E. R., and M. L. B. analyzed the magnetic data. S. P. and J. F. D. prepared the graphene/SiO2 substrate. M. M., A. B., and V. R. performed the sublimation on the substrate. M. M., A. T., A. B., V. R., M. A. A., E. O., T. M., and M. L. B. performed the XAS experiments. M. M. and A. B. analyzed the XAS data. Y. D. performed the DFT calculations and analysed the data. A. T., T. M., and A. B. wrote the paper from the contributions of all authors.Data availability
All data are available from the authors on request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Centre National de la Recherche Scientifique (CNRS, France), the French Ministry of Research, and ANR (ANR-21-CE09-0031, 2DSwitch project). We thank the team of DEIMOS beamline at Synchrotron SOLEIL and F. Leduc for the installation of the glove box and help with the vacuum suitcase. We thank S. Stanojevic for his contribution to the XAS experiments.References
- O. Kahn and C. J. Martinez, Science, 1998, 279, 44–48 CrossRef CAS.
- O. Kahn, Chem. Br., 1999, 35, 24–27 CAS.
- I.-R. Jeon, J. G. Park, C. R. Haney and T. D. Harris, Chem. Sci., 2014, 5, 2461–2465 RSC.
- G. Molnár, L. Salmon, W. Nicolazzi, F. Terki and A. Bousseksou, J. Mater. Chem. C, 2014, 2, 1360–1366 RSC.
- G. Molnár, S. Rat, L. Salmon, W. Nicolazzi and A. Bousseksou, Adv. Mater., 2018, 30, 1703862 CrossRef PubMed.
- P. J. von Ranke, B. P. Alho, R. M. Ribas, E. P. Nobrega, A. Caldas, V. S. R. de Sousa, M. V. Colaco, L. F. Marques, D. L. Rocco and P. O. Ribeiro, Phys. Rev. B, 2018, 98, 224408 CrossRef CAS.
- M. Romanini, Y. X. Wang, K. Gürpinar, G. Ornelas, P. Lloveras, Y. Zhang, W. K. Zheng, M. Barrio, A. Aznar, A. Gràcia-Condal, B. Emre, O. Atakol, C. Popescu, H. Zhang, Y. Long, L. Balicas, J. L. Tamarit, A. Planes, M. Shatruk and L. Mañosa, Adv. Mater., 2021, 33, 2008076 CrossRef CAS PubMed.
- Y. Zan, M. Piedrahita-Bello, S. E. Alavi, G. Molnár, B. Tondu, L. Salmon and A. Bousseksou, Adv. Intell. Syst., 2023, 5, 2200432 CrossRef.
- M. Seredyuk, R. X. Li, K. Znovjyak, Z. Zhang, F. J. Valverde-Muñoz, B. Li, M. C. Muñoz, Q. J. Li, B. B. Liu, G. Levchenko and J. A. Real, Adv. Funct. Mater., 2024, 2315487 CrossRef.
- M. Bernien, D. Wiedemann, C. F. Hermanns, A. Krüger, D. Rolf, W. Kroener, P. Müller, A. Grohmann and W. Kuch, J. Phys. Chem. Lett., 2012, 3, 3431–3434 CrossRef CAS PubMed.
- T. Miyamachi, M. Gruber, V. Davesne, M. Bowen, S. Boukari, L. Joly, F. Scheurer, G. Rogez, T. K. Yamada, P. Ohresser, E. Beaurepaire and W. Wulfhekel, Nat. Commun., 2012, 3, 938 CrossRef PubMed.
- B. Warner, J. C. Oberg, T. G. Gill, F. El Hallak, C. F. Hirjibehedin, M. Serri, S. Heutz, M.-A. Arrio, P. Sainctavit, M. Mannini, G. Poneti, R. Sessoli and P. Rosa, J. Phys. Chem. Lett., 2013, 4, 1546–1552 CrossRef CAS PubMed.
- M. Gruber, V. Davesne, M. Bowen, S. Boukari, E. Beaurepaire, W. Wulfhekel and T. Miyamachi, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 195415 CrossRef.
- M. Bernien, H. Naggert, L. M. Arruda, L. Kipgen, F. Nickel, J. Miguel, C. F. Hermanns, A. Krüger, D. Krüger, E. Schierle, E. Weschke, F. Tuczek and W. Kuch, ACS Nano, 2015, 9, 8960–8966 CrossRef CAS PubMed.
- K. Bairagi, O. Iasco, A. Bellec, A. Kartsev, D. Z. Li, J. Lagoute, C. Chacon, Y. Girard, S. Rousset, F. Miserque, Y. J. Dappe, A. Smogunov, C. Barreteau, M. L. Boillot, T. Mallah and V. Repain, Nat. Commun., 2016, 7, 12212 CrossRef CAS PubMed.
- S. Ossinger, H. Naggert, L. Kipgen, T. Jasper-Toennies, A. Rai, J. Rudnik, F. Nickel, L. M. Arruda, M. Bemien, W. Kuch, R. Berndt and F. Tuczek, J. Phys. Chem. C, 2017, 121, 1210–1219 CrossRef CAS.
- K. Bairagi, A. Bellec, C. Fourmental, O. Iasco, J. Lagoute, C. Chacon, Y. Girard, S. Rousset, F. Choueikani, E. Otero, P. Ohresser, P. Sainctavit, M. L. Boillot, T. Mallah and V. Repain, J. Phys. Chem. C, 2018, 122, 727–731 CrossRef CAS.
- S. Rohlf, J. Grunwald, T. Jasper-Toennies, S. Johannsen, F. Diekmann, M. Studniarek, R. Berndt, F. Tuczek, K. Rossnagel and M. Gruber, J. Phys. Chem. C, 2019, 123, 17774–17780 CrossRef CAS.
- Y. F. Tong, M. Kelaï, K. Bairagi, V. Repain, J. Lagoute, Y. Girard, S. Rousset, M. L. Boillot, T. Mallah, C. Enachescu and A. Bellec, J. Phys. Chem. Lett., 2021, 12, 11029–11034 CrossRef CAS PubMed.
- M. Kelai, A. Tauzin, A. Railean, V. Repain, J. Lagoute, Y. Girard, S. Rousset, E. Otero, T. Mallah, M. L. Boillot, C. Enachescu and A. Bellec, J. Phys. Chem. Lett., 2023, 14, 1949–1954 CrossRef CAS PubMed.
- M. Gavara-Edo, F. J. Valverde-Muñoz, M. C. Muñoz, S. Elidrissi Moubtassim, F. Marques-Moros, J. Herrero-Martín, K. Znovjyak, M. Seredyuk, J. A. Real and E. Coronado, Chem. Mater., 2023, 35, 9591–9602 CrossRef CAS PubMed.
- M. Gruber, T. Miyamachi, V. Davesne, M. Bowen, S. Boukari, W. Wulfhekel, M. Alouani and E. Beaurepaire, J. Chem. Phys., 2017, 146, 092312 CrossRef.
- L. Kipgen, M. Bernien, F. Nickel, H. Naggert, A. J. Britton, L. M. Arruda, E. Schierle, E. Weschke, F. Tuczek and W. Kuch, J. Phys.: Condens.Matter, 2017, 29, 394003 CrossRef PubMed.
- S. Ossinger, L. Kipgen, H. Naggert, M. Bernien, A. J. Britton, F. Nickel, L. M. Arruda, I. Kumberg, T. A. Engesser, E. Golias, C. Näther, F. Tuczek and W. Kuch, J. Phys.: Condens.Matter, 2020, 32, 114003 CrossRef CAS PubMed.
- L. Kipgen, M. Bernien, S. Ossinger, F. Nickel, A. J. Britton, L. M. Arruda, H. Naggert, C. Luo, C. Lotze, H. Ryll, F. Radu, E. Schierle, E. Weschke, F. Tuczek and W. Kuch, Nat. Commun., 2018, 9, 2948 CrossRef PubMed.
- L. Poggini, G. Londi, M. Milek, A. Naim, V. Lanzilotto, B. Cortigiani, F. Bondino, E. Magnano, E. Otero, P. Sainctavit, M.-A. Arrio, A. Juhin, M. Marchivie, M. M. Khusniyarov, F. Totti, P. Rosa and M. Mannini, Nanoscale, 2019, 11, 20006–20014 RSC.
- J. Dugay, M. Aarts, M. Giménez-Marqués, T. Kozlova, H. W. Zandbergen, E. Coronado and H. S. J. van der Zantt, Nano Lett., 2017, 17, 186–193 CrossRef CAS PubMed.
- M. Gavara-Edo, R. Córdoba, F. J. Valverde-Muñoz, J. Herrero-Martín, J. A. Real and E. Coronado, Adv. Mater., 2022, 34, 2202551 CrossRef CAS PubMed.
- N. Konstantinov, A. Tauzin, U. N. Noumbé, D. Dragoe, B. Kundys, H. Majjad, A. Brosseau, M. Lenertz, A. Singh, S. Berciaud, M. L. Boillot, B. Doudin, T. Mallah and J. F. Dayen, J. Mater. Chem. C, 2021, 9, 2712–2720 RSC.
- O. Iasco, E. Rivière, R. Guillot, M. Buron-Le Cointe, J.-F. Meunier, A. Bousseksou and M.-L. Boillot, Inorg. Chem., 2015, 54, 1791–1799 CrossRef CAS PubMed.
- Y. Li, J. Wu, X. Jin, J. Wang, S. Han, W. Wu, J. Xu, W. Liu, X. Yao and Y. Tang, Dalton Trans., 2014, 43, 1881–1887 RSC.
- J. P. Lewis, P. Jelínek, J. Ortega, A. A. Demkov, D. G. Trabada, B. Haycock, H. Wang, G. Adams, J. K. Tomfohr, E. Abad, H. Wang and D. A. Drabold, Phys. Status Solidi B, 2011, 248, 1989–2007 CrossRef CAS.
- J. Harris, Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 31, 1770–1779 CrossRef CAS PubMed.
- P. Jelínek, H. Wang, J. P. Lewis, O. F. Sankey and J. Ortega, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 235101 CrossRef.
- M. A. Basanta, Y. J. Dappe, P. Jelínek and J. Ortega, Comput. Mater. Sci., 2007, 39, 759–766 CrossRef CAS.
- V. E. Campbell, M. Tonelli, I. Cimatti, J.-B. Moussy, L. Tortech, Y. J. Dappe, E. Rivière, R. Guillot, S. Delprat, R. Mattana, P. Seneor, P. Ohresser, F. Choueikani, E. Otero, F. Koprowiak, V. G. Chilkuri, N. Suaud, N. Guihéry, A. Galtayries, F. Miserque, M.-A. Arrio, P. Sainctavit and T. Mallah, Nat. Commun., 2016, 7, 13646 CrossRef CAS PubMed.
- T. Knaak, C. González, Y. J. Dappe, G. D. Harzmann, T. Brandl, M. Mayor, R. Berndt and M. Gruber, J. Phys. Chem. C, 2019, 123, 4178–4185 CrossRef CAS.
- R. Ketkaew, Y. Tantirungrotechai, P. Harding, G. Chastanet, P. Guionneau, M. Marchivie and D. J. Harding, Dalton Trans., 2021, 50, 1086–1096 RSC.
- M. Seredyuk, M. C. Muñoz, M. Castro, T. Romero-Morcillo, A. B. Gaspar and J. A. Real, Chem. – Eur. J., 2013, 19, 6591–6596 CrossRef CAS PubMed.
- S. Schönfeld, C. Lochenie, P. Thoma and B. Weber, CrystEngComm, 2015, 17, 5389–5395 RSC.
- J. Weihermüller, S. Schlamp, B. Dittrich and B. Weber, Inorg. Chem., 2019, 58, 1278–1289 CrossRef PubMed.
- S. Kuppusamy, L. Kämmerer, B. Heinrich, S. Salamon, M. Gruber, H. Wende and M. Ruben, ChemRxiv, 2022 DOI:10.26434/chemrxiv-2022-9mtbs.
- T. D. Roberts, C. M. Pask, I. C. Berdiell, F. Tuna and M. A. Halcrow, J. Mater. Chem. C, 2022, 10, 16353–16362 RSC.
- R. Mičová, Z. Bielková, C. Rajnák, J. Titiš, M. Gembický, F. Renz, O. Malina, E. Samoľová, J. Nováčiková and R. Boča, New J. Chem., 2022, 46, 18083–18089 RSC.
- K. Bairagi, A. Bellec, C. Fourmental, O. Iasco, J. Lagoute, C. Chacon, Y. Girard, S. Rousset, F. Choueikani, E. Otero, P. Ohresser, P. Sainctavit, M. L. Boillot, T. Mallah and V. Repain, J. Phys. Chem. C, 2018, 122(1), 29080–29080 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Sample preparation and characterization, NMR, MS, XAS. CCDC 2350748 and 2350749. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01417k |
| ‡ Nim and Nq are nitrogen atoms of the imine and the quinoline groups of the qsal-NEt2, respectively. |
| § Assuming a χMT value for pure HS Fe(II) as large as 3.5 cm3 mol−1 K or as low as 3.0 cm3 mol−1 K leads to proportions of LS Fe(II) species of 7.5% and 0% above T = 120 K. Only a technique such as Mössbauer would allow confirmation of the presence or absence of LS Fe(II) species at high temperatures. |
| ¶ In the case of the absence of LS Fe(II) at high temperatures, which would correspond to a χMT of 3.0 cm3 mol−1 K for the pure HS Fe(II), the proportion of HS Fe(III) at room temperature would be equal to 18% in the sample, and a reasonable fit of the spectra would be obtained. |
| || The proportion of HS Fe(II) for the bulk at T = 4 K was taken from the very first XAS spectrum in order to minimize the soft X-ray induced excited spin-state trapping (SOXIESST) effect. |
| ** The energy difference between the centre of gravity of the occupied and empty d orbitals corresponds to the crystal field parameter (monoelectronic) of the pseudo-octahedral complex. A reduction of this parameter is expected to reduce the energy difference between the ground and excited electronic states responsible for the XAS signal. |
| This journal is © The Royal Society of Chemistry 2024 |