
Cellulose-complexing strategy induced surface regulation towards ultrahigh utilization rate of Zn†
Xin
Li
a,
Hong
Yao
a,
Yuhang
Li
a,
Xiangjie
Liu
a,
Du
Yuan
 *a,
Yingqian
Chen
b,
Ming Wah
Wong
b,
Yizhou
Zhang
*c and
Haitao
Zhang
*d
*a,
Yingqian
Chen
b,
Ming Wah
Wong
b,
Yizhou
Zhang
*c and
Haitao
Zhang
*d
aCollege of Materials Science and Engineering, Changsha University of Science and Technology, Changsha, Hunan 410004, P. R. China. E-mail: aduyuandu@outlook.com
bDepartment of Chemistry, National University of Singapore, Block S8, 3 Science Drive 3, Singapore 117543, Singapore
cSchool of Chemistry and Materials Science, Institute of Advanced Materials and Flexible Electronics (IAMFE), Nanjing University of Information Science and Technology, Nanjing 210044, China
dInstitute of Process Engineering, Chinese Academy of Sciences, Beijing, 100190, P. R. China
First published on 7th June 2023
Abstract
Electrolyte design provides a fundamental solution to address the irreversibility and instability of metallic Zn anodes for the fast-developing zinc-ion batteries, considering the increasing issue of their sustainability. Herein, a cellulose-complexing approach was developed for a ZnCl2-based eutectic electrolyte to reconstruct Zn2+ coordination, tune water activity and regulate the solid–electrolyte interface (SEI). In the case of deficient water, the oxygen atoms from the glucose unit were revealed to coordinate directly with Zn2+, resulting in the participation of cellulose in the solvation shell of Zn2+, with a change in the hydrogen-bond network, where water transformed into the bulk state. The reshaped Zn2+ coordination with sluggish water activity led to a widened electrochemical window and promising ion transport in the complex electrolyte. Endowed with a dissolution–regeneration induced in situ SEI with inorganic–organic characteristics, dendrite-free Zn stripping/plating were achieved at a high current density of 50 mA cm−2 and 50 mA h cm−2 for 2000 h, with a high depth-of-discharge of 85%. The complex electrolyte was demonstrated to be beneficial for the long-term cycling stability of the activated carbon/Zn cell compared to its ZnCl2 eutectic electrolyte counterpart. Further, an artificial SEI was fabricated via electrochemical deposition using the electrolyte, possessing the merits of organic-dominant characteristics. The developed approach provides a facile route to prepare novel zinc electrolytes towards a high utilization rate of Zn.
Introduction
Aqueous rechargeable zinc ion batteries (ZIBs) are fast-developing post-lithium alternatives with the merits of high safety, cost-effectiveness, promising capacity/kinetics, and environmental friendliness.1–4 Employing aqueous zinc electrolyte facilitates Zn stripping/plating, realizing the use of metallic Zn anodes. However, the reversibility of Zn stripping/plating in conventional aqueous electrolyte faces the deep-rooted issues of dendrite formation, competitive hydrogen evolution reaction (HER), and formation of a solid–electrolyte interface (SEI).5–12 Consequently, ZIBs adopting Zn anodes typically suffer from deteriorated coulombic efficiency (CE) and limited cycling lifespan. Hence, it is essential to achieve stable Zn stripping/plating during battery operation, especially under a high current density or fast charging scheme, towards a high utilization rate of Zn.Presently, the strategies employed to prepare stable Zn anodes include electrolyte regulation, surface protection/interface engineering, and Zn anode design.5,7,8,10,11,13–18 Through the rational design of the electrolyte, Zn2 coordination can be reconstructed from hexa-hydrated [Zn(H2O)6]2+, which influences the deposition of Zn, HER and formation of an SEI. Considering that both the free water and solvated water in the electrolyte can trigger water-induced side reactions,19,20 significant efforts have been devoted to regulating the content of water and its activity in the electrolyte. For example, “water-in-salt” electrolytes such as 21 m LiTFSI-1m Zn(TFSI)2 with an extremely high salt concentration possesses water molecules in their solvation shell, where the depressed water activity leads to a widened electrochemical window (EW) and suppresses the HER.21 However, the use of a high concentration of TFSI−-based salts increases the cost and fluorine content. Alternatively, ZnCl2-based deep eutectic solvents (DESs) have the advantage of easy preparation, low cost and environmental friendliness. [Zn(H2O)6]2+ is the predominant coordination in ZnCl2·RH2O for R = 3.22,23 For R < 3, the deviated solvation structure from hexa-coordination may allow the entry of anions in the solvation shell of Zn2+, while freeing up water molecules. Hence, simultaneous tuning of the water activity and Zn2+ coordination in ZnCl2·RH2O for R < 3 remains a challenge. To break through the solution limit of ZnCl2, inorganic salts were applied through bi-salt/multi-salt approaches.19,24–26 In ZnCl2–Zn(OAc)2-H2O, OAc− coordinates with Zn2+ and constructs H-bonds with H2O molecules, forming a cross-linking H-bond network, which suppresses the activity and side reactions of water.19 OAc−-capped water–salt oligomers bridged by Br−/Cl−–H and Br−/Cl−/O–Zn2+ interactions in ZnCl2–ZnBr2–Zn(OAc)2 led to super-solubility up to 75 m.25 In ZnCl2–LiCl, H2O and Cl− preferentially coordinate with Li+ and Zn2+, respectively, where Li2ZnCl4·9H2O achieved the optimal performance.24 In parallel, electrolyte additives, especially small organic molecules were introduced to alter the Zn2+ solvation shell, including ethylene glycol (EG), acetamide (Ace), and dimethyl sulfoxide (DMSO),27–30 where the hydrogen bond (H-bond) network was formed between the solvent/ligand/anion and water. In ZnCl2-EG DES, the EG molecules participate in the Zn2+ solvation via coordination and hydrogen-bond interactions to effectively eliminate the water-induced side reactions.27 Ace in ZnCl2-acetamide (Ace)-H2O DES can be involved in the cation complex [ZnCl(Ace)2(H2O)]+, which depresses the water activity by replacing the weak H2O–H2O hydrogen bond with the strong Ace–H2O counterpart.28 Also, DMSO can replace H2O in the Zn2+ solvation sheath.29 However, water molecules could still transit from the solvated to less-bonded or free states. Hence, it is essential for the electrolyte component to participate into the H-bond network and stabilize free water.
Herein, we propose a cellulose-complexing strategy to develop ZnCl2-based DES, i.e., ZnCl2–cellulose electrolytes (ZCEs), achieving stable Zn stripping/plating at a high current density with promising Zn utilization rate (Scheme 1). Inspired by the dissolution of cellulose in ZnCl2-based DES via the influence on the H-bond network by Zn2+, we explored the scenario for deficient water in the Zn2+ coordination. The underlying complexation interaction by spectroscopy analysis and DFT calculation revealed that when R is below 3, the oxygen atoms from the glucose unit can coordinate directly with Zn2+, and hence the glucose unit can participate in the solvation shell of Zn2+, meantime influencing the H-bond and tuning the water states. To the best of our knowledge, the incorporation of biomass macromolecules as green constituents in eutectic electrolytes has rarely been explored. We demonstrate the great potential of this approach of changing the H-bond network in the DES regime via the abundant functional groups. Also, the ability of an electrolyte to induce an effective SEI on Zn is highlighted.8,10,31,32 The protrusion of Zn5(OH)8Cl2·H2O flakes formed by the ZnCl2·RH2O DES could not protect the Zn anode, with a short lifespan of Zn stripping/plating under a high current density (see our discussion below). This hinders the application of the ZnCl2·RH2O DES, which makes the construction of a robust interphase necessary. Employing ZCEs, an inorganic–organic composite in situ SEI was unveiled, exhibiting the advantage of possessing an organic component. A dissolution–regeneration model for the formation of the SEI was proposed, distinct from either the hydroxide/phosphate precipitation or reduction/decomposition mechanisms. Importantly, ZCE-2 can facilitate stable Zn stripping/plating at a high current density of 50 mA cm−2 and 50 mA h cm−2 for a long duration of 2000 h, with a high depth-of-discharge (DOD) of 85%. ZCE-2 endowed the AC/Zn cell with long-term cycling stability at 5 A g−1 over 2000 cycles compared with only 273 cycles for its ZnCl2 DES counterpart. Based on the study on the SEI, we further demonstrated the design of an artificial organic-dominant SEI on Zn by ZCEs, leading to enhanced cycling stability in the V2O5/Zn cell. Hence, the developed approach presents promising triple-function of minimizing water content, stabilizing the solvation structure of Zn2+ and inducing the formation of an SEI to regulate the electrolyte–Zn interface.
 | ||
| Scheme 1 Developed cellulose complexing strategy for ZnCl2·RH2O (R < 3) towards high DOD under a high current density. | ||
Results and discussion
Synthesis of ZCE electrolytes and their physiochemical properties
To explore the situation of R below 3, a series of ZnCl2–H2O cellulose electrolytes were prepared based on ZnCl2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O = 1:2.2, namely, ZCE-x, where x represents the amount of cellulose added (x = 0, 0.2, 0.6, and 1.0 g) (Fig. 1a). The obtained clear and transparent solution state with a cellulose content of up to 1.0 g indicated that the cellulose was well dissolved in ZCEs, where the regeneration process to make a cellulose film from ZCEs confirmed its dissolution (Fig. S1a, ESI (ESI†)).
:H2O = 1:2.2, namely, ZCE-x, where x represents the amount of cellulose added (x = 0, 0.2, 0.6, and 1.0 g) (Fig. 1a). The obtained clear and transparent solution state with a cellulose content of up to 1.0 g indicated that the cellulose was well dissolved in ZCEs, where the regeneration process to make a cellulose film from ZCEs confirmed its dissolution (Fig. S1a, ESI (ESI†)).
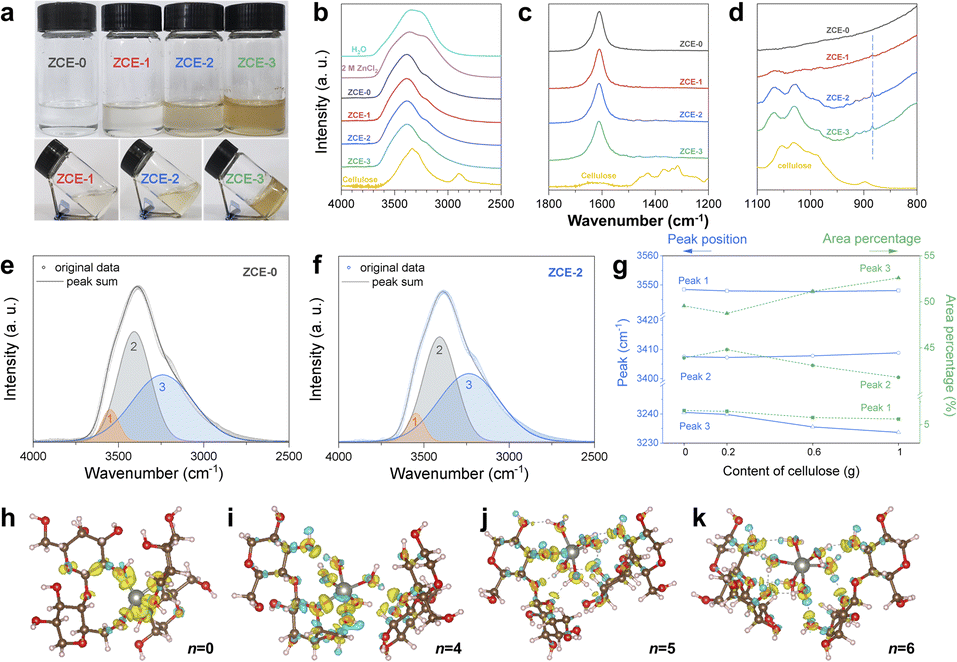 | ||
| Fig. 1 Synthesis of ZCE electrolytes. (a) Optical images of as-synthesized ZCE electrolytes. FTIR-ATR spectra of ZCEs for (b) 4000 to 2500 cm−1, (c) 1800 to 1200 cm−1, and (d) 1100 to 800 cm−1. Deconvolution on FTIR spectra for (e) ZCE-0 and (f) ZCE-2, with (g) peak shift and relative percentage change in different water states in ZCEs. Charge density difference maps for coordination of cellulose to [Zn(OH2)n]2+ in [Zn(OH2)n]2+·2 cellulose (h) n = 0, (i) n = 4, (j) n = 5, and (k) n = 6 clusters. The light turquoise corresponds to charge depletion, while light yellow shows charge accumulation, where isovalues = 0.01 e Å−3 for n = 0 and 0.005 e Å−3 for n = 4, 5, 6. Atom color scheme: C-brown, O-red, H-light pink, and Zn-grey. Visualization was done using VESTA.33 | ||
Fourier transform infrared (FTIR) spectra were obtained to characterize the solvation details of ZCEs (Fig. 1b–d). When cellulose was introduced, all the ZCEs presented distinctive –OH vibrational features from either the ZnCl2–H2O DES or cellulose (i.e., intramolecular O(2)H⋯O(6), O(3)H⋯O(5) and intermolecular O(6)H⋯O(3) stretching vibrations).34–37 This feature reflects the change in the H-bond network in ZCEs. Also, the intensity of the peak at 1430 cm−1 decreased, which is assigned to the OCH in-plane bending of the alcohol group, aromatic skeletal vibration with CH in-plane deformation, and CH2 scissoring.37 This suggests that the dissolution of cellulose in ZnCl2–H2O occurs when R < 3, accompanied with the loss of the crystalline structure of cellulose.35,38 In ZCEs, the C–O valence vibration mainly from C(3)–O(3)H blue-shifted (∼1065–1069 cm−1) with respect to that for cellulose (1052 cm−1), and the C–O stretching at C(6) red-shifted (∼1030–1027 cm−1) with respect to that for cellulose (1031 cm−1), which can be ascribed to the possible formation of HO⋯Zn2+.34,39 This was further verified by the emerging peak at 870 cm−1, which is assigned to the ZnCl2–cellulose complex.40 Deconvolution of –OH stretching was performed to differentiate the three types of water states in ZCEs (Fig. 1e and f), as follows: (1) ∼3540 cm−1, isolated water with weakly bonded amorphous state, (2) ∼3400 cm−1, cluster water with ice-like liquid state, and (3) ∼3240 cm−1, bulk water with ice-like state.41–45 When the cellulose content increased, the area percentages of the isolated and cluster water dropped from 6.5% to 5.6% and from 43.9% to 41.8%, respectively (Fig. 1g). Meanwhile, the peak corresponding to bulk water red-shifted from 3240 cm−1 to 3233 cm−1, and its area percentage increased from 49.6% to 52.6%. The above-mentioned results confirm the change in the H-bond network during the formation of ZCEs, where the isolated and cluster water may transform to the bulk state.
Although the vibrational characteristics of ZCEs strongly suggest the reconstructed Zn2+ coordination with the involved glucose unit, the coordination of Zn2+ with cellulose in the ZnCl2-based DES for R < 3 is still unknown. To provide insight into the Zn2+ coordination, DFT calculations were performed to study the correlation among Zn2+, water, and the glucose-unit. The molecular configuration of [Zn(OH2)n]2+·2 cellulose was considered, where the corresponding formation energy was calculated for each n (Fig. S2†). The evaluation of the convex hull plot (Fig. S2†) indicates the possible formation of stable Zn2+-(OH2)n cellulose clusters with n = 4, 5 besides n = 6 (i.e., R = 3). The selected charge density difference maps imply the possible bonding between [Zn(OH2)n]2+ and cellulose. At n = 6, Zn2+ is primarily coordinated with H2O and only the H of H2O exhibits hydrogen bonds with the O(H) of cellulose (Fig. 1k), consistent with the dissolution of cellulose by influencing the H-bond network by the ZnCl2 DES.46 Distinctively, bonding can occur between Zn2+ and the active sites in the glucose unit when n < 6, i.e., O(3) at n = 5 (Fig. 1j), and O(5), O(6)H at n = 4 (Fig. 1i). Meanwhile, the remaining oxygen sites in the unit, i.e., those unbonded with Zn2+, can form H-bonds with the solvated water. Hence, this clearly reveals that the glucose unit can participate in the Zn2+ coordination in ZnCl2:H2O (R < 3), reshaping the Zn2+ coordination and H-bond network with the altered water states in ZCEs.
Electrochemical properties of ZCEs
The fundamental properties of ZCEs were investigated to evaluate their potential as advanced electrolytes for ZIBs (Fig. 2). Although the EW of ZCE-1 is close to that of ZCE-0, the EWs of ZCE-2 and ZCE-3 are largely extended to 3.3 and 3.9 V, respectively (Fig. 2a). Specifically, the higher OER onset potential increased from 1.96 V (ZCE-0) to 3.872 V (ZCE-3) vs. Zn/Zn2+ (also see Table S1†). This indicates the greatly depressed water activity through ZnCl2–H2O–cellulose coordination, which can be attributed to the transformation of the water molecules from the free state into the bulk state (Fig. 1g) and the solvated water molecules form H-bonds with the unbonded O sites in the glucose unit (Fig. 1i and j). It should be noted that the close EWs of ZCE-0 and ZCE-1 can be attributed to either the insufficient cellulose content in ZCE-1 to effectively tune the water activity (Fig. 1g) or the formation of zinc chloride hydroxide on Zn in ZCE-0, which partially inhibits the evolution of oxygen. Alternatively, the onset potential of Zn deposition varied slightly across ZCEs (∼−0.02 to −0.03 V vs. Zn/Zn2+), suggesting that ZCEs may favor the deposition process. Also, a lower nucleation potential of ∼14 mV was achieved with ZCE-2 than that of ∼20 mV for ZCE-0 (Fig. 2b), suggesting the more uniform Zn deposition via ZCE-2. With the characteristics of Zn stripping/plating as in the cyclic voltammetry (CV) spectra (Fig. S3†), asymmetric Ti|Zn cells were tested to verify the advantage of the ZCE electrolytes on the coulombic efficiency (CE) during Zn stripping/plating under a constant current density and capacity density of 1 mA cm−2 and 1 mA h cm−2, respectively (Fig. 2c). Besides outperforming the reference electrolytes (e.g., 2 M ZnSO4 with a CE approaching ∼95% and 2 M ZnCl2 with CE fluctuating severely after 20 cycles) (Fig. S4†), ZCE-2 presented a stable CE of 100% after only the second cycle, which is superior to its counterparts (ZCE-0 stabilized after ∼20 cycles, while ZCE-1 and ZCE-3 approached CEs of ∼99% after 40 cycles but showed inflation during sequential cycles), implying the ability of quickly stabilizing the interface between electrolyte–Zn for ZCE-2 during stripping/plating, which can proceed up to 150 h. Further, ZCE-2 could facilitate stable stripping/plating even at high current densities of 10 mA cm−2, 10 mA h cm−2 and 50 mA cm−2 (Fig. S4†). In addition, a lower charge transfer resistance (RCT) of 5.3 Ω was observed in ZCE-2 compared with 8.1 Ω for ZCE-0 (Fig. 2f), which favors the interface kinetics. Tafel plots were applied to study the effects of ZCEs on the corrosion of the Zn surface (Fig. 2d and Table S2†). The corrosion voltages of ZCEs (−0.66–65 V vs. Ag/AgCl) showed significant positive shifts compared with that of 2 M ZnCl2 (−0.94 V vs. Ag/AgCl). Besides, ZCE-2 showed a reduced corrosion current of ∼0.19 mA cm−2 compared to its counterparts, indicating its effectiveness in hindering Zn corrosion. Furthermore, the ion transport behavior of ZCEs was studied. With the addition of cellulose, the ion conductivity followed a gradual decline trend from 8.7 mS cm−1 for ZCE-0 to 4.4 mS cm−1 for ZCE-3, where 5.4 mS cm−1 was obtained for ZCE-2 (Fig. 2e), which was confirmed by EIS (Fig. S5†). The decrease in ion conductivity can be attributed to the interaction between Zn2+ and the glucose unit. The transference number (tZn2+) was measured to study the contribution from Zn2+ to the ion transport (Fig. 2f). Compared with the tZn2+ of 0.51 for ZCE-0, the tZn2+ for ZCE-2 increased significantly to 0.70, indicating a higher contribution from Zn2+ to the ion transport. The improved transference number can be associated with the weaken cation–anion interaction or immobilized anion in ZCE-2, which can effectively reduce the concentration gradient and promote the interface kinetics.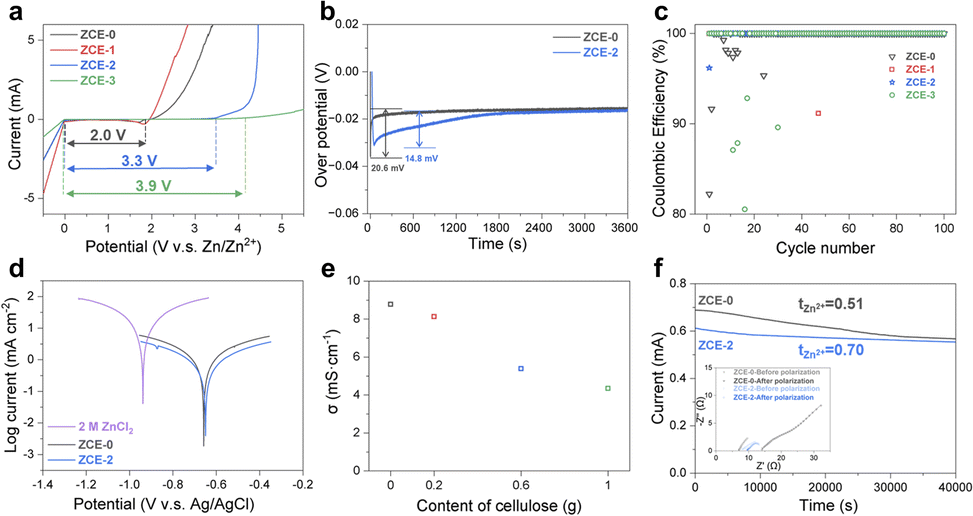 | ||
| Fig. 2 Fundamental properties of ZCE electrolytes: (a) electrochemical windows of ZCEs, (b) nucleation overpotentials of Zn deposition for ZCE-0 and ZCE-2, (c) coulombic efficiencies of Ti|Zn cells in ZCEs, (d) Tafel plots of Zn anodes in the electrolytes, (e) ion conductivities of ZCEs, and (f) transference numbers, where the inset presents the EIS spectra of the Zn|Zn symmetrical cell with ZCE-0 and ZCE-2 before and after polarization. | ||
Subsequently, the lifespan of the Zn metal anode with ZCEs was assessed by the Zn|Zn symmetric cells under a series of galvanostatic conditions (Fig. 3). Under 10 mA cm−2 and 10 mA h cm−2, although the improvement by ZCE-1 could be barely sensed (Fig. 3b), the cells with both ZCE-2 and ZCE-3 proceeded with stable Zn stripping/plating for over 2000 h at 10 mA cm−2 and 10 mA h cm−2, corresponding to a DOD of 17% (Fig. 3c and d, respectively). In addition, the cell with ZCE-2 possessed a much lower overpotential of ∼81 mV at 2000 h than that of ∼290 mV with ZCE-3 (Fig. S6†). Further, a stringent deep charging/discharging test was carried out for the cell with ZCE-2 under 50 mA cm−2 and 50 mA h cm−2. The highly reversible and stable stripping/plating lasted for over 2000 h, with a high DOD of 85% (Fig. 3e). This deep stripping/plating was verified by sequential cross-sectional SEM during cycling (Fig. 3m–o), where the estimation based on the thicknesses of the deposited and pristine Zn is consistent with the observed value. The stable cycling was further confirmed by EIS before and after cycling (Fig. S7†). This demonstrates the efficient utilization of Zn with ZCE-2, which is superior to the reported ZnCl2-based electrolytes (Table S3†) and among the current highest records.47,48
 | ||
| Fig. 3 Electrochemical performance of Zn plating/stripping in Zn|Zn symmetrical cells at 10 mA cm−2 and 10 mA h cm−2 for (a) ZCE-0, (b) ZCE-1, (c) ZCE-2, and (d) ZCE-3 and (e) at 50 mA cm−2 and 50 mA h cm−2 for ZCE-2, with a high DOD of 85%. Ex situ characterization of Zn foils after stripping/plating for 1000 cycles with ZCEs: optical images of the cycled Zn in (f) ZCE-0 and (g) ZCE-2, SEM images of the cycled Zn in (h) ZCE-0 with (i) the magnified view and in (j) ZCE-2 with (k) magnified view, and (l) FTIR spectra comparing the cycled Zn foils. (m–o) Ex situ cross-sectional SEM of stripping/plating of Zn|Zn with ZCE-2 under 50 mA h cm−2 and 50 mA cm−2, after (m) 1st cycle, (n) 10th cycle, and (o) 500th cycle, respectively. | ||
To explore the Zn surface after stripping/plating, ex situ characterization was conducted. Compared to the relatively rough and grey appearance with rugged and non-uniform flakes on the surface with ZCE-0 (Fig. 3f, h and i), the Zn surface after cycling in ZCE-2 appeared to be evenly covered with a dark layer, which was uniform, dense, and dendrite free (Fig. 3g, j and k). The EDX mapping comparing the cycled foils with ZCEs indicated the presence of a homogeneous distribution of C, O, Cl and Zn on the cycled Zn with ZCE-2 (Fig. S8†), suggesting the formation of organic constituents on the surface. According to FTIR-ATR, the peak at 3504 cm−1 (peak 1) for Zn with ZCE-0 (Fig. 3l) is assigned to the –OH stretching vibration at Zn5(OH)8Cl2·H2O.49 For Zn with ZCE-2, the peaks at 3330 (peak 2), 2908 (peak 3), 1375 (peak 4) and 1030 cm−1 (peak 5) can be assigned to the O(6)H⋯O(3) intermolecular stretching vibration, –CH stretching vibration, –CH deformation vibration and C(3)–O(3)H liberation vibration, respectively.34,35 Also, the emerging peak at ∼850 cm−1 (peak 6) can be assigned to the Zn2+–cellulose complex. These features confirm the presence of an inorganic–organic composite layer on the cycled Zn by ZCE-2 with the emerging Zn2+–cellulose complex, different from the inorganic surface by ZCE-0. To verify the surface structure, the XRD patterns of the samples were compared. The diffraction peaks of the cycled Zn with ZCE-0 are assigned to Zn5(OH)8Cl2·H2O with a strong peak at 11.13°, corresponding to the (003) diffraction (Fig. S9a†). When cycled in ZCE-2, the diffraction peak for zinc chloride hydroxide (003) became broader (Fig. S9a†), with an emerging broad diffraction feature peak at ∼12.8°, which is attributed to the possible formation of cellulose (Fig. S10†), consistent with the uniform distribution of C and the corresponding vibrational features. Also, the thickness of this surface layer increased from ∼12 to 43 μm during cycling (Fig. 3m–o). The formation of the Zn2+–cellulose complex can be further understood by the selected charge density difference map for Zn2+·2 cellulose (Fig. 1h), where Zn2+ can bond with the 5 O(H) of cellulose. Thus, compared to the loosely grown and unprotected surface by ZCE-0 (i.e., ZnCl2 DES), a uniform inorganic–organic composite surface by ZCE-2 was revealed, functioning as a robust SEI for the Zn anode. Meanwhile, the increase in the intensity ratio for Zn (002)/(101) by ZCE-2 compared to the ZnCl2-based electrolytes (Fig. S9b†) can be attributed to the reshaped Zn2+ coordination and preferred adsorption of glucose on Zn (002).50
Based on the above-mentioned results, the achieved long-term ultrahigh utilization rate of Zn during stripping/plating is attributed to the following factors via ZCEs: (1) by reshaping the Zn2+ coordination by cellulose with deficient water, ZCEs can depress the water activity to achieve a widened EW, with the depressed water-associated side reactions. (2) High ionic conductivity and the largely improved transference number can reduce the concentration gradient to hinder dendrite growth and promote the interface kinetics. (3) An inorganic–organic in situ SEI can be formed with the Zn2+–cellulose complex, functioning as a protective layer. (4) Besides, the Zn (002) texture is promoted, which benefits dendrite-free Zn deposition. To demonstrate the application of ZCEs in ZIBs, activated carbon (AC)/Zn full cells were assembled (Fig. 4). At 1 A g−1, stable cycling could proceed for 1500 cycles, with an initial discharge capacity of ∼57.8 mA h g−1 and ∼60.6 mA h g−1 in the 1500th cycle (Fig. S11†). In contrast, only ∼166 cycles were achieved with ZCE-0, where the short circuit is attributed to Zn dendrite growth. The long-term cycling of the AC/Zn cell with ZCE-2 was further displayed at 5 A g−1, with a promising capacity retention rate (47.2 mA h g−1 in the 1st cycle and 54.2 mA h g−1 in the 3000th cycle) and CE of 100% for over 3000 cycles compared with ∼273 cycles with ZCE-0. Thus, ZCE-2 presented a superior high-rate performance and cycling stability compared to its counterpart. Based on this, a cellulose-complexing strategy for the preparation of novel electrolytes was successfully developed. It should be noted that the developed approach did not lose its generality when using tissue paper (Fig. S1b†), which may further reduce the processing cost.
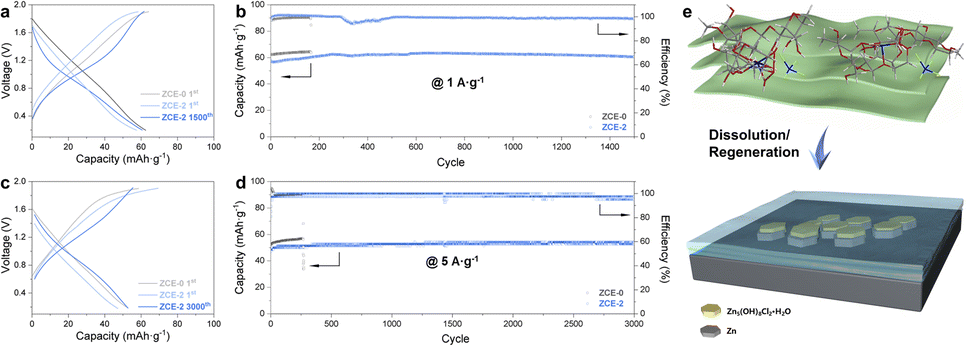 | ||
| Fig. 4 Charge/discharge curves of AC/Zn cells at (a) 1 A g−1 and (c) 5 A g−1, with their cycling stabilities at (b) 1 A g−1 and (d) 5 A g−1, comparing ZCE-0 and ZCE-2. (e) Scheme of constructing an organic-dominant artificial SEI by ZCEs for Zn anode. | ||
Artificial SEI by ZCEs
To further perceive the critical role of organic constituents in the ZCE-indued SEI as revealed by the ex situ study, an artificial layer on Zn foil was fabricated via an electrochemical deposition method using ZCEs (Fig. 4e). A uniform layer grew on Zn by ZCE-2 within 3 h (Zn-ZCE-2-3 h) on the scale of ∼20 cm2 (Fig. 5a, see also Fig. S12† for the samples deposited by different conditions). The microscale uniformity of this surface was confirmed by SEM (Fig. 5b and c) compared with the typical crystalline facets by ZCE-0 (Fig. S13†), with a homogeneous distribution of O, C, Cl, and Zn (Fig. 5d). Also, the thickness of this layer was determined to be ∼12.7 μm (Fig. 5e and f). The surface structure was differentiated by XRD spectra (Fig. 5g). By tuning the deposition time, the broad diffraction peaks at ∼9.89–14.90° became dominant over the signal of Zn5(OH)8Cl2·H2O (11.39°) from either ZCE-0 or 2 M ZnCl2 (see also Fig. S14† for Zn-ZCE-3-3 h), which can be attributed to the formation of crystalline cellulose with the Zn2+–cellulose complex (Fig. S10†). Meanwhile, FTIR spectra confirmed the growing trends of vibrational peaks for the C–O valence vibration and Zn2+-complex at 1044 and 828 cm−1 (peaks 1 and 3), over the –OH vibration at 904 cm−1 (peak 2) for Zn5(OH)8Cl2·H2O (Fig. 5h), respectively. The above-mentioned results confirm the dominance of the organic components over the inorganic ones in the surface layer. This organic-dominant feature suggests the presence of a stable electrolyte–Zn interface, given that the formation of zinc chloride hydroxide during electrodeposition resulted from the corrosion process. Further, XPS spectra were measured to determine the surface details (Fig. 5i and S15†). An intensity ratio I(C–O)/I(O–C–O) of 4 was taken as a measure on the characteristics of cellulose.51 Across the samples from 1 h to 3 h treatment, the I(C–O)/I(O–C–O) ratio was well-retained at ∼4, suggesting the presence of glucose units on the surface. Meanwhile, the peak for C–O red-shifted from 286.2 eV in cellulose to 286.0 eV for ZCE-Zn-1 h and 285.1 eV for ZCE-Zn-3 h. This is accompanied with a blue-shift in Zn–O during the growth of the surface layer (Fig. S16†). This can be ascribed to the participation of C–OH in the Zn2+ coordination. A time-dependent study was conducted to confirm the formation of an SEI during deposition (Fig. S17†). Moreover, XPS depth profiling on the artificial SEI confirmed the dominance of organic species on the surface and revealed a gradual increase in inorganic component towards the interior, suggesting a well-protected surface against corrosion (Fig. S18†). Therefore, the scheme in Fig. 4e is proposed for the growth of this organic-dominant SEI. During charging, ZCEs can be driven towards the electrode surface. When Zn deposition occurs, less Zn2+ is present in ZCE, where the regeneration of cellulose with the Zn2+–cellulose complex can be attributed to the change in the H-bond network and coordination of the glucose unit at the Zn surface. The organic-dominant surface feature further endows the gradient of organic (outer)–inorganic (inner) profiling in the SEI. This dissolution–regeneration mechanism driven by the change in H-bond network and coordination of the glucose unit is distinct from the current recognized precipitation or reduction/decomposition mechanism for in situ SEI formation on Zn. | ||
| Fig. 5 Investigation of the artificial SEI for Zn. (a) Optical image of the deposited layer. SEM images showing (b) its uniform morphology and (c) magnified view. (d) EDX mapping indicating the presence of O, C, Cl, and Zn. (e) Cross-section view and (f) the magnified view. (g) XRD spectra and (h) FTIR spectra of the surface layer deposited by different electrolytes. (i) XPS spectra of C 1s for Zn-ZCE-2-3 h. (j) LSV curves comparing the surface-protected Zn and bare Zn in 2 M NaCl. (k) Tafel plots comparing the corrosion resistance behaviors for the surface-protected Zn and bare Zn in 2 M Zn(OTf)2. (l) Transference number concerning the surface layer, where the inset shows the EIS spectra before and after polarization. | ||
This artificial SEI can effectively depress hydrogen evolution, where Zn-ZCE-2-3 h negatively shifts the HER onset potential to −1.55 V (vs. Ag/AgCl) compared with the bare Zn (−1.46 V vs. Ag/AgCl) in 2 M NaCl (Fig. 5j). Also, the surface presented a positive shift in corrosion potential to ∼0.88 V vs. Ag/AgCl with a lowered corrosion current of 0.37 mA cm−2 (∼1/2 of that for bare Zn) in 2 M Zn(OTf)2 (Fig. 5k), indicating its better corrosion resistance. The observed decrease in RCT with the growth of the surface layer by real-time EIS suggests that favored interface kinetics can be achieved through the surface layer (Fig. S19†). Moreover, its advantage may be understood by the improved transference number of ∼0.59 achieved for the cell assembled with Zn-ZCE-2-3 h in Zn(OTf)2 compared with ∼0.32 for that with bare Zn (Fig. 5i and S20†). This indicates the restriction of anion mobility of this organic-dominant surface, where electrostatic repulsion and steric hindrance may operate. With these merits, the protected Zn anode was applied in the V2O5/Zn full cell. The cell with Zn-ZCE-2-3 h showed a better retention of capacity of 97.0% compared to 90.6% with that of bare Zn (Fig. S21†). Also, at 0.1 A g−1, the V2O5/Zn-ZCE-2-3 h cell presented a promising capacity retention of 72.8% (279 mA h g− 1) compared with that of only ∼16.6% using bare Zn (Fig. S22†). The improved cycling stability can be attributed to the influenced balance of cathode dissolution by the retarded Zn corrosion.52 Therefore, the designed complexing electrolyte regulates the chemistry on the electrolyte–Zn interface, facilitating the development of ZIBs.
Conclusions
In summary, we demonstrated the synthesis of a series of ZnCl2–H2O-cellulose electrolytes concerning a scenario of R below 3. Spectroscopy analysis and DFT calculation revealed that glucose unit can participate in the solvation shell of Zn2+, where Zn2+ can be coordinated with either O(3) at n = 5, or O(5), O(6)H at n = 4 in the glucose unit. This underlying complexation interaction for deficient water is different from the case for R = 3 or above, where only the H-bond surrounding [Zn(H2O)6]2+ is influenced. Meanwhile, tuning of the water states was achieved in ZCEs, with a reduced free water content and enhanced bulk water content. ZCEs possessed a widened EW, fast ion transport and highly improved transference number. Stable and reversible Zn stripping/plating could be achieved even at a high current density of 50 mA cm−2 and 50 mA h cm−2 with a high utilization rate of 85%. In situ formation of an inorganic–organic composite SEI was unveiled with the Zn2+–cellulose complex, where a dissolution–regeneration mechanism was proposed. The enhanced long-term cycling stability of the AC/Zn cell with ZCE over that with ZnCl2 DES was demonstrated. Further, an artificial surface layer on Zn was prepared based on ZCEs with organic-dominant characteristics, favoring the interface kinetics and showing an advantage for the cycling stability of the V2O5/Zn cell. The developed approach provides a new and sustainable route towards the regulation of the solvation structure of electrolytes and building an effective SEI for aqueous rechargeable batteries.Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. Yuan would like to acknowledge the funding support from the 100 Talented Team of Hunan Province (XiangZu [2016] 91), and the Natural Science Foundation of Hunan Province (2022JJ30613). Y. Zhang would like to acknowledge the funding support from the National Natural Science Foundation of China (62174085), the Startup Foundation for Introducing Talent of NUIST (2021r091). H. Zhang would like to acknowledge the National Natural Science Foundation of China (No. 21878308).Notes and references
- N. Zhang, X. Chen, M. Yu, Z. Niu, F. Cheng and J. Chen, Chem. Soc. Rev., 2020, 49, 4203–4219 RSC.
- F. Wan, X. Zhou, Y. Lu, Z. Niu and J. Chen, ACS Energy Lett., 2020, 5, 3569–3590 CrossRef CAS.
- D. Chao, W. Zhou, F. Xie, C. Ye, H. Li, M. Jaroniec and S.-Z. Qiao, Sci. Adv., 2020, 6, eaba4098 CrossRef CAS PubMed.
- L. E. Blanc, D. Kundu and L. F. Nazar, Joule, 2020, 4, 771–799 CrossRef CAS.
- Q. Yang, Q. Li, Z. Liu, D. Wang, Y. Guo, X. Li, Y. Tang, H. Li, B. Dong and C. Zhi, Adv. Mater., 2020, 32, 2001854 CrossRef CAS PubMed.
- Y. Zhang, Z. Chen, H. Qiu, W. Yang, Z. Zhao, J. Zhao and G. Cui, NPG Asia Mater., 2020, 12, 4 CrossRef CAS.
- J. Hao, X. Li, X. Zeng, D. Li, J. Mao and Z. Guo, Energy Environ. Sci., 2020, 13, 3917–3949 RSC.
- W. Du, E. H. Ang, Y. Yang, Y. Zhang, M. Ye and C. C. Li, Energy Environ. Sci., 2020, 13, 3330–3360 RSC.
- Q. Zhang, J. Luan, Y. Tang, X. Ji and H. Wang, Angew. Chem., Int. Ed., 2020, 59, 13180–13191 CrossRef CAS PubMed.
- Z. Yi, G. Chen, F. Hou, L. Wang and J. Liang, Adv. Energy Mater., 2021, 11, 2003065 CrossRef CAS.
- V. Verma, S. Kumar, W. Manalastas and M. Srinivasan, ACS Energy Lett., 2021, 6, 1773–1785 CrossRef CAS.
- Q. Yang, G. Liang, Y. Guo, Z. Liu, B. Yan, D. Wang, Z. Huang, X. Li, J. Fan and C. Zhi, Adv. Mater., 2019, 31, 1903778 CrossRef CAS PubMed.
- J. Zheng and L. A. Archer, Sci. Adv., 2021, 7, eabe0219 CrossRef CAS PubMed.
- J. Zheng, Q. Zhao, T. Tang, J. Yin, C. D. Quilty, G. D. Renderos, X. Liu, Y. Deng, L. Wang, D. C. Bock, C. Jaye, D. Zhang, E. S. Takeuchi, K. J. Takeuchi, A. C. Marschilok and L. A. Archer, Science, 2019, 366, 645–648 CrossRef CAS PubMed.
- D. Yuan, J. Zhao, H. Ren, Y. Chen, R. Chua, E. T. J. Jie, Y. Cai, E. Edison, W. Manalastas Jr., M. W. Wong and M. Srinivasan, Angew. Chem., Int. Ed., 2021, 60, 7213–7219 CrossRef CAS PubMed.
- Z. Chen, J. Zhao, Q. He, M. Li, S. Feng, Y. Wang, D. Yuan, J. Chen, H. N. Alshareef and Y. Ma, ACS Energy Lett., 2022, 3564–3571, DOI:10.1021/acsenergylett.2c01920.
- Z. Shen, J. Mao, G. Yu, W. Zhang, S. Mao, W. Zhong, H. Cheng, J. Guo, J. Zhang and Y. Lu, Angew. Chem., Int. Ed., 2023, 135, e202218452 CrossRef.
- W. Yuan, X. Nie, G. Ma, M. Liu, Y. Wang, S. Shen and N. Zhang, Angew. Chem., Int. Ed., 2023, 135, e202218386 Search PubMed.
- M. Yang, J. Zhu, S. Bi, R. Wang and Z. Niu, Adv. Mater., 2022, 34, 2201744 CrossRef CAS PubMed.
- F. Yang, J. A. Yuwono, J. Hao, J. Long, L. Yuan, Y. Wang, S. Liu, Y. Fan, S. Zhao, K. Davey and Z. Guo, Adv. Mater., 2022, 34, 2206754 CrossRef CAS PubMed.
- F. Wang, O. Borodin, T. Gao, X. Fan, W. Sun, F. Han, A. Faraone, J. A. Dura, K. Xu and C. Wang, Nat. Mater., 2018, 17, 543–549 CrossRef CAS PubMed.
- R. J. Wilcox, B. P. Losey, J. C. W. Folmer, J. D. Martin, M. Zeller and R. Sommer, Inorg. Chem., 2015, 54, 1109–1119 CrossRef CAS PubMed.
- X. Ji, eScience, 2021, 1, 99–107 CrossRef.
- C. Yang, J. Xia, C. Cui, T. P. Pollard, J. Vatamanu, A. Faraone, J. A. Dura, M. Tyagi, A. Kattan, E. Thimsen, J. Xu, W. Song, E. Hu, X. Ji, S. Hou, X. Zhang, M. S. Ding, S. Hwang, D. Su, Y. Ren, X.-Q. Yang, H. Wang, O. Borodin and C. Wang, Nat. Sustain, 2023, 6, 325–335 CrossRef.
- S. Cai, X. Chu, C. Liu, H. Lai, H. Chen, Y. Jiang, F. Guo, Z. Xu, C. Wang and C. Gao, Adv. Mater., 2021, 33, 2007470 CrossRef CAS PubMed.
- H. Liu, C.-Y. Chen, H. Yang, Y. Wang, L. Zou, Y.-S. Wei, J. Jiang, J. Guo, W. Shi, Q. Xu and P. Cheng, Adv. Mater., 2020, 32, 2004553 CrossRef CAS PubMed.
- L. Geng, J. Meng, X. Wang, C. Han, K. Han, Z. Xiao, M. Huang, P. Xu, L. Zhang, L. Zhou and L. Mai, Angew. Chem., Int. Ed., 2022, 61, e202206717 CAS.
- J. Shi, T. Sun, J. Bao, S. Zheng, H. Du, L. Li, X. Yuan, T. Ma and Z. Tao, Adv. Funct. Mater., 2021, 31, 2102035 CrossRef CAS.
- L. Cao, D. Li, E. Hu, J. Xu, T. Deng, L. Ma, Y. Wang, X.-Q. Yang and C. Wang, J. Am. Chem. Soc., 2020, 142, 21404–21409 CrossRef CAS PubMed.
- D. Han, T. Sun, R. Zhang, W. Zhang, T. Ma, H. Du, Q. Wang, D. He, S. Zheng and Z. Tao, Adv. Funct. Mater., 2022, 32, 2209065 CrossRef CAS.
- J. Hao, L. Yuan, Y. Zhu, M. Jaroniec and S.-Z. Qiao, Adv. Mater., 2022, 34, 2206963 CrossRef CAS PubMed.
- S.-J. Zhang, J. Hao, H. Li, P.-F. Zhang, Z.-W. Yin, Y.-Y. Li, B. Zhang, Z. Lin and S.-Z. Qiao, Adv. Mater., 2022, 34, 2201716 CrossRef CAS PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- Q. Xu, C. Chen, K. Rosswurm, T. Yao and S. Janaswamy, Carbohydr. Polym., 2016, 149, 274–281 CrossRef CAS PubMed.
- S. Y. Oh, D. I. Yoo, Y. Shin and G. Seo, Carbohydr. Res., 2005, 340, 417–428 CrossRef CAS PubMed.
- T. Kondo and C. Sawatari, Polymer, 1996, 37, 393–399 CrossRef CAS.
- M. Schwanninger, J. C. Rodrigues, H. Pereira and B. Hinterstoisser, Vib. Spectrosc., 2004, 36, 23–40 CrossRef CAS.
- X.-F. Zhang, T. Hou, J. Chen, Y. Feng, B. Li, X. Gu, M. He and J. Yao, ACS Appl. Mater. Interfaces, 2018, 10, 24930–24936 CrossRef CAS PubMed.
- J. Han, C. Zhou, Y. Wu, F. Liu and Q. Wu, Biomacromolecules, 2013, 14, 1529–1540 CrossRef CAS PubMed.
- N. J. Richards and D. G. Williams, Carbohydr. Res., 1970, 12, 409–420 CrossRef CAS.
- Y. Wang, T. Wang, D. Dong, J. Xie, Y. Guan, Y. Huang, J. Fan and Y.-C. Lu, Matter, 2022, 5, 162–179 CrossRef CAS.
- J. B. Brubach, A. Mermet, A. Filabozzi, A. Gerschel and P. Roy, J. Chem. Phys., 2005, 122, 184509 CrossRef PubMed.
- T. Petit, L. Puskar, T. Dolenko, S. Choudhury, E. Ritter, S. Burikov, K. Laptinskiy, Q. Brzustowski, U. Schade, H. Yuzawa, M. Nagasaka, N. Kosugi, M. Kurzyp, A. Venerosy, H. Girard, J.-C. Arnault, E. Osawa, N. Nunn, O. Shenderova and E. F. Aziz, J. Phys. Chem. C, 2017, 121, 5185–5194 CrossRef CAS.
- Z. Wang, A. Pakoulev, Y. Pang and D. D. Dlott, J. Phys. Chem. A, 2004, 108, 9054–9063 CrossRef CAS.
- Z.-F. Wei, Y.-H. Zhang, L.-J. Zhao, J.-H. Liu and X.-H. Li, J. Phys. Chem. A, 2005, 109, 1337–1342 CrossRef CAS PubMed.
- S. Sen, B. P. Losey, E. E. Gordon, D. S. Argyropoulos and J. D. Martin, J. Phys. Chem. B, 2016, 120, 1134–1141 CrossRef CAS PubMed.
- Y. Qin, H. Li, C. Han, F. Mo and X. Wang, Adv. Mater., 2022, 34, 2207118 CrossRef CAS PubMed.
- Z. Zhao, J. Zhao, Z. Hu, J. Li, J. Li, Y. Zhang, C. Wang and G. Cui, Energy Environ. Sci., 2019, 12, 1938–1949 RSC.
- J. Winiarski, W. Tylus, K. Winiarska, I. Szczygieł and B. Szczygieł, J. Spectrosc., 2018, 2018, 2079278 Search PubMed.
- P. Sun, L. Ma, W. Zhou, M. Qiu, Z. Wang, D. Chao and W. Mai, Angew. Chem., Int. Ed., 2021, 60, 18247–18255 CrossRef CAS PubMed.
- L.-S. Johansson, J. M. Campbell and O. J. Rojas, Surf. Interface Anal., 2020, 52, 1134–1138 CrossRef CAS.
- Y. Kim, Y. Park, M. Kim, J. Lee, K. J. Kim and J. W. Choi, Nat. Commun., 2022, 13, 2371 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: experimental methods; regeneration of cellulose film, synthesis of ZCE-Px electrolytes; formation energy vs. number of water molecules; onset potentials of Zn/Zn2+ and OER; CV of ZCEs; CEs of ZCEs; corrosion potentials and corrosion currents for the electrolytes; EIS spectra to estimate the ionic conductivities of ZCEs; voltage profiles of ZCEs; EIS spectra of Zn|Zn before and after cycling; summary of DOD for ZnCl2-based electrolytes; EDX mapping for the cycled Zn; XRD for the cycled Zn; XRD literature for cellulose crystalline; rate performance of AC/Zn cells; optical image of the artificial SEI; SEM of deposited Zn by ZnCl2; XRD of the artificial SEI; XPS of C 1s, Zn 2p for the artificial SEI; time-dependent study of the SEI growth; real time ESI monitoring of the SEI growth; transference number; rate performance of V2O5/Zn cell, cycling stability of V2O5/Zn cell at 0.1 A g−1. See DOI: https://doi.org/10.1039/d3ta02117c |
| This journal is © The Royal Society of Chemistry 2023 |