
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rapid synthesis of magnetic microspheres and the development of new macro–micro hierarchically porous magnetic framework composites†
John Luke
Woodliffe
 a,
Jesús
Molinar-Díaz
a,
Md Towhidul
Islam
abc,
Lee A.
Stevens
a,
Matthew D.
Wadge
a,
Graham A.
Rance
d,
Rebecca
Ferrari
a,
Ifty
Ahmed
a and
Andrea
Laybourn
*a
a,
Jesús
Molinar-Díaz
a,
Md Towhidul
Islam
abc,
Lee A.
Stevens
a,
Matthew D.
Wadge
a,
Graham A.
Rance
d,
Rebecca
Ferrari
a,
Ifty
Ahmed
a and
Andrea
Laybourn
*a
aFaculty of Engineering, University of Nottingham, Nottingham, NG7 2RD, UK. E-mail: andrea.laybourn@nottingham.ac.uk
bSchool of Physical Sciences, University of Kent, Canterbury, UK
cDepartment of Applied Chemistry and Chemical Engineering, Faculty of Engineering, Noakhali Science and Technology University, Noakhali, Bangladesh
dNanoscale and Microscale Research Centre (nmRC), University of Nottingham, Cripps South Building, Nottingham, NG7 2RD, UK
First published on 30th May 2023
Abstract
Magnetic framework composites (MFCs) are a highly interesting group of materials that contain both metal–organic frameworks (MOFs) and magnetic materials. Combining the unique benefits of MOFs (tuneable natures, high surface areas) with the advantages of magnetism (ease of separation and detection, release of guests by induction heating), MFCs have become an attractive area of research with many promising applications. This work describes the rapid, scalable synthesis of highly porous magnetic microspheres via a flame-spheroidisation method, producing spheres with particle and pore diameters of 206 ± 38 μm and 12.4 ± 13.4 μm, respectively, with a very high intraparticle porosity of 95%. The MFCs produced contained three main iron/calcium oxide crystal phases and showed strong magnetisation (Ms: 25 emu g−1) and induction heating capabilities (≈80 °C rise over 30 s at 120 W). The microspheres were subsequently surface functionalised with molecular and polymeric coatings (0.7–1.2 wt% loading) to provide a platform for the growth of MOFs HKUST-1 and SIFSIX-3-Cu (10–11 wt% loading, 36–61 wt% surface coverage), producing macro–micro hierarchically porous MFCs (pores > 1 μm and <10 nm). To the best of our knowledge, these are the first example of MFCs using a single-material porous magnetic scaffold. The adaptability of our synthetic approach to novel MFCs is applicable to a variety of different MOFs, providing a route to a wide range of possible MOF–microsphere combinations with diverse properties and subsequent applications.
1 Introduction
Magnetic framework composites (MFCs) are an exciting group of materials comprising both metal–organic frameworks (MOFs) and magnetic materials.1 MOFs are porous coordination polymers containing metal ions or clusters as nodes, and linkers consisting of organic molecules.2 Their tuneable natures and high internal surface areas (up to 7800 m2 g−1)3 have enabled them to be explored for applications across a wide range of fields, including gas storage and separation,4,5 chemical sensing,6 biomedicine7 and many more.8,9 Sustainability and circularity considerations for MOFs are also currently advancing.10–12 By combining these unique benefits and applications with the advantages of magnetism, the resulting MFCs have become a highly attractive area of research.13 One such example is in the development of materials to capture carbon dioxide from flue gas. Hill et al. showed that attaching MOFs, such as Mg-MOF-74 or UiO-66, to magnetic nanoparticles provides an energy-efficient method to desorb captured CO2 from within the saturated MOF using rapid, localised magnetic induction heating.14 Another example is from Yan et al. who used MIL-101(Cr) with functionalised magnetic microparticles for the magnetic solid-phase extraction of polycyclic aromatic hydrocarbons from environmental water samples.15 Further applications of MFCs in environmental remediation, adsorption, sensing, biomedicine and catalysis, alongside preparation strategies for forming these composites are discussed at length in the literature.1,16–18Previous development work with MFCs has almost entirely been focused on nanosized iron oxides as the magnetic components,1 likely due to the high magnetisation and abundance of iron, and the ease of manufacture of the nanoparticles. This work, therefore, also starts with an iron oxide (magnetite), but describes an investigation into the single-stage synthesis of micro-sized low and high porosity magnetic microspheres (ranging from 50 to 300 μm in diameter). These are synthesised via a rapid, cost-effective and scalable flame-spheroidisation process, building on our recent work.19,20 These microspheres not only avoid the safety concerns over the use of nanoparticles,21,22 but also offer additional advantages over their nano-alternatives such as: improved flow properties/no particle agglomeration, beneficial for powder handling and scale-up; more sustainable synthesis route; increased ease of magnetic separation due to size;23,24 the potential to avoid the need for granulation/pelletisation in industrial applications; and the potential to form hierarchal porosity when combined with other porous materials such as MOFs, demonstrated to benefit many applications such as adsorption, separation and catalysis.25–27
Apart from their use in MFCs, magnetic micro- and nano-particles have also found various applications from biomedicine28 and catalysis13,29 to environmental remediation.30 Their advantages often come from their ease of detection, efficient separation by magnetic fields and induction heating capability.31,32 In addition to the magnetic properties, imparting porosity into the particles offers additional benefits due to the high surface areas and low densities produced, which can also enhance performance in applications involving the encapsulation of a substance, such as in drug delivery33 or separation/removal of molecules or heavy metals.34,35 These magnetic particles are typically surface functionalised to enhance interactions with other molecules and facilitate the formation of composite materials.36 One frequent route is using self-assembled monolayers such as mercaptoacetic acid,37 mercaptopyridine (MPYR),38 amino alkoxysilanes39 or tetraethyl orthosilicate.40 These routes affix functional groups such as –COOH or –NH2 to the surface of the particles, allowing them to form strong chemical bonds with other molecules during the formation of composites. Other common functionalisation agents are polymers, such as polydopamine (PDA),41 polyethylenimine,42 polystyrene sulfonate,43 polyvinylalcohol (PVA)44 and polyvinylpyrrolidone.45 These can adhere to the particles and act as binders for further composite development.
Alongside synthesis and optimisation studies of the highly porous magnetic microspheres (PMMs) made from iron and calcium oxides, we also describe their composition and morphology, induction heating capabilities and magnetisation, compared to low porosity counterparts. Surface functionalisation of the PMMs with PDA and MPYR was then conducted. Finally, the formation of novel macro–micro hierarchically porous MFCs (pores > 1 μm and <10 nm) with these PMMs and archetypal MOF HKUST-1 (ref. 46) and innovative carbon-capture MOF SIFSIX-3-Cu47 was demonstrated. To the best of our knowledge, these represent the first examples of MFCs using a single-material porous magnetic scaffold. A schematic representation of this process is shown in Fig. 1. These initial results with different MOFs highlight the potential suitability of the MFCs for a range of applications.
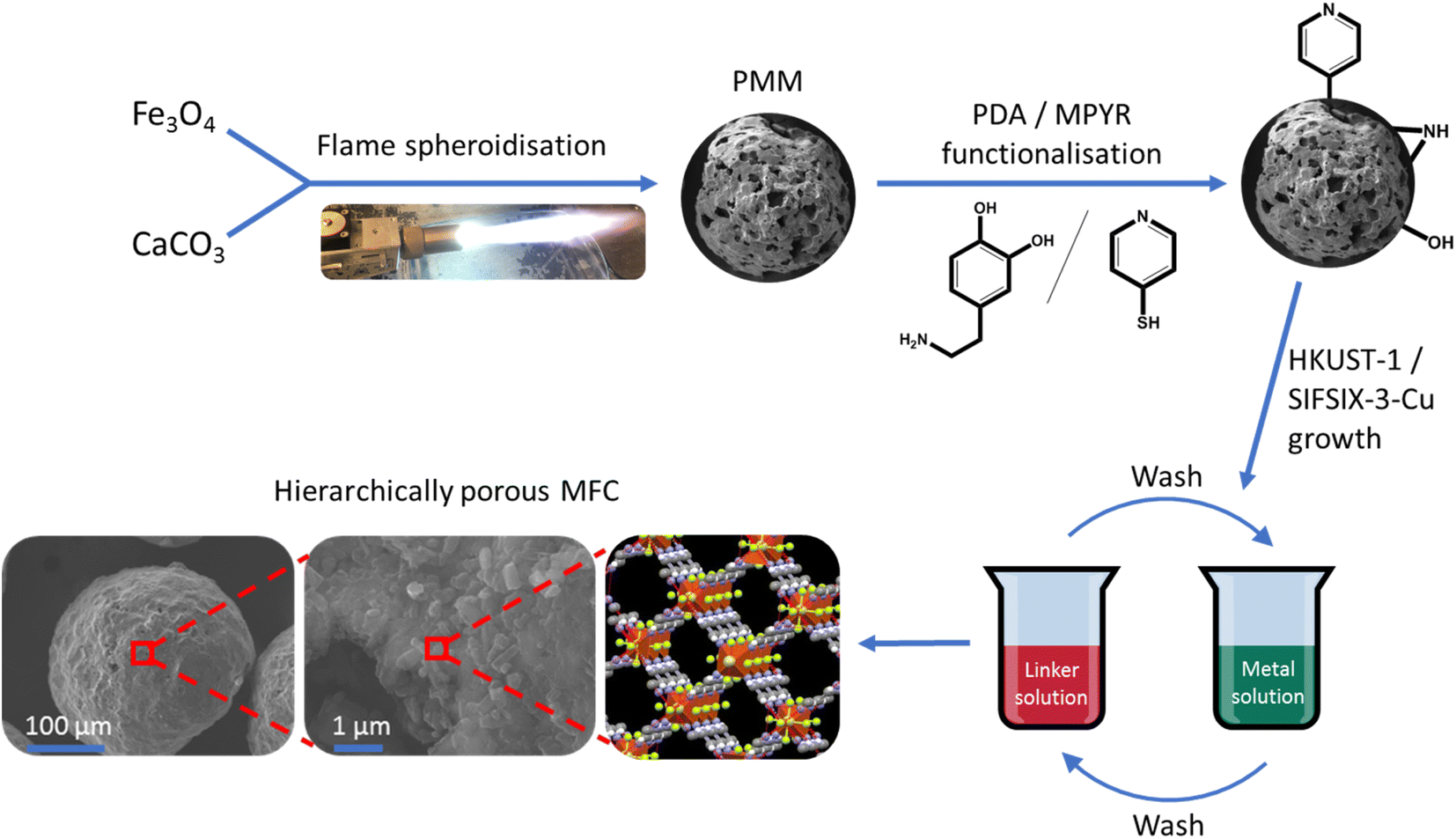 | ||
| Fig. 1 Schematic illustration of synthesis procedure for the macro–micro hierarchically porous magnetic framework composites. | ||
2 Experimental section
2.1 Materials
All chemicals and reagents were used as received. Fe3O4 was obtained from Inoxia; CaCO3 was purchased from Longcliffe. Dopamine hydrochloride, tris(hydroxymethyl)aminomethane (≥99.8%), copper(II) acetate monohydrate (ACS reagent, ≥98%), 4-mercaptopyridine (95%), pyrazine (≥99%) and trimesic acid (95%) were bought from Sigma-Aldrich, UK. Copper hexafluorosilicate hydrate was acquired from Fluorochem Ltd. Methanol (≥99.9%) was purchased from Fisher Scientific. Acetic acid (glacial) was obtained from VWR Chemicals. Polyvinyl alcohol (87.0–89.0% hydrolysed, MW ≈ 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–23000) was purchased from Acros Organics. Ethanol (absolute, SpS grade) was purchased from Scientific Laboratory Supplies Ltd.; all water used was deionised.
000–23000) was purchased from Acros Organics. Ethanol (absolute, SpS grade) was purchased from Scientific Laboratory Supplies Ltd.; all water used was deionised.
2.2 Preparation of pristine iron/calcium oxide magnetic microspheres
:1 mass ratio. The mixture was ground using a ball mill for 5 minutes at 500 rpm (Retsch PM100) and then granulated with the addition of PVA (2 wt% solution in deionised water) dropwise in a pestle and mortar. Once larger granules had formed, the mixture was sieved and granular particles between 125 and 212 μm were set aside for flame spheroidisation. Granules above 212 μm were ground further, whilst particles less than 125 μm were re-granulated to maximise the yield of particles between 125 and 212 μm. This process was again repeated until most of the starting material had been utilised. The total quantity of PVA added throughout was 4.0 mL. The set-aside granular particles were again sieved one last time, and those >125 μm were spheroidised using a thermal flame spray gun (MK74, Metallisation Ltd, UK) with an oxy-acetylene gas flow (C2H2:O2 1:1). The microsphere products were collected, acid washed with acetic acid (5 M (aq.), 1 minute), filtered (63 μm mesh) and rinsed extensively with deionised water before drying in an oven overnight (50 °C). The dry samples were then sieved into two fractions (>125 μm and <125 μm). Microspheres > 125 μm (≈60% of total microspheres produced) were taken as the final product. These microspheres were then stored in glass vials for analysis and further use.
2.3 Functionalisation of porous microsphere surfaces
2.4 Synthesis of magnetic framework composites (MFCs)
2.5 Characterisation
000 psia. The corresponding pore diameter range (210 μm to 6 nm) was calculated using the Washburn equation51 with a mercury surface tension of 480 mN m−1 and a contact angle of 130° (intrusion and extrusion). Bulk sample density was calculated at 9.39 psi for the low porosity microspheres and 2.21 psi for the high porosity microspheres, corresponding to the size of the largest pores by SEM (28 and 80 μm, respectively). Skeletal density was measured using a Quantachrome Pentapyc 5200e gas pycnometer, using helium as the intrusion gas at a pressure of 19.0 psig. Porosity (%) was calculated using the bulk and skeletal densities.
2.6 Induction heating
Induction heating measurements were undertaken using a 2 kW induction heater (204 kHz, Cheltenham Induction Heating Ltd.) and the temperature measured with a fibre optic thermocouple (Neoptix Reflex). The thermocouple was placed in a borosilicate glass tube (internal diameter: 11 mm) with 2.4 g of sample powder. The tube was then placed in a water-cooled copper coil (internal diameter: 27 mm, height: 38 mm) and various voltage settings were used to apply power to the sample. All measurements were recorded in triplicate, plotting averages with error bars showing ±1 standard deviation.3 Results and discussion
3.1 Synthesis and characterisation of magnetic microspheres
Highly porous magnetic microspheres (PMMs) were synthesised using a rapid and scalable flame spheroidisation process, expanding on previous work.20 PMMs have potential use in a variety of applications as previously discussed, including herein as scaffolds for further surface growth of MOFs in the production of MFCs with hierarchical porosity. During the optimisation process, parameters such as flame collection distance, starting material ratios, particle sizes, grinding durations and methods (puck and ring mill, ball mill) were investigated. The variable with the largest effect on porosity was found to be grinding of Fe3O4 with CaCO3 prior to flame processing, which was optimised at 5 min in a ball mill, as detailed in the Experimental section. Grinding has a significant effect as CaCO3 must be well mixed within the starting Fe3O4 granules during melting and decomposition to allow CO2 to form pores as it escapes while the microspheres cool and solidify. Without this level of mixing, CO2 can escape without leaving pores behind in the cooling microspheres. Less vigorous mixing techniques resulted in a decreased porosity (Fig. S1, ESI†). Low porosity solid microspheres were also produced using a similar process without porogen, with potential for use in applications where porosity is not required (e.g. in the production of core–shell composites for biocatalysis52), as well as for property comparisons with the high porosity microspheres.SEM micrographs of the high and low porosity microspheres and their cross-sections are shown in Fig. 2. The low porosity microspheres (Fig. 2A) show a high degree of shape and size uniformity, with solid fillings shown in the cross-sections (Fig. 2B). The high porosity microspheres (Fig. 2C) also show reasonable uniformity, with external pores opening to internal cavities. As shown in the cross-sectional image of the high porosity microspheres (Fig. 2D), some microspheres have pores which are large enough that the viscous resin used for embedding has entered the spheres (labelled ‘R’), and others where the pores are too small for the resin to enter (labelled ‘E’). It is considered that the microspheres are formed by the melting of the starting material granules in the flame (≈3100 °C),53 leading to droplet formation, followed by rapid cooling and solidification as they are ejected from the flame.20 The spherical shape is the result of minimising surface tension, and porosity is suggested to arise from the decomposition of the porogen material releasing CO2 gas, which is trapped in the molten particles and escapes during the cooling and solidification stage. The pore size distribution of the high porosity microspheres was first determined using ImageJ software measuring 268 pores (see Fig. S2, ESI†). The average microsphere was found to have approximately 45 pores, with a mean pore diameter of 12.4 μm and a standard deviation of 13.4 μm, demonstrating macro-porosity. Porosity was further quantified through mercury intrusion porosimetry analysis on both the high and low porosity microspheres.51Fig. 3C shows the decreasing pore diameters (from increasing mercury pressure) plotted against the cumulative volume of mercury intrusion (pore volume). The initial increase in gradient for both samples (point ‘X’) is from the filling of the interparticulate gaps/voids between the microspheres.54 The high porosity microspheres also exhibit a change in gradient (point ‘Y’) as a new size range of intraparticulate pores are filled.54Fig. 3D plots the pore diameter against the negative log differential volume intrusion for the high and low porosity microspheres up to the interparticle filling limits. These data correspond well to pore size distributions measured by analyses of SEM images (Fig. S2, ESI†). Intraparticle porosity was calculated as follows (eqn (1)):55
 | (1) |
 | ||
| Fig. 2 SEM micrographs of: (A) low porosity microspheres; (B) cross section of low porosity microspheres; (C) high porosity microspheres; (D) cross section of high porosity microspheres, showing both resin filled (R) and empty (E) examples; (E) singular high porosity microsphere; (F) image of a high porosity microsphere at higher magnification. | ||
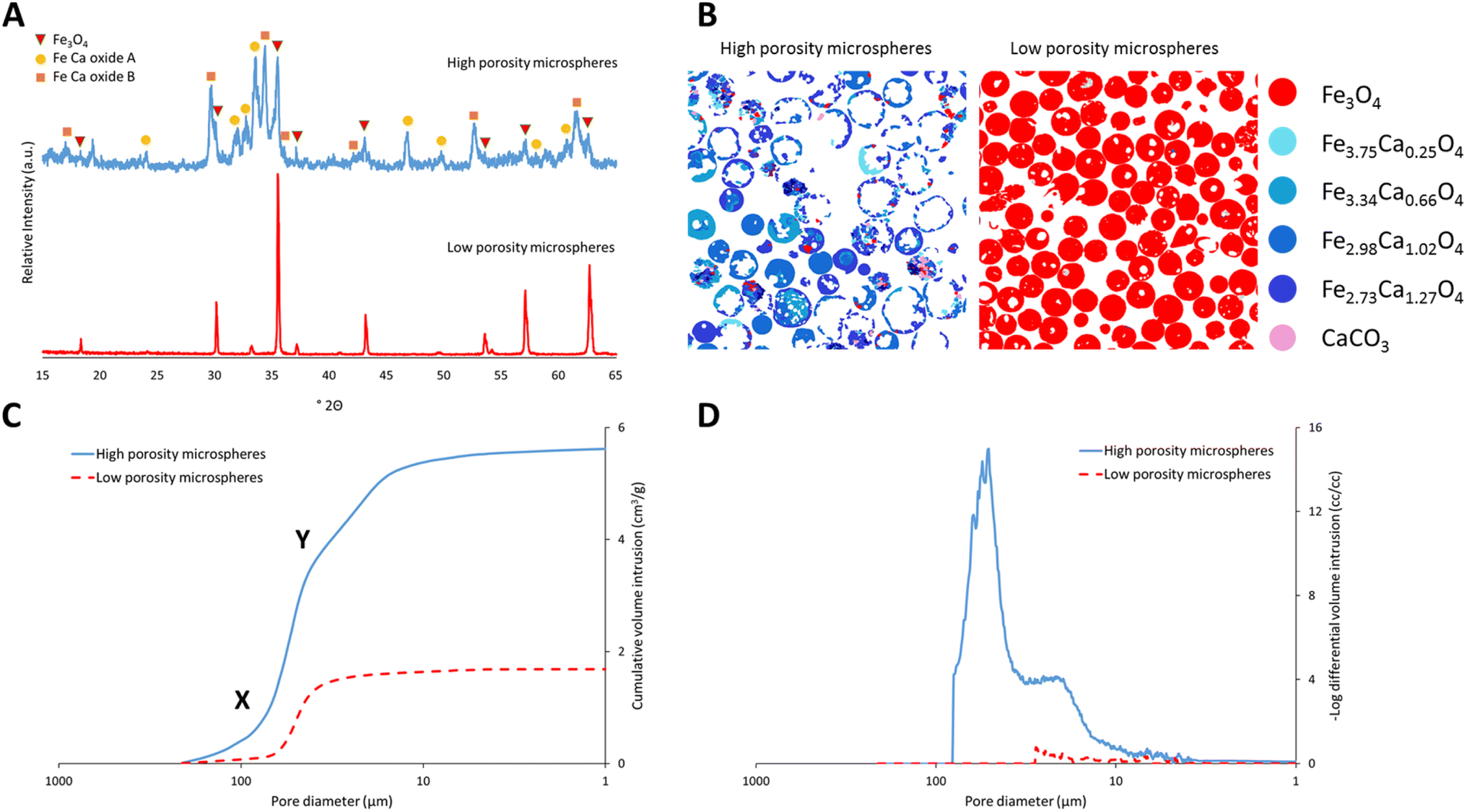 | ||
| Fig. 3 (A) Powder X-ray diffraction patterns for high (top) and low (bottom) porosity microspheres. (B) MLA images showing mineral mapping cross-sections of high (left) and low (right) porosity microspheres. Mineral phases are colour coded and their elemental compositions are shown in the figure legend (far right). (C) Mercury intrusion porosimetry data showing pore diameter against cumulative volume intrusion (pore volume) for the high and low porosity microspheres. Point ‘X’ shows the filling of interparticle voids, and point ‘Y’ shows a change in gradient as a new range of pore sizes are filled. (D) Mercury intrusion porosimetry data showing pore diameter against the −log differential volume intrusion for the high and low porosity microspheres up to the interparticle filling limits. | ||
The production yields of the spheroidisation and acid washing steps for the high and low porosity microspheres synthesised herein are shown in Table 1. Spheroidisation yield was calculated as the mass of microspheres produced (prior to sieving for the high porosity microspheres) divided by the mass of the granulated starting materials. Higher spheroidisation yields were observed with the low porosity microspheres, likely due to the mass loss of CO2 from the CaCO3 porogen used to generate pores in the high porosity microspheres.20 Other mass losses include particles missing the centre of the flame or falling outside of the collection trays. It was also observed that acid washing caused no mass loss for the low porosity microspheres, and a negligible loss for the high porosity microspheres, likely from the dissolution of any unreacted CaCO3 porogen. Image particle size analyses of the high and low porosity microspheres (N = 114 and 143, respectively) are also shown in Table 1. The larger sizes of the high porosity microspheres are evidence of the expanding process of the particles in the flame during synthesis, where porogen decomposition leads to CO2 gas release (generating pores). The higher size range and standard deviation of the high compared to the low porosity microspheres likely arises from slight variation in the exact amount of porogen present in each particle due to the granulation method.
| High porosity microspheres | Low porosity microspheres | |
|---|---|---|
| Spheroidization yield (%) | 76 | 84 |
| Acid washing yield (%) | 98 | 100 |
| Size range (μm) | 120–285 | 73–194 |
| Average size (μm) | 206 | 134 |
| Standard deviation (μm) | 38 | 24 |
To determine the crystalline mineral phases present, the high and low porosity microspheres were analysed using powder X-ray diffraction. As shown in Fig. 3A, the low porosity microspheres exhibited a peak pattern corresponding to a cubic magnetite phase (Fe3O4: PDF 00-019-0629), as identified from the DIFFRAC.EVA powder diffraction file database. The high porosity microspheres exhibited a pattern containing peaks corresponding to a cubic magnetite phase and two main calcium–iron oxide phases, identified from the database and marked in Fig. 3A as Fe–Ca oxide A and B (A: PDF-01-071-2108; B: PDF-00-013-0395). ‘Oxide A’ corresponds to the known orthorhombic structure srebrodolskite (Ca2Fe2O5), whereas ‘oxide B’ is a more complex calcium–iron oxide with a rhombohedral lattice. In order to quantify the mineral compositions present and obtain a better understanding of the chemical properties of the materials, a detailed compositional analysis of both the high and low porosity microspheres was undertaken using mineral liberation analysis (MLA). Fig. 3B shows a breakdown of the minerals present in the samples. Different minerals are colour-coded and their elemental compositions were determined via EDS analysis, as shown in the formulae on the right-hand side of the figure. The low porosity microspheres are composed entirely of Fe3O4, suggesting that no oxidation to Fe2O3 or higher oxides takes place during the flame process, corroborating the PXRD data. The high porosity microspheres are mainly composed of two calcium–iron oxide mineral compositions with Fe to Ca ratios of 2:1 and 3:1, though some other mineral compositions with higher ratios of Fe are also present. Table 2 shows the percentage by weight of the different mineral compositions present in the high porosity microspheres. EDS analyses show bands of locally averaged compositions, likely from multiple mineral phases. Assessment using the three-phase diagram for Ca–Fe–O on the highest wt% mineral compositions (Fe2.98Ca1.02O4 and Fe2.73Ca1.27O4) showed that the Ca2Fe2O5 phase (Fe–Ca oxide A) was thermodynamically favoured, consistent with the PXRD pattern.58 The other expected thermodynamic phase was a wustite–lime solid solution containing iron and calcium oxides. This phase may represent the Fe–Ca oxide B shown in the PXRD pattern (Fig. 3A), or the rapid cooling of the flame process could have resulted in the formation of a complex metastable calcium–iron oxide phase.
| Mineral | Fe/Ca molar ratio | High porosity microspheres (wt%) |
|---|---|---|
| Fe3O4 | — | 4.2 |
| Fe3.75Ca0.25O4 | 15 | 4.4 |
| Fe3.34Ca0.66O4 | 5 | 9.7 |
| Fe2.98Ca1.02O4 | 3 | 35.3 |
| Fe2.73Ca1.27O4 | 2 | 44.4 |
| CaCO3 | — | 0.7 |
3.2 Magnetisation and induction heating of microspheres
The magnetic properties of the microspheres at room temperature were measured and compared to the pure magnetite starting material by vibrating sample magnetometry (VSM), see Fig. 4A. All samples showed ferrimagnetic behaviour with low remnant magnetisation (<1.1 emu g−1) and intrinsic coercivity (<50 Oe), resulting in high permeability and low energy required to switch the spin directions. This result is beneficial for applications involving magnetic separation such as in environmental remediation, where only low energies would be required to magnetise the microspheres for collection, and they could also be easily demagnetised and re-dispersed by the removal of the magnetic field. The saturation magnetisation (Ms) values for the pure magnetite starting material, and the low and high porosity microspheres were 94, 91 and 25 emu g−1, respectively (Fig. 4A). The similarity of the magnetisation curve of low porosity microspheres to pure magnetite showed that there were no significant structural changes induced during flame spheroidization that would result in a decrease of Ms. The low porosity microspheres high Ms value of 91 emu g−1 is significant, with values between 75 and 90 emu g−1 for processed magnetite commonly noted for being “high” in the literature.59–61 The drop in Ms for high porosity magnetite was expected due to the introduction of non-magnetic calcite, resulting in the production of less magnetic mineral phases such as Ca2Fe2O5,62 as shown in the XRD and MLA data in Fig. 3. However, an Ms value of 25 emu g−1 was still sufficiently high for the microspheres to exhibit strong magnetic behaviour and benefits, as shown by the induction heating study below. | ||
| Fig. 4 Plots showing: (A) room-temperature magnetisation curves of high (blue) and low (red) porosity microspheres alongside the pure magnetite starting material (yellow); (B) induction heating rate of high (blue) and low (red) porosity microspheres alongside magnetite (yellow) and calcite (pink) starting materials at 120 W; (C) induction heating rate of high porosity microspheres at 30 W, 120 W and 250 W. Measurements in (B) and (C) were taken in triplicate and the averages plotted, the error bars show ±1 standard deviation. | ||
Induction heating is the process of energy deposition from an H-field applicator to a material placed in the magnetic field node. In conductive materials, heating occurs through ohmic losses from the resistance to induced eddy currents. In ferromagnetic materials, heating is additionally caused by magnetic hysteresis, an effect associated with losses from the magnetization–demagnetization cycle in magnetic domains.63 As discussed in the Introduction, the ability of magnetic microspheres to undergo induction heating is beneficial to many applications; for example, in the release of adsorbed or encapsulated guests or in medical treatments such as hyperthermia.64 Therefore, the induction heating ability of the high and low porosity magnetic microspheres were investigated and compared to the magnetite and calcite starting materials. Rapid heating was indeed observed for the microspheres, confirming their potential usefulness in such applications. Fig. 4B shows the change in temperature over 30 seconds at 120 W (applied magnetic field of 100 A m−1) for the high and low porosity microspheres, alongside the magnetite and porogen starting materials. As expected, calcite showed negligible heating under a magnetic field (0.4 ± 0.7 °C rise over 30 seconds), whilst pure magnetite powder heated very rapidly (116 ± 25 °C rise over 20 seconds). Interestingly, the high and low porosity microspheres exhibited similar heating profiles (81 ± 3 °C cf. 80 ± 7 °C rise over 30 seconds, respectively) (Fig. 4B), despite their differences in composition and magnetic susceptibility (Fig. 4A). We hypothesise that this effect could be due to the size and shape of the high and low porosity processed microspheres compared to the magnetite starting material. Both sets of microspheres have large spherical morphologies, resulting in low contact areas between particles and with the thermocouple in the sample tube. This low contact may limit the transfer of heat between neighbouring particles and to the fibre-optic thermocouple. This limit may be similar for both high and low porosity microspheres as both show similar spherical morphologies compared to the fine and dense powder magnetite starting material (<45 μm). For industrial applications, both energy and time must be balanced to achieve efficient heating. As such, we also briefly explored the effect of varying the power from the induction heater, where the high porosity microspheres were subjected to three different power levels from 30 W (applied magnetic field of 75 A m−1) to 250 W (applied magnetic field of 125 A m−1), each for 30 seconds. The results are shown in Fig. 4C. As anticipated, increasing the power resulted in a faster heating rate. However, the increase was not directly proportional to the input power due to the complex nature of the many factors affecting the heating mechanisms, including applicator efficiency.63Fig. 4C shows that reasonable heating rates can be achieved with even very low power (31 ± 2 °C rise over 30 seconds at 30 W), whereas increasing the power by over a factor of two from 120 to 250 W only increased the heating rate from an 81 ± 3 °C to a 102 ± 3 °C rise over 30 s.
3.3 Functionalisation of highly porous magnetic microspheres
Having compared the properties of high and low porosity magnetic microspheres, subsequent efforts focused on the highly porous magnetic microspheres (PMMs) due to the potential benefits of porosity such as high surface areas, low densities, and guest encapsulation capabilities as described in the Introduction. Firstly PDA was used as a surface functionalisation agent to coat the PMMs prior to MOF growth, due to literature precedent.65,66 PDA contains a mixture of catechol, quinone, amine and imine functional groups, which can provide good binding sites for composite formation. We further trialled MPYR on PDA65 for SIFSIX-3-Cu MFC, as the pendant pyridine functional group of MPYR was expected to have good compatibility with the MOF (which contains pyrazine linkers).The presence of PDA and MPYR on the PMMs was investigated using detailed XPS analyses. Fig. 5 shows the high-resolution spectra for the pristine PMMs alongside the surface functionalised PMMs. More detailed classifications and analyses can be found alongside the XPS survey spectra in Fig. S4, ESI.† XPS data of the pristine PMMs evidence metallic-bound oxygen in the O 1s spectrum at 530.2 eV and the expected Fe(III) and Ca(II) peaks in the Fe 2p and Ca 2p spectra, at 710.8 eV and 346.5 eV, respectively for the 3/2 peaks. The PDA and PDA/MYPR functionalised PMMs both show the anticipated nitrogen peak in the N 1s spectrum at 399.5 eV, corresponding to the polydopamine layer.67,68 Evidence of C–N is also seen in the C 1s deconvolution at 285.7 eV, and the O 1s spectrum shows the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–O peaks expected from the quinone and catechol moieties in polydopamine, at 288.8 eV and 286.9 eV respectively.69 Both the iron and calcium peaks are missing in these samples, indicating that the polydopamine layer must be thicker than the penetration depth of the XPS analysis technique, around 10 nm.70 Finally, a low intensity S 2p peak at 163.3 eV in the PDA/MPYR functionalised sample confirms the presence of the MPYR on the PDA layer on the PMMs. MPYR is possibly attached by a reaction between the terminal thiol on MPYR and the catechol/quinone groups of the polydopamine, through a thiol-nucleophile Michael addition type reaction.65,71,72
O and C–O peaks expected from the quinone and catechol moieties in polydopamine, at 288.8 eV and 286.9 eV respectively.69 Both the iron and calcium peaks are missing in these samples, indicating that the polydopamine layer must be thicker than the penetration depth of the XPS analysis technique, around 10 nm.70 Finally, a low intensity S 2p peak at 163.3 eV in the PDA/MPYR functionalised sample confirms the presence of the MPYR on the PDA layer on the PMMs. MPYR is possibly attached by a reaction between the terminal thiol on MPYR and the catechol/quinone groups of the polydopamine, through a thiol-nucleophile Michael addition type reaction.65,71,72
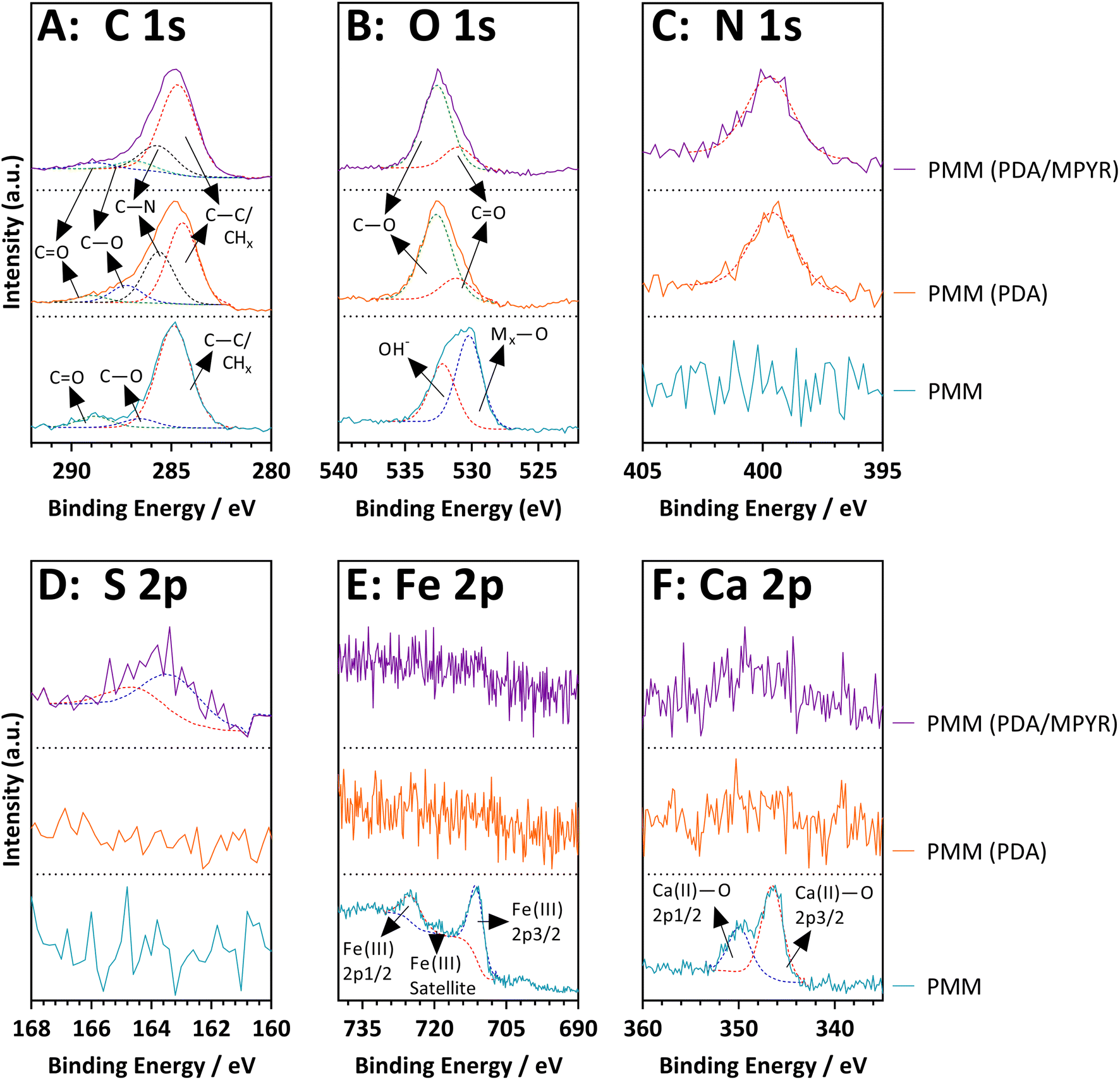 | ||
| Fig. 5 XPS high-resolution spectra of unfunctionalized, PDA and PDA/MPYR functionalised high porosity magnetic microspheres. | ||
Quantities of the functionalisation agents on the surfaces of the PMMs were determined by thermal gravimetric analyses, analysing the weight loss from decomposition of the organic agents on the inorganic PMMs (Fig. S3, ESI†). PDA functionalised PMMs showed a weight loss of around 0.7% at 500 °C, with an additional 0.5% loss seen for the PDA functionalised PMMs further functionalised with MPYR. These data support XPS analyses, evidencing successful functionalisation of the PMMs.
3.4 Preparation, structure, and particle attributes of magnetic framework composites (MFCs)
To assess the prospect of using functionalised PMMs to form MFCs, the archetypal MOF HKUST-1 was selected for initial experiments. Further trials were then conducted with a novel carbon-capture MOF, SIFSIX-3-Cu. HKUST-1 (ref. 46) is a copper-based MOF containing trimesic acid linkers and has been extensively studied for applications spanning the adsorption and separation of gases, organics and ions; to catalysis, sensing, and composites for wastewater treatment.73–75 SIFSIX-3-Cu has been more recently discovered and identified as a promising material for carbon capture and storage due to its high capacity and selectivity for CO2.47,76A layer-by-layer method (sequentially moving the functionalised PMMs between two solutions, one containing the MOF linker and another containing the metal salt precursor) was selected to prepare the MFCs due to the high level of control it offers for heterogeneous MOF growth on the PMM surfaces. PDA was selected since it has been reported to attach HKUST-1 owing to functional group binding sites.65 TGA results confirm successful loading of HKUST-1 on the PDA functionalised PMMs and enabled quantification of MOF loading from the percent weight loss of the sample upon thermal decomposition of the MOF (Fig. 6). A HKUST-1 loading of (10 ± 2) wt%, was calculated using a moisture normalised pure MOF decomposition as 45% between 280 and 400 °C.77 For the SIFSIX-3-Cu MOF, we first prepared PDA functionalised PMMs, and then employed a novel strategy which involved further reacting the PDA functionalised PMMs with MPYR to affix pyridine functional groups to the PMM surfaces.65 Pyridine functional groups provide nitrogen coordination sites for SIFSIX-3-Cu (which itself contains pyrazine linkers). Fig. 6B shows thermograms for PDA and PDA/MPYR functionalised PMMs and the resulting SIFSIX-3-Cu MFCs. A higher SIFSIX-3-Cu loading can clearly be identified for the PDA/MPYR functionalised PMMs (11 ± 1) wt% compared to PMMs functionalised solely with PDA (8 ± 1) wt%, calculated using a moisture normalised pure SIFSIX-3-Cu decomposition as 70.3 wt% between 125 and 250 °C (measured herein, Fig. S5, ESI†). This difference in SIFSIX-3-Cu loading leads us to conclude that the secondary functionalisation of PDA with MPYR is beneficial in promoting SIFSIX-3-Cu MOF nucleation and growth on the PMM surfaces, likely due to the enhanced interaction between the pyridine functional groups on the PMM surface and Cu(II) in the MOF.
 | ||
| Fig. 6 TGA data of: (A) PDA functionalised PMMs (orange) and HKUST-1@PMM (PDA) MFC (dark blue); (B) PDA (orange) and PDA/MPYR (purple) functionalised PMMs and SIFSIX-3-Cu@PMM (PDA) (light green) SIFSIX-3-Cu@PMM (PDA/MPYR) (dark green) MFCs. | ||
SEM and EDS analyses were used to examine the morphology, surface coverage and placement of MOF crystals on the MFCs with the highest MOF loadings. Images of HKUST-1@PMM (PDA) MFC are displayed in Fig. 7. These images show that the PMMs are coated in a discontinuous layer of irregular shaped HKUST-1 nano-crystals, which can also be seen inside some of the microsphere pores. The copper signal in the EDS analysis provides compelling evidence for the presence of HKUST-1 MOF on the surface of the PMMs, giving an approximately 36 wt% coverage of the MOF. Fig. 8 shows SIFSIX-3-Cu@PMM (PDA/MPYR), also showing MOF nano-crystals on the surfaces and in some of the pores of the PMMs. EDS mapping of SIFSIX-3-Cu@PMM (PDA/MPYR) MFC exhibits signals for copper, silicon, and fluoride ions present in SIFSIX-3-Cu. The red circles in Fig. 8 highlight an area of high MOF concentration. Using the copper EDS signal, the SIFSIX-3-Cu coverage on the surface of the PDA/MPYR functionalised PMM was determined as approximately 61 wt%. EDS elemental analyses of HKUST-1@PMM (PDA) and SIFSIX-3-Cu@PMM (PDA/MPYR) MFCs, show that MOFs can be loaded on functionalised PMMs. The substantially higher surface coverage of MOF on SIFSIX-3-Cu@PMM (PDA/MPYR) MFC compared to MOF on the HKUST-1@PMM (PDA) MFC (61% and 36%) with similar total weight loadings (11% and 10%) implies that the SIFSIX-3-Cu is more evenly spread over the functionalised PMM surface compared to HKUST-1. This observation is indicative of more concentrated areas of HKUST-1 where the MOF preferentially builds on itself, as in secondary growth type methods.78 This result is also supported by the copper EDS signals (green) in Fig. 7 and 8, which show more intensely bright areas in HKUST-1@PMM (PDA) MFC compared to SIFSIX-3-Cu@PMM (PDA/MPYR) MFC. These brighter areas in both MFCs show where more MOF has gathered/grown, possibly due to more protected anchoring points in the porous magnetic microsphere, or conceivably owing to initial MOF attachment and subsequent preferential growth.
 | ||
| Fig. 7 SEM images (left) and EDS elemental mapping (right) of HKUST-1@PMM (PDA) MFC. EDS mapping shows the presence of iron (yellow), oxygen (blue) and copper (green) in the MFC. | ||
 | ||
| Fig. 8 SEM images (top left) and EDS elemental mapping of SIFSIX-3-Cu@PMM (PDA/MPYR) MFC. EDS mapping shows the presence of iron (yellow), fluorine (turquoise), silicon (pink), oxygen (blue) and copper (green) in the MFC. Red circles highlight areas of high SIFSIX-3-Cu MOF concentration. | ||
FTIR and Raman spectroscopic analyses were used to confirm the chemical composition of the MOFs on/in the MFCs. Fig. 9A and B show the vibrational spectra of HKUST-1@PMM (PDA) and SIFSIX-3-Cu@PMM (PDA/MPYR). FTIR spectra for both MFCs show the expected absorbances for the MOFs as reported in the literature,47,79 including intense diagnostic peaks at 1371 cm−1 (assigned as a νip(COO) vibrational mode of the carboxylate linker in HKUST-1)79 and 1435 cm−1 (tentatively assigned as a ring bending mode of pyrazine47) in the FTIR spectra of HKUST-1@PMM (PDA) and SIFSIX-3-Cu@PMM (PDA/MPYR), respectively. Optical and Raman imaging (inset in Fig. 9B) confirmed the discontinuous surface coverage of MOFs on the MFCs, with higher, more uniform coverage observed on the SIFSIX-3-Cu@PMM (PDA/MPYR) MFC, relative to HKUST-1@PMM (PDA) MFC, consistent with EDS mapping. The Raman spectra collected from sky blue areas in the optical images of HKUST-1@PMM (PDA) match that expected for the HKUST-1 MOF.79 Interestingly, the intensity of the band at 1705 cm−1 (assigned as a νip(COOH) in benzene-1,3,5-tricarboxylic acid) is more intense in many of the Raman spectra captured from different sky blue domains on the MFCs than that observed in the reported pristine HKUST-1 MOF spectrum. This suggests a high density of structural defects where Cu–Cu moieties are not always included in the framework thus resulting in the transformation of carboxylates to carboxylic acids.79 However, hydrogen bonding between adjacent couples of COOH groups likely preserve continuity of the MOF network. Spectra obtained from white areas of the SIFSIX-3-Cu@PMM (PDA/MPYR) MFC are consistent with that obtained from the pristine SIFSIX-3-Cu MOF (Fig. S6, ESI†) and with the vibrational modes of the pyrazine linker (1044, 1519 and 1627 cm−1) and SiF6 structural motifs (677 and 705 cm−1), as reported previously in the Raman spectra of structural analogues.80 It is interesting to note the majority of MOF peaks in FTIR spectra of the MFCs are downshifted relative to those reported in the literature for the respective MOFs. This shift likely indicates that the MOF networks are subject to tensile strain, reflecting the complex surface topography and local curvature of the microsphere upon which the MOF is grown/attached. Raman maps of the MFCs with further analyses are detailed in Fig. S7 and S8 in the ESI.†
 | ||
| Fig. 9 Structural characterisation of SIFSIX-3-Cu@PMM (PDA/MPYR), and HKUST-1@PMM (PDA) MFCs and pristine PMMs. (A) FTIR absorbance spectra; (B) confocal Raman microscopy analysis obtained from MOF-rich regions of the respective MFCs, insets are false colour images depicting the distribution of SIFSIX-3-Cu (green, top) or HKUST-1 (blue, bottom) and PDA (red) in representative MFCs (see ESI† for further details), scale bars are 10 microns; (C) and (D), powder X-ray diffraction patterns. | ||
PXRD was used to examine the crystal structures present in the MFCs. Fig. 9C shows the diffractogram of HKUST-1@PMM (PDA) MFC, appearing as an approximate superposition of the patterns of the pristine PMM (experimental) and pristine HKUST-1 MOF (calculated). The sharp peak at 6° 2θ can be seen in both the MOF and the MFC evidencing crystallinity, though many of the other peaks for HKUST-1 are not observed due to the low MOF concentration present in the MFC. The high relative intensity of the 6° 2θ peak compared to the other peaks for HKUST-1 at higher ° 2θ could result from a highly ordered and preferentially oriented crystalline material, which has been observed elsewhere with HKUST-1 when using the layer-by-layer synthesis method.81 The small crystals produced by the layer-by-layer process and the slow crystal growth from the low synthesis temperature (room temperature then 50 °C) used herein could also result in some peak broadening, lowering the intensities of the HKUST-1 peaks in the HKUST-1@PMM (PDA) MFC diffraction pattern.82 This XRD result supports the stronger IR and Raman evidence for the HKUST-1 structure on the PMM surface. Fig. 9D shows the diffractogram of SIFSIX-3-Cu@PMM (PDA/MPYR) MFC against the pristine PMM and pristine SIFSIX-3-Cu MOF. Peaks corresponding to both SIFSIX-3-Cu and PMMs can be seen in the PXRD pattern of the MFC, confirming the presence and crystallinity of SIFSIX-3-Cu MOF on the PDA/MPYR functionalised PMMs. The relative intensities of peaks corresponding to SIFSIX-3-Cu (e.g. at 13 and 17° 2θ) change slightly in the MFC diffraction pattern, possibly owing to hydration of the MOF or slight changes in the crystal structure of the MOF when grown on the surface of the PMMs.76
The micro-porosity of the MFCs was evidenced by Kr gas sorption analyses. Mercury intrusion was not used on the MFCs as it cannot measure smaller pore sizes (<6 nm), it requires gram-scale quantities and is destructive. It is assumed that the macro-porosity of the PMMs is retained after MOF growth as the small (nm-scale) MOF crystals are seen to coat the PMM surfaces rather than block the large micron-scale pores (see Fig. 7). Kr was used as the gas sorbate (rather than N2 or CO2) owing to the thin coating of MOF present in the MFCs.83Fig. 10 shows the Kr sorption isotherms and the cumulative pore volume plots for HKUST-1@PMM (PDA) and SIFSIX-3-Cu@PMM (PDA/MPYR) MFCs for pore widths between 1.75 and 20 nm. Data labelled ‘total’ represents the MFCs, and data labelled ‘PMM subtracted’ correspond solely to the respective MOFs for each MFC, determined by subtracting the contribution of Kr sorption for PMMs from the MFCs, and accounting for the mass of MOF present in the MFCs. Both MFCs exhibit micro-porosity owing to the MOF pores, however the total pore volumes for the MOFs in the MFCs are lower than reported for their corresponding pristine MOF powders by N2/CO2 adsorption. This is possibly as pore widths less than 1.75 nm could not be mapped using the Kr sorption method as the instrument cannot start at <0.01P/P0. Nevertheless, micro-porosity is demonstrated in both samples due to the MOF attachment.
 | ||
| Fig. 10 Kr adsorption–desorption isotherms and cumulative pore volume plots for SIFSIX-3-Cu@PMM (PDA/MPYR) (A and B) and HKUST-1@PMM (PDA) MFCs (C and D). Data labelled ‘total’ represents the MFCs, and labelled ‘PMM subtracted’ represents the respective MOFs for each MFC, determined by subtracting the contribution of Kr sorption for PMMs from the MFCs, and accounting for the mass of MOF present in the MFCs. Data collected at −196 °C. Pore volume/size distribution (1.75–20 nm) was acquired from an extension of the BJH model by Chao et al.50 | ||
Overall, we have demonstrated a new route to producing MFCs with PMMs and MOFs HKUST-1 and SIFSIX-3-Cu, showing attachment and growth of the MOFs on the PMM surfaces. These demonstrate macro–micro hierarchical porosity from the iron/calcium oxide microsphere and MOF components respectively, and contain strong magnetic properties which could benefit many applications, as previously highlighted.
4 Conclusions
In this study a novel rapid, scalable and solvent-free flame-spheroidisation process was optimised for the sustainable production of both high porosity (iron/calcium oxides) and low porosity (magnetite) magnetic microspheres in the size range of 50 to 300 μm. The high porosity microspheres exhibited strong magnetisation (Ms: 25 emu g−1) alongside a very high intraparticle porosity of 95% (pore diameter: 12.4 ± 13.4 μm). The low porosity microspheres demonstrated extremely high magnetisation (Ms: 91 emu g−1) with an intraparticle porosity of 52%. Both materials showed ferrimagnetic behaviour with low remnant magnetisations. The crystalline and mineral compositions of the magnetic microspheres were elucidated through detailed XRD and MLA studies, followed by an investigation into their rapid induction heating capabilities, with both the high and low porosity microspheres showing an ≈80 °C rise over 30 s at only 120 W. These products represent a substantial advancement in the field of magnetic microspheres due to their simple and cost-effective manufacture and the high porosities and room-temperature magnetisations that can be obtained. The microspheres would be suitable to a range of applications, benefiting from ease of detection, magnetic field separation and efficient induction heating.An in-depth XPS analysis revealed the successful surface functionalisation of the PMMs with 0.7–1.2 wt% PDA and MPYR. A controlled layer-by-layer heterogeneous MOF growth strategy was used on the functionalised PMMs to form innovative magnetic framework composites with archetypal MOF HKUST-1 (10 wt% loading, 36 wt% surface coverage) and the advanced carbon-capture MOF SIFSIX-3-Cu (11 wt% loading, 61 wt% surface coverage). The tailored technique of further functionalising the PDA surface with MPYR was found to improve MOF loading for SIFSIX-3-Cu from 8 to 11 wt%. EDS and Raman mapping revealed the distribution of the MOFs on the PMM surfaces, while FTIR and XRD confirmed the MOF chemical compositions and crystallinities. These materials are the first example of MFCs using a single-material porous magnetic scaffold, in addition demonstrating macro–micro hierarchical porosity (pores > 1 μm and <10 nm) by Kr adsorption and mercury intrusion porosimetry. The MFCs have potential to be used in various applications from catalysis to the adsorption and separation of gases, organics and ions for environmental remediation. Our adaptable synthetic approach is applicable to a variety of different MOFs, opening the possibility of diverse properties and consequent applications.
Author contributions
John Luke Woodliffe: conceptualization, formal analysis, validation, funding acquisition (via Henry Royce Institute), investigation, methodology, writing – original draft, writing – review & editing, visualization. Jesús Molinar-Díaz: funding acquisition (via Henry Royce Institute), investigation, writing – review & editing, visualization. Md Towhidul Islam: methodology, writing – review & editing. Lee A. Stevens: investigation, methodology, validation, writing – review & editing, visualization. Matthew D. Wadge: formal analysis, investigation, writing – original draft, writing – review & editing, visualization. Graham A. Rance: investigation, validation, writing – original draft, writing – review & editing, visualization. Rebecca Ferrari: conceptualization, supervision, writing – review & editing. Ifty Ahmed: conceptualization, supervision, writing – review & editing. Andrea Laybourn: conceptualization, project administration, supervision, writing – review & editing, funding acquisition (EPSRC CDT).Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors give thanks to and acknowledge funding from the Engineering and Physical Sciences Research Council (EPSRC) and Science Foundation Ireland (SFI) Centre for Doctoral Training in Sustainable Chemistry: Atoms-2-Products an Integrated Approach to Sustainable Chemistry (Grant Number: EP/S022236/1). The authors acknowledge the support of the Henry Royce Institute for advanced materials for John Luke Woodliffe through the Student Equipment Access Scheme enabling access to QD MPMS 3 facilities at Royce Discovery Centre, the University of Sheffield; EPSRC Grant Number EP/R00661X/1 & EP/P02470X/1. The authors also thank Barry Reid at the SAXS/CNIE Facility in the Chemical Engineering Department, University College London for providing a mercury intrusion porosimetry analysis service. SEM analyses in this work were supported by the Engineering and Physical Sciences Research Council (EPSRC) [under grant EP/L022494/1] and the University of Nottingham. The authors thank the Nanoscale and Microscale Research Centre (nmRC) for providing access to SEM and Raman instrumentation and Hannah Constantin, Grace Belshaw and David Furniss for technical assistance.References
- Y. J. Ma, X. X. Jiang and Y. K. Lv, Chem.–Asian J., 2019, 14, 3515–3530 CrossRef CAS PubMed.
- O. M. Yaghi and H. Li, J. Am. Chem. Soc., 1995, 117, 10401–10402 CrossRef CAS.
- I. M. Hönicke, I. Senkovska, V. Bon, I. A. Baburin, N. Bönisch, S. Raschke, J. D. Evans and S. Kaskel, Angew. Chem., Int. Ed., 2018, 57, 13780–13783 CrossRef PubMed.
- J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- L. J. Murray, M. Dincă and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294 RSC.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232–1268 CrossRef CAS PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- R. Freund, O. Zaremba, G. Arnauts, R. Ameloot, G. Skorupskii, M. Dincă, A. Bavykina, J. Gascon, A. Ejsmont, J. Goscianska, M. Kalmutzki, U. Lächelt, E. Ploetz, C. S. Diercks and S. Wuttke, Angew. Chem., Int. Ed., 2021, 60, 23975–24001 CrossRef CAS PubMed.
- P. A. Julien, C. Mottillo and T. Friščić, Green Chem., 2017, 19, 2729–2747 RSC.
- E.-S. M. El-Sayed and D. Yuan, Green Chem., 2020, 22, 4082–4104 RSC.
- J. L. Woodliffe, R. S. Ferrari, I. Ahmed and A. Laybourn, Coord. Chem. Rev., 2021, 428, 213578 CrossRef CAS.
- S. Yadav, R. Dixit, S. Sharma, S. Dutta, K. Solanki and R. K. Sharma, Mater. Adv., 2021, 2, 2153–2187 RSC.
- M. M. Sadiq, K. Suzuki and M. R. Hill, Chem. Commun., 2018, 54, 2825–2837 RSC.
- S.-H. Huo and X.-P. Yan, Analyst, 2012, 137, 3445–3451 RSC.
- B. E. Meteku, J. Huang, J. Zeng, F. Subhan, F. Feng, Y. Zhang, Z. Qiu, S. Aslam, G. Li and Z. Yan, Coord. Chem. Rev., 2020, 413, 213261 CrossRef CAS.
- L. D. O'Neill, H. Zhang and D. Bradshaw, J. Mater. Chem., 2010, 20, 5720–5726 RSC.
- R. Ricco, L. Malfatti, M. Takahashi, A. J. Hill and P. Falcaro, J. Mater. Chem. A, 2013, 1, 13033–13045 RSC.
- K. M. Z. Hossain, U. Patel, A. R. Kennedy, L. Macri-Pellizzeri, V. Sottile, D. M. Grant, B. E. Scammell and I. Ahmed, Acta Biomater., 2018, 72, 396–406 CrossRef CAS.
- J. M. Díaz, S. A. Samad, E. Steer, N. Neate, H. Constantin, M. T. Islam, P. D. Brown and I. Ahmed, Mater. Adv., 2020, 1, 3539–3544 RSC.
- C. Buzea, I. I. Pacheco and K. Robbie, Biointerphases, 2007, 2, MR17–MR71 CrossRef PubMed.
- A. Nel, T. Xia, L. Mädler and N. Li, Science, 2006, 311, 622–627 CrossRef CAS PubMed.
- V. Schaller, U. Kräling, C. Rusu, K. Petersson, J. Wipenmyr, A. Krozer, G. Wahnström, A. Sanz-Velasco, P. Enoksson and C. Johansson, J. Appl. Phys., 2008, 104, 93918 CrossRef.
- N. Pamme and A. Manz, Anal. Chem., 2004, 76, 7250–7256 CrossRef CAS PubMed.
- L. Chen, X. Ding, J. Huo, S. El Hankari and D. Bradshaw, J. Mater. Sci., 2019, 54, 370–382 CrossRef.
- X.-Y. Yang, L.-H. Chen, Y. Li, J. C. Rooke, C. Sanchez and B.-L. Su, Chem. Soc. Rev., 2017, 46, 481–558 RSC.
- G. Srinivas, V. Krungleviciute, Z.-X. Guo and T. Yildirim, Energy Environ. Sci., 2014, 7, 335–342 RSC.
- A. K. Gupta and M. Gupta, Biomaterials, 2005, 26, 3995–4021 CrossRef CAS PubMed.
- M. Neamtu, C. Nadejde, V.-D. Hodoroaba, R. J. Schneider, L. Verestiuc and U. Panne, Sci. Rep., 2018, 8, 6278 CrossRef PubMed.
- A.-F. Ngomsik, A. Bee, M. Draye, G. Cote and V. Cabuil, C. R. Chim., 2005, 8, 963–970 CrossRef CAS.
- D. Xiao, T. Lu, R. Zeng and Y. Bi, Microchim. Acta, 2016, 183, 2655–2675 CrossRef CAS.
- M. Kawashita, M. Tanaka, T. Kokubo, Y. Inoue, T. Yao, S. Hamada and T. Shinjo, Biomaterials, 2005, 26, 2231–2238 CrossRef CAS PubMed.
- W. Zhao, H. Chen, Y. Li, L. Li, M. Lang and J. Shi, Adv. Funct. Mater., 2008, 18, 2780–2788 CrossRef CAS.
- F. Mou, J. Guan, Z. Xiao, Z. Sun, W. Shi and X. Fan, J. Mater. Chem., 2011, 21, 5414–5421 RSC.
- A. Ahmed, K. Skinley, S. Herodotou and H. Zhang, J. Sep. Sci., 2018, 41, 99–124 CrossRef CAS PubMed.
- W. Wu, Q. He and C. Jiang, Nanoscale Res. Lett., 2008, 3, 397 CrossRef CAS PubMed.
- W. Xie and F. Wan, Fuel, 2018, 220, 248–256 CrossRef CAS.
- S. Herrera, F. Tasca, F. J. Williams, E. J. Calvo, P. Carro and R. C. Salvarezza, Langmuir, 2017, 33, 9565–9572 CrossRef CAS PubMed.
- R. A. Bini, R. F. C. Marques, F. J. Santos, J. A. Chaker and M. Jafelicci, J. Magn. Magn. Mater., 2012, 324, 534–539 CrossRef CAS.
- Y. Hu, Z. Huang, J. Liao and G. Li, Anal. Chem., 2013, 85, 6885–6893 CrossRef CAS PubMed.
- Y. Deng, R. Zhang, D. Li, P. Sun, P. Su and Y. Yang, J. Sep. Sci., 2018, 41, 2046–2055 CrossRef CAS PubMed.
- N. Aeineh, F. Salehi, M. Akrami, F. Nemati, M. Alipour, M. Ghorbani, B. Nikfar, F. Salehian, N. R. Alam, S. E. S. Ebrahimi, A. Foroumadi, M. Khoobi, M. Rouini, M. Dibaei, I. Haririan, M. R. Ganjali and S. Safaei, J. Biomater. Sci., Polym. Ed., 2018, 29, 1109–1125 CrossRef CAS PubMed.
- M. Faustini, J. Kim, G.-Y. Jeong, J. Y. Kim, H. R. Moon, W.-S. Ahn and D.-P. Kim, J. Am. Chem. Soc., 2013, 135, 14619–14626 CrossRef CAS PubMed.
- S. Kayal and R. V. Ramanujan, Mater. Sci. Eng., C, 2010, 30, 484–490 CrossRef CAS.
- G. Lu, S. Li, Z. Guo, O. K. Farha, B. G. Hauser, X. Qi, Y. Wang, X. Wang, S. Han, X. Liu, J. S. DuChene, H. Zhang, Q. Zhang, X. Chen, J. Ma, S. C. J. Loo, W. D. Wei, Y. Yang, J. T. Hupp and F. Huo, Nat. Chem., 2012, 4, 310–316 CrossRef CAS PubMed.
- S. S.-Y. Chui, S. M.-F. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148–1150 CrossRef CAS PubMed.
- O. Shekhah, Y. Belmabkhout, Z. Chen, V. Guillerm, A. Cairns, K. Adil and M. Eddaoudi, Nat. Commun., 2014, 5, 4228 CrossRef CAS PubMed.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J.-Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
- CasaXPS, Version: 2.3.23PR1.0, https://www.casaxps.com/, accessed, 7 May 2021 Search PubMed.
- K. Chao, P. Liu and K. Huang, C. R. Chim., 2005, 8, 727–739 CrossRef CAS.
- J. Rouquerol, G. Baron, R. Denoyel, H. Giesche, J. Groen, P. Klobes, P. Levitz, A. V Neimark, S. Rigby, R. Skudas, K. Sing, M. Thommes and K. Unger, Pure Appl. Chem., 2011, 84, 107–136 CrossRef.
- Y. Zhang, Q. Yue, M. M. Zagho, J. Zhang, A. A. Elzatahry, Y. Jiang and Y. Deng, ACS Appl. Mater. Interfaces, 2019, 11, 10356–10363 CrossRef CAS PubMed.
- K. Weman, in Welding Processes Handbook, Woodhead Publishing, 2012, pp. 13–18 Search PubMed.
- S. Lowell and J. E. Shields, Interpretation of mercury porosimetry data, in, Powder Surface Area and Porosity, ed. S. Lowell and J. E. Shields, Powder Technology Series, Springer Netherlands, Dordrecht, 1991, pp. 99–120 Search PubMed.
- Poremaster Mercury Intrusion Pore Size Analyzer – Operating Manual, Quantachrome Instruments, Boynton Beach, FL 33426, USA, 2018 Search PubMed.
- Y. Tang, Y. Tang, C. Lv and Z. Zhou, Appl. Surf. Sci., 2008, 254, 5359–5362 CrossRef CAS.
- Y. Tai, L. Wang, J. Gao, W. A. Amer, W. Ding and H. Yu, J. Colloid Interface Sci., 2011, 360, 731–738 CrossRef CAS PubMed.
- T. Hidayat, D. Shishin, S. A. Decterov and E. Jak, Metall. Mater. Trans. B, 2016, 47, 256–281 CrossRef CAS.
- C. Han, J. Ma, H. Wu, Yaowei and K. Hu, J. Chil. Chem. Soc., 2015, 60, 2799–2802 CrossRef CAS.
- S. J. Kemp, R. M. Ferguson, A. P. Khandhar and K. M. Krishnan, RSC Adv., 2016, 6, 77452–77464 RSC.
- P. Hu, L. Kang, T. Chang, F. Yang, H. Wang, Y. Zhang, J. Yang, K. Wang, J. Du and Z. Yang, J. Alloys Compd., 2017, 728, 88–92 CrossRef CAS.
- I. Kagomiya, Y. Hirota, K. Kakimoto, K. Fujii, M. Shiraiwa, M. Yashima, A. Fuwa and S. Nakamura, Phys. Chem. Chem. Phys., 2017, 19, 31194–31201 RSC.
- M. Mehdizadeh, in Microwave/RF Applicators and Probes, Elsevier, 2015, pp. 219–268 Search PubMed.
- L. Chen, L. Li, H. Zhang, W. Liu, Y. Yang, X. Liu and B. Xu, RSC Adv., 2014, 4, 46806–46812 RSC.
- M. Zhou, J. Li, M. Zhang, H. Wang, Y. Lan, Y. Wu, F. Li and G. Li, Chem. Commun., 2015, 51, 2706–2709 RSC.
- J. Zhou, P. Wang, C. Wang, Y. T. Goh, Z. Fang, P. B. Messersmith and H. Duan, ACS Nano, 2015, 9, 6951–6960 CrossRef CAS PubMed.
- A. J. Parsons, R. M. Felfel, M. D. Wadge and D. M. Grant, Int. J. Appl. Glass Sci., 2020, 11, 35–45 CrossRef CAS.
- R. M. Felfel, A. J. Parsons, M. Chen, B. W. Stuart, M. D. Wadge and D. M. Grant, Composites, Part A, 2021, 146, 106415 CrossRef CAS.
- Z.-Y. Xi, Y.-Y. Xu, L.-P. Zhu, Y. Wang and B.-K. Zhu, J. Membr. Sci., 2009, 327, 244–253 CrossRef CAS.
- H. Hantsche, Scanning, 1989, 11, 257–280 CrossRef CAS.
- N. Huang, S. Zhang, L. Yang, M. Liu, H. Li, Y. Zhang and S. Yao, ACS Appl. Mater. Interfaces, 2015, 7, 17935–17946 CrossRef CAS PubMed.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed.
- T. Wang, H. Zhu, Q. Zeng and D. Liu, Adv. Mater. Interfaces, 2019, 6, 1900423 CrossRef.
- J. Moellmer, A. Moeller, F. Dreisbach, R. Glaeser and R. Staudt, Microporous Mesoporous Mater., 2011, 138, 140–148 CrossRef CAS.
- R. Sule and A. K. Mishra, Appl. Sci., 2019, 9, 4407 CrossRef CAS.
- D. G. Madden, H. S. Scott, A. Kumar, K.-J. Chen, R. Sanii, A. Bajpai, M. Lusi, T. Curtin, J. J. Perry and M. J. Zaworotko, Philos. Trans. R. Soc., A, 2017, 375, 20160025 CrossRef.
- N. Al-Janabi, P. Hill, L. Torrente-Murciano, A. Garforth, P. Gorgojo, F. Siperstein and X. Fan, Chem. Eng. J., 2015, 281, 669–677 CrossRef CAS.
- V. V. Guerrero, Y. Yoo, M. C. McCarthy and H.-K. Jeong, J. Mater. Chem., 2010, 20, 3938–3943 RSC.
- F. S. Gentile, M. Pannico, M. Causà, G. Mensitieri, G. Di Palma, G. Scherillo and P. Musto, J. Mater. Chem. A, 2020, 8, 10796–10812 RSC.
- P. Kanoo, S. K. Reddy, G. Kumari, R. Haldar, C. Narayana, S. Balasubramanian and T. K. Maji, Chem. Commun., 2012, 48, 8487–8489 RSC.
- O. Shekhah, Materials, 2010, 3, 1302–1315 CrossRef CAS.
- C. F. Holder and R. E. Schaak, ACS Nano, 2019, 13, 7359–7365 CrossRef CAS PubMed.
- T. Stassin, R. Verbeke, A. J. Cruz, S. Rodríguez-Hermida, I. Stassen, J. Marreiros, M. Krishtab, M. Dickmann, W. Egger, I. F. J. Vankelecom, S. Furukawa, D. De Vos, D. Grosso, M. Thommes and R. Ameloot, Adv. Mater., 2021, 33, 2006993 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta01927f |
| This journal is © The Royal Society of Chemistry 2023 |