
Copper–barium-decorated carbon-nanotube composite for electrocatalytic CO2 reduction to C2 products†
Feng-Yi
Wu
a,
Hsin-Jung
Tsai
a,
Tsung-Ju
Lee
a,
Zih-Yi
Lin
a,
Kang-Shun
Peng
a,
Pei-Hsuan
Chen
a,
Nozomu
Hiraoka
b,
Yen-Fa
Liao
b,
Chih-Wei
Hu
b,
Shao-Hui
Hsu
c,
Ying-Rui
Lu
*b and
Sung-Fu
Hung
 *a
*a
aDepartment of Applied Chemistry, National Yang Ming Chiao Tung University, Hsinchu 300, Taiwan. E-mail: sungfuhung@nycu.edu.tw
bNational Synchrotron Radiation Research Center, Hsinchu 300, Taiwan. E-mail: lu.yr@nsrrc.org.tw
cTaiwan Semiconductor Research Institute, National Applied Research Laboratories, Hsinchu 300, Taiwan
First published on 22nd May 2023
Abstract
A copper–barium-decorated carbon nanotube nanocomposite was designed to promote CO2 reduction to C2 products (ethylene and ethanol). We achieved a faradaic efficiency of 71% and partial current density of 355 mA cm−2 at −0.7 V vs. RHE. Operando methods revealed catalyst transformation and the catalytic mechanism.
 Sung-Fu Hung | Dr. Sung-Fu Hung is an Assistant Professor in the Department of Applied Chemistry at National Yang Ming Chiao Tung University (Taiwan). He received his doctorate in Chemistry from National Taiwan University in 2018. He is the winner of the 2019 IUPAC-Solvay International Award for Young Chemists. In 2019, he worked as a postdoctoral fellow in the research team of Professor Sargent at the University of Toronto. His research interests focus on the synthesis of nanomaterials for electrochemical catalysis, the design of catalytic reactors, and the development of in situ methods. |
The electrocatalytic carbon dioxide reduction reaction (CO2RR) has been regarded as a promising avenue to convert undesirable CO2 into valuable products.1–3 Among the possible products, C2 products, such as ethylene and ethanol, show a high market price and scale.3,4 It is highly attractive to create these products from air rather than to extract them from petroleum. Developing highly efficient electrocatalysts is essential in converting CO2RR to C2 products. Copper is the only catalyst that can reduce CO2 to hydrocarbons.5–10 However, the product selectivity of Cu is poor, and the catalytic current density of conversion into ethylene and alcohol at a high current density decreases, thereby limiting profitability. Therefore, these problems must be resolved for practical utilization of the CO2RR.
Recently, numerous strategies using foreign atoms have been employed to modify properties: defects created by Sn atoms;11,12 intermediate adsorption energies tuned by Ag or Au atoms;13–15 control of oxidation state or regulation of adsorption by rare-earth Ce atoms;16–18 p–d hybridization or interfacial engineering by main-group elements.19–22 These strategies enable the faradaic efficiency and catalytic current density toward the CO2RR to be obtained. Seitz et al. prepared a strontium copper oxide catalyst (SrCuO2) that exhibited a faradaic efficiency of 53% and a partial current density of 106 mA cm−2 toward C2+ products at −0.83 V vs. RHE.22 They found that Cu atoms were reduced to the metallic state with a low coordination number due to Sr incorporation, which improved the selectivity towards C2+ products. When the Sr content was increased further, the poor conductivity reduced the overall catalytic activity. Thus, improving the conductivity of a catalyst might boost the CO2RR performance towards multi-carbon products.
Carbon nanotubes are excellent candidates to improve the conductivity of a catalyst and charge transportation due to the π–electron conjugation, which benefits the catalytic current density of the CO2RR.23–25 Inspired by the strategies of foreign-atom doping and conductivity improvement, we designed a copper–barium-decorated carbon nanotube nanocomposite (CuBaCNT) to promote the activity of the CO2RR towards C2 products, including ethylene and ethanol (Fig. 1a). The CuBaCNT electrocatalyst enabled a faradaic efficiency of 71% and a partial current density of 355 mA cm−2 to C2 products at −0.7 V vs. RHE, which was superior to the benchmark sputtered Cu at the same catalytic current density (faradaic efficiency of 48% and partial current density of 241.65 mA cm−2). Unveiled by two-dimensional resonant inelastic X-ray scattering (RIXS) and operando spectroscopy, this remarkable enhancement was attributed to the regulation of 3d orbitals of Cu by doped barium atoms, rapid reduction of Cu cations owing to the conductive carbon nanotube composite, and the appearance of extra reaction intermediates.
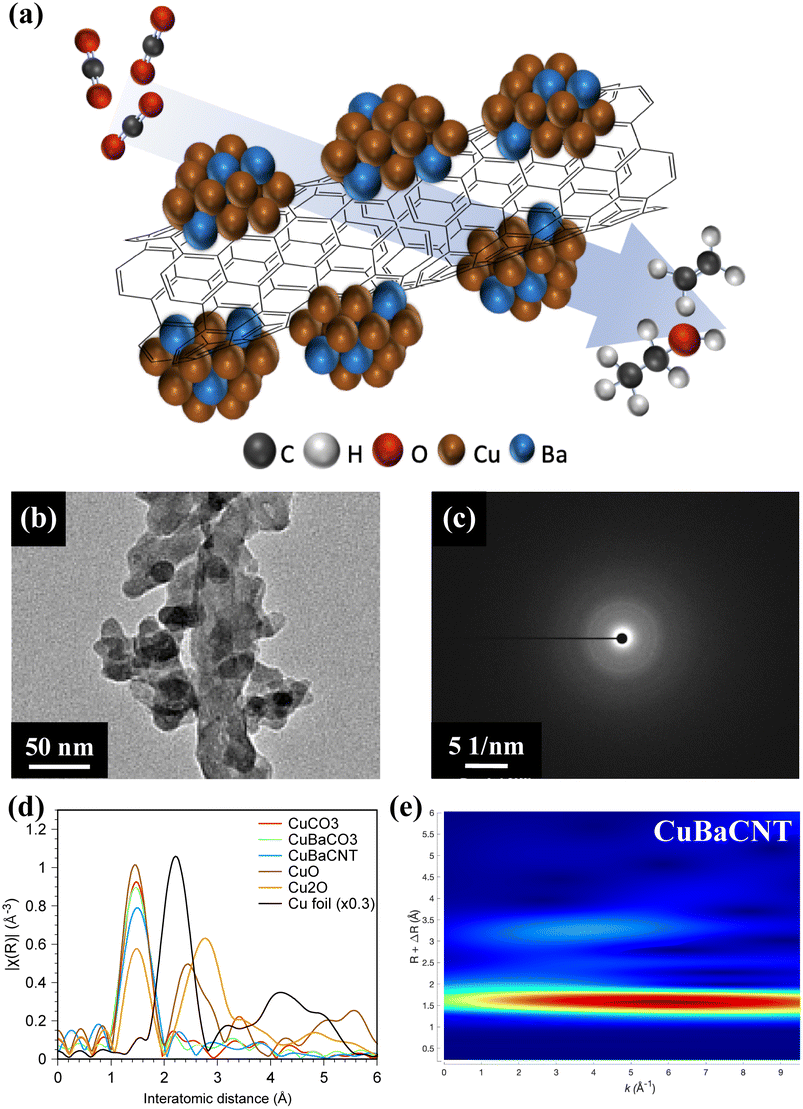 | ||
| Fig. 1 (a) Copper–barium-decorated carbon nanotube nanocomposite (CuBaCNT) for the carbon dioxide reduction reaction (schematic). (b) Transmission electron micrograph and (c) selected area electron diffraction pattern of CuBaCNT. (d) Fourier-transformed extended X-ray absorption fine structures of CuCO3, CuBaCO3, and CuBaCNT. The interatomic distance is presented without phase correction. (e) Wavelet analyses of CuBaCNT. | ||
We synthesized CuBaCNT via co-precipitation of Cu and Ba ions by sodium carbonate onto functionalized carbon nanotubes in an ice bath. Sodium carbonate was utilized because the Ksp of CuCO3 and BaCO3 are both very low (1.4 × 10−10 and 5.1 × 10−9, respectively) to prevent redissolution of precipitates. We also prepared CuCO3 and CuBaCO3 as control groups. The microstructure in Fig. S1† shows CuCO3 and CuBaCO3 to be nanoparticles of diameter ∼50 nm, and the nanoparticles adhered to carbon nanotubes for CuBaCNT. Energy dispersive X-ray (EDX) microanalysis displayed the uniformity of Cu and Ba in these electrocatalysts, but no Ba distribution in CuCO3 (Fig. S2†). Inductively coupled plasma-mass spectrometry (ICP-MS) revealed the percent Ba/Cu of CuBaCO3 and CuBaCNT was 8.6% and 9.5%, respectively, consistent with the ratio of the precursor. These data indicated that sodium carbonate could restrain the cation redissolution effectively (especially for Ba2+).
Fig. 1b and S3† show high-magnification transmission electron microscopy (TEM) images. CuBaCO3 nanoparticles were anchored on the carbon nanotubes in CuBaCNT, which should facilitate the interfacial interaction among Cu, Ca, and CNT and regulate the catalytic behavior. Fig. 1c revealed diffuse rings in the selected area of the electron diffraction image of CuCO3, CuBaCO3, and CuBaCNT, showing the amorphous nature of these electrocatalysts, which was confirmed by synchrotron X-ray diffraction patterns (Fig. S4†). To identify the amorphous structure,26,27 we analyzed the Cu K edge-extended X-ray absorption fine structure (EXAFS) (Fig. 1d) of these electrocatalysts: only the Cu–O path, without the second Cu–Cu path, was shown (2.4–2.8 Å). This observation implied that the surrounding large carbonate anions increased the interatomic Cu–Cu distance. The lower intensity of the first peak in CuBaCNT could be attributed to the coordination between Cu ions and CNT surface. Wavelet analyses (Fig. 1e) verified the peak around 1.42 Å to be the Cu–O path rather than the metallic Cu–Cu path because the middle point of the first region was located at less than 7 Å−1,28,29 and the atomic structure of CuBaCNT was entirely dissimilar to that of Cu2O and CuO (Fig. S5†).
Next, we examined the valence state of these electrocatalysts using X-ray absorption near-edge structure (XANES) (Fig. 2a). The near edge of these electrocatalysts was located between Cu2O and CuO, suggesting the oxidation state to be between 1 and 2. We compared the inflection points of the spectra with the standards and obtained the exact valence state of +1.9 for the three electrocatalysts (Fig. S6†). A binding energy of 934.5 eV for Cu 2p3/2 and 953.8 eV for Cu 2p1/2, accompanied by shake-up satellite peaks (942.5 and 962.5 eV), in X-ray photoelectron spectroscopy (XPS) (Fig. 2b) inferred a valence state of +2.0, consistent with XANES results.30,31 The Cu 2p satellite peaks (which originated from the electron transfer from the surrounding ligands to the 3d orbitals of Cu32,33) of Ba-containing electrocatalysts were divergent from that of CuCO3. This finding implied that the 3d orbitals of Cu had hybridized with Ba atoms, and that the catalytic behavior would be altered. Hence, we studied the 3d conduction band of Cu using Cu L-edge XAS spectra (2p → 3d) (Fig. S7†).34,35 The coordination environment of copper in CuCO3 was a distorted square pyramidal (Fig. S8†). Hence, the white-line-height augmentation of Ba-containing electrocatalysts meant that the electron transfer from dx2–y2 of copper to Ba ions was due to orbital hybridization.36 Next, we conducted 1s3p RIXS to explore the 3d valence band of Cu.37,38 After exciting a 1s core electron, we measured Kβ fluorescence to analyze the energy transfer from 3d to 3p orbitals of the two-dimensional 1s3p RIXS plane (Fig. 2c), which provided more information regarding the divergence of 3d orbitals.39,40 The RIXS plane of CuCO3 showed an energy-transfer peak at 90 eV for an incident energy of 8991 eV, whereas CuBaCO3 and CuBaCNT exhibited an energy-transfer peak at 95 eV for an incident energy of 8997 eV. These data unveiled the distinctive 3d orbitals of these electrocatalysts and revealed hybridization of the 3d orbitals of Cu with Ba atoms. Redox behavior in cyclic voltammetry reflects the valence band of an electrocatalyst. Their redox peaks differed from each other (Fig. S9†), which corresponded to XPS, XAS, and RIXS results. We believe the intense interaction among Cu, Ba, and CNT could regulate and improve the selectivity and activity of the CO2RR significantly.
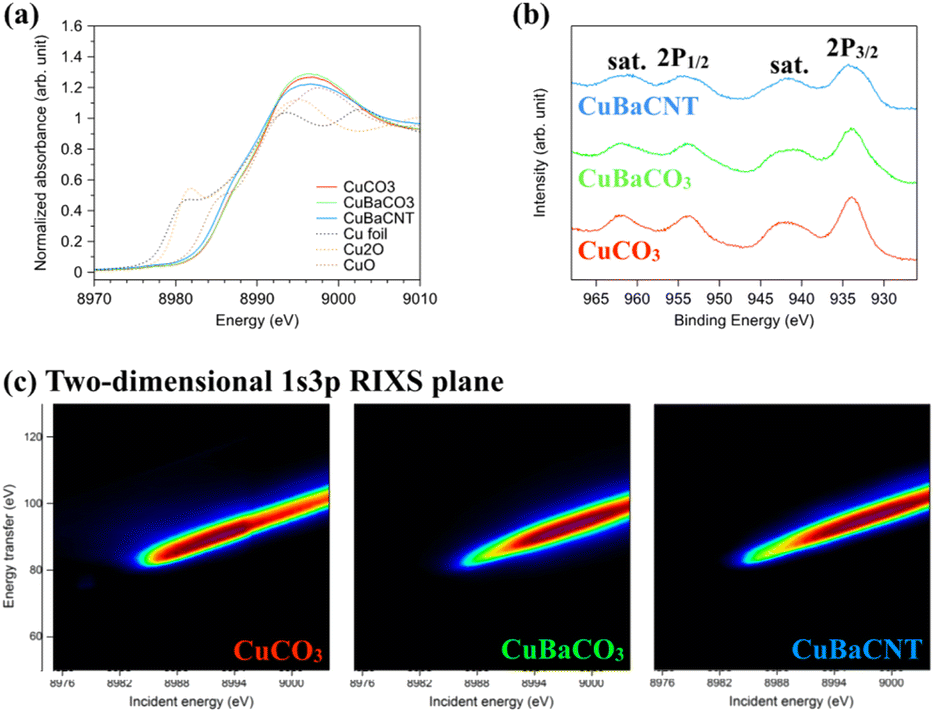 | ||
| Fig. 2 (a) X-ray absorption near-edge structures and (b) X-ray photoelectron spectroscopy of CuCO3, CuBaCO3, and CuBaCNT. (c) Two-dimensional 1s3p RIXS plane of CuCO3, CuBaCO3, and CuBaCNT (energy transfer from 3d to 3p of Cu orbitals). | ||
Next, we evaluated the CO2RR performance of these electrocatalysts in KOH (1 M) in a flow cell. CuCO3 electrocatalysts showed an optimal faradaic efficiency of 67.8% and partial current density of 203.3 mA cm−2 to C2 products at 300 mA cm−2 (Fig. 3a). Hydrogen evolution increased at a current density > 500 mA cm−2. Incorporation of Ba atoms into CuCO3 as the CuBaCO3 electrocatalyst and optimizing the Ba content (Fig. S10†) led to superior activity: faradaic efficiency of 73.1% and partial current density of 292.4 mA cm−2 to C2 products at 400 mA cm−2 (Fig. 3b). These data suggested that barium atoms regulate the 3d orbitals of Cu and enhance the CO2RR activity. However, when we attempted to increase the catalytic current density to >500 mA cm−2, the catalytic performance of CuBaCO3 decreased markedly. This phenomenon was associated with poor conductivity due to addition of an alkaline-earth element. As we increased the catalytic current density to 600 mA cm−2, the GDE flooded rapidly. To resolve this conductivity issue for augmenting the catalytic current density, we deposited CuBaCO3 onto the functionalized carbon nanotubes. After optimizing addition of the carbon nanotubes and Ba content (Fig. S11 and S12†), we achieved a faradaic efficiency of 70.9% and partial current density of 354.6 mA cm−2 to C2 products at 500 mA cm−2 (Fig. 3c). The partial current density to C2 products of CuBaCNT was 74.4% higher than that of CuCO3, 21.3% higher than that of CuBaCO3, and 40.8% higher than the benchmark sputtered Cu and numerous electrocatalysts in the literature (Fig. S13, 3d, and Table S1†).16,17,22,35,41–45 CuBaCNT exhibited this outstanding activity only at −0.7 V vs. RHE, which was also lower than the benchmark sputtered Cu (Fig. 3d). CuBaCNT maintained acceptable stability at a continuous operating current density of 500 mA cm−2 (Fig. S14†). Thus, CuBaCNT was an outstanding electrocatalyst showing superb selectivity and activity toward C2 products.
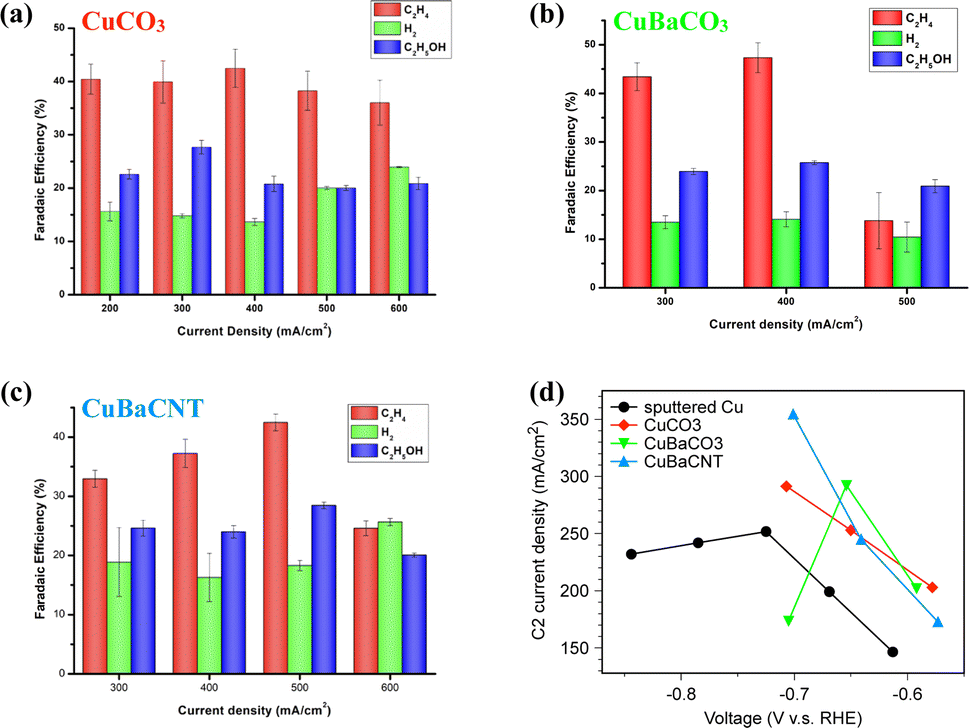 | ||
| Fig. 3 Faradaic efficiency of (a) CuCO3, (b) CuBaCO3, and (c) CuBaCNT at various current densities for the CO2RR in a flow cell. Error bars represent 1 standard deviation on the basis of three independent measurements. (d) Partial current density to C2 products (ethylene and ethanol) vs. applied potential. | ||
Operando spectroscopy allowed us to investigate material evolution and reaction intermediates during the CO2RR. We conducted operando Cu K-edge X-ray absorption spectroscopy (XAS) of electrocatalysts using our specially designed flow cell, which provided the equivalent catalytic environment with the electrochemical reactor.28 CuCO3 maintained a similar chemical state of Cu at −0.5 V vs. RHE compared with the unbiased situation (Fig. 4a), and the valence state was fitted as 1.87. When we increased the voltage further, CuCO3 transformed to metallic copper swiftly, and an evident Cu–Cu metallic bond formed (Fig. S14†). In contrast, the chemical state of Cu in CuBaCNT was reduced promptly from +1.9 to +0.5 at −0.5 V vs. RHE, and it became nearly 0 at a higher negative voltage (Fig. 4b). The Cu–Cu metallic bond also appeared speedily (Fig. S15†). This was because addition of the carbon nanotubes promoted the overall conductivity and formed a conductive network, so electrons were transported to electrocatalysts efficiently, thereby accelerating the reduction of copper and the catalytic rate. This finding also implied that improving conductivity was crucial to highly efficient CO2RR systems. We also investigated the change in morphology and structure of CuBaCNT after the CO2RR (Fig. S16 and S17†). Copper in CuBaCNT reduced to a polycrystalline metallic state, consistent with the operando XAS results.
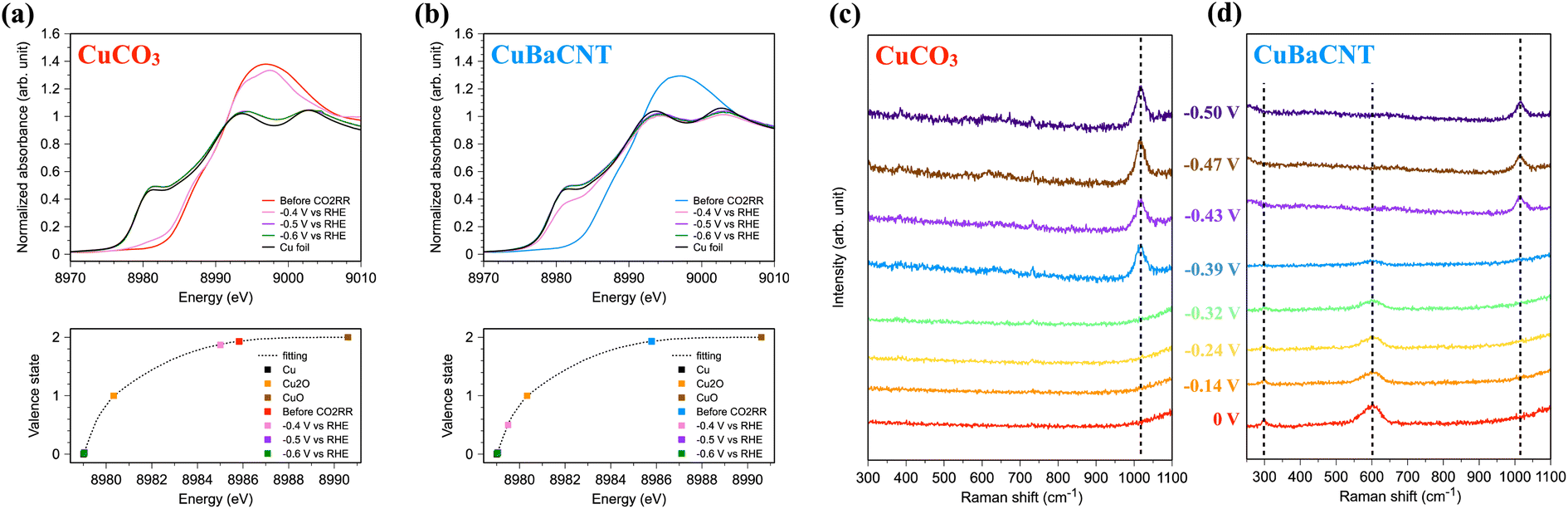 | ||
| Fig. 4 Operando X-ray absorption near-edge structure and valence-state analyses of (a) CuCO3 and (b) CuBaCNT during the CO2RR in a flow cell. Operando Raman spectroscopy of (c) CuCO3 and (d) CuBaCNT. The voltage is vs. RHE. | ||
We were also interested in whether the catalytic mechanism was changed in these systems. Hence, we undertook operando Raman spectroscopy using a specially designed flow cell in which the working electrode was separated by an anion-exchange membrane. First, we analyzed the Raman spectra of CuCO3 (Fig. 4c). We did not observe any intermediates at a negative voltage lower than −0.32 V vs. RHE, but a prominent peak at 1017 cm−1, representing the C–OH stretch of HCO32− on the Cu surface,46,47 appeared at a higher negative voltage. Interestingly, we did not detect the symmetric stretch of the intermediates of CO32−, which is commonly seen in copper/copper oxide systems.35,48 This finding might indicate that CuCO3 continued to follow a different catalytic CO2RR mechanism compared with typical copper catalysts. For CuBaCNT, we monitored the peak at 297 cm−2 (assigned as the restricted rotation of adsorbed CO on the Cu surface49) and the peak at 600 cm−2 (identified as the Cu–Oad stretch50) at a negative voltage lower than −0.39 V vs. RHE (Fig. 4d). In this initial stage of the CO2RR, the Cu 3dz2 orbital interacted with the carbon 2pz orbital in adsorbed CO (Fig. S18†). Electron transfer from Cu 3dx2–y2 to barium ions reduced the electron repulsion of carbon 2px and 2py, which is covalent with oxygen as a triple bond. Thus, the adsorbed CO would coordinate with Cu more readily than CuCO3. At a higher negative voltage, the peaks at 297 cm−2 and 600 cm−2 disappeared, and a peak at 1017 cm−1 appeared. Thus, the adsorption behaviors between CuCO3 and CuBaCNT were divergent.
Based on the discoveries proffered by operando XAS and Raman spectroscopy, the different catalyst transformations and intermediate adsorption might be related to the different 3d electron configurations of Cu between them. We believe that the distinctive intermediates of adsorbed CO on the Cu surface and Cu–Oad stretch led to the excellent catalytic activity of CuBaCNT.
In conclusion, we designed CuBaCNT that achieved a faradaic efficiency of 71% and partial current density of 355 mA cm−2 to C2 products at −0.7 V vs. RHE. We undertook RIXS, operando XAS, and Raman spectroscopy to ascertain if conductive nanotubes could reduce Cu cations promptly and facilitate the catalytic rate, and if regulation of the d orbital of Cu by doped barium atoms altered the catalytic behavior; these features led to the superb CO2RR activity of CuBaCNT. This strategy for material design of CuBaCNT to boost the selectivity and activity for the CO2RR could accelerate the progress towards its practical application.
Author contributions
S.-F. H. and Y.-R. L. supervised the project. S.-F. H. and F.-Y. W. conceived the idea and carried out experiments. S.-F. H. wrote the manuscript. F.-Y. W., H.-J. T., T.-J. L., and Z.-Y. L. carried out electrochemical experiments and operando measurements. T.-J. L., Z.-Y. L., and K.-S. P. conducted the X-ray emission spectroscopy. P.-H. C. and S.-H. H. characterized the materials and carried out data analyses. N. H., Y.-F. L., and C.-W. H. analyzed data from X-ray absorption spectroscopy and X-ray emission spectroscopy. All authors discussed the results and assisted during manuscript preparation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the National Science and Technology Council, Taiwan (NSTC 111-2628-M-A49-007 and 110-2112-M-213-019-MY3). We also acknowledge support from the Yushan Young Scholar Program, Ministry of Education, Taiwan. We thank Ms C.-Y. Chien of the Ministry of Science and Technology (National Taiwan University) for assistance in FE-TEM and EDS experiments.Notes and references
- T. N. Nguyen and C. T. Dinh, Chem. Soc. Rev., 2020, 49, 7488–7504 RSC.
- S. Overa, B. H. Ko, Y. Zhao and F. Jiao, Acc. Chem. Res., 2022, 55, 638–648 CrossRef CAS PubMed.
- S.-F. Hung, Pure Appl. Chem., 2020, 92, 1937–1951 CrossRef CAS.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- Y. Wang, Z. Wang, C.-T. Dinh, J. Li, A. Ozden, M. Golam Kibria, A. Seifitokaldani, C.-S. Tan, C. M. Gabardo, M. Luo, H. Zhou, F. Li, Y. Lum, C. McCallum, Y. Xu, M. Liu, A. Proppe, A. Johnston, P. Todorovic, T.-T. Zhuang, D. Sinton, S. O. Kelley and E. H. Sargent, Nat. Catal., 2020, 3, 98–106 CrossRef CAS.
- Z. Zhao, X. Peng, X. Liu, X. Sun, J. Shi, L. Han, G. Li and J. Luo, J. Mater. Chem. A, 2017, 5, 20239–20243 RSC.
- A. Ozden, F. Li, F. P. García de Arquer, A. Rosas-Hernández, A. Thevenon, Y. Wang, S.-F. Hung, X. Wang, B. Chen, J. Li, J. Wicks, M. Luo, Z. Wang, T. Agapie, J. C. Peters, E. H. Sargent and D. Sinton, ACS Energy Lett., 2020, 5, 2811–2818 CrossRef CAS.
- X. Wang, A. Xu, F. Li, S.-F. Hung, D.-H. Nam, C. M. Gabardo, Z. Wang, Y. Xu, A. Ozden, A. S. Rasouli, A. H. Ip, D. Sinton and E. H. Sargent, J. Am. Chem. Soc., 2020, 142, 3525–3531 CrossRef CAS PubMed.
- P. Li, J. Bi, J. Liu, Q. Zhu, C. Chen, X. Sun, J. Zhang, Z. Liu and B. Han, Chem. Sci., 2023, 14, 310–316 RSC.
- C. Zhu, Y. Song, X. Dong, G. Li, A. Chen, W. Chen, G. Wu, S. Li, W. Wei and Y. Sun, Energy Environ. Sci., 2022, 15, 5391–5404 RSC.
- X. Zhong, S. Liang, T. Yang, G. Zeng, Z. Zhong, H. Deng, L. Zhang and X. Sun, ACS Nano, 2022, 16, 19210–19219 CrossRef CAS PubMed.
- Z. Wei, J. Ding, X. Duan, G.-L. Chen, F.-Y. Wu, L. Zhang, X. Yang, Q. Zhang, Q. He, Z. Chen, J. Huang, S.-F. Hung, X. Yang and Y. Zhai, ACS Catal., 2023, 13, 4711–4718 CrossRef CAS.
- J. Li, H. Xiong, X. Liu, D. Wu, D. Su, B. Xu and Q. Lu, Nat. Commun., 2023, 14, 698 CrossRef CAS PubMed.
- L. Xiong, X. Zhang, H. Yuan, J. Wang, X. Yuan, Y. Lian, H. Jin, H. Sun, Z. Deng, D. Wang, J. Hu, H. Hu, J. Choi, J. Li, Y. Chen, J. Zhong, J. Guo, M. H. Rümmerli, L. Xu and Y. Peng, Angew. Chem., Int. Ed., 2021, 60, 2508–2518 CrossRef CAS PubMed.
- Y. C. Li, Z. Wang, T. Yuan, D. H. Nam, M. Luo, J. Wicks, B. Chen, J. Li, F. Li, F. P. G. de Arquer, Y. Wang, C. T. Dinh, O. Voznyy, D. Sinton and E. H. Sargent, J. Am. Chem. Soc., 2019, 141, 8584–8591 CrossRef CAS PubMed.
- S. Hong, H. G. Abbas, K. Jang, K. K. Patra, B. Kim, B.-U. Choi, H. Song, K.-S. Lee, P.-P. Choi, S. Ringe and J. Oh, Adv. Mater., 2023, 35, 2208996 CrossRef CAS PubMed.
- M. Luo, Z. Wang, Y. C. Li, J. Li, F. Li, Y. Lum, D.-H. Nam, B. Chen, J. Wicks, A. Xu, T. Zhuang, W. R. Leow, X. Wang, C.-T. Dinh, Y. Wang, Y. Wang, D. Sinton and E. H. Sargent, Nat. Commun., 2019, 10, 5814 CrossRef CAS PubMed.
- Y. Jiang, K. Mao, J. Li, D. Duan, J. Li, X. Wang, Y. Zhong, C. Zhang, H. Liu, W. Gong, R. Long and Y. Xiong, ACS Nano, 2023, 17, 2620–2628 CrossRef CAS PubMed.
- P. Li, J. Bi, J. Liu, Y. Wang, X. Kang, X. Sun, J. Zhang, Z. Liu, Q. Zhu and B. Han, J. Am. Chem. Soc., 2023, 145, 4675–4682 CrossRef CAS PubMed.
- A. Xu, S.-F. Hung, A. Cao, Z. Wang, N. Karmodak, J. E. Huang, Y. Yan, A. Sedighian Rasouli, A. Ozden, F.-Y. Wu, Z.-Y. Lin, H.-J. Tsai, T.-J. Lee, F. Li, M. Luo, Y. Wang, X. Wang, J. Abed, Z. Wang, D.-H. Nam, Y. C. Li, A. H. Ip, D. Sinton, C. Dong and E. H. Sargent, Nat. Catal., 2022, 5, 1081–1088 CrossRef CAS.
- A. Sedighian Rasouli, X. Wang, J. Wicks, C.-T. Dinh, J. Abed, F.-Y. Wu, S.-F. Hung, K. Bertens, J. E. Huang and E. H. Sargent, Chem Catal., 2022, 2, 908–916 CrossRef CAS.
- X. K. Lu, B. Lu, H. Li, K. Lim and L. C. Seitz, ACS Catal., 2022, 12, 6663–6671 CrossRef CAS.
- G. Valenti, M. Melchionna, T. Montini, A. Boni, L. Nasi, E. Fonda, A. Criado, A. Zitolo, S. Voci, G. Bertoni, M. Bonchio, P. Fornasiero, F. Paolucci and M. Prato, ACS Appl. Energy Mater., 2020, 3, 8509–8518 CrossRef CAS.
- S. Liu, H. B. Yang, S.-F. Hung, J. Ding, W. Cai, L. Liu, J. Gao, X. Li, X. Ren, Z. Kuang, Y. Huang, T. Zhang and B. Liu, Angew. Chem., Int. Ed., 2020, 59, 798–803 CrossRef CAS PubMed.
- G. Wang, J. Pan, S. P. Jiang and H. Yang, J. CO2 Util., 2018, 23, 152–158 CrossRef CAS.
- S.-F. Hung, Pure Appl. Chem., 2020, 92, 733–749 CrossRef CAS.
- S.-F. Hung, Y. Zhu, G.-Q. Tzeng, H.-C. Chen, C.-S. Hsu, Y.-F. Liao, H. Ishii, N. Hiraoka and H. M. Chen, ACS Energy Lett., 2019, 4, 2813–2820 CrossRef CAS.
- S. F. Hung, F. Y. Wu, Y. H. Lu, T. J. Lee, H. J. Tsai, P. H. Chen, Z. Y. Lin, G. L. Chen, W. Y. Huang and W. J. Zeng, Catal. Sci. Technol., 2022, 12, 2739–2743 RSC.
- Y.-R. Lu, H.-C. Chen, K. Liu, M. Liu, T.-S. Chan and S.-F. Hung, ACS Sustainable Chem. Eng., 2022, 10, 6736–6742 CrossRef CAS.
- Q. Li, K. Li, C. Sun and Y. Li, J. Electroanal. Chem., 2007, 611, 43–50 CrossRef CAS.
- B. Li, X. Luo, Y. Zhu and X. Wang, Appl. Surf. Sci., 2015, 359, 609–620 CrossRef CAS.
- S. Larsson, Chem. Phys. Lett., 1976, 40, 362–366 CrossRef CAS.
- T. M. Ivanova, K. I. Maslakov, A. A. Sidorov, M. A. Kiskin, R. V. Linko, S. V. Savilov, V. V. Lunin and I. L. Eremenko, J. Electron Spectrosc., 2020, 238, 146878 CrossRef CAS.
- M. Grioni, J. B. Goedkoop, R. Schoorl, F. M. F. de Groot, J. C. Fuggle, F. Schäfers, E. E. Koch, G. Rossi, J. M. Esteva and R. C. Karnatak, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 39, 1541–1545 CrossRef CAS PubMed.
- S.-F. Hung, A. Xu, X. Wang, F. Li, S.-H. Hsu, Y. Li, J. Wicks, E. G. Cervantes, A. S. Rasouli, Y. C. Li, M. Luo, D.-H. Nam, N. Wang, T. Peng, Y. Yan, G. Lee and E. H. Sargent, Nat. Commun., 2022, 13, 819 CrossRef CAS PubMed.
- T. Jurca, A. Farghal, P.-H. Lin, I. Korobkov, M. Murugesu and D. S. Richeson, J. Am. Chem. Soc., 2011, 133, 15814–15817 CrossRef CAS PubMed.
- A. Bagger, T. Haarman, A. Puig Molina, P. G. Moses, H. Ishii, N. Hiraoka, Y. H. Wu, K. D. Tsuei, I. Chorkendorff and F. De Groot, J. Synchrotron Radiat., 2017, 24, 296–301 CrossRef CAS PubMed.
- U. Bergmann and P. Glatzel, Photosynth. Res., 2009, 102, 255–266 CrossRef CAS PubMed.
- S. F. Hung, Y. T. Chan, C. C. Chang, M. K. Tsai, Y. F. Liao, N. Hiraoka, C. S. Hsu and H. M. Chen, J. Am. Chem. Soc., 2018, 140, 17263–17270 CrossRef CAS PubMed.
- M. Al Samarai, M. U. Delgado-Jaime, H. Ishii, N. Hiraoka, K.-D. Tsuei, J. P. Rueff, B. Lassale-Kaiser, B. M. Weckhuysen and F. M. F. de Groot, J. Phys. Chem. C, 2016, 120, 24063–24069 CrossRef CAS.
- X. Wang, Z. Wang, F. P. García de Arquer, C.-T. Dinh, A. Ozden, Y. C. Li, D.-H. Nam, J. Li, Y.-S. Liu, J. Wicks, Z. Chen, M. Chi, B. Chen, Y. Wang, J. Tam, J. Y. Howe, A. Proppe, P. Todorović, F. Li, T.-T. Zhuang, C. M. Gabardo, A. R. Kirmani, C. McCallum, S.-F. Hung, Y. Lum, M. Luo, Y. Min, A. Xu, C. P. O'Brien, B. Stephen, B. Sun, A. H. Ip, L. J. Richter, S. O. Kelley, D. Sinton and E. H. Sargent, Nat. Energy, 2020, 5, 478–486 CrossRef CAS.
- F. Li, Y. C. Li, Z. Wang, J. Li, D.-H. Nam, Y. Lum, M. Luo, X. Wang, A. Ozden, S.-F. Hung, B. Chen, Y. Wang, J. Wicks, Y. Xu, Y. Li, C. M. Gabardo, C.-T. Dinh, Y. Wang, T.-T. Zhuang, D. Sinton and E. H. Sargent, Nat. Catal., 2020, 3, 75–82 CrossRef CAS.
- Y. Liang, J. Zhao, Y. Yang, S.-F. Hung, J. Li, S. Zhang, Y. Zhao, A. Zhang, C. Wang, D. Appadoo, L. Zhang, Z. Geng, F. Li and J. Zeng, Nat. Commun., 2023, 14, 474 CrossRef CAS PubMed.
- D.-S. Huang, H.-L. Zhu, Z.-H. Zhao, J.-R. Huang, P.-Q. Liao and X.-M. Chen, ACS Catal., 2022, 12, 8444–8450 CrossRef CAS.
- Z.-Z. Wu, X.-L. Zhang, Z.-Z. Niu, F.-Y. Gao, P.-P. Yang, L.-P. Chi, L. Shi, W.-S. Wei, R. Liu, Z. Chen, S. Hu, X. Zheng and M.-R. Gao, J. Am. Chem. Soc., 2022, 144, 259–269 CrossRef CAS PubMed.
- D. A. Henckel, M. J. Counihan, H. E. Holmes, X. Chen, U. O. Nwabara, S. Verma, J. Rodríguez-López, P. J. A. Kenis and A. A. Gewirth, ACS Catal., 2021, 11, 255–263 CrossRef CAS.
- S. Jiang, K. Klingan, C. Pasquini and H. Dau, J. Chem. Phys., 2019, 150, 041718 CrossRef PubMed.
- I. V. Chernyshova, P. Somasundaran and S. Ponnurangam, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E9261 CrossRef CAS PubMed.
- C. Zhan, F. Dattila, C. Rettenmaier, A. Bergmann, S. Kühl, R. García-Muelas, N. López and B. R. Cuenya, ACS Catal., 2021, 11, 7694–7701 CrossRef CAS PubMed.
- N. Bodappa, M. Su, Y. Zhao, J.-B. Le, W.-M. Yang, P. Radjenovic, J.-C. Dong, J. Cheng, Z.-Q. Tian and J.-F. Li, J. Am. Chem. Soc., 2019, 141, 12192–12196 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, SEM images, XRD patterns, and CO2RR activity. See DOI: https://doi.org/10.1039/d3ta01912h |
| This journal is © The Royal Society of Chemistry 2023 |