DOI:
10.1039/D3PY00158J
(Paper)
Polym. Chem., 2023,
14, 2494-2509
Solid state chemical transformation provides a fully benzoxazine-linked porous organic polymer displaying enhanced CO2 capture and supercapacitor performance†
Received
13th February 2023
, Accepted 24th April 2023
First published on 25th April 2023
Abstract
In this study, we synthesized a fully benzoxazine (BZ)-linked porous organic polymer (POP) comprising triphenylamine (TPA) and dihydroxyterephthalaldehyde (DHPT) units through Sonogashira–Hagihara coupling of TPA- and DHTP-functionalized BZ monomers, prepared through multistep sequences involving the Schiff base formation, reduction, and Mannich reactions. The chemical structure of this fully BZ-linked POP (TPA–DHTP–BZ POP) was validated using Fourier transform infrared (FTIR) and solid-state nuclear magnetic resonance (NMR) spectroscopy. The Brunauer–Emmett–Teller surface area and total pore volume of the TPA–DHTP–BZ POP were 195 m2 g−1 and 0.53 cm3 g−1, respectively. The poly(TPA–DHTP–BZ) POP showed an impressive CO2 capture performance of 3.29 mmol g−1 and a specific capacitance of 67.1 F g−1 at 0.5 A g−1. After thermal ring-opening polymerization, a solid-state chemical transformation, the resulting poly(TPA–DHTP–BZ) POP featured Mannich bridges and phenolic groups that formed strong inter- and intramolecular hydrogen bonds, thereby enhancing the electrochemical and CO2 capture properties. Therefore, poly(TPA–DHTP–BZ) POP has the potential to be employed in practical applications for CO2 capture and as an efficient electrode for energy storage.
Introduction
Porous organic polymers (POPs), including covalent organic frameworks (COFs), hyper cross-linked polymers (HCPs), and conjugated microporous polymers (CMPs), are promising materials featuring high surface areas and porous properties; they have been used in many applications, including energy storage, iodine (I2) uptake, photocatalysis, catalysis, gas capture/separation, and H2 evolution.1–8 For example, Jiang and Tan et al. revealed that POPs exhibited the highest capacitance for CO2 or I2 uptake.9,10 Wang and his group constructed a 3D COF membrane for sieving multi- and monovalent ions.11 Pan et al. constructed a series of CMP materials containing dibenzothiophene-S,S-dioxide for H2 production.12 Mai et al. successfully prepared a CMP tube based on a porphyrin unit with a high surface area for photocatalytic application.13 The energy shortage, global warming, and emission of carbon dioxide (CO2) cause several environmental concerns; hence CO2 capture and the development of efficient energy storage devices like supercapacitors (SCs) are very important. POPs have emerged as excellent candidates for CO2 capture and active electrode materials in SCs due to their high surface areas, adequate pore size, low cost, and high stability.14,15 Several organic reactions (e.g., Suzuki, Yamamoto, Friedel couplings, and Schiff base formation) have been used to develop POP structures with various covalently bonded linkages (e.g., boroxine, hydrazine, imide, triazine, and imine units).3,16,17 For example, Li et al. prepared imine-based 2D-CPPs (TAPA–CPPs) through the condensation reaction of tris(4-aminophenyl)amine with p-phthalaldehyde.18 Our group used Sonogashira–Hagihara cross-couplings to prepare three pyrene-based CMPs for H2 production19 and also prepared three ThTh–CMPs through the condensation reaction of dithiooxamide with different aromatic aldehydes.20 The addition of new chemical functionalities in POP frameworks can influence their surface areas and their possible applications.21–23 For example, POPs with imine linkages have been converted into materials containing secondary amide/amine, oxazole, quinolone, and thiazole units.24–31 Furthermore, Yaghi et al. used multistep solid-state post-modification processes to transform an imine-COF into carbamate- and thiocarbamate-linked COFs of a high surface area.32 In addition, Ma et al. converted an imine-COF into a benzoxazine (BZ) derivative through reduction and cyclization reactions with CH2O and benzyl aldehyde, obtaining materials of a high surface area (up to 651 m2 g−1).33 BZ-linked resins are attractive materials because they can feature significant degrees of intra- and intermolecular hydrogen bonding after thermal ring-opening polymerization (ROP) of their oxazine rings. BZ resins are generally synthesized through Mannich condensations of primary amines, aromatic phenols, and paraformaldehyde; when subjected to ROP, they can form polybenzoxazines (PBZs) with thermal stability higher than that of other thermosetting resins.34–42 BZ units can undergo ROP without needing any external stimuli or catalyst, proceeding with minimal shrinkage; the syntheses of BZ-linked POPs having high surface areas and pore volumes have been investigated recently.43–48 For example, we have previously prepared a nitrile-functionalized BZ (CN–BZ) from 4-cyanolphenol, CH2O, and 1,3,5-tris(4-aminophenoxy)benzene (TPhA) that displayed a high surface area after cyclotrimerization.43 We have also prepared a BZ-linked porous material through a one-pot Mannich reaction of 1,3,5-tris(4-aminophenyl)triazine, 2,4,6-tris(p-hydroxyphenyl) triazine, and CH2O; it also exhibited a low surface area (ca. 71.8 m2 g−1).44 Xu et al. reacted 1,3,5-trihydroxybenzene and CH2O with three diamines to form BZ-linked POPs of a high surface area (231 m2 g−1).45 Sun et al. followed Sonogashira–Hagihara coupling to form BZ-based POPs having a high surface area (623 m2 g−1).46 In previous studies,47,48 we also prepared BZ-based POPs with a high surface area (325 m2 g−1).47 In these cases, however, the BZ-based POPs were prepared through Sonogashira–Hagihara couplings in which only the brominated partner was a BZ monomer; because the other monomer presenting the ethynyl group (HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–) was not a BZ derivative, the content of BZ units in the BZ-linked POPs was relatively low, thereby limiting the number of functional phenolic OH and N atoms after thermal ROP.42–48 To the best of our knowledge, fully BZ-linked POPs have never been prepared previously through Sonogashira–Hagihara couplings in which both monomer units were BZ derivatives. In this study, we synthesized two BZ monomers, one brominated unit (TPA–Br BZ) and the other presenting ethynyl units (DHTP–Ea BZ). We prepared the TPA–Br BZ monomer by reacting a triamine containing a triphenylamine unit (TPA–NH2) with 4-bromo-2-hydroxybenzaldehyde [Fig. 1(a)], and we obtained the DHTP–Ea BZ monomer from the reaction of 2,5-dihydroxyterephthalaldehyde (DHTP) with 4-ethynylaniline [Fig. 2(a)]; in both cases, the transformations involved a condensation reaction, reduction, and Mannich condensation. Finally, we synthesized a TPA–DHTP–BZ POP, with an abundance of BZ-linkages, through Sonogashira–Hagihara coupling of TPA–Br BZ and DHTP–Ea BZ monomers at 110 °C for 3 days, using tetrakis(triphenylphosphine)palladium(0) [Pd(PPh3)4] as the catalyst. Finally, we exposed this TPA–DHTP–BZ POP to thermal ROP in the solid state, leading to the emergence of Mannich bridges and phenolic OH units capable of strong intermolecular hydrogen bonding; therefore, the resulting fully-BZ-linked POPs were endowed with excellent electrochemical and CO2 capture applications.
C–) was not a BZ derivative, the content of BZ units in the BZ-linked POPs was relatively low, thereby limiting the number of functional phenolic OH and N atoms after thermal ROP.42–48 To the best of our knowledge, fully BZ-linked POPs have never been prepared previously through Sonogashira–Hagihara couplings in which both monomer units were BZ derivatives. In this study, we synthesized two BZ monomers, one brominated unit (TPA–Br BZ) and the other presenting ethynyl units (DHTP–Ea BZ). We prepared the TPA–Br BZ monomer by reacting a triamine containing a triphenylamine unit (TPA–NH2) with 4-bromo-2-hydroxybenzaldehyde [Fig. 1(a)], and we obtained the DHTP–Ea BZ monomer from the reaction of 2,5-dihydroxyterephthalaldehyde (DHTP) with 4-ethynylaniline [Fig. 2(a)]; in both cases, the transformations involved a condensation reaction, reduction, and Mannich condensation. Finally, we synthesized a TPA–DHTP–BZ POP, with an abundance of BZ-linkages, through Sonogashira–Hagihara coupling of TPA–Br BZ and DHTP–Ea BZ monomers at 110 °C for 3 days, using tetrakis(triphenylphosphine)palladium(0) [Pd(PPh3)4] as the catalyst. Finally, we exposed this TPA–DHTP–BZ POP to thermal ROP in the solid state, leading to the emergence of Mannich bridges and phenolic OH units capable of strong intermolecular hydrogen bonding; therefore, the resulting fully-BZ-linked POPs were endowed with excellent electrochemical and CO2 capture applications.
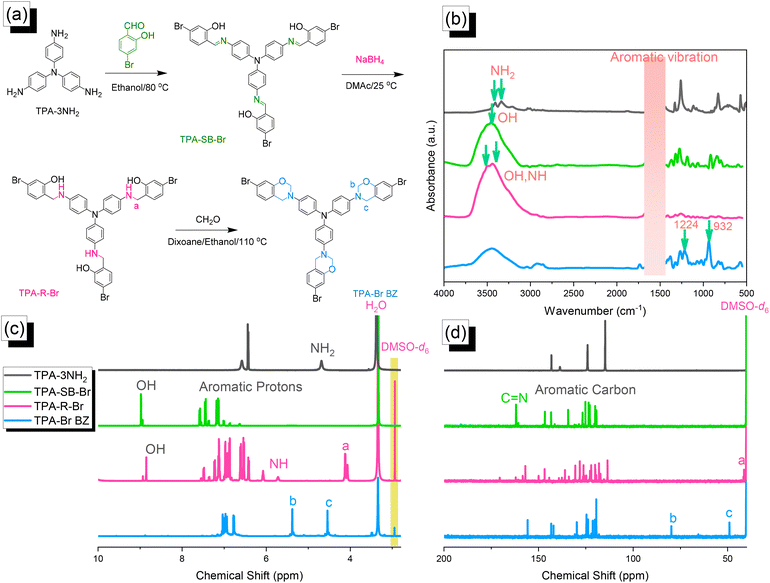 |
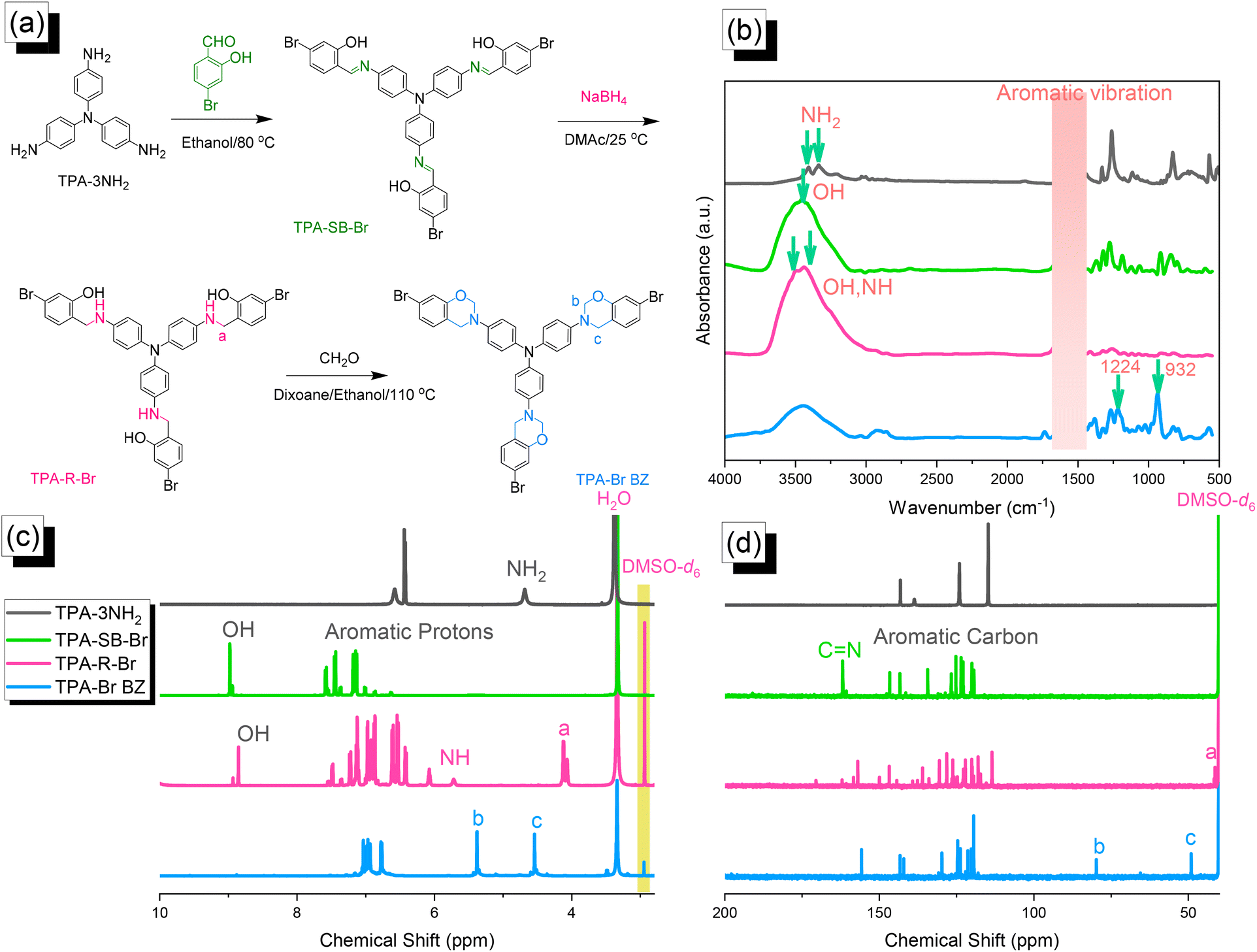
| | Fig. 1 (a) Synthesis of TPA–Br BZ from TPA–3NH2, TPA–SB–Br, and TPA–R–Br.(b) FTIR, (c) 1H NMR, and (d) 13C NMR spectra. | |
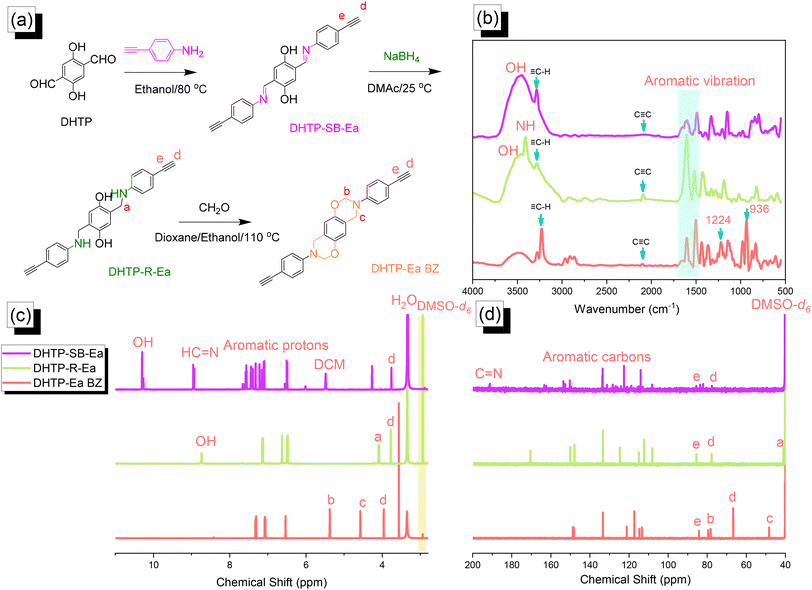 |
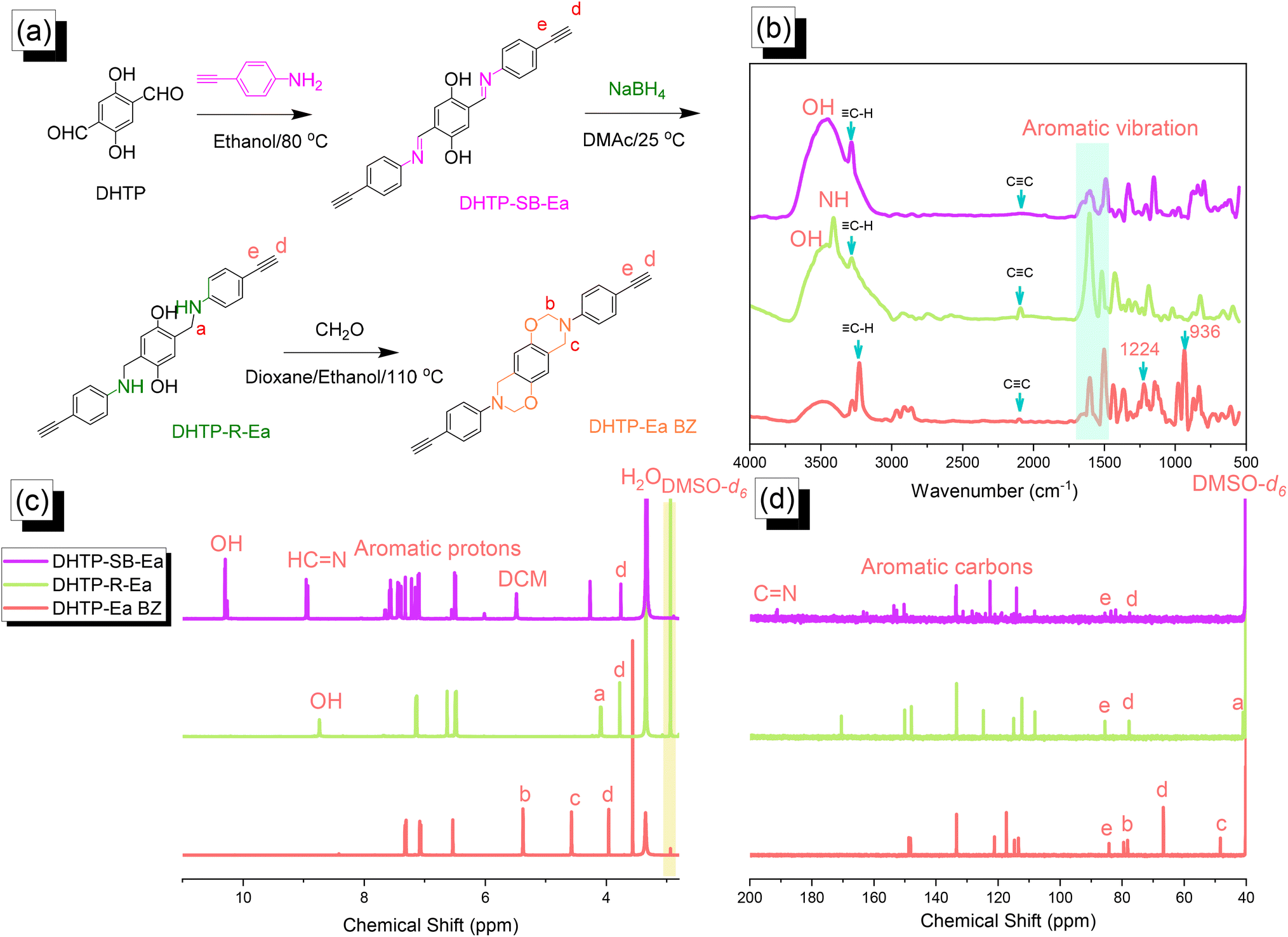
| | Fig. 2 (a) Synthesis of DHTP–Ea BZ from DHTP, DHTP–SB–Ea, and DHTP–R–Ea. (b–d) Corresponding (b) FTIR, (c) 1H NMR, and (d) 13C NMR spectra. | |
Experimental section
Materials
4-Bromo-2-hydroxybenzaldehyde, 1,4-dioxane (DO), N,N-dimethylacetamide (DMAc), sodium borohydride (NaBH4), absolute EtOH, MeOH, N,N-dimethylformamide (DMF), CH2Cl2, and CHCl3 were obtained from Alfa Aesar. Pd(PPh3)4, copper(I) iodide (CuI), sodium bicarbonate (NaHCO3), paraformaldehyde (CH2O), 4-ethynylaniline (Ea), 2,5-dihydroxyterephthalaldehyde (DHTP), and potassium carbonate (K2CO3) were purchased from Sigma–Aldrich. TPA–3NH2 was synthesized according to a previously published procedure.49
TPA–SB–Br
A solution of TPA–3NH2 (8.00 g, 27.55 mmol) and 4-bromo-2-hydroxybenzaldehyde (16.61 g, 82.65 mmol) in EtOH (300 mL) was refluxed overnight at 90 °C. Then, the orange solid was washed with EtOH collected and dried overnight at 60 °C. Yield: 88%. FTIR: 3460 (OH), 1614 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 600 (C–Br). 1H NMR (DMSO-d6, ppm): δ = 11.83 (OH), 8.94 (NCH), 7.6–7.0 (aromatic protons). 13C NMR (DMSO-d6, ppm): δ = 162.60 (NCH), 133.6–107.5 (aromatic carbon nuclei). High-resolution FT-MS (m/z): calcd for (C39H27Br3N4O3), 839.38; found, 838.84 (Fig. S1†).
N), 600 (C–Br). 1H NMR (DMSO-d6, ppm): δ = 11.83 (OH), 8.94 (NCH), 7.6–7.0 (aromatic protons). 13C NMR (DMSO-d6, ppm): δ = 162.60 (NCH), 133.6–107.5 (aromatic carbon nuclei). High-resolution FT-MS (m/z): calcd for (C39H27Br3N4O3), 839.38; found, 838.84 (Fig. S1†).
TPA–R–Br
TPA–SA–Br (4.00 g, 4.76 mmol), NaBH4 (1.44 g, 38.12 mmol), and DMAc (60 mL) were stirred together for 24 h at 25 °C, and then the mixture was added to ice water.
The brown solid was filtered off, washed three times in water, and dried. Yield: 90%. FTIR (KBr, cm−1): 3500 (OH stretching), 3392 (NH). 1H NMR (DMSO-d6, ppm): δ = 10.05 (OH), 8.84 (NH), 7.5–5.7 (aromatic protons), 4.11 (HNCH2). 13C NMR (DMSO-d6, ppm): δ = 42.23 (NHCH2). High-resolution FT-MS (m/z): calcd for (C39H33Br3N4O3), 845.45; found, 845.01 (Fig. S2†).
TPA–Br BZ
A mixture of TPA–R–Br (2.40 g, 2.88 mmol), CH2O (0.34 g, 11.33 mmol), and EtOH/DO (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) was heated for 24 h at 110 °C under a N2 atmosphere. The solvent was removed using a rotary evaporator.
:2) was heated for 24 h at 110 °C under a N2 atmosphere. The solvent was removed using a rotary evaporator.
The residue was treated with hexane, and the mixture was placed in a fridge for 24 h. The brown solid was filtered off, washed three times in EtOH, and dried. Yield: 80%. FTIR (KBr, cm−1): 1223 (C–O–C), 932 (oxazine ring). 1H NMR (DMSO-d6, ppm): δ = 7.50–6.11 (aromatic protons), 5.32 (OCH2N), 4.52 (ArCH2N). 13C NMR (DMSO-d6, ppm): δ = 83.7 (OCH2N), 47.8 (ArCH2N). High-resolution FT-MS (m/z): calcd for (C42H33Br3N4O3), 860.46; found, 860.01 (Fig. S3†).
DHTP–SB–Ea
A solution of DHTP (6 g, 36.145 mmol) and 4-ethynylaniline (8.46 g, 72.21 mmol) in EtOH (150 mL) was kept heating overnight at 90 °C. The orange solid was collected, washed three times with EtOH, and dried for two days at 50 °C under a vacuum. Yield: 85%. FTIR: 3443 (OH stretching), 3222 (CH), 1597 (CN). 1H NMR (DMSO-d6, ppm): δ = 11.83 (OH), 8.94 (NCH), 7.60–6.50 (aromatic protons), 3.75 (CH). 13C NMR (DMSO-d6, ppm): δ = 163.80 (NCH), 134–113.5 (aromatic nuclei). High-resolution FT-MS (m/z): calcd for (C24H16N2O2), 364.46; found, 365.13 (Fig. S4†).
DHTP–R–Ea
DMAc (30 mL), DHTP–SB–Ea (2.00 g, 5.43 mmol), and NaBH4 (1.02 g, 26.96 mmol) were kept heating overnight at room temperature. Then the mixture was added to ice water, and the brown solid was dried. Yield: 90%. FTIR: 3500 (OH stretching), 3392 (NH). 1H NMR (ppm): δ = 10.05 (OH), 8.84 (NH), 7.50–5.70 (aromatic protons), 4.11 (HNCH2), 3.73 (CH). 13C NMR (DMSO-d6, ppm): δ = 42.23 (NHCH2). High-resolution FT-MS (m/z): calcd for (C24H20N2O2), 368.44; found, 369.16 (Fig. S5†).
DHTP–Ea BZ
A mixture of DHTP–R–Ea (0.600 g, 1.52 mmol), CH2O (0.190 g, 6.33 mmol), and EtOH/DO (1:2) was heated for 24 h at 110 °C under a N2 atmosphere. The EtOH/DO was removed using reduced pressure. The residue was treated with hexane and placed in a fridge overnight. The brown solid was washed with EtOH and dried. Yield: 79%. FTIR (KBr, cm−1): 1228 (C–O–C), 936. 1H NMR (ppm): δ = 7.30–6.50 (aromatic protons), 5.34 (OCH2N), 4.55 (ArCH2N), 3.9 (CH). 13C NMR (DMSO-d6, ppm): δ = 84.0 (OCH2N), 48.3 (ArCH2N). High-resolution FT-MS (m/z): calcd for (C26H20N2O2), 392.46; found, 393.16 (Fig. S6†).
TPA–DHTP–BZ POP
A mixture of Pd(PPh3)4 (0.030 g, 0.020 mmol), TPA–Br BZ (0.2 g, 0.23 mmol), DHTP–Ea BZ (0.13 g, 0.34 mmol), PPh3 (0.01 g, 0.023 mmol), CuI (0.001 g, 0.032 mmol), Et3N (10 mL), and DMF (10 mL) was heated at 100 °C under N2 for 72 h. A dark brown powder of TPA–DHTP–BZ POP was obtained.
Results and discussion
Synthesis of the TPA–Br BZ monomer
We reacted TPA–3NH2 with 4-bromo-2-hydroxybenzaldehyde to form the Schiff base TPA–SB–Br. Subsequent NaBH4-mediated reduction provided TPA–R–Br, which underwent Mannich condensation in the presence of CH2O and EtOH/dioxane (1:2) for 24 h at 110 °C to give the TPA–Br BZ monomer [Fig. 1(a)]. The FTIR spectrum of TPA–3NH2 [Fig. 1(b)] featured signals at 3460 and 3334 cm−1 for asymmetric and symmetric NH2 stretching. The spectrum of TPA–SB–Br contained absorption peaks at 3460, 1614, and 600 cm−1, representing its OH, CN, and C–Br units, respectively. After reduction, broad peaks appeared at 3500 and 3392 cm−1, corresponding to OH and NH units, respectively. Finally, after Mannich condensation, absorption peaks were observed at 1224 and 932 cm−1, demonstrating the C–O–C and oxazine units, respectively. Fig. 1(c) and 1(d) display the corresponding 1H and 13C NMR spectra of each TPA derivative. The 1H NMR spectrum of TPA–3NH2 featured a signal at 4.69 ppm for the NH2 unit [Fig. 1(c)]. For TPA–SB–Br, this signal was absent, but other signals were present at 8.94 and 7.6–7.0 ppm, due to the phenolic OH and C–H aromatics, respectively. The 1H NMR spectrum of TPA–R–Br contained signals at 8.73, 5.72, and 4.11 ppm for its OH, NH, and NCH2 protons, respectively. The spectrum of TPA–Br BZ featured signals at 5.32 and 4.52 ppm representing OCH2N and ArCH2N units, respectively, confirming the formation of an oxazine ring. The 13C NMR spectrum [Fig. 1(d)] of TPA–SB–Br contained signals at 162.60 and 133.6–107.5 ppm assigned to its NC and aromatic carbon nuclei, respectively. After reduction and the formation of TPA–R–Br, a signal appeared at 42.23 ppm corresponding to the NCH2 unit, while the signal for the NCH unit had disappeared. The spectrum of TPA–Br BZ featured signals at 83.7 and 47.8 ppm for OCH2N and ArCH2N units, respectively. The presence of signals for an oxazine ring and the absence of a signal for an NHCH2 unit confirmed the synthesis of the TPA–Br BZ monomer. Furthermore, the high-resolution mass spectrum of each TPA derivative (Fig. S1–S3†) provided consistent theoretical and experimental molecular weights, confirming the formation purity of the TPA–Br BZ monomer.
Synthesis of the DHTP–Ea BZ monomer
We synthesized the DHTP–Ea BZ monomer by following the same steps as those for the TPA–Br BZ monomer. First, we reacted DHTP with 4-ethynylaniline (Schiff base formation) to obtain DHTP–SB–Ea. Next, NaBH4-mediated reduction provided DHTP–R–Ea, with ring closure (Mannich condensation) in the presence of CH2O yielding DHTP–Ea BZ [Fig. 2(a)]. The FTIR spectrum of DHTP–SB–Ea featured peaks at 3463, 3269, and 1597 cm−1 representing OH, C–H, and CN groups, respectively. After reduction led to the formation of DHTP–R–Ea, the peaks appeared at 3509, 3399, 3284, and 2091 cm−1, due to OH, NH, C–H, and CC groups, respectively. Mannich condensation led to the formation of the DHTP–Ea BZ monomer, with peaks observed at 3222, 2098, 1228, and 936 cm−1 for its C–H, CC, C–O, and oxazine units, respectively [Fig. 2(b)]. Fig. 2(c) and (d) display the corresponding 1H and 13C NMR spectra of each DHTP derivative. The 1H NMR spectrum of DHTP–SB–Ea showed peaks at 10.29, 8.94, and 7.60–6.50 ppm representing OH, NCH, and aromatic rings, respectively. The spectrum of DHTP–R–Ea contained signals at 8.72 (OH) and 3.98 (N–CH2) ppm. For the DHTP–Ea BZ monomer, the spectrum featured signals at 5.34 (OCH2N) and 4.55 (ArCH2N) ppm [Fig. 2(c)]. The 13C NMR spectrum of DHTP–SB–Ea featured signals at 163.80, 134.0–113.5, and 85.56/77.60 ppm for the CN bond, aromatic carbon nuclei, and CC bond (peaks d and e), respectively [Fig. 2(d)]. After reduction to give DHTP–R–Ea, a signal appeared at 42.15 ppm corresponding to the NCH2 unit. The spectrum of DHTP–Ea BZ contained signals at 84.0 and 48.3 ppm for the OCH2N and ArCH2N units, respectively, validating the synthesis of this monomer. High-resolution mass spectrometry supported the formation of DHTP derivatives and the DHTP–Ea BZ monomer (Fig. S4–S6†).
Thermal polymerization of TPA–Br BZ and DHTP–Ea BZ monomers
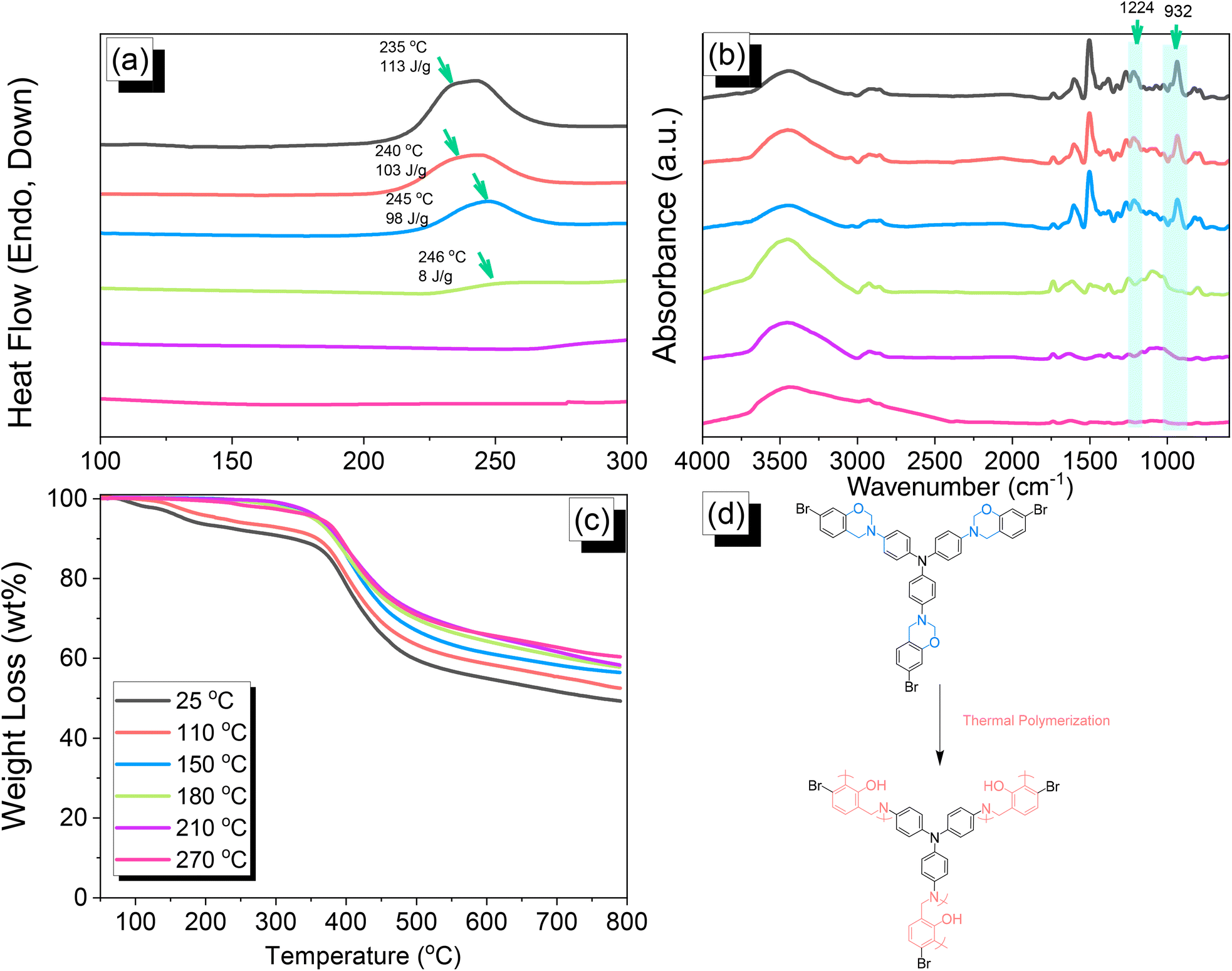
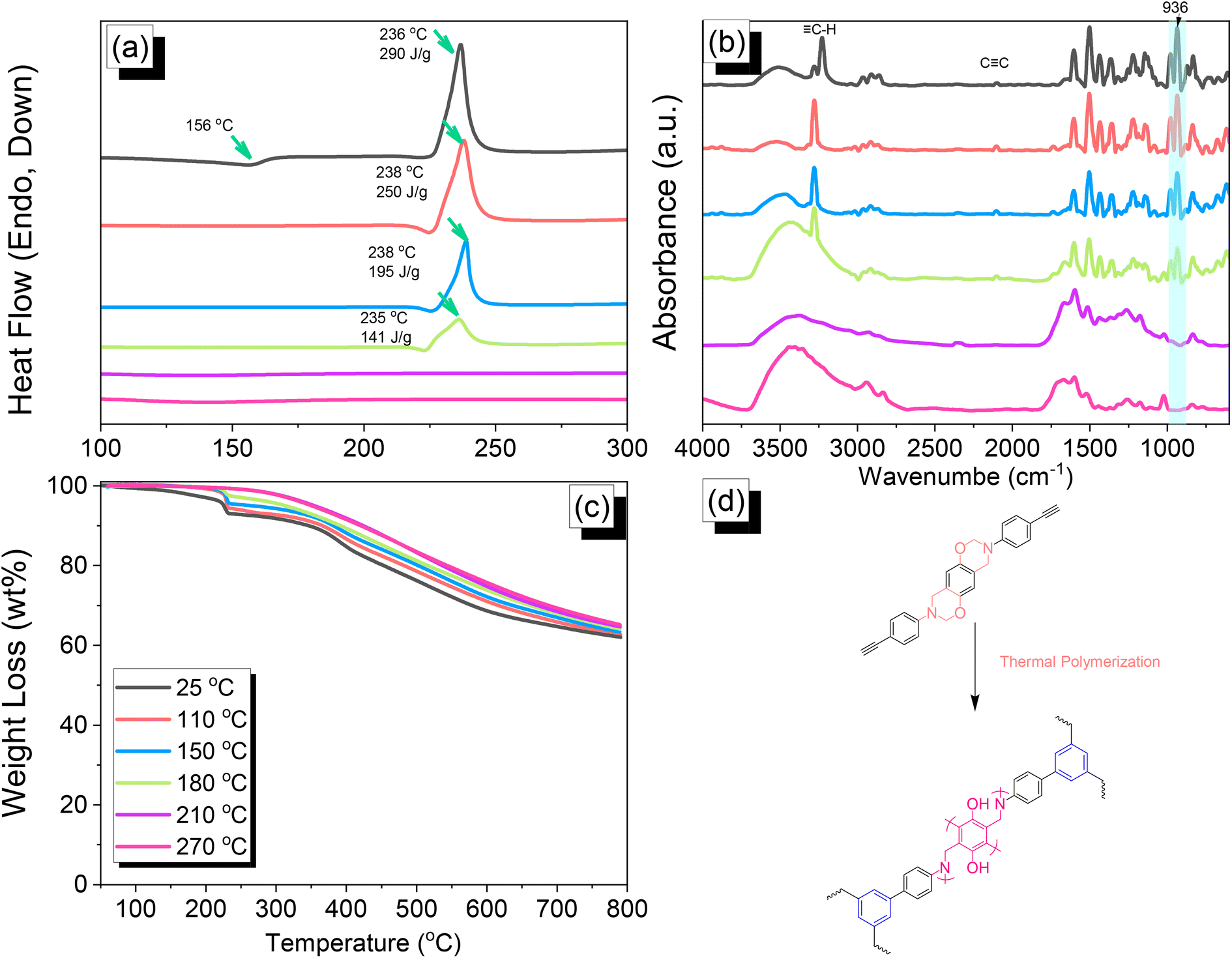
We employed differential scanning calorimetry (DSC), FTIR spectroscopy, and thermogravimetric analyses (TGA) at various temperatures to investigate the thermal polymerization behavior of the TPA–Br BZ monomer [Fig. 3(a)–(c)]. The DSC curve of the uncured TPA–Br BZ monomer featured a thermal polymerization peak at 235 °C with a heat of enthalpy of 113 J g−1. After curing the TPA–Br BZ monomer at temperatures of 110, 150, and 180 °C, curing peaks appeared at 240, 245, and 246 °C, respectively, with heats of enthalpy of 103, 98, and 8 J g−1, respectively [Fig. 3(a)]. The ROP of the oxazine unit, the absence of the thermal polymerization peak of the TPA–Br BZ monomer, and the development of poly(TPA–Br BZ) are shown in Fig. 3(d) after thermal polymerizations at 210 and 270 °C for 2 h. Moreover, we recorded FTIR spectra at various temperatures from 25 to 270 °C to observe the thermal polymerization of the TPA–Br BZ monomer [Fig. 3(b)]. We focused on the peak of the oxazine ring to evaluate the ROP response of the TPA–Br BZ monomer. The intensity of the peaks at 1224 and 932 cm−1 decreased upon increasing the thermal temperature to 180 °C; further increasing the temperature caused the oxazine peak to disappear, suggesting complete ROP. We also used TGA to examine the thermal stability of the TPA–Br BZ monomer at various temperatures from 25 to 270 °C [Fig. 3(c)]. The uncured TPA–Br BZ monomer had a degradation temperature (Td10) and char yield of 323 °C and 49.3 wt%, respectively. The thermal stability and char yield of the TPA–Br BZ monomer increased to 392 °C and 60.3 wt%, respectively, after thermal ROP at 270 °C, due to the existence of a rigid TPA structure and the high crosslinking density of poly(TPA–Br BZ), as displayed in Fig. 3(d) and in comparison, with the TGA trace of a typical Pa-type PBZ (char yield = 35 wt%).50 In addition, as revealed in DTG curves (Fig. S7†), the curing peak of the TPA–Br BZ was near 237 °C before and after thermal treatments at 110 and 150 °C, consistent with DSC and FTIR data. The weight loss before and after thermal treatments is consistent with the overhanging groups degrading and the main chain Mannich bridge breaking, releasing amines and phenolic compounds, and these events occur at degradation temperatures of about 400 °C.51,52 We also used DSC, FTIR spectroscopy, and TGA to investigate the thermal polymerization behavior and thermal stability of the DHTP–Ea BZ monomer at various temperatures from 25 to 270 °C (Fig. 4). The uncured DHTP–Ea BZ exhibited a sharp exothermic curing peak at 236 °C with a heat of enthalpy of 290 J g−1. Thus, the TPA–Br BZ and DHTP–Ea BZ monomers both displayed their thermal ROP peaks at temperatures lower than that of the typical Pa-type BZ monomer (255–263 °C).50 The electron-withdrawing Br unit presumably affected the electron delocalization over the aromatic ring and thus, the electron density of the phenolic OH unit may have been enhanced by a strong hydrogen-bonded donor to enhance the oxazine ring opening of the TPA–Br BZ monomer.39,40,47,48 Furthermore, the introduction of alkynyl units into the DHTP–Ea BZ monomer might have decreased the thermal ROP temperature due to the highly exothermic reactions of alkynyl units and the possible synergistic reaction with the oxazine ring.53 After increasing the temperature to 110, 150, and 180 °C, the resulting samples revealed curing peaks at 238, 238, and 235 °C, respectively, with heats of enthalpy of 250, 195, and 141 J g−1, respectively [Fig. 4(a)]. The ROP of the oxazine unit, the absence of the thermal polymerization peak of the DHTP–Ea BZ monomer, and the development of poly(DHTP–Ea BZ) were evident after thermal polymerizations at 210 and 270 °C for 2 h. FTIR spectral analysis of the DHTP–Ea BZ monomer at various temperatures supported the notion of ROP of the oxazine ring and the synergistic reaction of the alkynyl units with the oxazine ring [Fig. 4(b)]. The intensity of the signal at 936 cm−1 for the oxazine ring decreased upon increasing the thermal temperature to 180 °C; further increasing the temperature beyond 210 °C caused the oxazine peak at 936 cm−1 to disappear completely, confirming the ROP of the oxazine ring. The 1H-NMR results revealed the presence of proton's signal for aromatic rings, oxazine ring, and the CHC unit for the DHTP–Ea BZ monomer after thermal treatment at 110, 150, and 180 °C, respectively, as presented in Fig. S8.† Unfortunately, the 1H-NMR data of DHTP–Ea Bz after thermal curing at 210 and 270 °C could not be obtained due to the poor solubility of the obtained products at these two temperatures. Interestingly, the CHC and CC bond vibrations at around 3222 and 2100 cm−1 completely disappeared after thermal curing at 210 and 270 °C, respectively [Fig. 4(b)], confirming the acetylene polymerization and cyclic trimerization reaction of the alkynyl units in the DHTP–Ea Bz structure.53,54 Furthermore, the thermal stability of DHTP–Ea BZ also increased upon increasing the thermal polymerization temperature [Fig. 4(c)]. The uncured DHTP–Ea BZ provided a thermal degradation temperature (Td10) of 365 °C and a char yield of 62.7 wt%. After thermal polymerization at a temperature of 270 °C, the value of Td10 and the char yield both increased (to 420 °C and 65.1 wt%, respectively), indicating that a highly crosslinked structure of poly(DHTP–Ea BZ) had formed from both the BZ and alkynyl units [Fig. 4(d)] and that this structure was more highly crosslinked than that of poly(TPA–Br BZ) in Fig. 3(d).
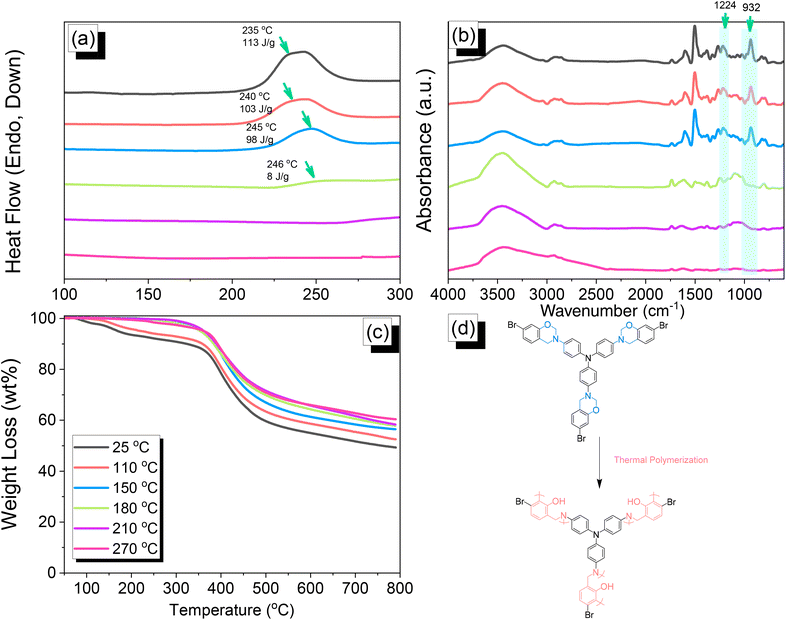 |
| | Fig. 3 (a) DSC, (b) FTIR, and (c) TGA analyses of TPA–Br BZ after thermal ROP at different temperatures. (d) Possible thermal ROP of TPA–Br BZ to form poly(TPA–Br BZ). | |
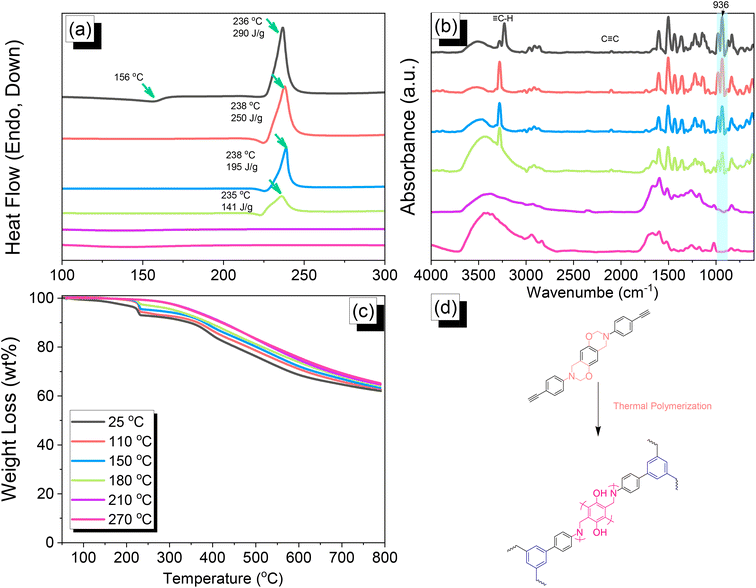 |
| | Fig. 4 (a) DSC, (b) FTIR spectral, and (c) TGA analyses of DHTP–Ea BZ after thermal ROP at various temperatures. (d) Possible thermal ROP of DHTP–Ea BZ to form poly(DHTP–Ea BZ). | |
Synthesis and thermal polymerization of TPA–DHTP–BZ POP
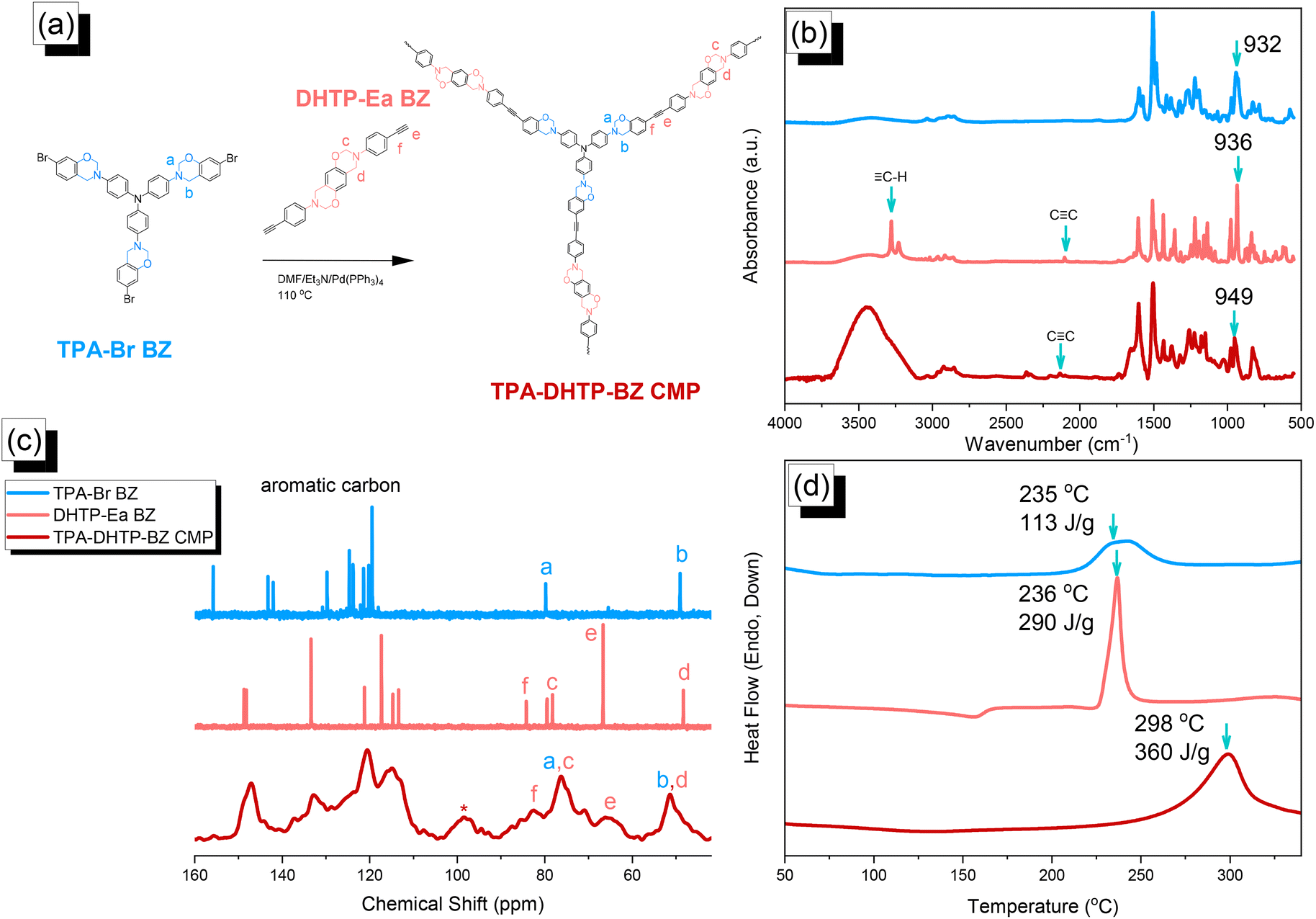
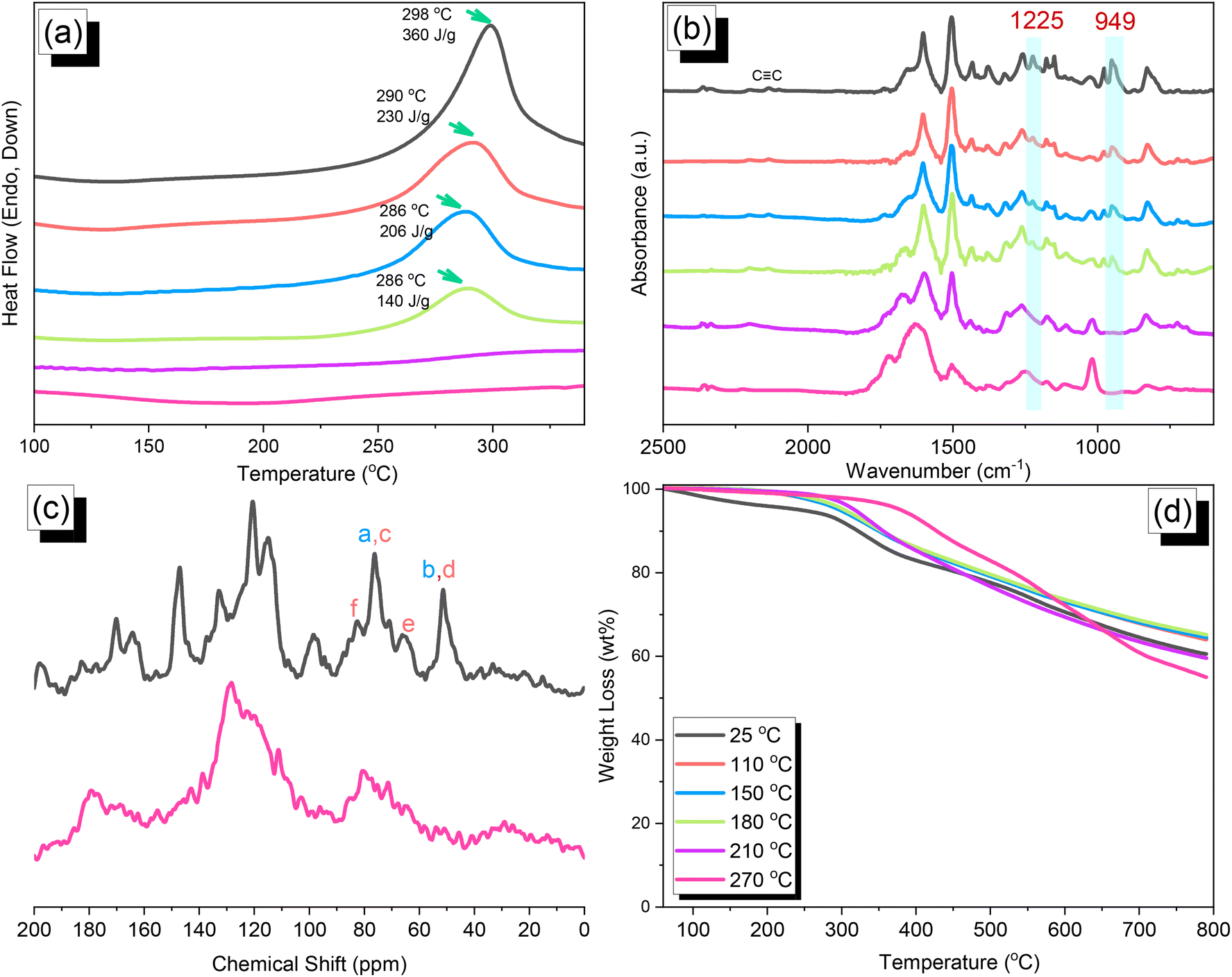
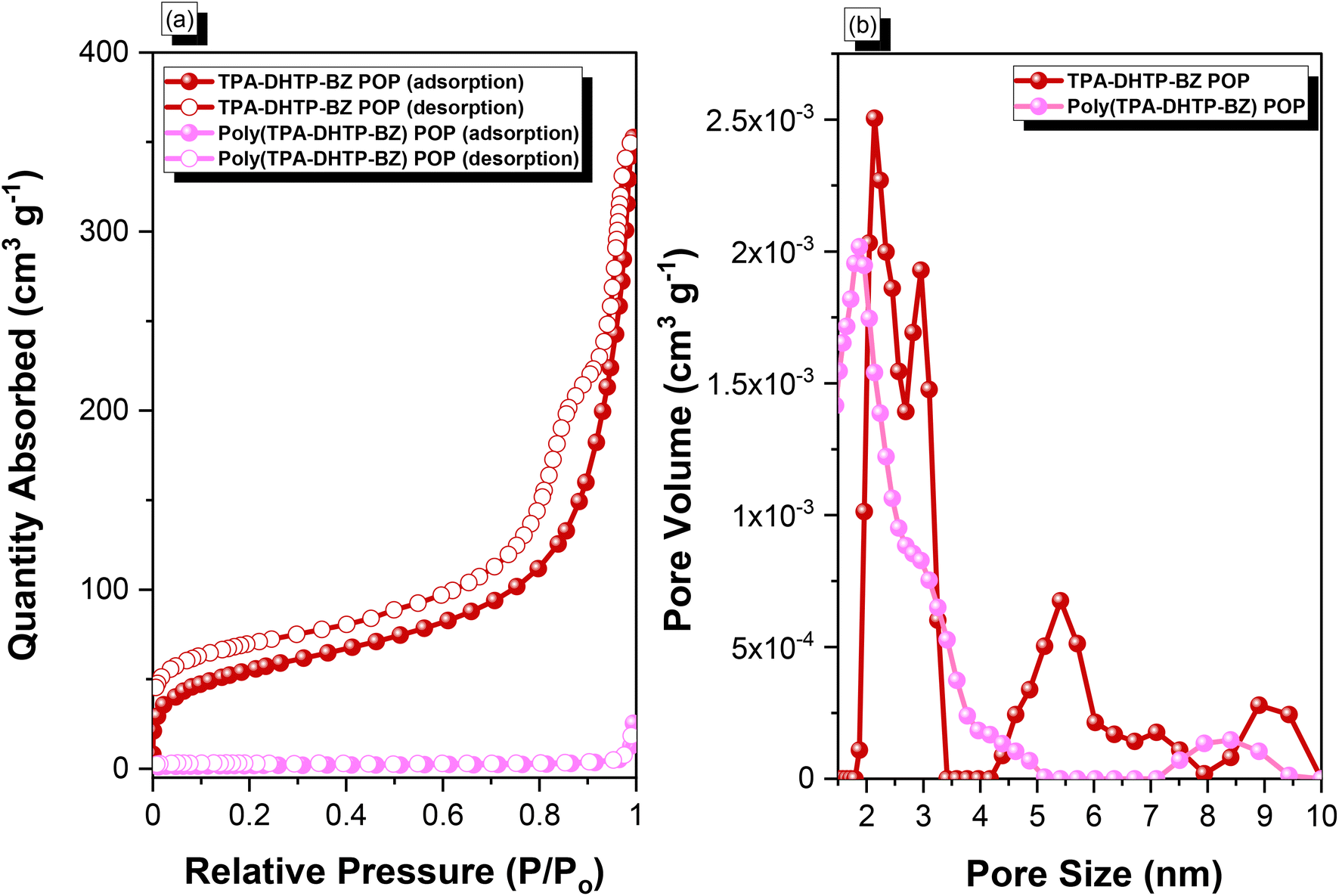
We synthesized TPA–DHTP–BZ POP through Sonogashira–Hagihara coupling of TPA–Br BZ with DHTP–Ea BZ, using Pd(PPh3)4 as the catalyst and Et3N/DMF as the solvent, at 110 °C for 3 days [Fig. 5(a)]. We used FTIR spectroscopy, solid-state NMR spectroscopy, and DSC thermal analyses to confirm the presence of a fully-BZ-linked TPA–DHTP–BZ POP [Fig. 5(b)–(d)]. For example, Fig. 5(b) shows the FTIR spectra of the TPA–Br BZ monomer and the DHTP–Ea BZ monomer, and their corresponding TPA–DHTP–BZ POP formed through Sonogashira–Hagihara coupling. The signal at 3222 cm−1 corresponding to the C–H unit had disappeared, but those of the CC and oxazine units near 2100 and 949 cm−1, respectively, remained after Sonogashira–Hagihara coupling to form the TPA–DHTP–BZ POP. In addition, Fig. 5(c) displays the solution 13C NMR spectra of the TPA–Br BZ and DHTP–Ea BZ monomers [as discussed in Fig. 1(d) and 2(d)] and the solid-state 13C NMR spectrum of their corresponding TPA–DHTP–BZ POP; the crosslinked structure of this POP meant that it could not be dissolved in any kind of organic solvent. In the spectrum of the TPA–DHTP–BZ POP [Fig. 5(c)], the signals for the aromatic carbon nuclei appeared in the range 115–139 ppm, the signals for the OCH2N (a, c) and ArCH2N (b, d) units appeared near 76.3 and 51.4 ppm, respectively, and the signals for the CC unit (peaks e and f) appeared at 66.1 and 82.6 ppm. We used DSC to examine the thermal polymerization behavior of the TPA–DHTP–BZ POP [Fig. 5(d)]. A sharp thermal polymerization peak appeared at 298 °C with a heat of reaction of 360 J g−1. The thermal polymerization temperature of the TPA–DHTP–BZ POP was higher than that of the TPA–Br BZ and DHTP–Pa BZ monomers because the crosslinking structure formed after Sonogashira–Hagihara coupling inhibited the ROP of the oxazine ring. Furthermore, the heat of enthalpy of the TPA–DHTP–BZ POP was larger than those of both the TPA–Br BZ and DHTP–Ea BZ monomers, indicating that this new fully-BZ-linked POP have a higher content of BZ rings relative to those in our previously reported TPE–TPE BZ, Py–TPE–BZ, and CE–BZ–TPE POPs.47,48 As a result, these two BZ-linked monomers formed a POP that could provide a higher crosslinking density during the thermal ROP. We also used DSC, FTIR spectroscopy, solid-state NMR spectroscopy, and TGA to investigate the thermal polymerization behavior and thermal stability of the TPA–DHTP–BZ POP at various temperatures from 25 to 270 °C (Fig. 6). The DSC traces of the TPA–DHTP–BZ POP after thermal ROP at 110, 150, and 180 °C [Fig. 6(a)] featured thermal polymerization peaks at 290, 286, and 286 °C, respectively, with heats of enthalpy of 230, 206, and 140 J g−1, respectively. No thermal polymerization peaks appeared for the TPA–DHTP–BZ POP after thermal polymerization at 210 or 270 °C, indicating that complete ROP of the oxazine rings had occurred at these temperatures. FTIR spectral analyses confirmed this behavior [Fig. 6(b)], with the intensity of the peak at 949 cm−1 of the oxazine ring decreasing upon increasing the thermal ROP temperature to 180 °C; further increasing the temperature beyond 210 °C caused the signals for the oxazine ring at 949 cm−1 and the alkynyl unit at 2098 cm−1 to disappear completely, confirming the ROP of the oxazine ring and the reaction of the alkynyl units. Furthermore, solid-state 13C NMR spectra revealed that the intensities of the signals for the oxazine ring and CC unit in the range from 51.4 to 82.6 ppm [Fig. 6(c)] had decreased significantly after thermal polymerization at 270 °C, consistent with the ROP of the oxazine ring and the acetylene polymerization and cyclic trimerization reaction of the alkynyl units in the TPA–DHTP–BZ POP structure.53,54 The thermal stability of the TPA–DHTP–BZ POP was evidenced through TGA analyses performed at various thermal polymerization temperatures [Fig. 6(d)]. After thermal treatment at various temperatures, the TPA–DHTP–BZ POP exhibited excellent thermal decomposition temperatures (from 321 to 427 °C), because of the large number of intra- and intermolecular [OH⋯O] hydrogen bonds formed after thermal ROP; its char yield decreased, however, from 60.5 wt% at 25 °C to 55.0 wt% at 270 °C, because the aliphatic methylene (CH2) units possessed weaker thermal stability after the thermal ROP and the crosslinking density did not increase.55,56 Thus, the char yield decreased after the ROP of TPA–DHTP–BZ POP in this study. We used N2 adsorption/desorption isotherms at 77 K to examine the porosities of the TPA–DHTP–BZ POP and poly(TPA–DHTP–BZ) POP after their ROPs [Fig. 7(a) and (b), respectively]. The pattern of the TPA–DHTP–BZ POP displayed sharp N2 uptake at lower relative pressure due to the presence of a microporous structure and higher relative pressure, corresponding to the presence of a mesoporous structure in the POP framework; this pattern may be classified as a type I isotherm, based on the IUPAC definition. The higher pressure for the sharp N2 uptake was also due to the presence of a mesoporous structure or interparticulate voids in this POP [Fig. 7(a)]. The TPA–DHTP–BZ POP has a BET-specific surface area of 195 m2 g−1 and a total pore volume of 0.53 cm3 g−1. This fully-BZ-linked POP is the first example of such a POP displaying a high surface area. Furthermore, its average pore size was approximately 2.14 nm, based on nonlocal density functional theory (NLDFT) [Fig. 7(b)]. In addition, Fig. 7(a) presents the N2 adsorption/desorption isotherms at 77 K for the poly(TPA–DHTP–BZ) POP. The BET surface area (40 m2 g−1) was much lower than that of the TPA–DHTP–BZ POP, with the total pore volume also having decreased to 0.12 cm3 g−1; the average pore size was 1.87 nm, with a much greater content of micropores [Fig. 7(b)]. This result is consistent with our findings from previous studies,47,48 where the solid-state ROP of the BZ rings resulted in a new functionality (e.g., inter- or intramolecular hydrogen-bonded phenolic OH and Mannich bridges) that shrunk the framework and, thus, provided lower surface areas and pore volumes. Fig. 8 and S9–S11† display the transmission electron microscopy (TEM) and scanning electron microscopy (SEM) images, their corresponding C-, N-, and O-mappings, and energy dispersive X-ray (EDX) spectra of the TPA–DHTP–BZ POP and poly(TPA–DHTP–BZ) POP, respectively, after their ROPs. All images of both POPs indicated the presence of aggregated and irregular spherical clusters of nanoparticles with microporous structures. In addition, the SEM elemental mapping images indicated the presence of C, N, and O atoms on the surfaces of the TPA–DHTP–BZ POP and poly(TPA–DHTP–BZ) POP. We used TEM-EDX analyses to calculate the weight percent of each element, including terminated Br atoms [Fig. S11(a) and (b)†].
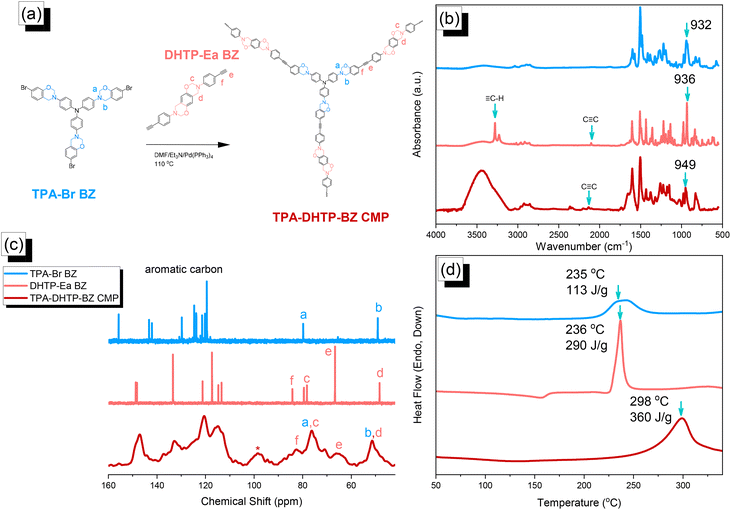 |
| | Fig. 5 (a) Synthesis of the TPA–DHTP–BZ POP from the TPA–Br BZ and DHTP–Ea BZ monomers through Sonogashira–Hagihara coupling. (b–d) Corresponding (b) FTIR spectral, (c) solution and solid-state NMR spectral, and (d) DSC analyses. | |
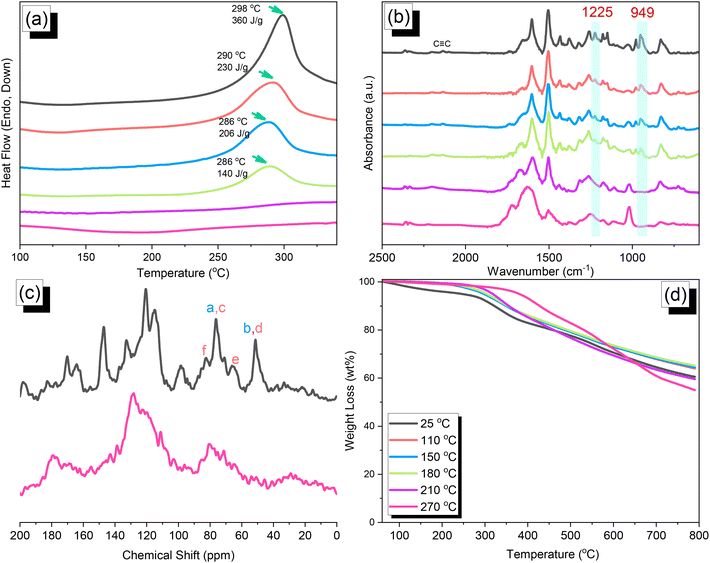 |
| | Fig. 6 (a) DSC, (b) FTIR spectral, (c) solid-state NMR spectral, and (d) TGA analyses of the TPA–DHTP–BZ POP after thermal ROP at various temperatures. | |
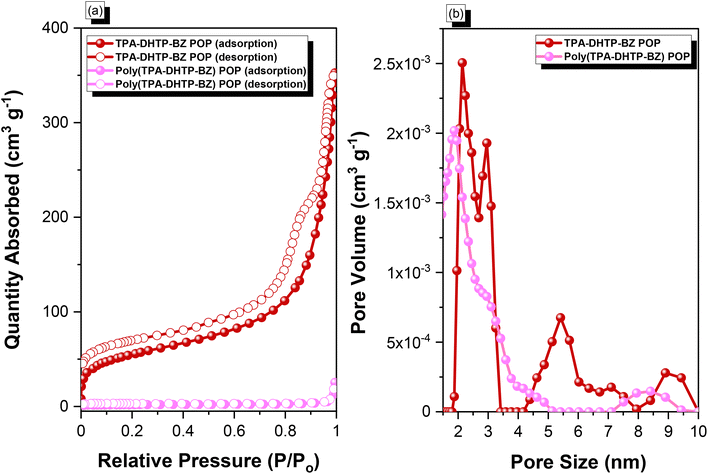 |
| | Fig. 7 (a) N2 adsorption/desorption isotherms at 77 K and (b) pore size distributions of the TPA–DHTP–BZ POP and poly(TPA–DHTP–BZ) POP. | |
 |
| | Fig. 8 SEM and TEM images of the TPA–DHTP–BZ POP (a and c) and poly(TPA–DHTP–BZ) POP (b and d). | |
CO2 capture of the TPA–DHTP–BZ POP
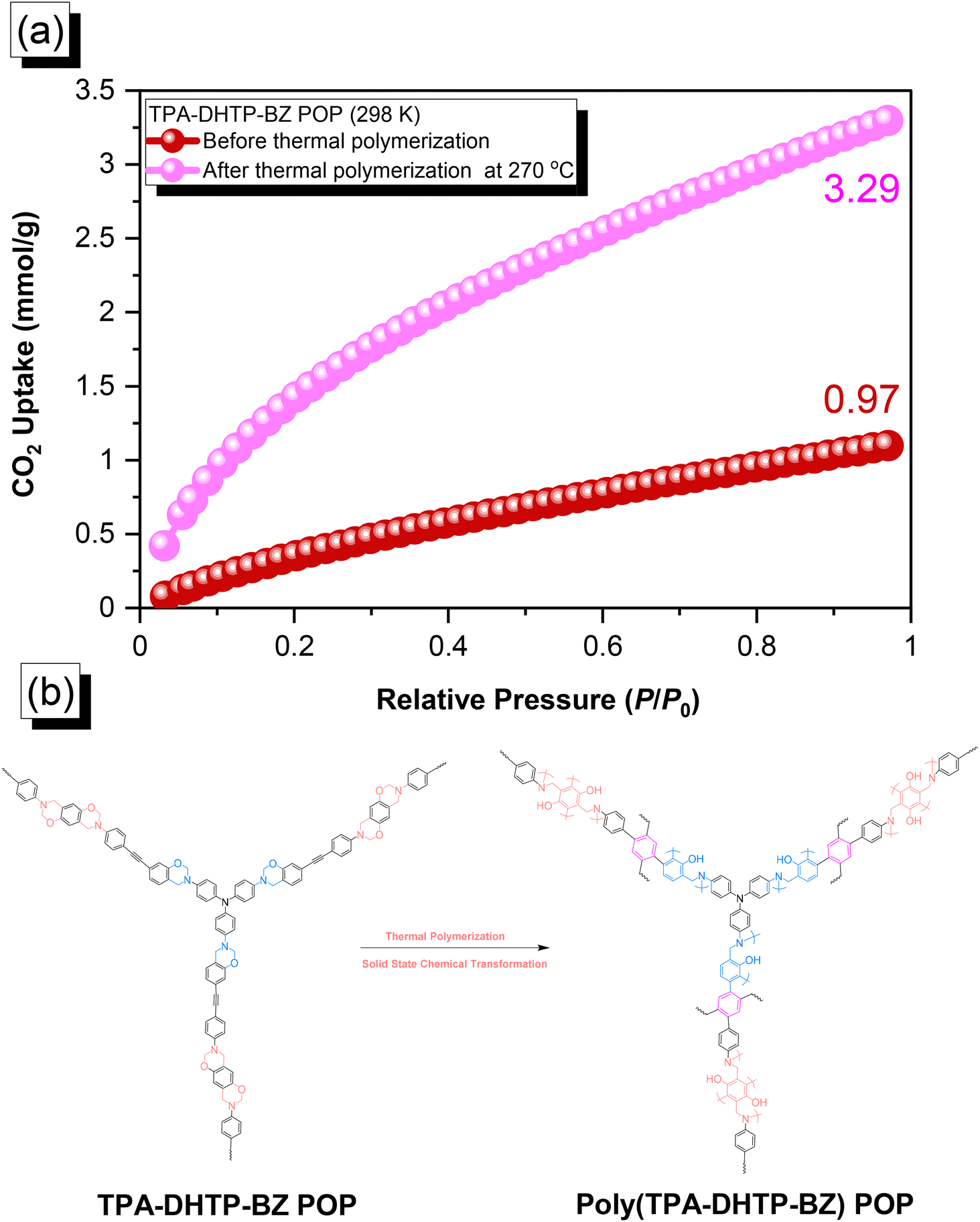
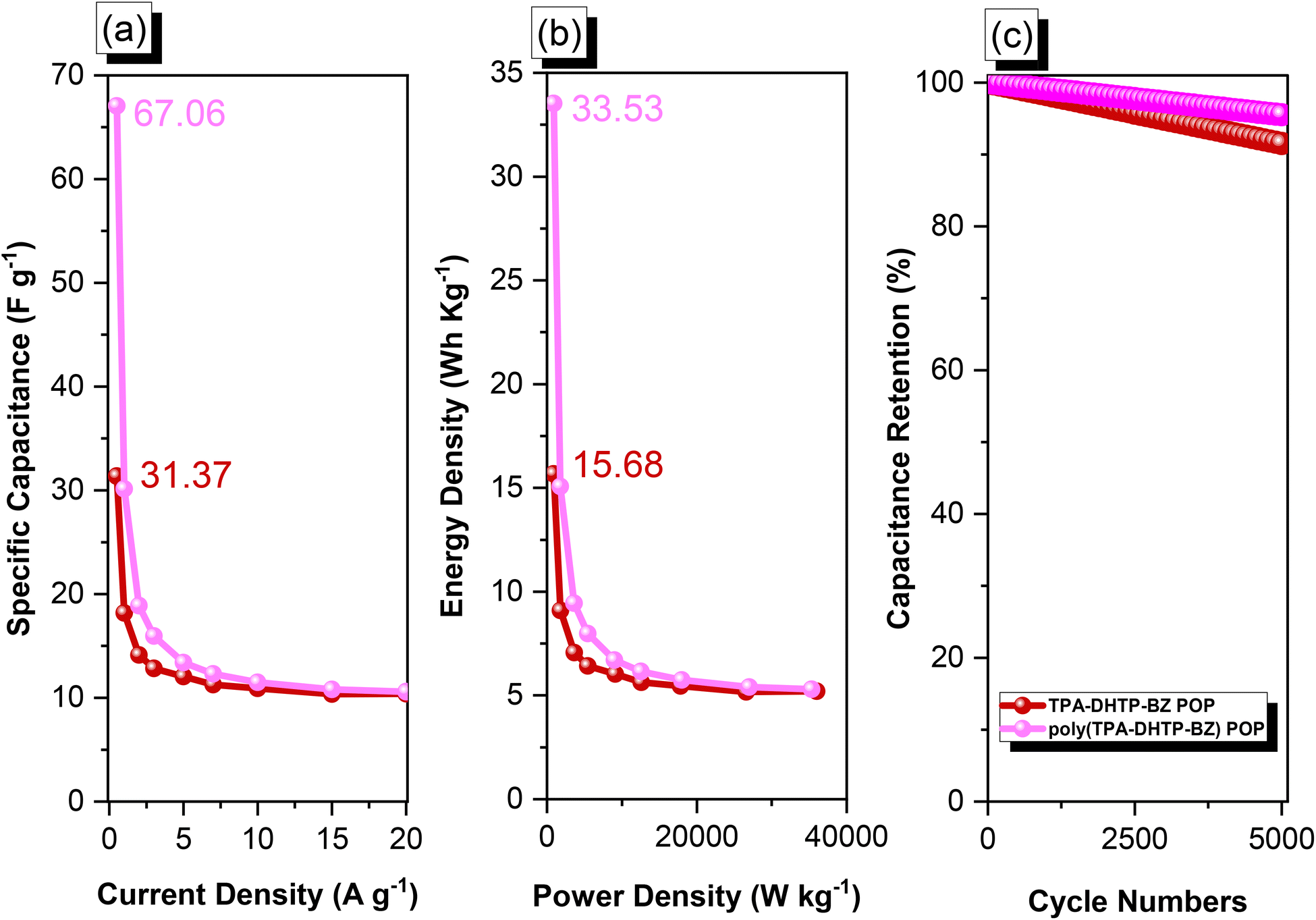
The amounts of CO2 capture of the TPA–DHTP–BZ POP and poly(TPA–DHTP–BZ) POP after their ROPs [Fig. 9(a)] were 0.97 and 3.29 mmol g−1, respectively, at 1 bar and 298 K. In previous studies,43,45–48 we found that the CO2 capture performance in porous materials was dependent on a balance of several factors, including the surface area, pore size, volume, and—most importantly—the nature of the chemical functionalization on the surface. Here, the surface area and pore volume both decreased for the poly(TPA–DHTP–BZ) POP after its ROP, potentially unfavorable for CO2 capture; the solid-state chemical transformation, however, formed highly abundant phenolic OH and N atoms on the common surface, thereby enhancing the CO2 capture performance [Fig. 9(b)]. These transformed units were capable of hydrogen bonding or acid–base interactions through [OH⋯OC] or [N⋯CO] interactions to enhance the CO2 capture ability. The CO2 capture performance [Table S1†] of this fully-BZ-linked poly(TPA–DHTP–BZ) POP (3.29 mmol g−1) was superior to those of other BZ-linked POPs, including poly(Py–TPE–BZ) (2.20 mmol g−1),47 poly(TPE–TPE–BZ) (2.21 mmol g−1),47 BPOP-1 (0.98 mmol g−1),46 BPOP-2 (0.67 mmol g−1),46 BoxPOP-1 (0.91 mmol g−1),45 BoxPOP-2 (1.04 mmol g−1),45 and BoxPOP-3 (0.29 mmol g−1),45 all measured at 298 K and 1 bar, because the greater content of phenolic OH and N atoms improved its CO2 capture ability.
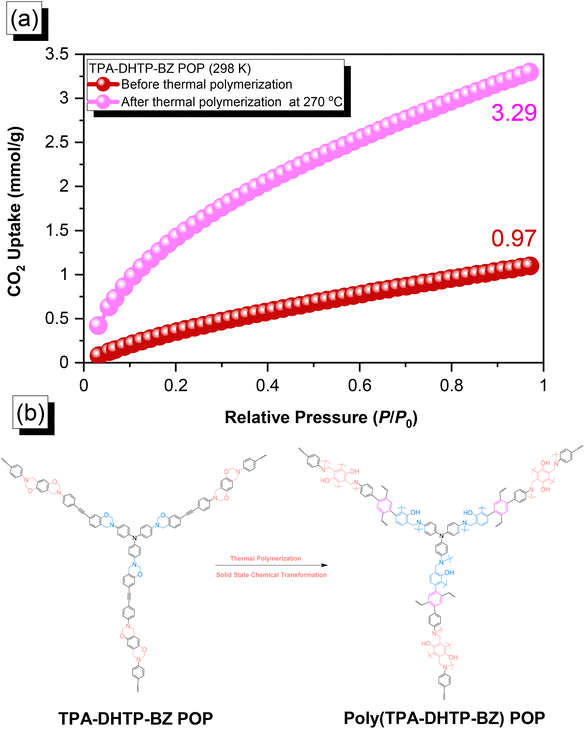 |
| | Fig. 9 (a) CO2 uptake of the TPA–DHTP–BZ POP and poly(TPA–DHTP–BZ) POP. (b) Corresponding chemical structures before and after the thermal ROP. | |
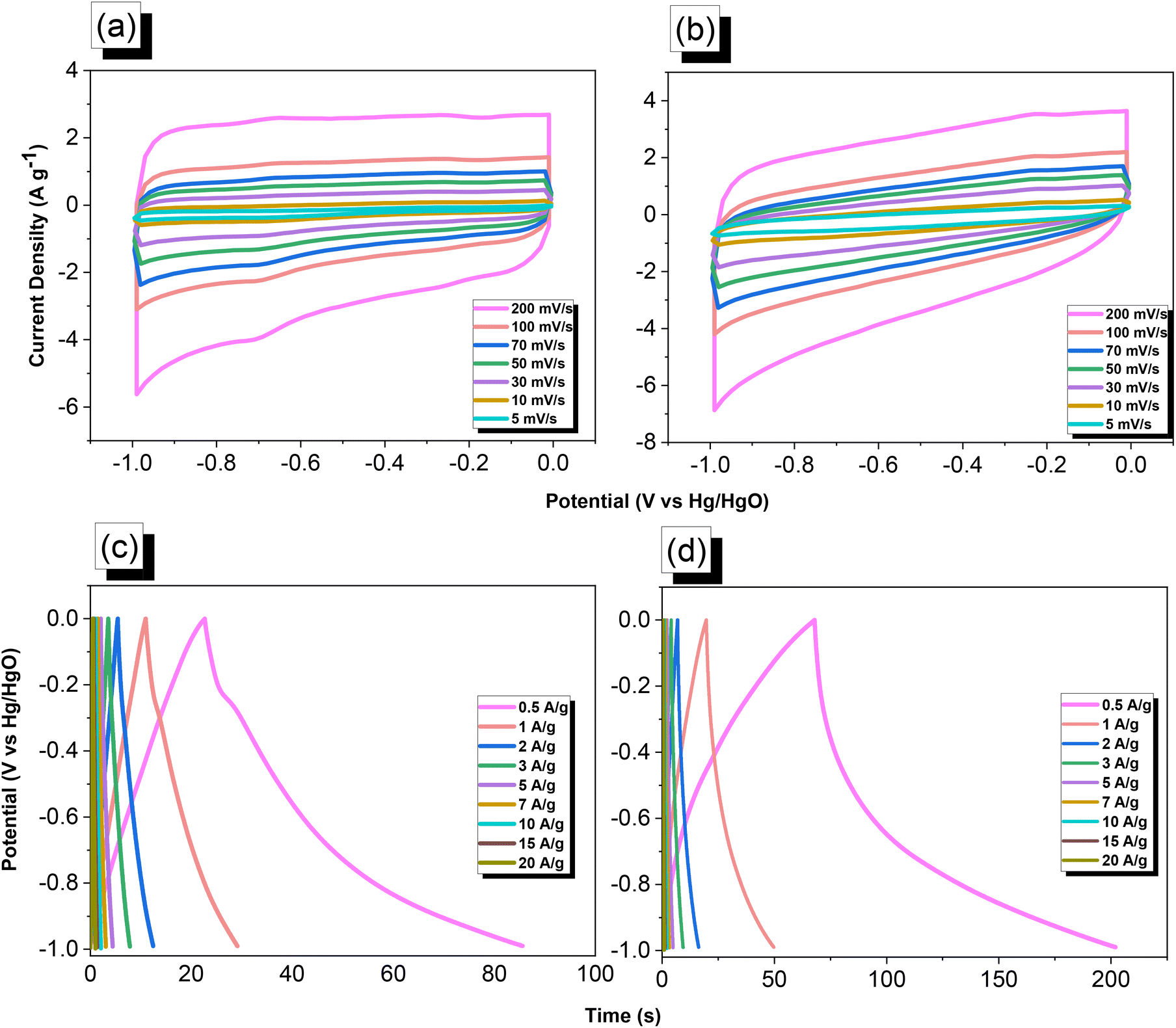
Electrochemical performance of the TPA–DHTP–BZ POP
We used cyclic voltammetry (CV) and the galvanostatic charge–discharge (GCD) method in 1 M aqueous KOH with a three-electrode system to evaluate the electrochemical performance of TPA–DHTP–BZ POP before and after its ROP. We measured the CV traces at various scan rates over the potential range from −1 to 0 V [Fig. 10(a) and (b)]. The CV curves of the TPA–DHTP–BZ POP before and after ROP were almost rectangular, with small humps caused by the electric double-layer capacitance (EDLC) and pseudocapacitance responses.55–58 The pseudocapacitance behavior was due to the presence of O and N heteroatoms in the TPA–DHTP–BZ POP and OH and N heteroatoms in the cured poly(TPA–DHTP–BZ) POP interacting with free electrons [Fig. 9(b)]. These heteroatoms permitted effective electron transmission by developing good interactions with the electrolyte.59–63 The current densities increased upon increasing the scan rate without altering the morphologies of the CV curves, validating the high-rate capabilities, stabilities, and kinetic properties of the TPA–DHTP–BZ POP before and after ROP. Furthermore, we examined the charge/discharge trends and capacitances of these compounds at various current densities [Fig. 10(c) and (d)]. The GCD curves of the poly(TPA–DHTP–BZ) POP had a bent-triangle appearance, indicative of pseudocapacitive and EDLC properties; these bent curves were due to the abundance of heteroatoms.59 In all of the GCD curves, the discharge time was higher than the charging time, suggesting enhanced capacitance.55–58 The discharging profile of the poly(TPA–DHTP–BZ) POP was more significant than that of the TPA–DHTP–BZ POP, demonstrating the high capacitance of the former. The specific capacities of the TPA–DHTP–BZ POP and poly(TPA–DHTP–BZ) POP were 31.4 and 67.1 F g−1, respectively, measured at a current density of 0.5 A g−1 [Fig. 11(a)]. In addition, both the TPA–DHTP–BZ POP and the poly(TPA–DHTP–BZ) POP demonstrated remarkable electrochemical performance (Table S2†) when evaluated with previously reported porous polymers. We attribute the improved capacitance of the poly(TPA–DHTP–BZ) POP to the free OH groups and N heteroatoms in its chemical structure permitting the electrolyte to move quickly. Moreover, the energy density of the poly(TPA–DHTP–BZ) POP (33.5 W h kg−1) was also higher than that of the TPA–DHTP–BZ POP (15.7 W h kg−1) [Fig. 11(b)]. Fig. 11(c) presents the long-term cycling stability measured at a relatively high current density of 5 A g−1 over 2000 cycles. These two POPs as electrode materials both displayed excellent capacity retentions, with the poly(TPA–DHTP–BZ) POP (95.5%) still exhibiting better performance than that of the TPA–DHTP–BZ POP (91.5%). To the best of our knowledge, this study is the first to test a BZ-linked POP for its suitability for supercapacitor applications. Because of its good specific capacitance, reasonable surface area, tunable morphology, and facile preparation, this fully-BZ-linked POP appears to be an outstanding candidate material for energy storage applications.
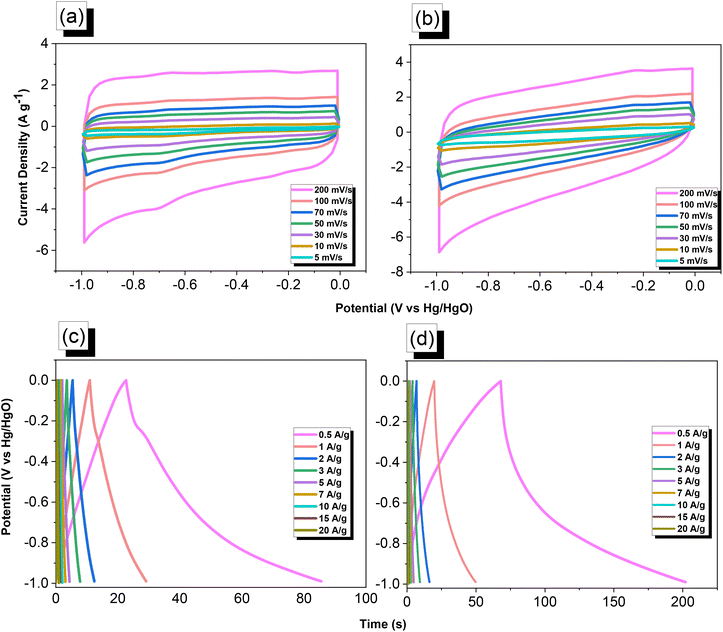 |
| | Fig. 10 (a and b) CV and (c and d) GCD curves of the (a and c) TPA–DHTP–BZ POP and (b and d) poly(TPA–DHTP–BZ) POP. | |
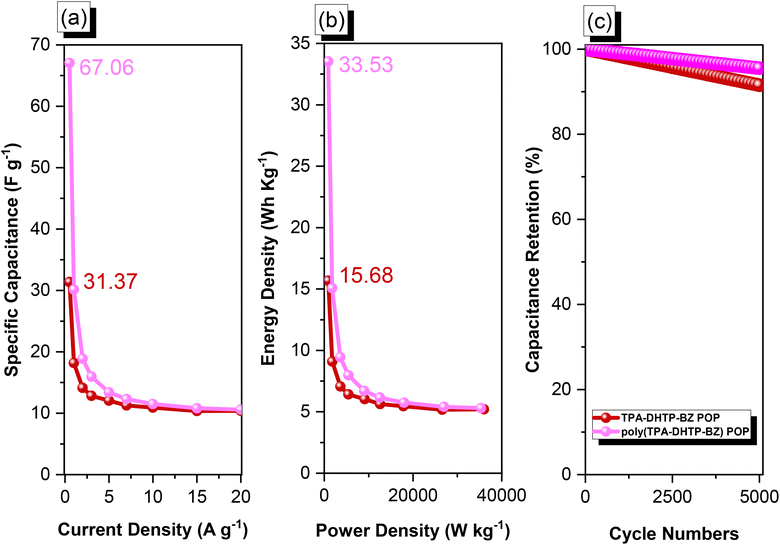 |
| | Fig. 11 (a) Capacitance performance, (b) Ragone plots, and (c) capacitance retention of the TPA–DHTP–BZ POP and poly(TPA–DHTP–BZ) POP. | |
Conclusions
We have synthesized a fully-BZ-linked POP through a multistep process of Schiff base formations, reductions, Mannich reactions, and Sonogashira–Hagihara coupling and validated each of the intermediates and products through FTIR spectral, solid-state NMR spectral, DSC, and TGA analyses. The resulting TPA–DHTP–BZ POP displayed good thermal stability (Td10 = 321 °C; char yield = 60.5 wt%) and porosity (BET surface area = 195 m2 g−1; total pore volume = 0.53 cm3 g−1). The TPA–DHTP–BZ POP underwent solid-state transformations through ROP without the interference of any curing agent; although its surface area and char yield both decreased, the resulting Mannich bridges and phenolic groups experienced strong intermolecular hydrogen bonding that enhanced the electrochemical and CO2 capture properties. This study provides a new approach for synthesizing fully-BZ-linked POPs through solid-state transformations, with potentially enhanced applications in gas capture and supercapacitors.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This study was supported financially by the National Science and Technology Council, Taiwan, under contracts NSTC 110-2124-M-002-013 and 111-2223-E-110-004. The authors thank the staff at National Sun Yat-sen University for their assistance with the TEM (ID: EM022600) experiments.
References
- Y. Zhu, P. Xu, X. Zhang and D. Wu, Emerging Porous Organic Polymers for Biomedical Applications, Chem. Soc. Rev., 2022, 51, 1377–1414, 10.1039/D1CS00871D.
- S. Das, P. Heasman, T. Ben and S. Qiu, Porous organic materials: strategic design and structure–function correlation, Chem. Rev., 2017, 117, 1515–1563, DOI:10.1021/acs.chemrev.6b00439.
- M. G. Mohamed, A. F. M. El-Mahdy, M. G. Kotp and S. W. Kuo, Advances in Porous Organic Polymers: Syntheses, Structures, and Diverse Applications, Mater. Adv., 2022, 3, 707–733, 10.1039/D1MA00771H.
- T. Zhang, G. Xing, W. Chen and L. Chen, Porous organic polymers: a promising platform for efficient photocatalysis, Mater. Chem. Front., 2020, 4, 332–353, 10.1039/C9QM00633H.
- X. Guan, F. Chen, Q. Fang and S. Qiu, Design and Applications of Three Dimensional Covalent Organic Frameworks, Chem. Soc. Rev., 2020, 49, 1357–1384, 10.1039/C9CS00911F.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. I. Matzger and O. M. Yaghi, Porous, Crystalline, Covalent Organic Frameworks, Science, 2005, 310, 1166–1170, DOI:10.1126/science.1120411.
- D. Yuan, W. Lu, D. Zhao and H. C. Zhou, Highly Stable Porous Polymer Networks with Exceptionally High Gas-Uptake Capacities, Adv. Mater., 2011, 23, 3723–3725, DOI:10.1002/adma.201101759.
- M. Ejaz, M. M. Samy, Y. Ye, S. W. Kuo and M. G. Mohamed, Design Hybrid Porous Organic/Inorganic Polymers Containing Polyhedral Oligomeric Silsesquioxane/Pyrene/Anthracene Moieties as a High-Performance Electrode for Supercapacitor, Int. J. Mol. Sci., 2023, 24, 2501, DOI:10.3390/ijms24032501.
- C. Liu, Y. Jin, Z. Yu, L. Gong, H. Wang, B. Yu, W. Zhang and J. Jiang, Transformation of Porous Organic Cages and Covalent Organic Frameworks with Efficient Iodine Vapor Capture Performance, J. Am. Chem. Soc., 2022, 144, 12390–12399, DOI:10.1021/jacs.2c03959.
- A. Abid, S. Razzaque, I. Hussain and B. Tan, Eco-Friendly Phosphorus and Nitrogen-Rich Inorganic–Organic Hybrid Hypercross-linked Porous Polymers via a Low-Cost Strategy, Macromolecules, 2021, 55, 5848–5855, DOI:10.1021/acs.macromol.1c00385.
- X. Shi, Z. Zhang, M. Wei, X. Wang, J. Wang, Y. Zhang and Y. Wang, Three-Dimensional Covalent Organic Framework Membranes: Synthesis by Oligomer Interfacial Ripening and Application in Precise Separations, Macromolecules, 2022, 55, 3259–3266, DOI:10.1021/acs.macromol.1c02333.
- C. Ru, T. Zhou, J. Zhang, X. Wu, P. Sun, P. Chen, L. Zhou, H. Zhao, J. Wu and X. Pan, Introducing Secondary Acceptors into Conjugated Polymers to Improve Photocatalytic Hydrogen Evolution, Macromolecules, 2021, 54, 8839–8848, DOI:10.1021/acs.macromol.1c00705.
- P. Zhang, Y. Yin, Z. Wang, C. Yu, Y. Zhu, D. Yan, W. Liu and Y. Mai, Porphyrin-Based Conjugated Microporous Polymer Tubes: Template-Free Synthesis and A Photocatalyst for Visible-Light-Driven Thiocyanation of Anilines, Macromolecules, 2021, 54, 3543–3553, DOI:10.1021/acs.macromol.1c00190.
- H. Gao, Q. Li and S. Ren, Progress on CO2 capture by porous organic polymers, Curr. Opin. Green Sustainable Chem., 2019, 16, 33–38, DOI:10.1016/j.cogsc.2018.11.015.
- X. Liu, C. F. Liu, S. Xu, T. Cheng, S. Wang, W. Y. Lai and W. Huang, Porous organic polymers for high-performance supercapacitors, Chem. Soc. Rev., 2022, 51, 3181–3225, 10.1039/D2CS00065B.
- K. Y. Geng, V. Arumugam, H. J. Xu, Y. N. Gao and D. L. Jiang, Covalent Organic Frameworks: Polymer Chemistry and Functional Design, Prog. Polym. Sci., 2020, 108, 101288, DOI:10.1016/j.progpolymsci.2020.101288.
- X. Yan, H. Liu, Y. Li, W. Chen, T. Zhang, Z. Zhao, G. Xing and L. Chen, Ultrastable Covalent Organic Frameworks via Self-Polycondensation of an A2B2 Monomer for Heterogeneous Photocatalysis, Macromolecules, 2019, 52, 7977–7983, DOI:10.1021/acs.macromol.9b01600.
- D. Li, F. Li, H. Yu, L. Guo, J. Huang, J. Li and C. Li, Nickel-modified triphenylamine-based conjugated porous polymers as precatalyst for ethylene oligomerization, Inorg. Chim. Acta, 2022, 544, 121228, DOI:10.1016/j.ica.2022.121228.
- M. G. Mohamed, M. H. Elsayed, A. M. Elewa, A. F. M. El-Mahdy, C. Yang, A. A. K. Mohammed, H. H. Chou and S. W. Kuo, Pyrene-Containing Conjugated Organic Microporous Polymers for Photocatalytic Hydrogen Evolution from Water, Catal. Sci. Technol., 2021, 11, 2229–2241, 10.1039/D0CY02482A.
- M. M. Samy, I. M. A. Mekhemer, M. G. Mohamed, M. H. Elsayed, K. H. Lin, Y. K. Chen, T. L. Wu, H. H. Chou and S. W. Kuo, Conjugated Microporous Polymers Incorporating Thiazolo[5,4-d]thiazole Moieties for Sunlight-Driven Hydrogen Production From Water, Chem. Eng. J., 2022, 446, 137158, DOI:10.1016/j.cej.2022.137158.
- J. H. Kim, D. W. Kang, H. Yun, M. Kang, N. Singh, J. S. Kim and C. S. Hong, Post-Synthetic Modifications in Porous Organic Polymers for Biomedical and Related Applications, Chem. Soc. Rev., 2022, 51, 43–56, 10.1039/D1CS00804H.
- Y. Zhang, X. Hong, X. M. Cao, X. Q. Huang, B. Hu, S. Y. Ding and H. Lin, Functional Porous Organic Polymers with Conjugated Triaryl Triazine as the Core for Superfast Adsorption Removal of Organic Dyes, ACS Appl. Mater. Interfaces, 2021, 13, 6359–6366, DOI:10.1021/acsami.0c21374.
- R. Ullah, M. Atilhan, B. Anaya, S. Al-Muhtaseb, S. Aparicio, H. Patel, D. Thirion and C. T. Yavuz, Investigation of Ester- and Amide-Linker-Based Porous Organic Polymers for Carbon Dioxide Capture and Separation at Wide Temperatures and Pressures, ACS Appl. Mater. Interfaces, 2016, 8, 20772–20785, DOI:10.1021/acsami.6b05927.
- V. M. Suresh, S. Bonakala, S. Balasubramanian and T. K. Maji, Amide Functionalized Microporous Organic Polymer (Am-MOP) for Selective CO2 Sorption and Catalysis, ACS Appl. Mater. Interfaces, 2014, 6, 4630–4637, DOI:10.1021/am500057z.
- P. J. Waller, S. J. Lyle, T. O. Popp, C. S. Diercks, J. A. Reimer and O. M. Yaghi, Chemical Conversion of Linkages in Covalent Organic Frameworks, J. Am. Chem. Soc., 2016, 138, 15519–15522, DOI:10.1021/jacs.6b08377.
- H. Liu, J. Chu, Z. Yin, X. Cai, L. Zhuang and H. Deng, Covalent Organic Frameworks Linked by Amine Bonding for Concerted Electrochemical Reduction of CO2, Chem, 2018, 4, 1696–1709, DOI:10.1016/j.chempr.2018.05.003.
- X. Li, C. Zhang, S. Cai, X. Lei, V. Altoe, F. Hong, J. J. Urban, J. Ciston, E. M. Chan and Y. Liu, Facile Transformation of Imine Covalent Organic Frameworks into Ultrastable Crystalline Porous Aromatic Frameworks, Nat. Commun., 2018, 9, 2998, DOI:10.1038/s41467-018-05462-4.
- P. J. Waller, Y. S. Alfaraj, C. S. Diercks, N. N. Jarenwattananon and O. M. Yaghi, Conversion of Imine to Oxazole and Thiazole Linkages in Covalent Organic Frameworks, J. Am. Chem. Soc., 2018, 140, 9099–9103, DOI:10.1021/jacs.8b05830.
- P. F. Wei, M. Z. Qi, Z. P. Wang, S. Y. Ding, W. Yu, Q. Liu, L. K. Wang, H. Z. Wang, W. K. An and W. Wang, Benzoxazole-Linked Ultrastable Covalent Organic Frameworks for Photocatalysis, J. Am. Chem. Soc., 2018, 140, 4623–4631, DOI:10.1021/jacs.8b00571.
- C. Zhao, H. Lyu, Z. Ji, C. Zhu and O. M. Yaghi, Ester-Linked Crystalline Covalent Organic Frameworks, J. Am. Chem. Soc., 2020, 142, 14450–14454, DOI:10.1021/jacs.0c07015.
- F. Haase, P. Hirschle, R. Freund, S. Furukawa, Z. Ji and S. Wuttke, Beyond Frameworks: Structuring Reticular Materials Across Nano-, Meso-, and Bulk Regimes, Angew. Chem., Int. Ed., 2020, 59, 22350–22370, DOI:10.1002/anie.201914461.
- S. J. Lyle, T. M. O. Popp, P. J. Waller, X. Pei, J. A. Reimer and O. M. Yaghi, Multistep Solid-State Organic Synthesis of Carbamate-Linked Covalent Organic Frameworks, J. Am. Chem. Soc., 2019, 141, 11253–11258, DOI:10.1021/jacs.9b04731.
- Q. Ma, X. Liu, H. Wang, Q. Zhuang and J. Qian, Construction of novel benzoxazine-linked covalent organic framework with antimicrobial activity via postsynthetic cyclization, Mater. Today Chem., 2022, 23, 100707, DOI:10.1016/j.mtchem.2021.100707.
- M. M. Samy, M. G. Mohamed, T. H. Mansoure, T. S. Meng, M. A. R. Khan, C. C. Liaw and S. W. Kuo, Solid state chemical transformations through ring-opening polymerization of ferrocene-based conjugated microporous polymers in host–guest complexes with benzoxazine-linked cyclodextrin, J. Taiwan Inst. Chem. Eng., 2022, 132, 104110, DOI:10.1016/j.jtice.2021.10.010.
- M. G. Mohamed, C. J. Li, M. A. R. Khan, C. C. Liaw, K. Zhang and S. W. Kuo, Formaldehyde-Free Synthesis of Fully Bio-Based Multifunctional Bisbenzoxazine Resins from Natural Renewable Starting Materials, Macromolecules, 2022, 55, 3106–3115, DOI:10.1021/acs.macromol.2c00417.
- M. G. Mohamed, M. M. Samy, T. H. Mansoure, C. J. Li, W. C. Li, J. H. Chen, K. Zhang and S. W. Kuo, Microporous Carbon and Carbon/Metal Composite Materials Derived from Bio-Benzoxazine-Linked Precursor for CO2 Capture and Energy Storage Applications, Int. J. Mol. Sci., 2022, 23, 347, DOI:10.3390/ijms23010347.
- Y. Lu, X. Yu, C. J. Evans, S. Yang and K. Zhang, Elucidating the role of acetylene in ortho-phthalimide functional benzoxazines: Design, synthesis, and structure–property investigations, Polym. Chem., 2021, 12, 5059–5068, 10.1039/D1PY00850A.
- K. Zhang, Y. Liu and H. Ishida, Polymerization of an AB-Type Benzoxazine Monomer toward Different Polybenzoxazine Networks: When Diels–Alder Reaction Meets Benzoxazine Chemistry in a Single-Component Resin, Macromolecules, 2019, 52, 7386–7395, DOI:10.1021/acs.macromol.9b01581.
- I. Machado, I. Hsieh, E. Rachita, M. L. Salum, D. Iguchi, N. Pogharian, A. Pellot, P. Froimowicz, V. Calado and H. Ishida, A truly bio-based benzoxazine derived from three natural reactants obtained under environmentally friendly conditions and its polymer properties, Green Chem., 2021, 23, 4051–4064, 10.1039/D1GC00951F.
- K. Zhang, Y. Liu, M. Han and P. Froimowicz, Smart and sustainable design of latent catalyst-containing benzoxazine-bio-resins and application studies, Green Chem., 2020, 22, 1209–1219, 10.1039/C9GC03504D.
- Z. Deliballi, B. Kiskan and Y. Yagci, Light induced crosslinking of main chain polybenzoxazines, Polym. Chem., 2021, 12, 5781–5786, 10.1039/D1PY01080H.
- M. G. Mohamed and S. W. Kuo, Crown Ether-Functionalized Polybenzoxazine for Metal Ion Adsorption, Macromolecules, 2020, 53, 2420–2429, DOI:10.1021/acs.macromol.9b02519.
- J. Y. Wu, M. G. Mohamed and S. W. Kuo, Directly synthesized nitrogen-doped microporous carbons from polybenzoxazine resins for carbon dioxide capture, Polym. Chem., 2017, 8, 5481–5489, 10.1039/c7py01026e.
- H. R. Abuzeid, A. F. M. El-Mahdy, M. M. M. Ahmed and S. W. Kuo, Triazine-Functionalized Covalent Benzoxazine Framework for Direct Synthesis of N-Doped Microporous Carbon, Polym. Chem., 2019, 10, 6010–6020, 10.1039/C9PY01231A.
- S. Xu, J. He, S. Jin and B. Tan, Heteroatom-Rich Porous Organic Polymers Constructed by Benzoxazine Linkage with High Carbon Dioxide Adsorption Affinity, J. Colloid Interface Sci., 2018, 509, 457–462, DOI:10.1016/j.jcis.2017.09.009.
- X. Sun, J. Li, W. Wang and Q. Ma, Constructing Benzoxazine-Containing Porous Organic Polymers for Carbon Dioxide and Hydrogen Sorption, Eur. Polym. J., 2018, 107, 89–95, DOI:10.1016/j.eurpolymj.2018.07.043.
- M. G. Mohamed, T. C. Chen and S. W. Kuo, Solid-State Chemical Transformations to Enhance Gas Capture in Benzoxazine-Linked Conjugated Microporous Polymers, Macromolecules, 2021, 54, 5866–5877, DOI:10.1021/acs.macromol.1c00736.
- M. G. Mohamed, W. C. Chang and S. W. Kuo, Crown Ether– and Benzoxazine–Linked Porous Organic Polymers Displaying Enhanced Metal Ion and CO2 Capture through Solid State Chemical Transformation, Macromolecules, 2022, 55, 7879–7892, DOI:10.1021/acs.macromol.2c01216.
- R. C. Lin, M. G. Mohamed and S. W. Kuo, Benzoxazine/Triphenylamine-Based Dendrimers Prepared through Facile One-Pot Mannich Condensations, Macromol. Rapid Commun., 2017, 38, 1700251, DOI:10.1002/marc.201700251.
-
H. Ishida and P. Froimowicz, Advanced and Emerging Polybenzoxazine Science and Technology, Elsevier, Amsterdam, 2017 Search PubMed.
- Y. Liu, Q. Ran and Y. Gu, Preparation and properties of benzoxazine blends with intumescent. flame retardancy, Polym. Degrad. Stab., 2019, 163, 15–24, DOI:10.1016/j.polymdegradstab.2019.02.022.
- P. Wang, M. Liu and Q. Ran, The study on curing and weight-loss mechanisms of benzoxazine during thermal curing process, Polym. Degrad. Stab., 2020, 179, 109279, DOI:10.1016/j.polymdegradstab.2020.109279.
- Y. Liu, W. Sheng, R. Yin and K. Zhang, Propargylamine: an attractive amine source for designing high-performance benzoxazine resins with low polymerization temperatures, Polym. Chem., 2021, 12, 65194–66704, 10.1039/D1PY01166A.
- M. Muraoka, M. Goto, M. Minami, D. Zhou, T. Suzuki, T. Yajima, J. Hayashi, H. Sogawa and F. Sanda, Ethynylene-linked multifunctional benzoxazines: the effect of the ethynylene group and packing on thermal behavior, Polym. Chem., 2022, 13, 5590–5596, 10.1039/D2PY00840H.
- M. G. Mohamed, S. V. Chaganti, M. S. Li, M. M. Samy, S. U. Sharma, J. T. Lee, M. H. Elsayed, H. H. Chou and S. W. Kuo, Ultrastable Porous Organic Polymers Containing Thianthrene and Pyrene Units as Organic Electrode Materials for Supercapacitors, ACS Appl. Energy Mater., 2022, 5, 6442–6452, DOI:10.1021/acsaem.2c00942.
- H. Zhou, L. Hou, W. Zhang and H. J. Zhai, A dual-doped strategy to enhance the electrochemical performances of electropolymerized polyaniline electrodes for flexible energy storage, Mater. Chem. Phys., 2020, 240, 122259, DOI:10.1016/j.matchemphys.2019.122259.
- A. Ehsani, M. Bigdeloo, F. Assefi, M. Kiamehr and R. Alizadeh, Ternary nanocomposite of conductive polymer/chitosan biopolymer/metal-organic framework: synthesis, characterization and electrochemical performance as effective electrode materials in pseudocapacitors, Inorg. Chem. Commun., 2020, 115, 107885, DOI:10.1016/j.inoche.2020.107885.
- W. Wu, D. Wei, J. Zhu, D. Niu, F. Wang, L. Wang, L. Yang, P. Yang and C. Wang, Enhanced electrochemical performances of organ-like Ti3C2 MXenes/polypyrrole composites
as supercapacitors electrode materials, Ceram. Int., 2019, 45, 7328–7337, DOI:10.1016/j.ceramint.2019.01.016.
- M. M. Samy, M. G. Mohamed and S. W. Kuo, Pyrene-functionalized tetraphenylethylene polybenzoxazine for dispersing single-walled carbon nanotubes and energy storage, Compos. Sci. Technol., 2020, 199, 108360, DOI:10.1016/j.compscitech.2020.108360.
- M. G. Mohamed, H. Y. Hu, M. Madhu, M. M. Samy, I. M. A. Mekhemer, W. L. Tseng, H. H. Chou and S. W. Kuo, Ultrastable two-dimensional fluorescent conjugated microporous polymers containing pyrene and fluorene units for metal ion sensing and energy storage, Eur. Polym. J., 2023, 189, 111980, DOI:10.1016/j.eurpolymj.2023.111980.
- M. M. Samy, M. G. Mohamed, S. U. Sharma, S. V. Chaganti, J. T. Lee and S. W. Kuo, An Ultrastable Tetrabenzonaphthalene-Linked conjugated microporous polymer functioning as a high-performance electrode for supercapacitors, J. Taiwan Inst. Chem. Eng., 2023, 104750, DOI:10.1016/j.jtice.2023.104750.
- M. G. Mohamed, M. H. Elsayed, Y. Ye, M. M. Samy, A. E. Hassan, T. H. Mansoure, Z. Wen, H. H. Chou, K. H. Chen and S. W. Kuo, Construction of Porous Organic/Inorganic Hybrid Polymers Based on Polyhedral Oligomeric Silsesquioxane for Energy Storage and Hydrogen Production from Water, Polymers, 2023, 15, 182, DOI:10.3390/polym15010182.
- M. G. Mohamed, S. U. Sharma, N. Y. Liu, T. H. Mansoure, M. M. Samy, S. V. Chaganti, Y. L. Chang, J. T. Lee and S. W. Kuo, Ultrastable Covalent Triazine Organic Framework Based on Anthracene Moiety as Platform for High-Performance Carbon Dioxide Adsorption and Supercapacitors, Int. J. Mol. Sci., 2022, 23, 3174, DOI:10.3390/ijms23063174.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Mohamed Gamal
Mohamed
a,
Mohamed Gamal
Mohamed