
Depolymerisation–hydrogenation of condensed tannins as a strategy for generating flavan-3-ol monomers†
Thijs
Vangeel
 a,
Ruben
Smets
b,
Mik
Van Der Borght
b and
Bert
Sels
*a
a,
Ruben
Smets
b,
Mik
Van Der Borght
b and
Bert
Sels
*a
aDepartment of Microbial and Molecular Systems, Center Center for Sustainable Catalysis and Engineering (CSCE), KU Leuven – Chem&Tech, Celestijnenlaan 200F 3001 Leuven, Belgium. E-mail: bert.sels@kuleuven.be
bDepartment of Microbial and Molecular Systems, Research Group for Insect production and Processing, KU Leuven – Campus Geel, Kleinhoefstraat 4, 2240 Geel, Belgium
First published on 1st February 2023
Abstract
Flavan-3-ol monomers are interesting, functionalised phenolic building blocks. Here, a strategy is presented for producing these flavan-3-ol monomers, starting from condensed tannins present in biomass waste streams such as tree bark. Using purified procyanidin condensed tannins as a reference substrate, a one pot depolymerisation–hydrogenation process was developed. Briefly, procyanidin condensed tannins were processed in methanol at elevated temperature (125–175 °C) in the presence of hydrogen gas and a heterogeneous hydrogenation catalyst (Pd/C). The elevated temperature enables a solvolytic depolymerisation of the condensed tannins, forming reactive intermediates which are immediately stabilised through catalysed hydrogenation. The flavan-3-ol monomers catechin and epicatechin are the two main products, with a combined yield of up to 34 wt%. Furthermore, the role of the hydrogenation catalyst is elucidated, and the effect of reaction temperature, hydrogen pressure and catalyst loading is studied. Finally, the successful depolymerisation–hydrogenation of a crude methanolic larch bark extract demonstrated the feasibility of this process and its robustness against impurities.
Introduction
Generating chemicals from renewable sources has been of growing interest over the last decades.1,2 With respect to the production of phenolic or aromatic compounds, lignin – the world's largest source of renewable phenolics – has received immense attention, and many advances toward its effective utilisation have been made.3,4 Next to lignin, condensed tannins are the second-most abundant source of renewable phenolics, and thus also an interesting feedstock for producing phenolic building blocks.5–8Condensed tannins, or proanthocyanidins, are ubiquitous in the plant kingdom, playing a role in the plant's defence mechanism.9,10 Condensed tannins can be found in different parts of the plant, e.g. tree bark, fruit seeds or peels, and wood. The condensed tannin content and distribution within the plant varies strongly between species. Some species, such as mimosa bark (Acacia mearnsii) and quebracho wood (Schinopsis lorentzii), are commercially used for extracting condensed tannins, which are in turn used for e.g. leather tanning, among others.7,11 Nevertheless, vast sources of unexploited condensed tannins still exist, often in biomass waste streams such as pine bark – a forest industry residue – or grape pomace – a winery side product.12,13
Condensed tannins, or proanthocyanidins, have a highly functionalised chemical structure. The structure consists of flavanol monomers that are linked through interflavonoid bonds forming oligomers or short polymers (Fig. 1). Two types of bonds exist: (i) the B-type bond (most common, shown in Fig. 1), having one C–C bond (from position 4 to 8, i.e. 4 → 8 bond), and (ii) the A-type bond, having the same 4 → 8 C–C bond, but additionally also a 2 → 7 C–O–C (ether bond) (less common, not shown).5,14,15
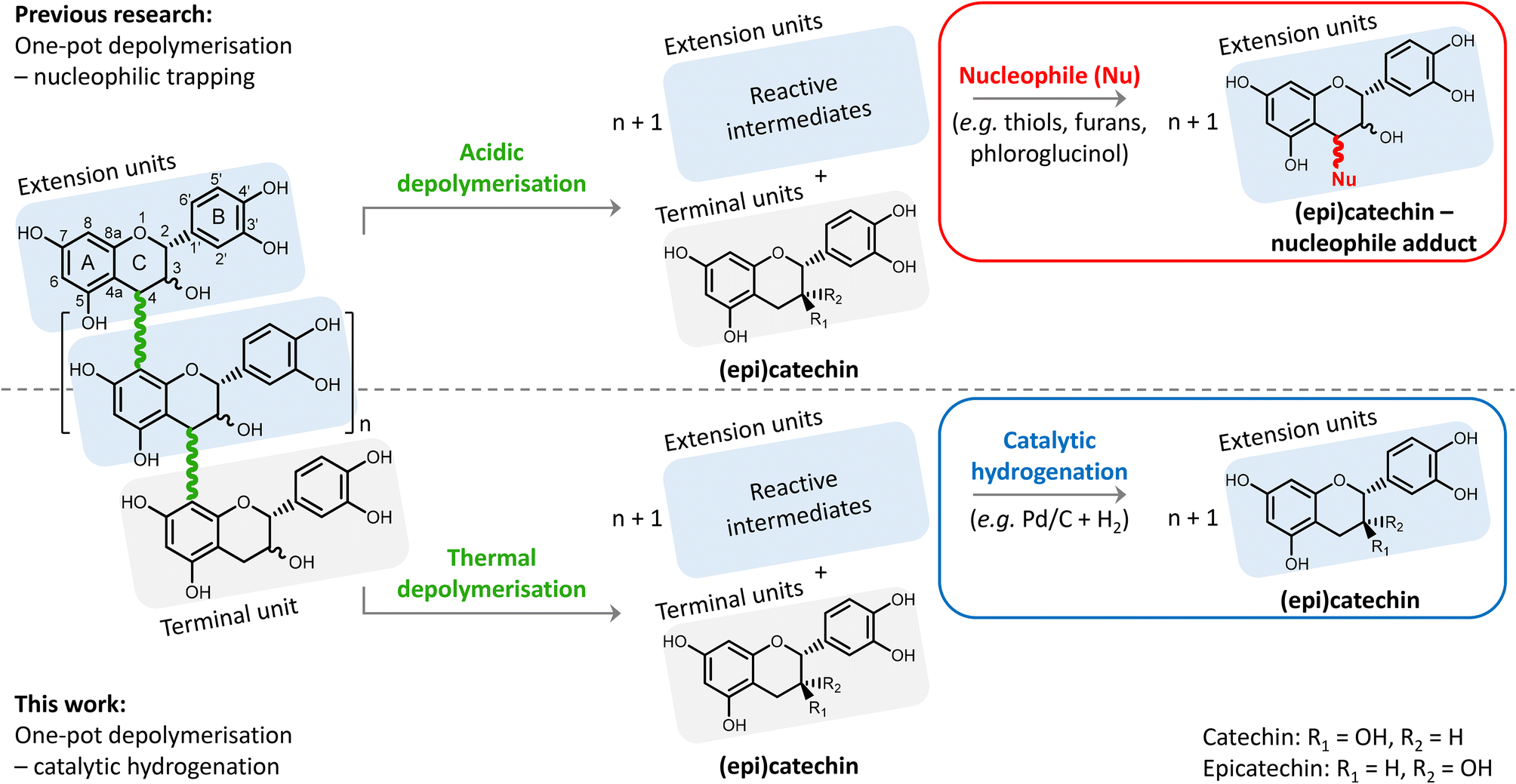 | ||
| Fig. 1 Comparison of the proposed one-pot depolymerisation–hydrogenation of condensed tannins, with the state-of-the-art one-pot depolymerisation – nucleophilic trapping. Above: common acidic depolymerisation combined with nucleophilic trapping forming adducts from (epi)catechin rather than (epi)catechin itself. Below: thermal depolymerisation combined with catalytic hydrogenation, leading to (epi)catechin. A general procyanidin structure is shown on the left, with the numbering indicated on one of its monomers. | ||
The general structure of a flavanol monomer consists of two aromatic rings (A and B), a pyran ring (C) and a hydroxyl group on position 3 (see Fig. 1 for numbering, and an example of a flavanol monomer). The hydroxylation pattern on the A- and B-rings of the flavanol may vary depending on the source of condensed tannins. Based on the hydroxylation pattern, subgroups of condensed tannins are constructed (e.g. procyanidins, prodelphinidins, prorobinetinidins), having different chemical reactivity (e.g. tendency towards depolymerisation).5,15
One frequently occurring hydroxylation pattern is depicted in Fig. 1, with two hydroxyl groups on the A-ring at position 5 and 7, and two hydroxyl groups on the B-ring at position 3′ and 4′. This type of condensed tannins is referred to as procyanidins, and has catechin or epicatechin, which are diastereomers, as monomeric unit. Procyanidins are typically present in grape seeds and skins, and pine bark. For simplicity, only this procyanidin type of condensed tannins is considered and studied in this contribution.5,11,15
Depolymerisation is an interesting route for the valorisation of procyanidins. It can funnel the complex, sterically hindered, polyphenolic polymers into only a few flavanol monomers.8 These flavanol monomers in turn have the potential to be used as phenolic building block for e.g. the synthesis of new polymers.6,16
Depolymerisation of procyanidins has already been widely investigated, but typically for analytical (i.e. structure elucidation) purposes only.17 In acidic media, position 8 on the electron-rich A-ring can be protonated, resulting in the cleavage of the 4 → 8 interflavonoid bonds (B-type bonds) (see Fig. 1). This results in the formation of terminal units (i.e. (epi)catechin) and extension units having a positive charge on position 4 (i.e. a carbocation). Despite the resonance stabilisation, these carbocations are prone to repolymerisation and rearrangement reactions that lead to a degraded structure of low value. To avoid this repolymerisation, a nucleophilic agent is typically added to chemically trap the formed carbocations, resulting in (epi)catechin-nucleophile adducts. Typical nucleophiles include various thiols, phloroglucinol, and – more recently – furans.8,18–23
This nucleophilic trapping strategy for depolymerisation has a few drawbacks. Firstly, the nucleophile itself is costly, and needed in excess, requiring downstream separation. Secondly, the use of an acid requires downstream neutralisation, yielding salts as by-product. Thirdly, the products from the extension units (i.e. (epi)catechin-nucleophile adducts) are different from those from the terminal units (i.e. (epi)catechin), requiring additional downstream separation. Because of these drawbacks, this strategy has been mainly limited to analytical purposes and tannin structure unravelling.
Inspired by the recently developed “reductive catalytic fractionation” process24–27 – also originally a lignin structure analytical tool,28 but now adopted for lignin valorisation – we herein propose a depolymerisation–hydrogenation strategy for upgrading the procyanidin condensed tannins. We hypothesise that a thermal depolymerisation of the procyanidin is possible, yielding unstable intermediates that can be reductively stabilised via heterogeneously catalysed hydrogenation (see Fig. 1). This strategy thus avoids the otherwise common use of an acid catalyst and a nucleophilic trapping agent. Furthermore, the obtained monomeric phenolic products are all (epi)catechin, instead of the more complicated (epi)catechin and (epi)catechin-nucleophile adducts, facilitating future downstream purification. The use of a heterogeneous catalyst, being a metal activating hydrogen and chemically stabilizing intermediates and preventing polymerisation reactions, also enables facile catalyst separation and its reuse.
Earlier work has already demonstrated the reductive cleavage of the interflavonoid bond in dimers using sodium cyanoborohydride in trifluoroacetic acid.29 Also catalysed hydrogenolysis has been studied as a means of condensed tannin depolymerisation,30–35 but detailed analysis and quantification of the reaction products has remained scarce. Most closely related but different from the work here, hydrogenolysis has also been used in synthetic chemistry for the removal of protecting benzyl groups from benzylated flavonoid molecules.36–38
This study aims to advance the depolymerisation–hydrogenation strategy for procyanidin condensed tannin valorisation, by (i) studying both model compounds as well as real procyanidin-rich extracts (ii) combining various analytical tools to gain deeper insight in the reactions at hand, (iii) elucidating the role of the hydrogenation catalyst, and (iv) demonstrating the feasibility of selectively producing (epi)catechin from procyanidins, and comparing it with the state-of-the-art nucleophilic trapping process.
Results and discussion
Proof of concept
To demonstrate the concept of condensed tannin depolymerisation–hydrogenation, reactions were carried out using Oligopin as substrate. Oligopin is a purified maritime pine bark extract, consisting mainly of oligomeric procyanidin compounds.39 It will be used as model substrate throughout this study. Its composition, as mentioned in a study by Segal et al., can also be found in the ESI (Table S1†).40As proof-of-concept reaction, 200 mg of Oligopin, 25 mg Pd/C catalyst powder (5 wt% Pd) and 60 mL of methanol were loaded in a 100 mL Parr autoclave batch reactor. The reactor was pressurised with 30 bar of H2, stirred at 450 rpm and heated to 150 °C for 3 h. The reactor content was filtered after cooling by a membrane filter to remove the solid catalyst powder and to prepare the sample for HPLC analysis. For comparison, a blank reaction was also carried out under similar reaction conditions, but in absence of the catalyst and by using 6 bar N2 instead of H2.
In Fig. 2, (i) the Oligopin substrate, (ii) the proof-of-concept catalysed reaction, and (iii) the blank reaction are compared. A first observation of importance is the different colour of the three solutions (at roughly equal concentration). The starting substrate solution has a light brown colour. After the catalysed reaction, the solution becomes almost transparent – hinting at the reduction of the oxidised anthocyanidin chromophore structures. In contrast, the blank reaction clearly provides a darker product solution suggesting repolymerisation reactions have taken place.
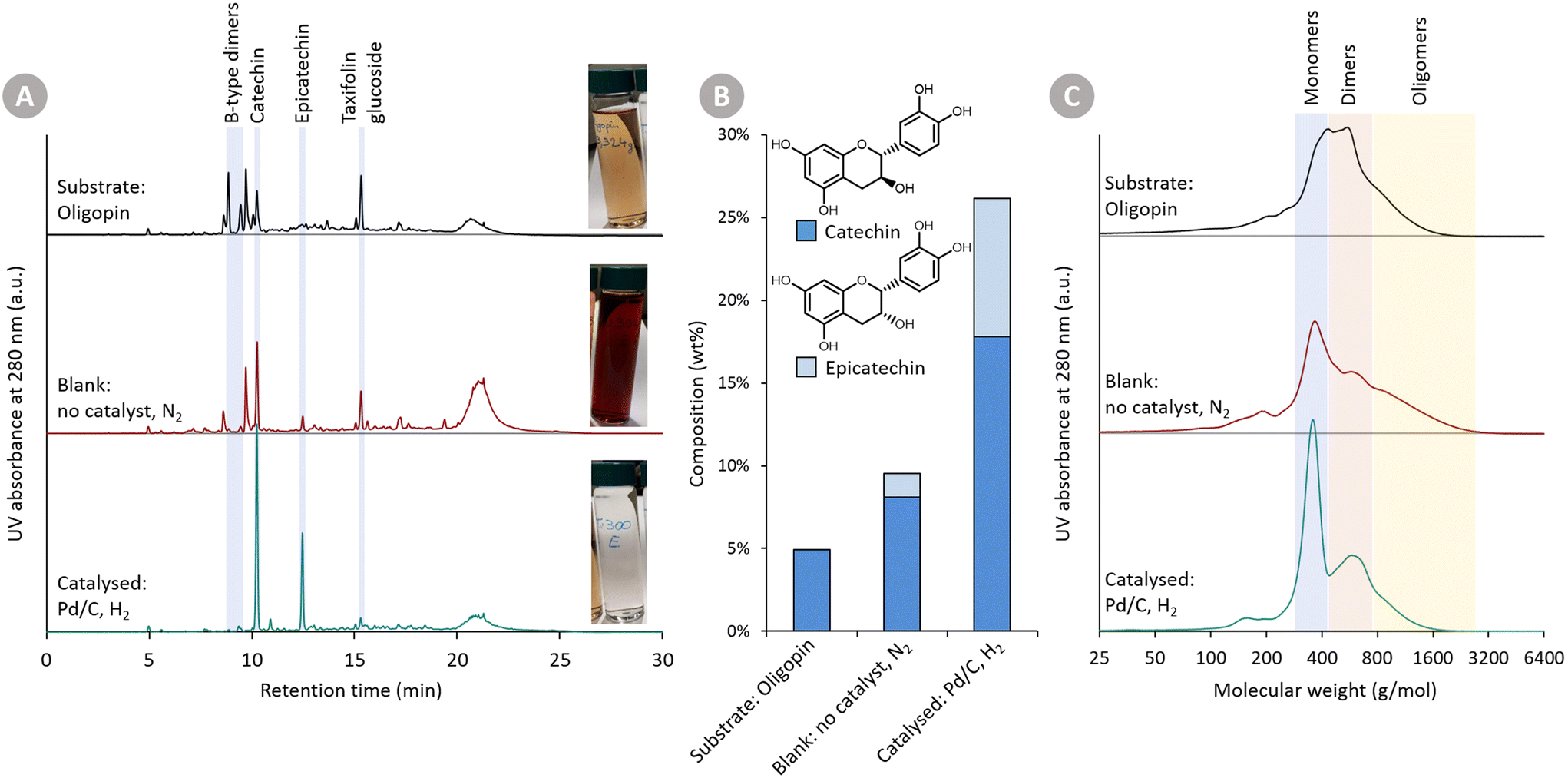 | ||
| Fig. 2 Proof-of-concept of the depolymerisation–hydrogenation strategy for condensed tannins, showing three samples: (i) the Oligopin substrate, (ii) the product mixture after a blank reaction, and (iii) the product mixture after a catalysed hydrogenation reaction. Reaction conditions: 200 mg Oligopin, 60 mL methanol, 150 °C, 6 bar N2 and no catalyst (blank reaction) or 30 bar H2 and 25 mg Pd/C (5 wt% Pd) (catalysed reaction). (A) HPLC-UV analysis at 280 nm, as well as visual comparison of substrate or product solutions. The catalysed reaction provides an almost transparent product solution, and much cleaner chromatogram with only two main products. (B) Quantification of catechin and epicatechin. (C) GPC analysis with indications of flavonoid mono-, di- and oligomers. Chromatograms are normalised to have the same area under the curve. The catalysed reaction shows a large and clear monomer signal, whereas the blank reaction displays more oligomers due to repolymerisation. | ||
Visual inspection of the chromatograms of the HPLC-UV analysis at 280 nm (Fig. 2A) already reveals stark differences. The Oligopin substrate shows a lot of different signals – and a slightly elevated baseline – likely due to a variety of oligomeric procyanidin structures present in the sample.8 Using UPLC-MS, some additional peaks were tentatively identified (Fig. S1†). The tentatively identified B-type dimers and taxifolin glucoside, known components in Oligopin,40 are indicated on the chromatograms as well. After the blank reaction, we see several signals disappearing at the expense of an increasing catechin peak. Also the large hump at the end of the chromatogram increases, suggesting the formation of larger structures through repolymerisation. The chromatogram of the catalysed reaction, by contrast, looks cleaner with only two dominant signals, assigned to the diastereomers catechin and epicatechin, appearing. Contrary to the blank reaction, the hump at the end of the chromatogram remains unchanged, suggesting larger structures remain present but do not increase after reaction. Quantifying the (epi)catechin in these three samples is also done via HPLC-UV (Fig. 2B). It can be seen that some catechin (4.9 wt%) is already present in the Oligopin substrate. After a blank reaction, more catechin is formed (8.1 wt%). This originates likely from the terminal units, which do not form reactive species upon depolymerisation. As terminal units of procyanidins in maritime pine bark are typically catechin, the observed epicatechin (1.4 wt%) is most likely formed via epimerisation.41 The catalysed reaction results in a much higher yield of catechin (17.8 wt%) and epicatechin (8.3 wt%), already demonstrating the effect of the catalytic hydrogenation on the monomer yield.
NMR analysis (1H–13C HSQC NMR) revealed the presence of B-type interflavonoid bonds in the Oligopin substrate (Fig. S3†). Interestingly, those bonds are (almost) completely cleaved in both the catalysed as well as the blank depolymerisation reaction (Fig. S4†). This illustrates that the catalyst is not required for cleaving those bonds, but merely serves to stabilise the formed reactive depolymerisation products.
Gel permeation chromatography (GPC) (Fig. 2C) was used to assess the molecular size of the substrate and formed products. This analysis confirms – in line with earlier GPC analysis – that the Oligopin substrate consists of a mixture of flavonoid mono-, di- and oligomers.42 After the blank reaction, the share of both monomers as well as larger oligomers seems to be increasing. This can be explained by a depolymerisation and repolymerisation mechanism: the original oligomers are depolymerised, yielding relatively stable extension units (catechin), but the unstable, depolymerised extension units may repolymerise forming new C–C bonds and larger structures (Fig. 1).
GPC analysis of the product mixture, obtained in presence of the catalyst, shows a clear increase in share of monomers (in line with HPLC-UV analysis). Furthermore, there is a decrease in the share of di- and oligomers, illustrating that depolymerisation has occurred, and that the catalytic hydrogenation prevented the (extensive) repolymerisation reactions.
Lastly, the Oligopin substrate was subjected to a state-of-the-art depolymerisation – nucleophilic trapping procedure, in order to compare it to the above proof-of-concept catalysed reaction. Recent work has demonstrated that various furans (e.g. furan, 2-methyl furan, menthofuran) are excellent nucleophiles for this purpose.22,23 For this study, 2-methyl furan was selected as nucleophile, because of its promising results in an earlier study and its availability from biomass.22 Previously identified optimal reaction conditions were used (30 °C, 0.1 M HCl). The reaction was monitored over time using HPLC-UV analysis. The product yield over time and an HPLC-UV chromatogram are displayed in Fig. S5.† The highest yield of monomeric products was achieved after 30 min of reaction: 13.5 wt% catechin and 0.5 wt% epicatechin (originating from the terminal units), and 16.8 wt% of the two isomers of 4-(2-methyl furan)-(epi)catechin (originating from the extension units; expressed in weight of catechin equivalents, to ignore the incorporation of the mass of the nucleophile).
Overall, the total monomer yield of 26.1 wt% for the proof-of-concept catalysed reaction, is thus close to the total, maximal monomer yield of 30.8 wt% obtained via an optimised, state-of-the-art of depolymerisation – nucleophilic trapping procedure. This may indicate the same interflavonoid bonds are being broken in both these processes, and that the stabilisation of intermediates is already promising in the proof-of-concept catalysed reaction.
Molecular and kinetic insights
At all three temperatures, the major products are catechin and epicatechin. Their yield over time is displayed in Fig. 3A. Firstly, it is clear that a higher temperature leads to a faster formation rate of (epi)catechin. After 1 h of reaction, for example, 14.8 wt% (epi)catechin is formed at 125 °C, whereas 20.5 wt% is formed at 150 °C. Secondly, a substantially higher (epi)catechin yield can be achieved by working at a lower temperature and longer reaction time. For instance, the maximum obtained yield at 125 °C is 33.9 wt% (epi)catechin, whereas this is only 26.5 wt% at 150 °C and 22.2 wt% at 175 °C. At all three temperatures, a plateau in yield is reached (or even started already to decrease), indicating that the reactions leading to (epi)catechin (e.g. depolymerisation of cleavable bonds in procyanidin) are completed.
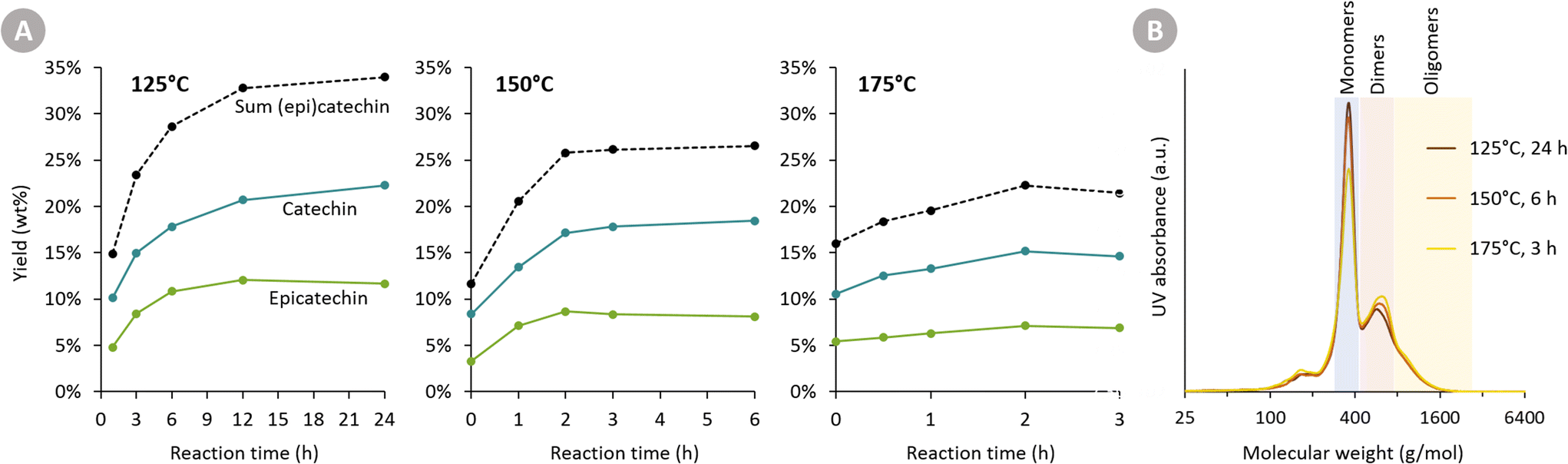 | ||
| Fig. 3 Depolymerisation–hydrogenation of Oligopin at different temperatures (125, 150 and 175 °C). The formed products are monitored over time. Reaction conditions: 200 mg Oligopin, 60 mL methanol, 30 bar H2 and 25 mg Pd/C (5 wt% Pd), 20 min heating to reaction temperature. (A) Yield of (epi)catechin at different temperatures and at varying reaction time. (B) GPC analysis of the samples taken at the final reaction time of each reaction temperature. Lower reaction temperatures and longer reaction times result in a lower molecular weight compared to higher reaction temperatures and shorter reaction times. | ||
The difference in maximum yield at different temperatures can have two explanations. Either (epi)catechin itself might degrade as the reaction progresses, or the reactive intermediates formed upon cleavage of procyanidins might undergo faster repolymerisation reactions compared to reductive stabilisation reactions at higher temperatures. To assess the stability of catechin, reactions with pure catechin were carried out at 150 °C (see following section). The negligible catechin degradation (only epimerisation) thus suggests that the second explanation for having a reduced maximum yields at a higher temperature is most plausible.
GPC analysis (Fig. 3B) also corroborates the higher maximum monomer yield at lower temperatures. Furthermore, it shows that at higher temperatures more di- and oligomers are present, suggesting repolymerisation has occurred.
The Oligopin substrate does not have any epicatechin initially present. However, already at the first sampling point for each temperature, some epicatechin is formed (e.g. 4.8 wt% after 1 h at 125 °C). If we consider the ratio of epicatechin/catechin for the reaction at 125 °C, we find that this ratio first increases to reach a maximum of 0.61 at 6 h of reaction, and afterwards decreases again to 0.52 at 24 h. This proves that epicatechin is formed directly from the Oligopin, and that it is not solely a product of the epimerisation reaction of catechin.
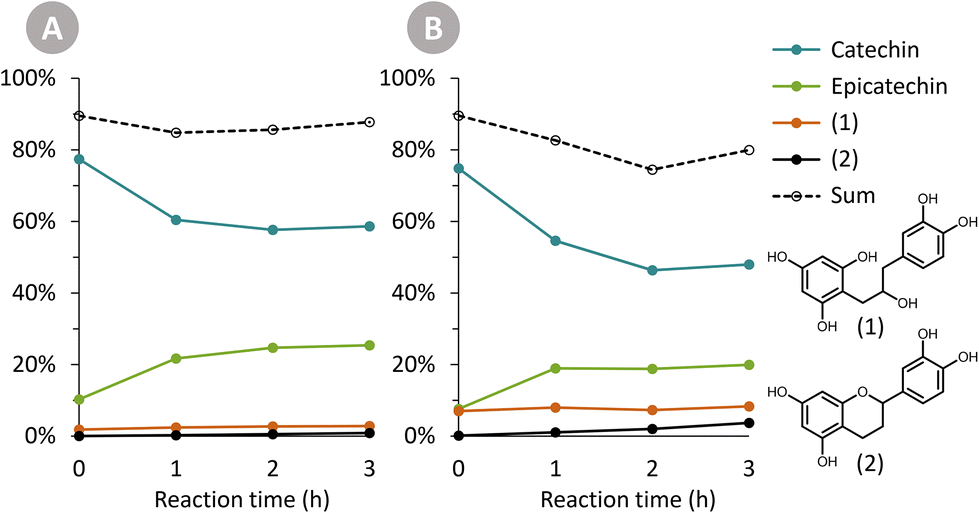 | ||
| Fig. 4 Product composition over time for the hydrogenation of catechin. Reaction conditions: 200 mg catechin, 60 mL methanol, 150 °C, 30 bar H2 and either (A) 25 mg Pd/C or (B) 100 mg Pd/C. Two hydrogenation products (1) and (2) were detected, especially at a higher catalyst loading. | ||
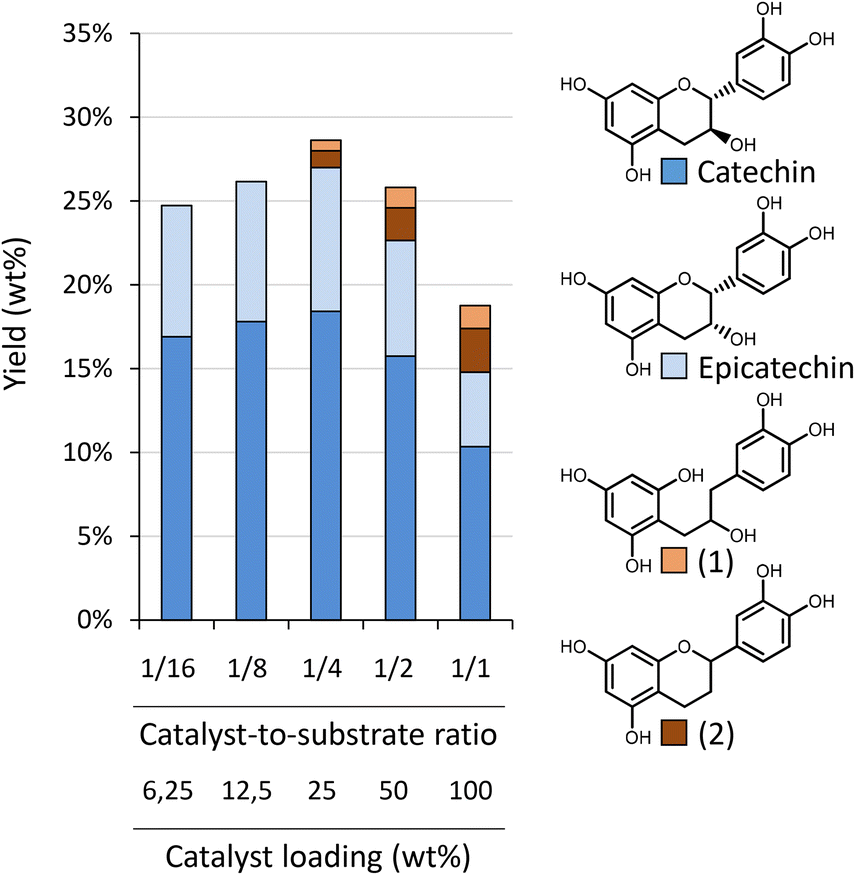 | ||
| Fig. 5 Influence of catalyst loading (with respect to substrate) on the depolymerisation–hydrogenation of procyanidins. Reaction conditions: 200 mg Oligopin, 60 mL methanol, 30 bar H2, varying catalyst loading (from 12.5 to 200 mg Pd/C), 3 h reaction time. Next to (epi)catechin, also hydrogenation products were detected: a ring opened catechin (1) and a dehydroxylated catechin (2). | ||
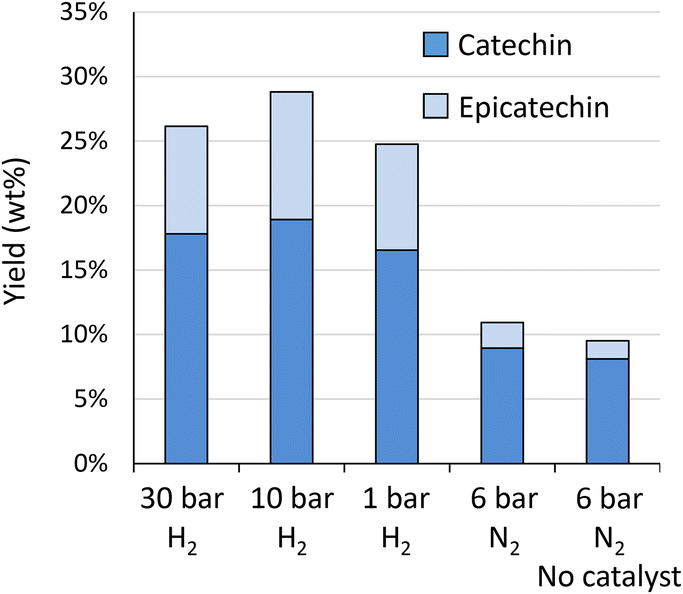 | ||
| Fig. 6 Influence of H2 pressure (at RT) on the depolymerisation–hydrogenation of procyanidins. Reaction conditions: 200 mg Oligopin, 60 mL methanol, 1–30 bar H2 or 6 bar N2, 25 mg Pd/C (or no catalyst), 3 h reaction time. | ||
The highest yield is obtained at 10 bar H2 (28.8 wt%), whereas slightly lower yields were obtained at 30 bar (26.1 wt%) and 1 bar (24.7 wt%). The original HPLC-UV chromatograms are displayed in Fig. S12A.† This figure illustrates that chromatograms are most clear for reaction outcomes at 30 and 10 bar of H2. At 1 bar of H2, more products are observed, possibly due to incomplete hydrogenation of initial components, such as taxifolin glucoside. More importantly, at 1 bar of H2, already a slight increase in the “hump” at the end of the chromatogram can be noted, illustrative of repolymerisation. Without any hydrogen (6 bar N2) repolymerisation has occurred even more. GPC analysis (Fig. S12B†) confirms the presence of larger molecular structures in absence of hydrogen. This analysis further corroborates the highest share in monomers for reaction at 10 bar of H2.
Towards a real feedstock
In the previous sections, Oligopin was consistently used as substrate for our research. Because of its high procyanidin content, high solubility in various solvents and previously determined composition, it serves as an excellent benchmark for future reference. Nevertheless, as Oligopin is produced via several purification steps (S/L extraction, L/L extraction, crystallisation and dehydration, i.e. the CrystalPure process), the actual mass yield starting from pine bark is very low (0.1 wt%).44In an attempt to obtain higher overall mass yields (based on the bark), and avoid laborious purification steps, we set out to use a crude bark extract as substrate. Larch (Larix decidua) bark – of which the composition was determined in an earlier study – was selected because of its high amount of ethanol and water soluble extractives.45 To obtain a crude extract, we performed a Soxhlet extraction of this ground bark (<1 mm) using methanol as solvent (solid/liquid ratio of 21.4 g L−1). After exhaustive extraction (4 h of extraction, ∼4 cycles per h), the extraction yield was 22.5 wt% (relative to the larch bark). The crude methanolic extract (4.8 g extractives per L) was used without further workup in a catalytic experiment. Similar reaction conditions were used as described in previous sections (150 °C, 30 bar H2, 200 min reaction time). A catalyst loading of 25 mg was used, corresponding to a catalyst loading of 9.2 wt% with respect to the substrate. Both the crude methanolic extract and the product solution were analysed using HPLC-UV (Fig. 7). It has to be noted that, as always, samples were filtered prior to analysis. In this case, however, it resulted in the removal of a significant insoluble part (at RT) of the crude extract, whereas the product mixture was fully soluble in methanol at RT. The chromatogram of the crude mixture has an elevated “baseline” (in line with that of Oligopin), indicating a large number of unresolved components. Furthermore, a lot of smaller signals are present – two of which corresponding to (epi)catechin. After the depolymerisation–hydrogenation process, the chromatogram appears much cleaner, with a lower “baseline”, fewer product signals, and an apparent increase in (epi)catechin signal intensity. Indeed, the amount of (epi)catechin in the product mixture after reaction is 16.2 wt%, compared to only 2.9 wt% in the original crude extract.
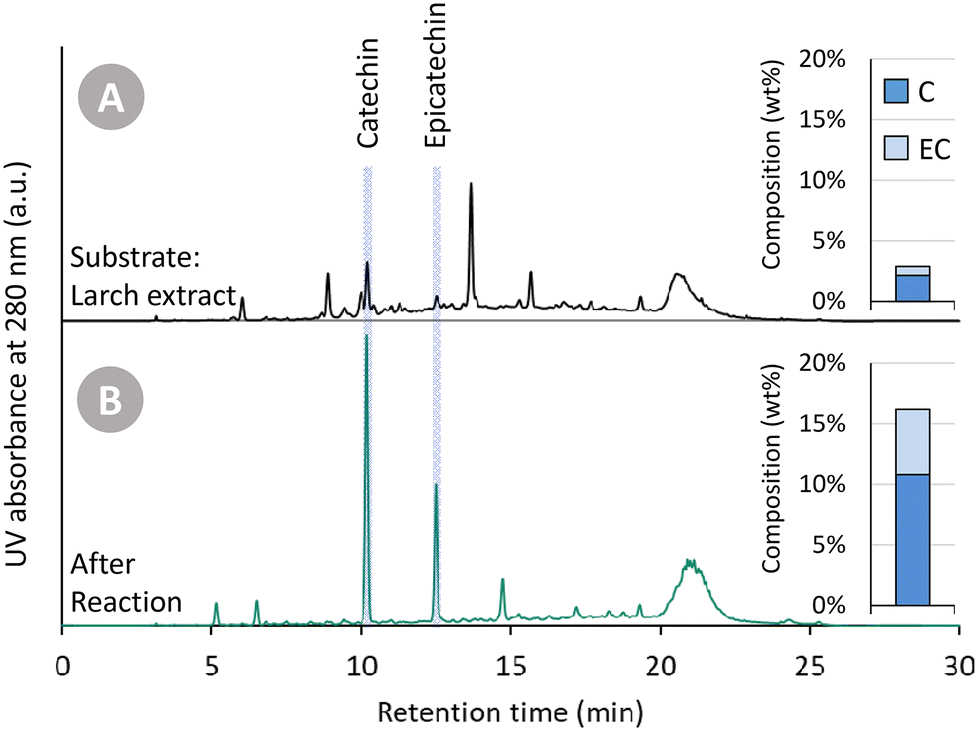 | ||
| Fig. 7 HPLC-UV analysis and quantification of catechin (C) and epicatechin (EC) of two solutions: (A) crude larch bark extract, as obtained via Soxhlet extraction with methanol, and (B) the product mixture after depolymerisation–hydrogenation of a crude larch bark extract. Reaction conditions: 60 mL crude larch bark extract in methanol (4.8 g extractives per L), 30 bar H2, 25 mg Pd/C, 200 min reaction time. | ||
The yield of (epi)catechin from the crude larch extract (16.2 wt%) is clearly lower than the obtained yield starting from Oligopin under similar conditions (26.1 wt%, after 3 h at 150 °C). Nevertheless, the share of procyanidins in the crude extract is likely much lower (as no purifications steps were applied) resulting in a lower theoretical maximum yield. Furthermore, the larger share of impurities in the crude extract doesn't seem to affect the production of (epi)catechin very strongly, judging from the chromatogram. This thus suggests that crude procyanidin rich extracts can be a good starting point for producing (epi)catechin, thus circumventing costly purification steps and at the same time obtaining a higher yield with respect to the original bark.
Conclusions
In this contribution, a depolymerisation–hydrogenation strategy is proposed for converting procyanidin condensed tannins into its flavanol monomers. A mixture of oligomeric procyanidins (Oligopin) was processed in methanol at elevated temperatures (125–175 °C) in presence of H2 and a heterogeneous hydrogenation catalyst (Pd/C). At these temperatures, thermal cleavage of the interflavonoid bonds occurs, while the catalyst reductively stabilises the generated intermediates. The precise identity of the formed intermediates is not yet clear, but should be subject of further investigation such as more advanced (e.g., in situ NMR) kinetic studies. The result of this process is an effective depolymerisation into mainly two main products, catechin and epicatechin (up to 33.9 wt% yield). Furthermore, it was demonstrated that crude larch bark extract can also be depolymerised into mainly (epi)catechin. On the one hand, this contribution thus presents a route for valorising tree bark or other procyanidin-rich residues, while on the other hand, it opens up possibilities for using (epi)catechin as phenolic building blocks. Future research in this respect should focus on different methods to separate these interesting products from the product mixture.Experimental
Chemicals and materials
All commercial chemicals were analytic reagents and were used without further purifications. 5% Pd on carbon (Pd/C), tetrahydrofuran (>99%, stabilised with 250 ppm BHT), dimethylsulfoxide-d6 (99.9% atom D), N-methyl-N-(trimethylsilyl)trifluoroacetamide (>98.5%), anhydrous pyridine (99.8%), (+)-catechin hydrate (>98%) were purchased from Merck. Acetonitrile (99.9%), methanol (99.9%), hydrochloric acid (37% in water), sodium acetate (>99%), phosphoric acid (85% in water) were purchased from Acros Organics. (−)-Epicatechin (>97%) was purchased from TCI Europe. Procyanidin A2 (>98%) and procyanidin B2 (>90%) were purchased from Extrasynthese. 2-Methylfuran (>99%) was purchased from Carl Roth. Oligopin® was kindly provided by DRT (les Dérivés Résiniques et Terpéniques). Larch (Larix decidua) bark was obtained from a tree located in Hamont-Achel (Belgium), and provided by the division ‘Forest, Nature and Landscape’ (KU Leuven). The chemical composition of this bark sample had been determined previously.45High pressure batch reactions
The depolymerisation–hydrogenation experiments were performed in a 100 mL stainless steel batch reactor (Parr Instruments & Co.). In a typical reaction, 200 mg of Oligopin were loaded into the reactor together with the catalyst powder (typically 25 mg Pd/C) and methanol (60 mL). The reactor was sealed, flushed threefold with N2, and pressurised with H2 (typically 30 bar at room temperature). Next, the reaction mixture was stirred (450 rpm) and heated to 125–175 °C (20 min heating time). If samples were taken during reaction, they were quickly cooled in an ice bath. After the reaction, the reactor was cooled and depressurised at room temperature. Afterwards, a part (±20 mL) of the product liquor was filtered through a 0.2 μm PTFE membrane to remove any particulate matter (e.g. Pd/C powder), and stored in the fridge for future analysis.Depolymerisation – nucleophilic trapping with 2-methylfuran
The procedure had been described by Fulcrand et al.,22 and was slightly adapted here. The depolymerisation reagent was prepared by mixing the nucleophile (5 mL of 2-methylfuran) and methanol (5 mL) in equal volumes, and it was then acidified with fuming HCl (167 μL). Next, a substrate solution (10 mL) of Oligopin in methanol (∼7 mg mL−1) was prepared. A vial was loaded with 0.5 mL of depolymerisation reagent and 0.5 mL of substrate solution, and was immediately placed in a copper block at 30 °C to perform the reaction. After a certain reaction time (0–90 min), the reaction was stopped by cooling the vial in an ice bath and immediately adding 0.25 mL of a 0.4 M sodium acetate in methanol solution (equimolar amounts of NaOAc and HCl). Afterwards, the product solution was filtered through a 0.2 μm PTFE membrane and analysed using HPLC-UV.HPLC-UV analysis
Prior to HPLC-UV analysis, samples were filtered through a 0.2 μm PTFE membrane to remove any particulate matter and prevent clogging of the column. The high pressure liquid chromatography (HPLC) system used was a Waters e2695 Separations Module, equipped with a Waters 2489 UV/Visible Detector. The column was a Supelcosil LC-18 HPLC Column (5 μm particle size, 25 cm × 4.6 mm), and column temperature was 40 °C. The solvents that were used are: solvent A (100% acetonitrile) and solvent B (95% Milli-Q water, 5% acetonitrile, with 1 mL L−1 85% phosphoric acid). The flow rate was 1 mL min−1, with following linear solvent gradients: 0–16 min, 0% to 20% A; 16–20 min, 20% to 70% A; 20–22 min, 70% A (isocratic); 22–23 min, 70% to 0% A; 23–30 min, 0% A (isocratic). Detection of the eluting compounds was done at 280 nm.Molar response factors of (+)-catechin and (−)-epicatechin at 280 nm were determined using calibration curves with commercial standards. The commercially unavailable compounds (1) and (2) were assigned the same molar response factor as the one for (−)-epicatechin. The furylated products (i.e. 4-(5-methylfuran-2-yl)-(epi)catechin) were also given the same molar response factor as (−)-epicatechin, based on literature data.22,23
UPLC-MS
UPLC-MS was used for the identification of peaks that weren't verified based on commercial standards. The reaction mixture was analysed using an ACQUITY UPLC H-Class System (Waters Co.) equipped with and ACQUITY QDa Mass detector (Waters Co.), controlled by the Empower 3 software (Waters Co.). An injection volume of 2 μL was used. The separation was achieved on an ACQUITY BEH C18 column (1.7 μm, 2.1 mm × 50 mm; Waters Co.), which was kept at 45.0 ± 0.5 °C. The mobile phase consisted of 0.1% formic acid in ultrapure water (A) and 0.1% formic acid in acetonitrile (B) and was delivered at a flow rate of 0.3 mL min−1. Gradient elution was performed as follows: 5% B at 0–0.5 min, 5–15% at 0.5–4 min, 15–70% B at 4–8 min, 70% B at 8–9 min, 70–95% B at 9–10 min, 95% B at 10–11 min, 95–5% B at 11–11.1 min, and then equilibration with 5% B until 13 min. The QDa Detector operated in electrospray (ESI) negative mode with the ESI-probe maintained at 600 °C. Capillary and cone voltages of the QDa Detector were 0.8 kV and 10 V, respectively. Data were collected in SCAN mode (m/z 160.0–950.0) at an acquisition rate of 8 Hz.GC analysis
The monomeric products of the depolymerisation–hydrogenation of procyanidins were qualitatively analysed by GC. Therefore, 1 mL of product mixture (∼3 mg mL−1 of products, dissolved in methanol) was evaporated using a N2 flow. The remaining solid products were derivatised by trimethylsilylation (TMS). Therefore, they were dissolved in 0.5 mL of pyridine and 0.1 mL of N-methyl-N-(trimethylsilyl)trifluoroacetamide. The vial was sealed and put in an oven at 80 °C for 20 min to allow the reaction to complete.Afterwards, the samples were analysed on a GC (Agilent 6890 series) equipped with a HP5-column and a flame ionisation detector (FID). The following operating conditions were used: split ratio (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25), injection temperature of 300 °C, column temperature program: 90 °C, 20 °C min−1 to 200 °C, 10 °C min−1 to 305 °C (5 min hold), with a detection temperature of 310 °C.
:25), injection temperature of 300 °C, column temperature program: 90 °C, 20 °C min−1 to 200 °C, 10 °C min−1 to 305 °C (5 min hold), with a detection temperature of 310 °C.
The identification of the trimethylsilylated monomeric products was performed with GC-MS using an Agilent 6890 series GC equipped with a HP5-MS capillary column and an Agilent 5973 series Mass Spectroscopy detector. The scanning range of the MS was set between 65 and 800 g mol−1.
GPC analysis
The distribution of the molar mass of the procyanidin products was investigated using gel permeation chromatography (GPC). Therefore, a sample of the Oligopin substrate or product mixture was solubilised in THF (∼3 mg mL−1) and subsequently filtered with a 0.2 μm PTFE membrane to remove any particulate matter to prevent plugging of the column. GPC analyses were performed at 40 °C on a Waters E2695 equipped with a PL-Gel 3 μm Mixed-E column with a length of 300 mm, using THF as a solvent with a flow of 1 mL min−1. The detection was UV based at a wavelength of 280 nm. Calibration was based on commercial polystyrene standards of Agilent.1H–13C 2D HSQC NMR analysis
Approximately 60 mg of Oligopin or product mixture was dissolved in 0.5 mL DMSO-d6 and loaded in an NMR tube. For (epi)catechin the amount was ∼20 mg, and for procyanidin A2 and B2, this was ∼1 mg. The two-dimensional 1H–13C HSQC NMR experiment was conducted at 298 K using a Bruker Avance III HD 400 MHz console with a Bruker Ascend™ 400 Magnet, equipped with a 5 mm PABBO probe. A Bruker standard pulse sequence (‘hsqcetgpsp.3’) was used with the following parameters: spectral width in F2 dimension (1H) of 13 ppm using 2048 data points, a spectral width in F1 dimension (13C) of 165 ppm, using 256 data points, a total of 6 scans were recorded with a 2s interscan delay (D1). Bruker's Topspin 4.0.9 software was used for data processing. The solvent peak of DMSO was used as the internal reference (δC/δH: 39.5 ppm/2.49 ppm) following by manually phasing and automatic baseline correction.Conflicts of interest
There are no conflicts to declare.Acknowledgements
R.S. Thanks KULeuven for a posdoctorale PDM mandate. B.S. and M.V.D.B thanks KULeuven for financial support. Especially, Biowood (SBO, Vlaio), Biofact (Excellence of Science, FWO) and NEXTBIOref (iBOF) are acknowledged.References
- A. Corma, S. Iborra and A. Velty, Chemical routes for the transformation of biomass into chemicals, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed
.
- I. Delidovich, P. J. C. Hausoul, L. Deng, R. Pfützenreuter, M. Rose and R. Palkovits, Alternative Monomers Based on Lignocellulose and Their Use for Polymer Production, Chem. Rev., 2016, 116, 1540–1599 CrossRef CAS PubMed
- W. Schutyser, T. Renders, S. Van den Bosch, S. F. Koelewijn, G. T. Beckham and B. F. Sels, Chemicals from lignin: an interplay of lignocellulose fractionation, depolymerisation, and upgrading, Chem. Soc. Rev., 2018, 47, 852–908 RSC
- Z. Sun, B. Fridrich, A. de Santi, S. Elangovan and K. Barta, Bright Side of Lignin Depolymerization: Toward New Platform Chemicals, Chem. Rev., 2018, 118, 614–678 CrossRef CAS PubMed
-
R. W. Hemingway, J. J. Karchesy and S. J. Branham, Chemistry and Significance of Condensed Tannins, Plenum Press, New York and London, 1989 Search PubMed
- A. Arbenz and L. Averous, Chemical modification of tannins to elaborate aromatic biobased macromolecular architectures, Green Chem., 2015, 17, 2626–2646 RSC
- D. E. Garcia, W. G. Glasser, A. Pizzi, S. P. Paczkowski and M.-P. Laborie, Modification of condensed tannins: from polyphenol chemistry to materials engineering, New J. Chem., 2016, 40, 36–49 RSC
- L. Roumeas, C. Aouf, E. Dubreucq and H. Fulcrand, Depolymerisation of condensed tannins in ethanol as a gateway to biosourced phenolic synthons, Green Chem., 2013, 15, 3268–3275 RSC
- A. Scalbert, Antimicrobial properties of tannins, Phytochemistry, 1991, 30, 3875–3883 CrossRef CAS
- R. V. Barbehenn and C. P. Constabel, Tannins in plant–herbivore interactions, Phytochemistry, 2011, 72, 1551–1565 CrossRef CAS PubMed
- Y. Shirmohammadli, D. Efhamisisi and A. Pizzi, Tannins as a sustainable raw material for green chemistry: A review, Ind. Crops Prod., 2018, 126, 316–332 CrossRef CAS
- S. Bianchi, I. Kroslakova, R. Janzon, I. Mayer, B. Saake and F. Pichelin, Characterization of condensed tannins and carbohydrates in hot water bark extracts of European softwood species, Phytochemistry, 2015, 120, 53–61 CrossRef CAS PubMed
- C. Beres, G. N. Costa, I. Cabezudo, N. K. da Silva-James, A. S. Teles, A. P. Cruz, C. Mellinger-Silva, R. V. Tonon, L. M. Cabral and S. P. Freitas, Towards integral utilization of grape pomace from winemaking process: A review, Waste Manage., 2017, 68, 581–594 CrossRef PubMed
-
L. J. Porter, Plant Polyphenols, Springer, 1992, pp. 245–258 Search PubMed
-
D. Ferreira, J. P. J. Marais, C. M. Coleman and D. Slade, in Comprehensive Natural Products II, ed. H.-W. Liu and L. Mander, Elsevier, Oxford, 2010, pp. 605–661 Search PubMed
- H. Nouailhas, C. Aouf, C. Le Guerneve, S. Caillol, B. Boutevin and H. Fulcrand, Synthesis and properties of biobased epoxy resins. part 1. Glycidylation of flavonoids by epichlorohydrin, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2261–2270 CrossRef CAS
- P. Schofield, D. Mbugua and A. Pell, Analysis of condensed tannins: a review, Anim. Feed Sci. Technol., 2001, 91, 21–40 CrossRef CAS
- B. R. Brown and M. R. Shaw, Reactions of flavanoids and condensed tannins with sulphur nucleophiles, J. Chem. Soc., Perkin Trans. 1, 1974, 2036–2049 RSC
- S. Matthews, I. Mila, A. Scalbert, B. Pollet, C. Lapierre, C. L. Hervé du Penhoat, C. Rolando and D. M. Donnelly, Method for estimation
of proanthocyanidins based on their acid depolymerization in the presence of nucleophiles, J. Agric. Food Chem., 1997, 45, 1195–1201 CrossRef CAS
- R. W. Hemingway and G. W. McGraw, Kinetics of acid-catalyzed cleavage of procyanidins, J. Wood Chem. Technol., 1983, 3, 421–435 CrossRef CAS
- J. A. Kennedy and G. P. Jones, Analysis of proanthocyanidin cleavage products following acid-catalysis in the presence of excess phloroglucinol, J. Agric. Food Chem., 2001, 49, 1740–1746 CrossRef CAS PubMed
- L. Rouméas, G. Billerach, C. Aouf, E. r. Dubreucq and H. l. n. Fulcrand, Furylated flavonoids: fully biobased building blocks produced by condensed tannins depolymerization, ACS Sustainable Chem. Eng., 2018, 6, 1112–1120 CrossRef
- G. Billerach, L. Rouméas, E. Dubreucq and H. Fulcrand, Furanolysis with menthofuran: A new depolymerization method for analyzing condensed tannins, J. Agric. Food Chem., 2020, 68(10), 2917–2926 CrossRef CAS PubMed
- T. Renders, G. Van den Bossche, T. Vangeel, K. Van Aelst and B. Sels, Reductive catalytic fractionation: state of the art of the lignin-first biorefinery, Curr. Opin. Biotechnol., 2019, 56, 193–201 CrossRef CAS PubMed
- S. Van den Bosch, W. Schutyser, R. Vanholme, T. Driessen, S. F. Koelewijn, T. Renders, B. De Meester, W. J. J. Huijgen, W. Dehaen, C. M. Courtin, B. Lagrain, W. Boerjan and B. F. Sels, Reductive lignocellulose fractionation into soluble lignin-derived phenolic monomers and dimers and processable carbohydrate pulps, Energy Environ. Sci., 2015, 8, 1748–1763 RSC
- E. M. Anderson, M. L. Stone, M. J. Hülsey, G. T. Beckham and Y. Román-Leshkov, Kinetic studies of lignin solvolysis and reduction by reductive catalytic fractionation decoupled in flow-through reactors, ACS Sustainable Chem. Eng., 2018, 6, 7951–7959 CrossRef CAS
- I. Kumaniaev, E. Subbotina, J. Sävmarker, M. Larhed, M. V. Galkin and J. S. Samec, Lignin depolymerization to monophenolic compounds in a flow-through system, Green Chem., 2017, 19, 5767–5771 RSC
- J. M. Pepper and H. Hibbert, Studies on lignin and related compounds. LXXXVII. High pressure hydrogenation of Maple Wood, J. Am. Chem. Soc., 1948, 70, 67–71 CrossRef CAS PubMed
- P. J. Steynberg, J. P. Steynberg, B. C. Bezuidenhoudt and D. Ferreira, Oligomeric flavanoids. Part 19. Reductive cleavage of the interflavanyl bond in proanthocyanidins, J. Chem. Soc., Perkin Trans. 1, 1995, 3005–3012 RSC
- L. Y. Foo, Polymeric proanthocyanidins of Photinia glabrescens, modification of molecular weight and nature of products from hydrogenolysis, Phytochemistry, 1982, 21, 1741–1746 CrossRef CAS
- G.-q. Jiang, G.-z. Fang, L.-l. Li, Z.-x. Shi and Z.-r. Zhang, Study on antioxidant activity of catalyzed hydrogen degradation product of polymeric proanthocyanidins (LPPC) from larix gmelinii bark, BioResources, 2014, 9, 662–672 CAS
- Z. Li, J. Zeng, Z. Tong, Y. Qi and L. Gu, Hydrogenolytic depolymerization of procyanidin polymers from hi-tannin sorghum bran, Food Chem., 2015, 188, 337–342 CrossRef CAS PubMed
- H. Zhu, P. Li, S. Ren, W. Tan and G. Fang, Low-Cost Ru/C-Catalyzed Depolymerization of the Polymeric Proanthocyanidin-Rich Fraction from Bark To Produce Oligomeric Proanthocyanidins with Antioxidant Activity, ACS Omega, 2019, 4, 16471–16480 CrossRef CAS PubMed
- H. Zhu, L. Ni, S. Ren, G. Fang and S. Li, A composite carbon-based solid acid-supported palladium catalyst (Pd/C-SO 3 H) for hydrogenolysis of plant-derived polymeric proanthocyanidins, RSC Adv., 2020, 10, 20665–20675 RSC
- M. Zhou, X. Chen, C. Gao, L. Ni, X. Wang, W. Zhang and S. Ren, Catalytic hydrogenolysis of larix bark proanthocyanidins in ionic liquids produces UV blockers with potential for use in cosmetics, RSC Adv., 2021, 11, 30078–30087 RSC
- A. P. Kozikowski, W. Tückmantel and C. George, Studies in Polyphenol Chemistry and Bioactivity. 2.1 Establishment of Interflavan Linkage Regio-and Stereochemistry by Oxidative Degradation of an O-Alkylated Derivative of Procyanidin B2 to (R)-(−)-2, 4-Diphenylbutyric Acid, J. Org. Chem., 2000, 65, 5371–5381 CrossRef CAS PubMed
- D. Ferreira and X.-C. Li, Oligomeric proanthocyanidins: naturally occurring O-heterocycles, Nat. Prod. Rep., 2000, 17, 193–212 RSC
- K. Ohmori, N. Ushimaru and K. Suzuki, Oligomeric catechins: An enabling synthetic strategy by orthogonal activation and C (8) protection, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 12002–12007 CrossRef CAS PubMed
- Oligopin® – Pine bark extract by DRT, DRT, https://www.drt.fr/en/markets/oligopin/, 18/11/2022.
- L. Segal, M. Penman and Y. Piriou, Evaluation of the systemic toxicity and mutagenicity of OLIGOPIN®, procyanidolic oligomers (OPC) extracted from French Maritime Pine Bark extract, Toxicol. Rep., 2018, 5, 531–541 CrossRef CAS PubMed
- M. Jerez, S. Touriño, J. Sineiro, J. L. Torres and M. J. Núñez, Procyanidins from pine bark: Relationships between structure, composition and antiradical activity, Food Chem., 2007, 104, 518–527 CrossRef CAS
- G. Cleaver, Improved GPC/SEC analysis of low molecular weight polyphenols with Agilent OligoPore columns, Agilent Technologies, https://www.agilent.com/cs/library/applications/5991-0487EN.pdf, 18/11/2022.
- D. Jacques, E. Haslam, G. R. Bedford and D. Greatbanks, Plant proanthocyanidins. Part II. Proanthocyanidin-A2 and its derivatives, J. Chem. Soc., Perkin Trans. 1, 1974, 2663–2671 RSC
- Unique process for the extraction of OPCs (procyanidins of low molecular weight), DRT, https://www.theoriginalpinebarkextract.com/02-crystal-pure-2/, 18/11/2022.
- T. Vangeel, D. M. Neiva, T. Quilho, R. A. Costa, V. Sousa, B. F. Sels and H. Pereira, Tree bark characterization envisioning an integrated use in a biorefinery, Biomass Convers. Biorefin., 2021, 1–15 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc04470f |
| This journal is © The Royal Society of Chemistry 2023 |