
Reversing Mg suppression effect on Co-site water oxidation of MgCo2O4 based on vanadium-atom electronic affinity synergy with Mg sites toward electronic redistribution†
Hui
Zhang‡
a,
Hui
Han‡
a,
Xuan
Yang
a,
Hongyu
Ma
a,
Zhifei
Song
ab and
Xuqiang
Ji
 *a
*a
aCollege of Materials Science and Engineering, Institute for Graphene Applied Technology Innovation, Qingdao University, Qingdao 266071, Shandong, China. E-mail: xuqianglucky@163.com
bSchool of Electromechanic Engineering, Qingdao University, Qingdao 266071, China
First published on 31st October 2023
Abstract
Active-site regeneration from d-band-center control engineering and electronic redistribution based on target-atom implantation is of enormous importance for efficient water oxidation. Here, low-activity Co catalytic sites of MgCo2O4 are reactivated for large-current (500 mA cm−2) and high-efficiency water oxidation through vanadium-atom electronic affinity synergy with Mg sites toward charge rearrangement around Co sites. Interestingly, very little vanadium (1.3 wt%) was implanted into MgCo2O4 to achieve atom-scale structure tailoring and electronic redistribution, reversing the Mg suppression effect on Co-site water oxidation of MgCo2O4. Vanadium-implantation-tailored MgCo2O4 coupled with MgO (V–MgCo2O4@MgO) exhibits significantly enhanced oxygen evolution reaction performance, with low overpotentials of 240 and 290 mV for 100 and 500 mA cm−2, respectively, in 1 M KOH. Operating at 500 mA cm−2, V–MgCo2O4@MgO has good catalytic stability for at least 20 hours. This work constructs excellent catalysts through atomic-level structural regulation and provides a new perspective for the principle of electronic-affinity synergy between V and Mg species toward Co–O bond optimization.
1. Introduction
The increasing global energy crisis and serious environmental pollution have forced researchers to develop sustainable clean energy without secondary pollution.1–3 Hydrogen (H2) as an ideal substitute for traditional fossil fuels with high energy density can be produced via electrochemical water splitting driven by electricity from wind or solar energy.4–6 The process involves the cathodic two-electron hydrogen evolution reaction (HER) and the anodic four-electron oxygen evolution reaction (OER), and serves as the most promising route among various H2-production strategies.7–9 The OER counterpart suffers from slow kinetics and high overpotentials, enormously limiting the efficiency of water splitting (2H2O → 2H2 + O2). Tremendous effort has been devoted to exploring stable and efficient electrocatalysts10 to facilitate water splitting. Noble-metal materials such as Pt, Ir and Ru are still considered as the most advanced catalytic electrodes,11,12 but high cost and scarcity severely restrict their scaled-up utilization.Non-precious-metal-based catalysts with advantages of large abundance, easy synthesis and low price were explored to catalyze water decomposition including oxides,13 phosphides,14 nitrides,15 hydroxides,16etc. Among them, the AB2O4-type spinel structure has attracted more attention whose tetrahedral and octahedral geometry can better accommodate various transition-metal cations17–20 for excellent structural flexibility. Cobalt-relevant spinel materials, e.g. NiCo2O4,21 CuCo2O4,22 MnCo2O4,19 FeCo2O4,23 ZnCo2O422 and LiCo2O4,24 are widely utilized as water-splitting catalysts.
Spinel-type oxides formed by Mg and transition-metal elements often demonstrate poor electrocatalytic performance.25,26 For example, Kumar et al. reported that MgFe2O4 is a promising material for semiconductor photocatalytic water splitting, but its low electro-conductivity hinders the electrocatalytic application.27 Even if changing Fe sites into the Co element toward MgCo2O4, the as-synthesized urea- and (nitrilotriacetic acid)-stabilized MgCo2O4 catalysts for electrochemical water oxidation require overpotentials of 463 and 573 mV, respectively, for 10 mA cm−2 catalytic current density in 1 M KOH.28 Indeed, associated electrochemical performances are extremely unsatisfactory, necessitating abundant scientific input to promote fundamental understanding of catalytic activity enhancement.29,30 Qiu et al. constructed a MgCo2O4@WO3 core–shell heterostructure by a simple one-step hydrothermal coordination for rapid electron transfer of MgCo2O4.31
The lower electronegativity of Mg in the Mg-relevant spinel structure may cause large electron density of adjacent transition-metal sites toward undesirable intermediate binding.32 Compared with Mg, however, the smaller atomic size and higher electron-affinity ability of V make it an intriguing atom which can be implanted into a spinel platform, destroying the lattice periodicity of the catalysts33,34 and balancing the electronic distribution. Notably, V-atom-utilization was also proven as a pivotal way to regulate the d-band center of active sites toward appropriate d-band intensity and bandwidth.35,36 Moreover, the synergistic protocol of V and Mg for electronic structure regulation of adjacent metal sites is also ambiguous and must be interesting.37,38
Based on the above consideration, we achieved the co-occurrence of the vanadium and Mg atoms in the MgCo2O4 platform as V–MgCo2O4@MgO (vanadium-doping-tailored MgCo2O4 coupled with MgO) to influence the adjacent Co active sites for optimizing electronic structure. Unlike the low catalytic activity of the MgCo2O4 sample, the appearance of few V atoms (1.3 wt%) endows MgCo2O4 with superhigh water-oxidation activity and long-term large-current catalytic stability. V–MgCo2O4@MgO exhibits significantly reinforced OER performance, with low overpotentials of 240 and 290 mV for 100 and 500 mA cm−2, respectively, in 1 M KOH. Operating at 500 mA cm−2, V–MgCo2O4@MgO presents good catalytic stability for at least 20 hours.
2. Experimental
2.1 Materials
CoCl2·6H2O, MgCl2·6H2O, VCl3 and urea were purchased from Aladdin Co Ltd. (China). All chemical reagents are used without further purification. Deionized water was manufactured with a Millipore system and used in all experiments.2.2 Synthesis of V–MgCo2O4@MgO and MgCo2O4 on Ni foam
Ni foam (2 cm × 4 cm) was ultrasound-treated in 5 wt% hydrochloric acid for 10 minutes, and subsequently washed with distilled water and alcohol. CoCl2·6H2O (8 mmol), MgCl2·6H2O (4 mmol), VCl3 (1 mmol) and urea (16 mmol) were dissolved in 30 mL water to form a light red solution. Then, the mixed solution was transferred to a Teflon-lined autoclave with NF immersed in the solution. The autoclave was sealed and kept at 120 °C for 10 h. The as-obtained V–CoOOH@MgCO3 was pyrolyzed at 400 °C in an Ar atmosphere for 4 h to obtain V–MgCo2O4@MgO.The synthesis process of CoOOH@MgCO3 was similar to that of V–CoOOH@MgCO3 without V species. CoOOH@MgCO3 was annealed at 400 °C to obtain MgCo2O4@MgO.
2.3 Characterization
X-ray diffraction (XRD) data were collected on a LabX XRD-6100 X-ray diffractometer equipped with a Cu Kα radiation source. X-ray photoelectron spectroscopy (XPS) results are recorded on an ESCALABMK II ray photoelectron spectrometer (Mg is the excitation source). Scanning electron microscopy (SEM) measurements were performed on a Hitachi S-4800 field emission scanning electron microscope with an accelerating voltage of 20 kV. Transmission electron microscopy (TEM) measurements were carried out with a Zeiss Libra 200FE transmission electron microscope operated at 200 kV. Inductively coupled plasma optical emission spectrometry (ICP-OES) was performed on a PlasmaPro100 Cobra300, Oxford.Electrochemical data were collected on a CHI 760e electrochemical analyzer with a scan rate of 5 mV s−1 in 1 M KOH, utilizing the as-prepared sample (0.5 × 0.5 cm2) as the working electrode, platinum as the counter electrode, and Hg/HgO as the reference electrode. The equation of “E vs. RHE = E vs. Hg/HgO + (0.098 + 0.0591 pH) V” was used to convert IR corrected LSV curves. The overpotential for the OER was obtained via η = E vs. RHE − 1.23 V.
3. Results and discussion
Fig. 1a shows multi-step preparation processes of V–MgCo2O4@MgO and MgCo2O4@MgO, which are described in detail in the Experimental section. Combining the hydrothermal coordination strategy and calcination treatment, very few vanadium atoms entered the MgCo2O4@MgO system as V–MgCo2O4@MgO, alleviating the Mg suppression for Co-site water oxidation. Unlike the low water-oxidation activity of MgCo2O4@MgO, Co sites in V–MgCo2O4@MgO are activated with the optimized electronic structure and Co–O bond. V–MgCo2O4@MgO was characterized by transmission electron microscopy (TEM) and scanning electron microscopy (SEM). The TEM image in Fig. 1b reveals the rod-like morphology of V–MgCo2O4@MgO constituted by nanoparticles (with a diameter ca. 60 nm), which is beneficial for active-site exposure and electrolyte permeation. The selected area electron diffraction (SAED) pattern of V–MgCo2O4@MgO (inset in Fig. 1b) well displays typical diffraction rings corresponding to the (220) and (111) planes of spinel MgCo2O4 and the (200) plane of MgO, consistent with X-ray powder diffraction (XRD) results (Fig. 2a). The related high-resolution TEM (HRTEM) image of V–MgCo2O4@MgO also reveals the existence of the (220) and (111) planes of spinel MgCo2O4 and the (200) plane of MgO (Fig. 1f) with plane spacings of 0.28, 0.46 and 0.22 nm, respectively. For SEM characterization, V–MgCo2O4@MgO (Fig. 1d and h) demonstrates a rod-like feature with a rough surface. In addition, the energy dispersive X-ray (EDX) element mapping is shown in Fig. 1j, confirming the elemental composition of Mg, Co, O and V. Abundant V–MgCo2O4@MgO was deposited on Ni foam (Fig. S1†). For comparison, the SEM and EDX element mapping images of MgCo2O4@MgO and V–CoOOH@MgCO3 are shown in Fig. S2.† As the precursor to prepare V–MgCo2O4@MgO, vanadium-containing CoOOH@MgCO3 (V–CoOOH@MgCO3) presents a smooth nanowire morphology (Fig. 1c and g), uniformly grown on nickel foam. MgCo2O4@MgO (Fig. 1e and i) presents a curved nanowire morphology with a zigzag surface.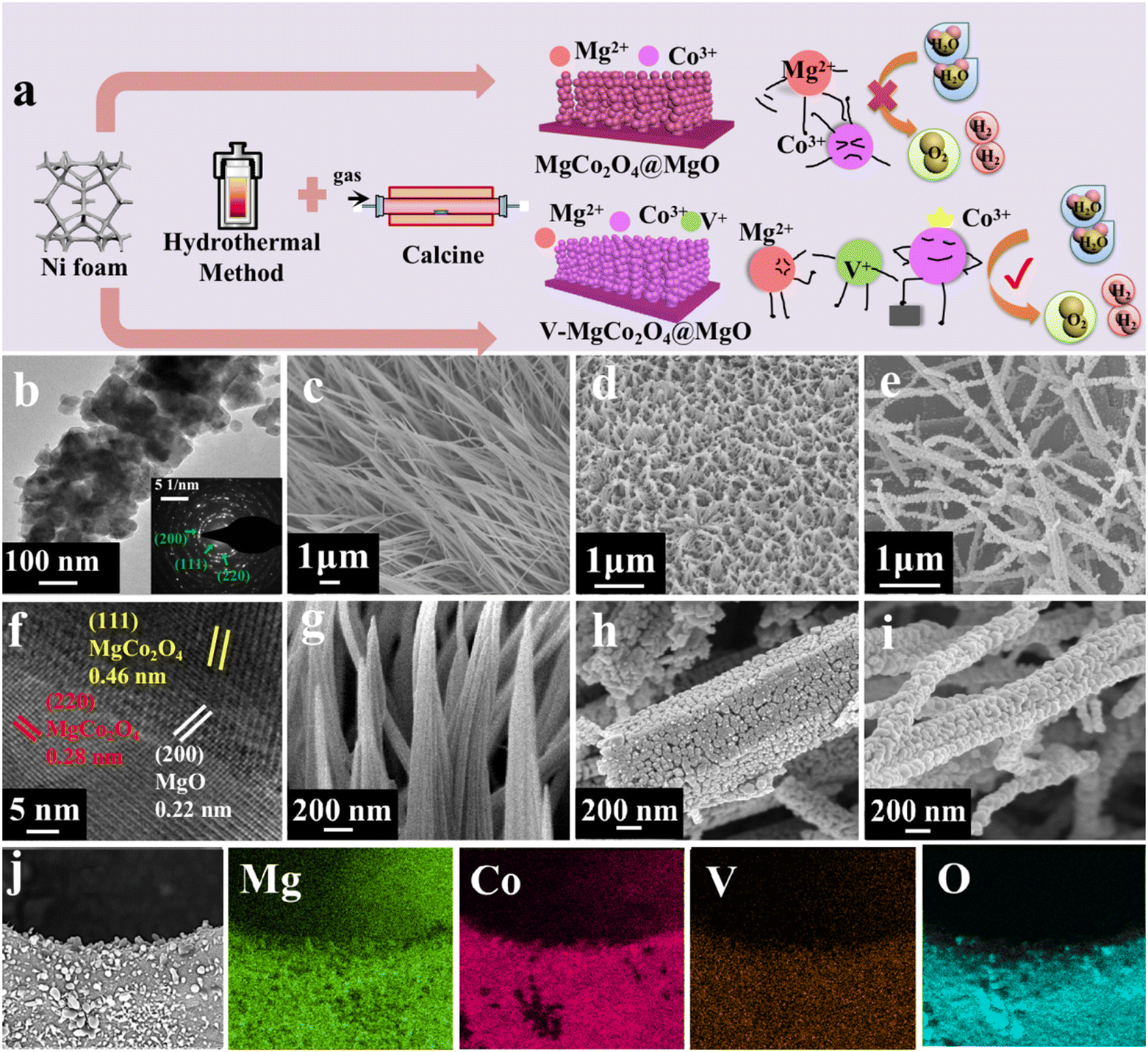 | ||
| Fig. 1 Material preparation and characterization. (a) Schematic diagram for V–MgCo2O4@MgO and MgCo2O4@MgO preparation. (b) TEM image and SAED pattern of V–MgCo2O4@MgO. (f) HRTEM image of V–MgCo2O4@MgO. (c and g) V–CoOOH@MgCO3, (d and h) V–MgCo2O4@MgO, (e and i) MgCo2O4@MgO SEM images. (j) EDX elemental mapping of Mg, Co, V and O for V–MgCo2O4@MgO. | ||
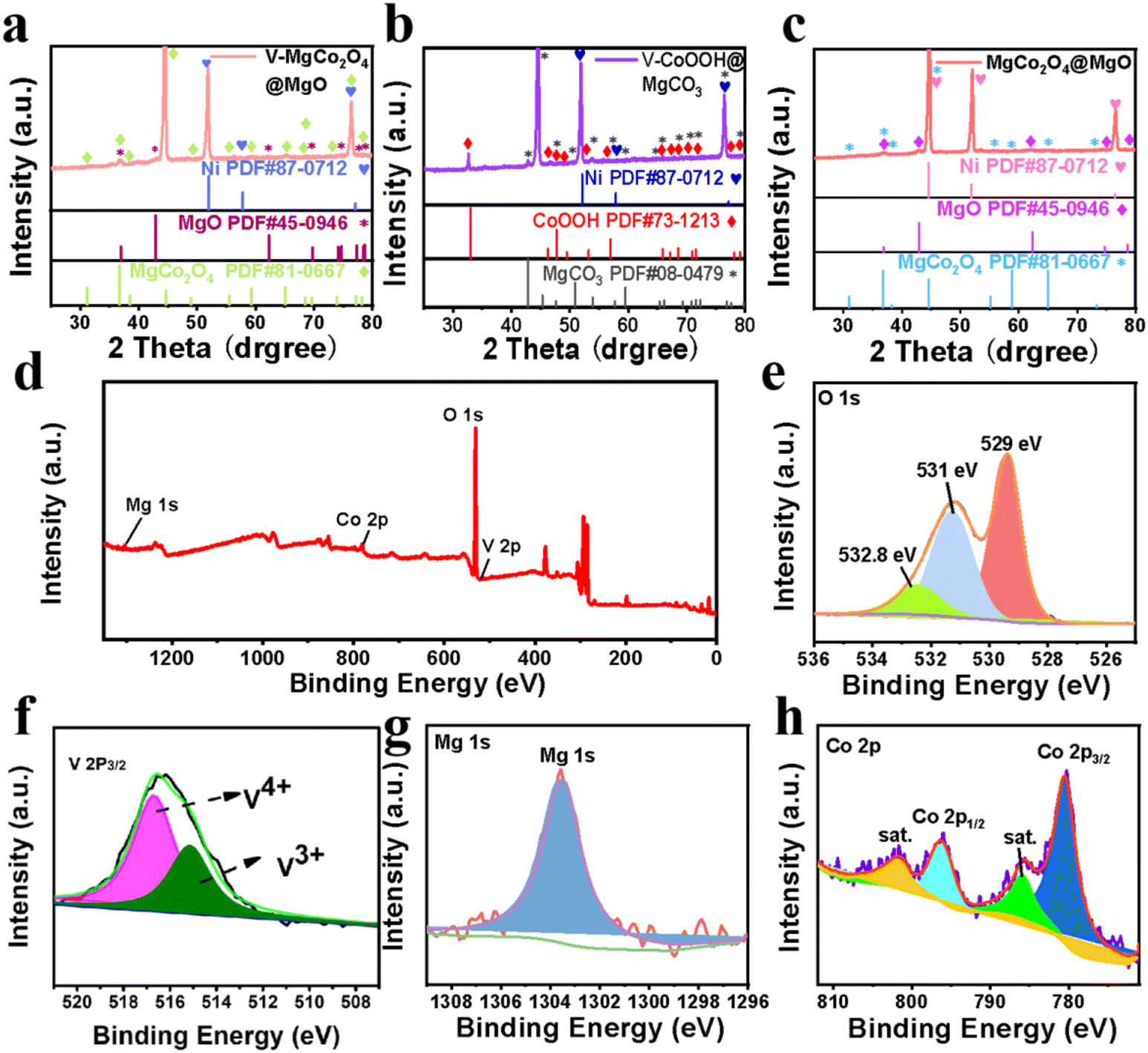 | ||
| Fig. 2 XRD and XPS analyses. XRD spectra of (a) V–MgCo2O4@MgO, (b) V–CoOOH@MgCO3 and (c) MgCo2O4@MgO. (d) XPS survey spectrum of V–MgCo2O4@MgO. XPS spectra of (e) O 1s, (f) V 2p, (g) Mg 1s, and (h) Co 2p for V–MgCo2O4@MgO. | ||
Fig. 2a and c show the XRD patterns of V–MgCo2O4@MgO and MgCo2O4@MgO, respectively. For V–MgCo2O4@MgO, XRD characteristic peaks of MgCo2O4 (JCPDS NO. 81-0667) and MgO (JCPDS No. 45-0946) are detected, (Fig. 2a) which are similar to that of MgCo2O4@MgO (Fig. 2c). In the V–MgCo2O4@MgO XRD curve, the characteristic signal of vanadium species was not found, indicating vanadium-atom doping into MgCo2O4@MgO as V–MgCo2O4@MgO. The mass content of V in V–MgCo2O4@MgO is measured as 1.3 wt% using an inductively coupled plasma optical emission spectrometer (ICP-OES). The content of Mg, Co and O elements in V–MgCo2O4@MgO was 13.7, 53.9 and 31.1 wt%, respectively. As the precursor to prepare V–MgCo2O4@MgO, V–CoOOH@MgCO3 (Fig. 2b) presents XRD peaks of CoOOH (JCPDS No. 73-1213) and MgCO3 (JCPDS No. 08-0479). Fig. 2d shows the X-ray photoelectron spectroscopy (XPS) survey spectrum of V–MgCo2O4@MgO, evidencing the existence of Mg, Co, O and V elements, consistent with EDX results (Fig. 1j). The measured O 1s XPS spectrum of V–MgCo2O4@MgO presents three peaks at ∼532.8, ∼531 and ∼529 eV (Fig. 2e). The 532.8 eV peak is related to water on the V–MgCo2O4@MgO surface. The 531 eV signal is ascribed to the oxygen vacancy. The binding energy at 529 eV is due to the typical metal–oxygen bond.39 The V 2p2/3 peaks involve two subpeaks at 515.7 and 516.6 eV (Fig. 2f), which may be attributed to distinct valence states of V (V3+ and V4+).40 The large electronegativity of high-valence-state V4+ in V–MgCo2O4@MgO can achieve strong electronic affinity synergy with Mg/Co sites. For the MgCo2O4@MgO sample, the V 2p XPS signal is absent (Fig. S3†). The characteristic XPS peak of Mg 1s for V–MgCo2O4@MgO is observed at 1303.5 eV (Fig. 2g).41 Two main peaks of Co 2p at 780.6 and 796.6 eV (Fig. 2h) are assigned to Co 2p3/2 and Co 2p1/2, respectively, suggesting the existence of Co3+.42 Differing from MgCo2O4@MgO (Co 2p3/2: 780.38 eV; Co 2p1/2: 796.58 eV, Fig. S3†), the larger Co 2p binding energies of V–MgCo2O4@MgO reveal the higher oxidation state of Co atoms in V–MgCo2O4@MgO.
The OER performance of V–MgCo2O4@MgO was studied using a standard three-electrode system in 1 M KOH at room temperature. The precursor to prepare V–MgCo2O4@MgO (vanadium-containing CoOOH@MgCO3, denoted as V–CoOOH@MgCO3), and MgCo2O4@MgO are also used as comparative samples (Fig. 3a). It can be seen that the overpotential of V–MgCo2O4@MgO for 100 mA cm−2 is about 240 mV, which is significantly lower than that of V–CoOOH@MgCO3 (η100 = 361 mV) and MgCo2O4@MgO (η100 = 394 mV), indicating that the introduction of vanadium is of great benefit for improving OER performance. The OER kinetics of the catalyst can be evaluated using the Tafel slope (Fig. 3b). The smaller Tafel slope for V–MgCo2O4@MgO (108.13 mV dec−1) than V–CoOOH@MgCO3 (113.24 mV dec−1) and MgCo2O4@MgO (134.5 mV dec−1) reveals the faster OER kinetics and higher charge transfer rate of V–MgCo2O4@MgO. V–MgCo2O4@MgO only needs overpotentials of 261.5 and 274.7 mV for 150 and 200 mA cm−2 (Fig. 3c), respectively, significantly lower than V–CoOOH@MgCO3 (η150 = 379.5 mV; η200 = 393 mV) and MgCo2O4@MgO (η150 = 419.2 mV; η200 = 437 mV). Cyclic voltammetry (CV) test was conducted to measure the double-layer capacitance (Cdl) with scan rates ranging from 10 to 100 mV s−1 (Fig. S4a–c†). Based on Cdl values (Fig. 3d), the electrochemical surface area (ECSA) can be calculated (Fig. S4d†). The Cdl (51.27 mF cm−2) and ECSA (320.44 cm2) of V–MgCo2O4@MgO were larger than those of V–CoOOH@MgCO3 (Cdl: 42.08 mF cm−2; ECSA: 263 cm2) and MgCo2O4@MgO (Cdl: 32.66 mF cm−2; ECSA: 204.13 cm2), indicating the better active-site exposure of V–MgCo2O4@MgO. In order to comprehensively evaluate V–MgCo2O4@MgO, we also investigated the catalytic stability of the OER by multistep chronopotentiometry (Fig. 3e), setting the current from 10 mA cm−2 to 100 mA cm−2. It can be seen that the potential of V–MgCo2O4@MgO responds rapidly to a certain value and can be stable for 500 s. Finally, we drove a long-term chronoamperometric test for 24 h at 500 mA cm−2 (Fig. 3f). Clearly, after 24 h, V–MgCo2O4@MgO still presents 400 mA cm−2 catalytic current density (80% of the initial current), suggesting the long-term stability of V–MgCo2O4@MgO and its potential in practical application. V–MgCo2O4@MgO after 24 h stability test still maintains its original morphology with almost no nanowire breakage or shedding (Fig. S5a–f†). The OER performance of V–MgCo2O4@MgO is comparable to some excellent catalysts reported recently (Fig. 3g and Table S1†).
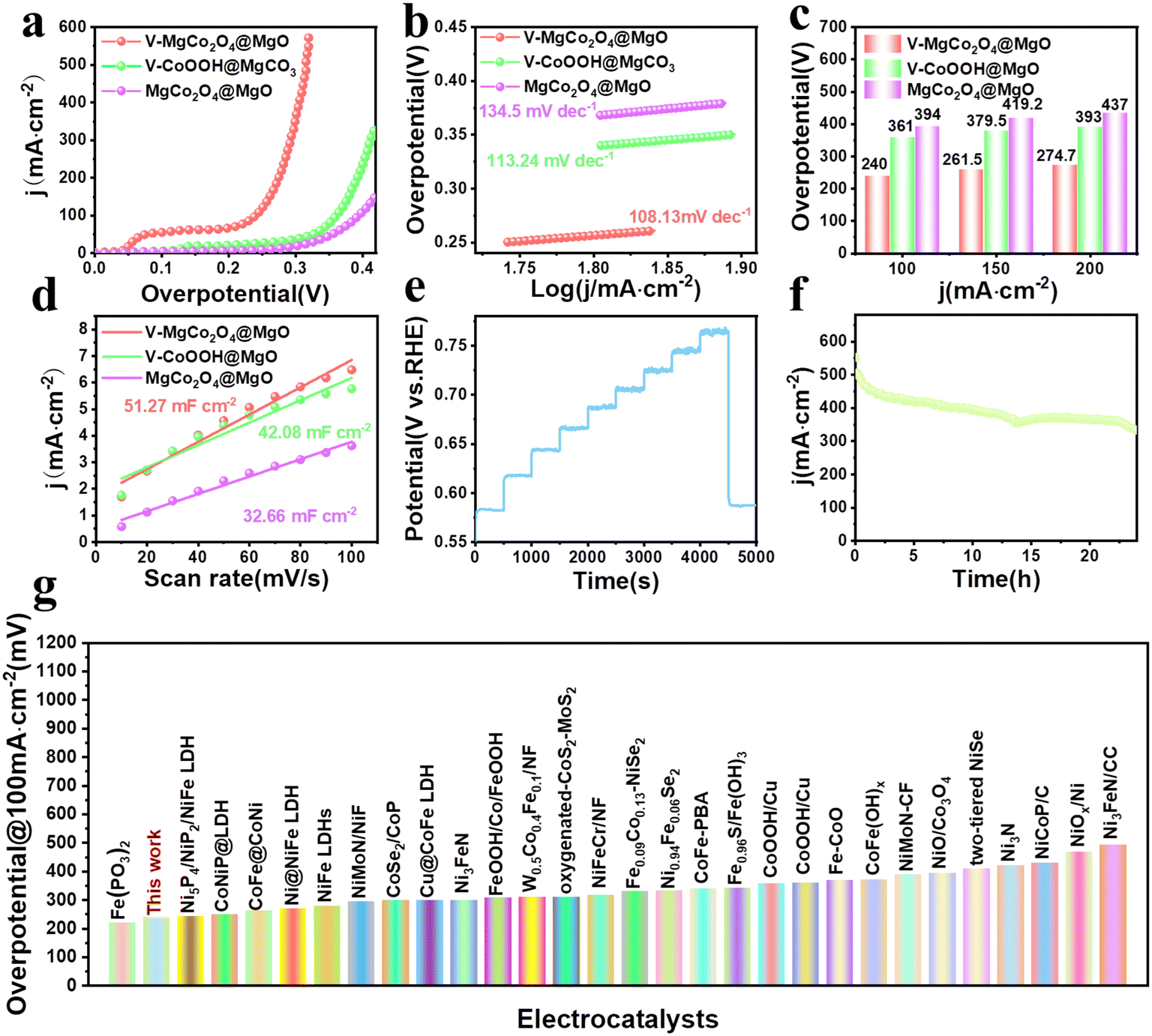 | ||
| Fig. 3 Evaluation of OER performance. (a) LSV curves and (b) Tafel plots of V–MgCo2O4@MgO, MgCo2O4@MgO and V–CoOOH@MgCO3 at a scan rate of 5 mV s−1. (c) The required overpotentials for V–MgCo2O4@MgO, V–CoOOH@MgCO3 and MgCo2O4@MgO to drive 100, 150, and 200 mA cm−2. (d) Linear plot of capacitive current dependent on scan rate for V–MgCo2O4@MgO (R2 = 0.9887), V–CoOOH@MgCO3 (R2 = 0.9856) and MgCo2O4@MgO (R2 = 0.9912). (e) Multistep chronopotentiometry test for the V–MgCo2O4@MgO electrode in 1.0 M KOH without IR correction. The current density ranges from 10 mA cm−2 to 100 mA cm−2, with an increment of 10 mA cm−2 per 500 s. (f) Time-dependent current density curve for V–MgCo2O4@MgO. (g) Comparison of overpotential for V–MgCo2O4@MgO at 100 mA cm−2 with other electrocatalysts recently reported in 1 M KOH. | ||
In view of the good OER performance of V–MgCo2O4@MgO, we also evaluate the associated HER activity. As shown in Fig. 4a, V–MgCo2O4@MgO requires an overpotential of 222 mV for 50 mA cm−2, lower than that of V–CoOOH@MgCO3 (η50 = 267 mV) and MgCo2O4@MgO (η50 = 342 mV). To drive large current densities of 100 and 200 mA cm−2 (Fig. 4c), V–MgCo2O4@MgO demands overpotentials of 262 and 303 mV, respectively, outperforming V–CoOOH@MgCO3 (η100 = 312 mV; η200 = 362 mV) and MgCo2O4@MgO (η100 = 379 mV; η200 = 422 mV). As observed in Fig. 4b, the Tafel slope of V–MgCo2O4@MgO (122.61 mV dec−1) is smaller than that of V–CoOOH@MgCO3 (147.01 mV dec−1) and MgCo2O4@MgO (160.16 mV dec−1), indicating the faster HER kinetics of V–MgCo2O4@MgO. The LSV curve for V–MgCo2O4@MgO after 2000 consecutive CV scanning is similar to the initial polarization curve (Fig. 4d), revealing good catalytic stability. As shown in Fig. 4e, the multi-step chronopotentiometry measurement curve of V–MgCo2O4@MgO shows that it has a rapid and stable response to the change of HER current density. In addition, the 24 h chronoamperometric measurement curve of V–MgCo2O4@MgO at 10 mA cm−2 (Fig. 4f) showed only small fluctuations. The HER performance of V–MgCo2O4@MgO is better than many recently reported non-noble metal electrocatalysts (Fig. 4g and Table S2†).
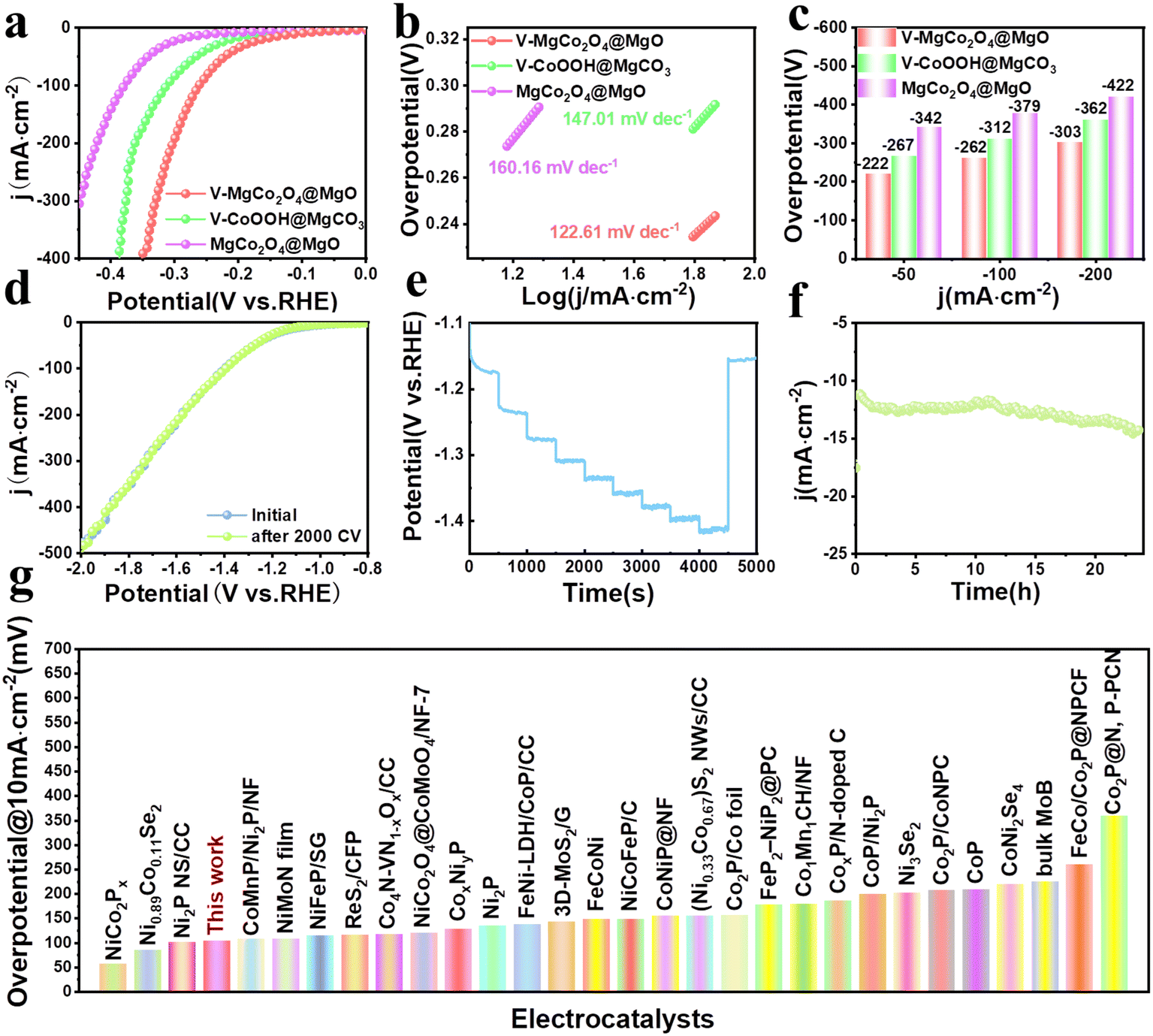 | ||
| Fig. 4 Evaluation of HER performance. (a) LSV curves and (b) Tafel plots of V–MgCo2O4@MgO, V–CoOOH@MgCO3 and MgCo2O4@MgO at a scan rate of 5 mV s−1. (c) The required overpotentials for V–MgCo2O4@MgO, V–CoOOH@MgCO3 and MgCo2O4@MgO to drive −50, −100, and −200 mA cm−2. (d) LSV curves of V–MgCo2O4@MgO before and after 2000 CV cycles. (e) Multicurrent process obtained with the V–MgCo2O4@MgO electrode in 1.0 M KOH without IR correction. The current density ranges from 10 mA cm−2 to 100 mA cm−2, with an increment of 10 mA cm−2 per 500 s. (f) Time-dependent current density curve for V–MgCo2O4@MgO. (g) Comparison of overpotential for V–MgCo2O4@MgO at 10 mA cm−2 with other electrocatalysts recently reported in 1 M KOH. | ||
After the evaluation of HER and OER catalytic activities of V–MgCo2O4@MgO, we assemble a V–MgCo2O4@MgO‖V–MgCo2O4@MgO alkaline electrolyzer to examine its catalytic ability (Fig. 5a). As shown in Fig. 5b, at a current density of 50 mA cm−2, the voltage of the V–MgCo2O4@MgO catalyst is 1.69 V, which is lower than that of V–CoOOH@MgCO3 (1.82 V) and MgCo2O4@MgO (1.9 V), and very close to the potential difference (△V) between the OER and HER of 1.64 V (Fig. 5c). The overall water decomposition process of V–MgCo2O4@MgO can be described in Fig. 5d. Upon continuous water electrolysis after 2000 CV cycles for the V–MgCo2O4@MgO‖V–MgCo2O4@MgO alkaline electrolyzer, the LSV curve is similar to the original curve (Fig. 5e). For the 24 h chronoamperometric measurement of the V–MgCo2O4@MgO‖V–MgCo2O4@MgO alkaline electrolyzer, the recorded curve fluctuates up and down only in a small range (Fig. 5f), evidencing the excellent operation stability of the V–MgCo2O4@MgO‖V–MgCo2O4@MgO electrolyzer.
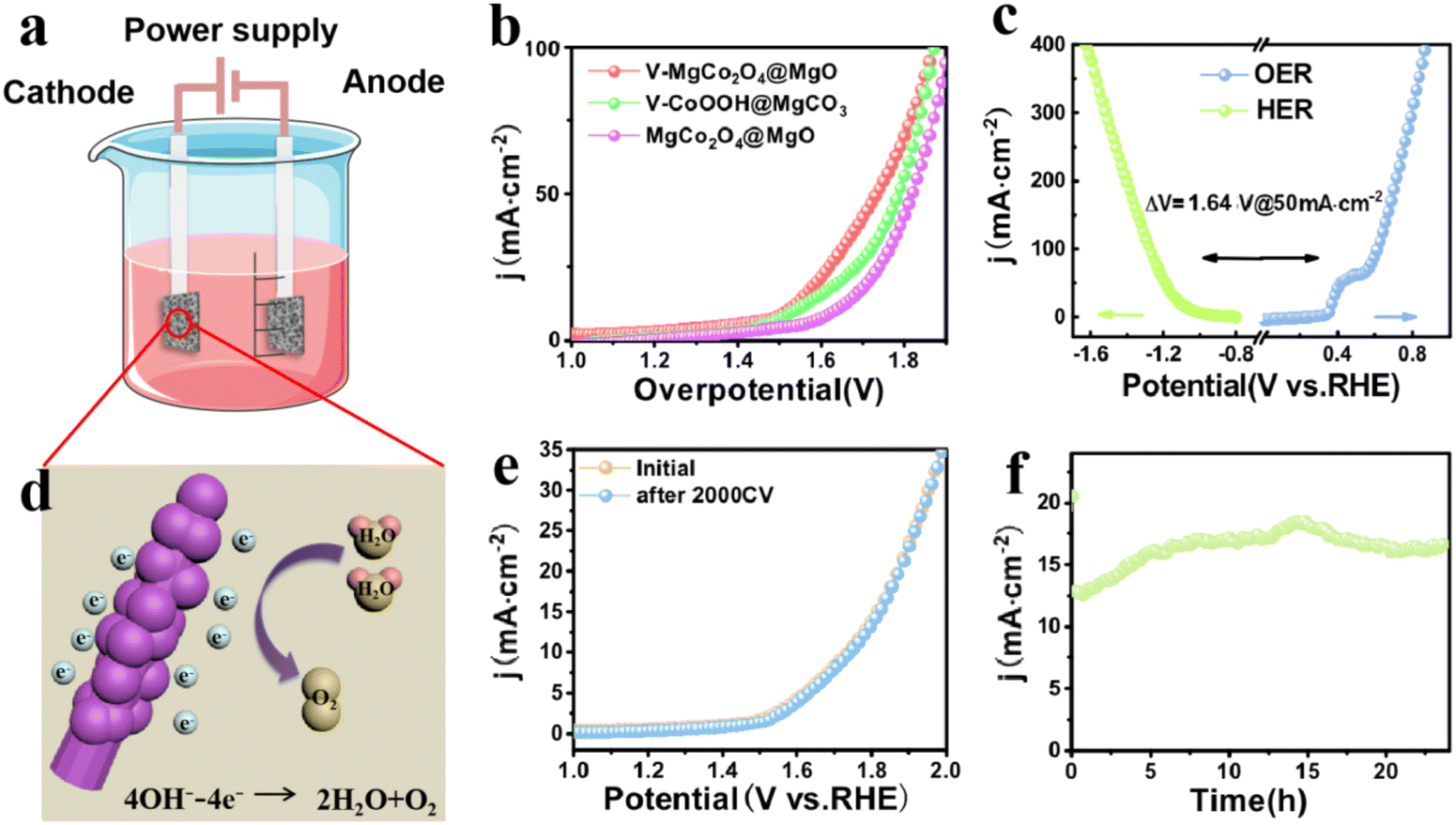 | ||
| Fig. 5 Evaluation of overall water splitting performance. (a) Schematic of the two-electrode water-splitting electrolyzer with V–MgCo2O4@MgO as both the anode and cathode. (b) LSV curves of V–MgCo2O4@MgO‖V–MgCo2O4@MgO, MgCo2O4@MgO‖MgCo2O4@MgO, and V–CoOOH@MgCO3‖V–CoOOH@MgCO3 two-electrode configurations at a scan rate of 5 mV s−1. (c) The polarization curves of V–MgCo2O4@MgO for the HER and OER. (d) Schematic diagram of V–MgCo2O4@MgO for water splitting performance in 1 M KOH. (e) LSV curves of V–MgCo2O4@MgO‖V–MgCo2O4@MgO before and after 2000 CV cycles. (f) Time-dependent current density curve for the two-electrode configuration of V–MgCo2O4@MgO‖V–MgCo2O4@MgO. | ||
4. Conclusions
In summary, target-atom vanadium was implanted into the MgCo2O4@MgO system to cooperate with Mg sites, optimizing the Co-site electronic structure as the V–MgCo2O4@MgO sample through hydrothermal coordination and calcination strategies. The introduction of vanadium reverses the Mg suppression effect on Co-site water oxidation. Low-activity Co catalytic sites of MgCo2O4 are reactivated for large-current (500 mA cm−2) alkaline water oxidation with low overpotentials of 240 and 290 mV for 100 and 500 mA cm−2, respectively, in 1 M KOH. Operating at 500 mA cm−2, V–MgCo2O4@MgO has good catalytic stability for at least 20 hours. This work not only presents excellent catalysts through atomic-level structural tailoring, but also throws light on superior electronic affinity synergy between V and Mg species toward Co–O bond optimization.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was financially supported by the Natural Science Foundation of Shandong Province (ZR2020QB068). We also appreciate Jian Zhou from Measurement and Control Lab for assistance with the experiment.Notes and references
- D. Wu, Y. Wei, X. Ren, X. Ji, Y. Liu, X. Guo, Z. Liu, A. Asiri, Q. Wei and X. Sun, Adv. Mater., 2018, 30, 1705366 CrossRef PubMed.
- K. Tang, X. Wang, M. Wang, Y. Xie, J. Zhou and C. Yan, ChemElectroChem, 2017, 4, 2150–2157 CrossRef CAS.
- N. Dalai, B. Mohanty, A. Mitra and B. Jena, ChemistrySelect, 2019, 4, 7791–7796 CrossRef CAS.
- T. Gao, Z. Jin, M. Liao, J. Xiao, H. Yuan and D. Xiao, J. Mater. Chem. A, 2015, 3, 17763–17770 RSC.
- X. Li, Y. Fang, F. Li, M. Tian, X. Long, J. Jin and J. Ma, J. Mater. Chem. A, 2016, 4, 15501–15510 RSC.
- H. Jin, S. Liu, L. Pei, G. Li, Z. Ma, W. Bai, S. Wu, Y. Yuan and J. Zhong, RSC Adv., 2021, 11, 22467–22472 RSC.
- Y. Wang, Y. Zhang, Z. Liu, C. Xie, S. Feng, D. Liu, M. Shao and S. Wang, Angew. Chem., Int. Ed., 2017, 56, 5867–5871 CrossRef CAS PubMed.
- C. Sathiskumar, S. Ramakrishnan, M. Vinothkannan, A. Kim, S. Karthikeyan and D. Yoo, Nanomaterials, 2019, 10, 76 CrossRef PubMed.
- X. Liu, J. Zang, L. Chen, L. Chen, X. Chen, P. Wu, S. Zhou and Y. Wang, J. Mater. Chem. A, 2017, 5, 5865–5872 RSC.
- R. Liu, Y. Wang, D. Liu, Y. Zou and S. Wang, Adv. Mater., 2017, 29, 1701546 CrossRef PubMed.
- Y. Wang, B. Zhang, W. Pan, H. Ma and J. Zhang, ChemSusChem, 2017, 10, 4170–4177 CrossRef CAS PubMed.
- C. Li and J. Baek, ACS Omega, 2019, 5, 31–40 CrossRef PubMed.
- G. Ou, P. Fan, H. Zhang, K. Huang, C. Yang, W. Yu, H. Wei, M. Zhong, H. Wu and Y. Li, Nano Energy, 2017, 35, 207–214 CrossRef CAS.
- P. Xiao, W. Chen and X. Wang, Adv. Energy Mater., 2015, 5, 1500985 CrossRef.
- Y. Zhu, G. Chen, X. Xu, G. Yang, M. Liu and Z. Shao, ACS Catal., 2017, 7, 3540–3547 CrossRef CAS.
- G. Hutchings, Y. Zhang, J. Li, B. Yonemoto, X. Zhou, K. Zhu and F. Jiao, J. Am. Chem. Soc., 2015, 137, 4223–4229 CrossRef CAS PubMed.
- C. Wei, Z. Feng, G. Scherer, J. Barber, Y. Shao-Horn and Z. Xu, Adv. Mater., 2017, 29, 1606800 CrossRef PubMed.
- Y. Zhou, S. Sun, J. Song, S. Xi, B. Chen, Y. Du, A. Fisher, F. Cheng, X. Wang, H. Zhang and Z. Xu, Adv. Mater., 2018, 30, 1802912 CrossRef PubMed.
- J. Rajesh, B. Min, J. Kim, H. Kim and K. Ahn, J. Electrochem. Soc., 2016, 163, A2418–A2427 CrossRef CAS.
- X. Ge, Y. Liu, F. Goh, T. Hor, Y. Zong, P. Xiao, Z. Zhang, S. Lim, B. Li, X. Wang and Z. Liu, ACS Appl. Mater. Interfaces, 2014, 6, 12684–12691 CrossRef CAS PubMed.
- X. Yu, Z. Sun, Z. Yan, B. Xiang, X. Liu and P. Du, J. Mater. Chem. A, 2014, 2, 20823–20831 RSC.
- Y. Zhao, X. Zhou, Y. Ding, J. Huang, M. Zheng and W. Ye, J. Catal., 2016, 338, 30–37 CrossRef CAS.
- D. Zhao, M. Dai, Y. Zhao, H. Liu, Y. Liu and X. Wu, Nano Energy, 2020, 72, 104715 CrossRef CAS.
- T. Maiyalagan, K. Jarvis, S. Therese, P. Ferreira and A. Manthiram, Nat. Commun., 2014, 5, 3949 CrossRef CAS PubMed.
- X. Cao, T. Wang and L. Jiao, Adv. Fiber Mater., 2021, 3, 210–228 CrossRef CAS.
- C. Li, X. Han, F. Cheng, Y. Hu, C. Chen and J. Chen, Nat. Commun., 2015, 6, 7345 CrossRef CAS PubMed.
- G. Kumar, H. Cho, D. Lee, J. Kumar, C. Siva, P. Ilanchezhiyan, D. Kim and T. Kang, Chemosphere, 2021, 283, 131134 CrossRef CAS PubMed.
- E. Ekebas, A. Cetin, A. Önao and E. Esenturk, J. Appl. Electrochem., 2019, 49, 315–325 CrossRef CAS.
- B. You, M. Tang and C. Tsai, Adv. Mater., 2019, 31, 1807001 CrossRef PubMed.
- T. Zhao, Y. Wang, S. Karuturi, K. Catchpole, Q. Zhang and C. Zhao, Carbon Energy, 2020, 2, 582–613 CrossRef CAS.
- Y. Qiu, J. Zhou, Z. Liu, X. Zhang, H. Han, X. Ji and J. Liu, Appl. Surf. Sci., 2022, 578, 152049 CrossRef CAS.
- K. Kang and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 094105 CrossRef.
- P. Zhai, M. Xia, Y. Wu, G. Zhang, J. Gao, B. Zhang, S. Cao, Y. Zhang, Z. Li, Z. Fan, C. Wang, X. Zhang, J. Miller, L. Sun and J. Hou, Nat. Commun., 2021, 12, 4587 CrossRef CAS PubMed.
- D. Yan, Y. Li, J. Huo, R. Chen, L. Dai and S. Wang, Adv. Mater., 2017, 29, 1606459 CrossRef PubMed.
- Z. Luo, Q. Peng, Z. Huang, L. Wang, Y. Yang, J. Dong, T. Isimjan and X. Yang, J. Colloid Interface Sci., 2023, 629, 111–120 CrossRef CAS PubMed.
- Y. Sun, Z. Zhao, S. Wu, W. Li, B. Wu, G. Liu, G. Chen, B. Xu, B. Kang, Y. Li and C. Li, ChemSusChem, 2020, 13, 2671–2676 CrossRef CAS PubMed.
- Y. Pan, K. Sun, Y. Lin, X. Cao, Y. Cheng, S. Liu, L. Zeng, W. Cheong, D. Zhao and K. Wu, Nano Energy, 2019, 56, 411–419 CrossRef CAS.
- B. Cao, M. Hu, Y. Cheng, P. Jing, B. Liu, B. Zhou, X. Wang, R. Gao, X. Sun, Y. Du and J. Zhang, NPG Asia Mater., 2021, 13, 1–14 CrossRef CAS.
- H. Gao, X. Wang, G. Wang, C. Hao, S. Zhou and C. Huang, Nanoscale, 2018, 10, 10190–10202 RSC.
- Q. Li, X. Zhang, J. Shen, X. Ji and J. Liu, J. Colloid Interface Sci., 2022, 628, 467–476 CrossRef CAS PubMed.
- Z. Zhu, R. Zhang, J. Lin, K. Zhang, N. Li, C. Zhao, G. Chen and C. Zhao, J. Power Sources, 2019, 437, 226941 CrossRef CAS.
- Y. Zhu, S. Chen, X. Quan and Y. Zhang, RSC Adv., 2013, 3, 520–525 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cy01085f |
| ‡ These authors contribute equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |