
Access to the meta position of arenes through transition metal catalysed C–H bond functionalisation: a focus on metals other than palladium
Madalina T.
Mihai
,
Georgi R.
Genov
and
Robert J.
Phipps
 *
*
Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK. E-mail: rjp71@cam.ac.uk
First published on 26th October 2017
Abstract
The elaboration of simple arenes in order to access more complex substitution patterns is a crucial endeavor for synthetic chemists, given the central role that aromatic rings play in all manner of important molecules. Classical methods are now routinely used alongside stoichiometric organometallic approaches and, most recently, transition metal catalysis in the range of methodologies that are available to elaborate arene C–H bonds. Regioselectivity is an important consideration when selecting a method and, of all those available, it is arguably those that target the meta position that are fewest in number. The rapid development of transition metal-catalysed C–H bond functionalisation over the last few decades has opened new possibilities for meta-selective C–H functionalisation through the diverse reactivity of transition metals and their compatibility with a wide range of directing groups. The pace of discovery of such processes has grown rapidly in the last five years in particular and it is the purpose of this review to examine these but in doing so to place the focus on metals other than palladium, the specific contributions of which have been very recently reviewed elsewhere. It is hoped this will serve to highlight to the reader the breadth of current strategies and mechanisms that have been used to tackle this challenge, which may inspire further progress in the field.
 Madalina T. Mihai | Madalina Mihai obtained her MSci Chemistry with Pharmacology degree from the University of Birmingham in 2014. During her final year, she investigated organocatalytic reactions of triketopiperazines under the supervision of Prof. Nigel Simpkins. She is currently pursuing her PhD in the group of Dr Robert Phipps at the University of Cambridge. Her current research focusses on ion pair-directed borylation of arenes and heteroarenes. |
 Georgi R. Genov | Georgi Genov completed his MChem in Medicinal and Biological Chemistry at the University of York in 2015 and spent his Masters year as a placement student in Medical Research Council Technology in London. He is now pursuing his PhD under the supervision of Dr Robert Phipps at the University of Cambridge. His research interest lies in the design of new catalysts employing non-covalent interactions for regio- and stereoselective methodologies. His current project is on hydrogen bond-directed regioselective iridium catalysed borylation of arenes. |
 Robert J. Phipps | Robert Phipps obtained his undergraduate degree from Imperial College, London in 2006 before moving to the University of Cambridge where he completed his PhD studies with Prof. Matthew Gaunt in 2010. He spent two years working with Prof. F. Dean Toste at UC Berkeley as a Marie Curie Postdoctoral Fellow. In 2014 he commenced his independent career at Cambridge as a Royal Society University Research Fellow. His research group is interested in applying non-covalent interactions to control regioselectivity and site selectivity in catalysis. |
1. Introduction
Ever more efficient and selective methods for the elaboration of simple aromatic rings into those bearing multiple substituents are in constant demand in synthetic organic chemistry.1 Until relatively recently the predominant approach used to achieve this was electrophilic aromatic substitution (SEAr) chemistry (nitration, sulfonation, halogenation, Friedel–Crafts etc.) in which the electronics of the arene generally define both the reactivity and the regioselectivity outcome of the ensuing reaction. In some cases, with careful planning, these classical methods can lead quickly to the desired product. However, there are many substitution patterns which would be incompatible with the relatively limited reactivity/selectivity rules of SEAr and may require exhaustive manipulations to access. A revolutionary technique was developed in the 1970s and 1980s which relied on strong organometallic bases being guided by an existing functional group on the aromatic ring to deprotonate at the ortho position. In this ‘Directed ortho Metalation’ (DoM) the use of proximity as the selectivity determinant and a high tolerance of a range of electronic properties of the arene resulted in a somewhat complementary approach which revolutionised arene functionalisation.2 One disadvantage of DoM is the requirement for stoichiometric organometallic bases which can present limitations with regard to functional group tolerance. In addition, electrophiles are not generally available for sp2 carbon functionality such as alkenes and arenes. In addressing these issues, transition metal catalysis has played a defining role, triggered by the pioneering report from Murai in 1993 which demonstrated that a catalytic amount of a transition metal could be guided to functionalise the arene ortho position in an analogous manner to DoM, but using the unique reactivity of the transition metal centre to achieve C–H activation.3 Enabled by the diversity of mechanistic pathways accessible to transition metals, a huge variety of catalytic processes have subsequently been developed which hinge on this cyclometalation mechanism.4–9 This has led to a wealth of transformations using a variety of different directing groups and has provided a third general and practical approach to the functionalisation of arene C–H bonds, in addition to aforementioned SEAr-type processes and DoM chemistry (Fig. 1). Whilst a variety of metals have been utilised in cyclometalation approaches, including Ru, Rh and Pt, it is Pd that has been most broadly applied. This has been attributed to a number of factors, including the compatibility of Pd(II) catalysts with a variety of oxidants allowing ready access to high-valent mechanisms, the ease with which Pd undergoes cyclopalladation with a variety of groups and the numerous ways which these intermediates can be further functionalised.4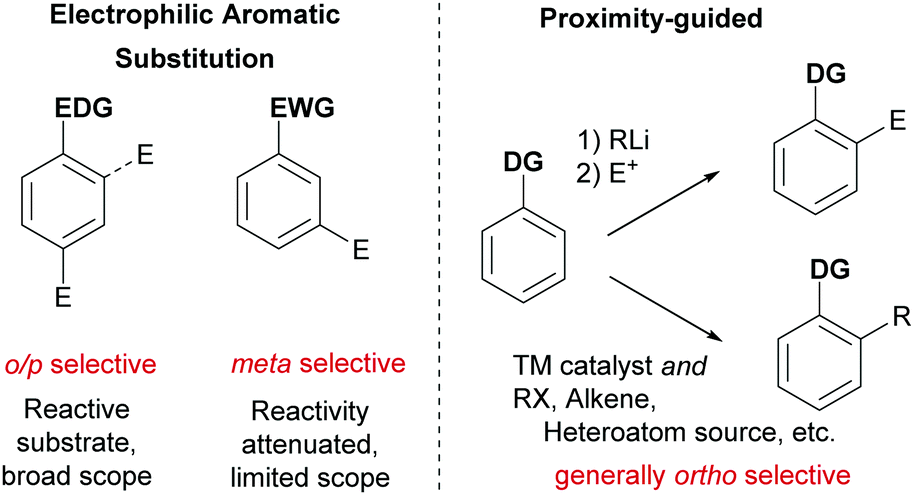 | ||
| Fig. 1 Overview of selectivity outcomes resulting from common arene functionalisation approaches. | ||
In considering the unaddressed challenges in arene functionalisation chemistry, directly accessing the meta position is an obvious one. The ortho position is well served by both DoM and catalytic cyclometalation chemistry. In addition, there are methodologies that depend on the most acidic C–H bond being activated by a transition metal, which in most cases occurs ortho to an electron-withdrawing substituent. The para position is far less well served by transition metals and poses a formidable challenge due to the remote location from a potential directing group.10 However, the fact that ‘activated’ electron rich arenes possess intrinsic para selectivity in SEAr means that this mode of functionalisation is often viable and can be used to selectively install a handle for further elaboration, such as a halogen (e.g. for cross coupling or lithium halogen exchange) or a nitro group (e.g. for reduction to amines and possible diazotisation). The arene meta position is arguably the most problematic as it is still sufficiently remote from a directing group to preclude traditional cyclometalation through coordination to a conventional directing group. Furthermore, meta-selective SEAr chemistry only occurs on electron deficient arenes which, by their very nature, possess severely diminished reactivity. Hence more forcing and strongly acidic conditions are commonly required which limits substrate scope and reduces practical application. Furthermore, the presence of other substituents on the ring can often lead to isomeric mixtures due to conflicting directing effects.
Of special note at this point are several remarkable stoichiometric directed metalation reactions that occur at the arene meta-position, as pioneered by Mulvey and co-workers. These include selective meta-deprotonation of toluene11 and N,N-dimethylaniline12 using complex metalation reagents composed of alkali metals in combination with a non-alkali metal such as magnesium or zinc.13 This has recently been extended in elegant work by Mulvey, O’Hara and co-workers to ortho–meta′ and meta–meta′ dimetalation in which a disodium–monomagnesium alkyl-amide base acts as a template to direct metalation to the traditionally remote position on a selection of monosubstituted arenes (Fig. 2).14
 | ||
| Fig. 2 (a) meta–meta′ Dimetalation of N,N-dimethylaniline followed by quenching with iodine. (b) Molecular structure of [Na4Mg2(TMP)6(C6H3NMe2-3,5)]. From Science, 2014, 346, 834–837. Reprinted with permission from AAAS. | ||
Due to the above considerations, developing catalytic methodology for regioselectively elaborating the arene meta position has been something of an elusive goal relative to developments in ortho functionalisation. However, what began as a handful of examples has now accelerated into a rich and varied area of cutting-edge chemical research involving a variety of transition metal catalysts. Amongst these, palladium has played a leading role and is responsible for the largest body of published works addressing this challenge. An authoritative review on the use of palladium for meta C–H functionalisation was published by Maiti and co-workers in 2016.15 We will summarise these advances briefly in Section 2, in order to provide context for the bulk of the review. We direct the interested reader to Maiti's review for a full literature survey and more detailed discussions. In contrast, this Review will place the focus on progress thus far with metals other than palladium, to provide a balance and to highlight the unique and sometimes complementary opportunities presented by considering more diverse metal catalysts. The broad utility of palladium as a catalyst in a variety of synthetic transformations is remarkable but its success should not dissuade chemists from exploring other metals, due to the potential for enhanced or complementary reactivity or selectivity. A previous review from Yang of meta-selective C–H functionalisation in general covered the literature until 2014 but is now somewhat dated given the numerous recent advances in this rapidly developing field.16 Additionally, a book chapter from Ackermann and co-workers covered literature until May 2015.17
2. Summary of advances in use of palladium for meta-selective C–H functionalisation
As mentioned, palladium catalysis has supplied a number of the most significant advances in the area of meta-functionalisation. This is perhaps not surprising given the wealth of chemistry developed using palladium in a cyclometalation context, which has led to a deep understanding of how to productively employ organopalladium intermediates.4,18 In this section, we will give a brief overview of these key advances using palladium for the purposes of context and comparison. For full details and complete literature survey, the reader is directed to the recent review from Maiti and co-workers.15In 2012, Yu and co-workers reported the first example of a novel strategy which emulated the basic cyclometalation mechanism that enables so many ortho-selective reactions, but crucially incorporated a significantly longer-reaching directing group.19 This carefully-designed group acts as a template and when complexed with palladium enables cyclopalladation resulting in a much larger ring than the typical five or six membered one that would conventionally occur. This medium ring size allows the complexed palladium to reach around to the meta position via what the authors describe as cyclophane-like transition states. The initially developed templates possessed nitrile groups, based on the hypothesis that these would have a weak interaction with the Pd(II) metal centre.18 This interaction should ideally be sufficient to direct the cyclopalladation but weak enough to allow high reactivity of the arylpalladium(II) intermediate. Using this approach, highly meta-selective alkenylation was achieved (Fig. 3a).
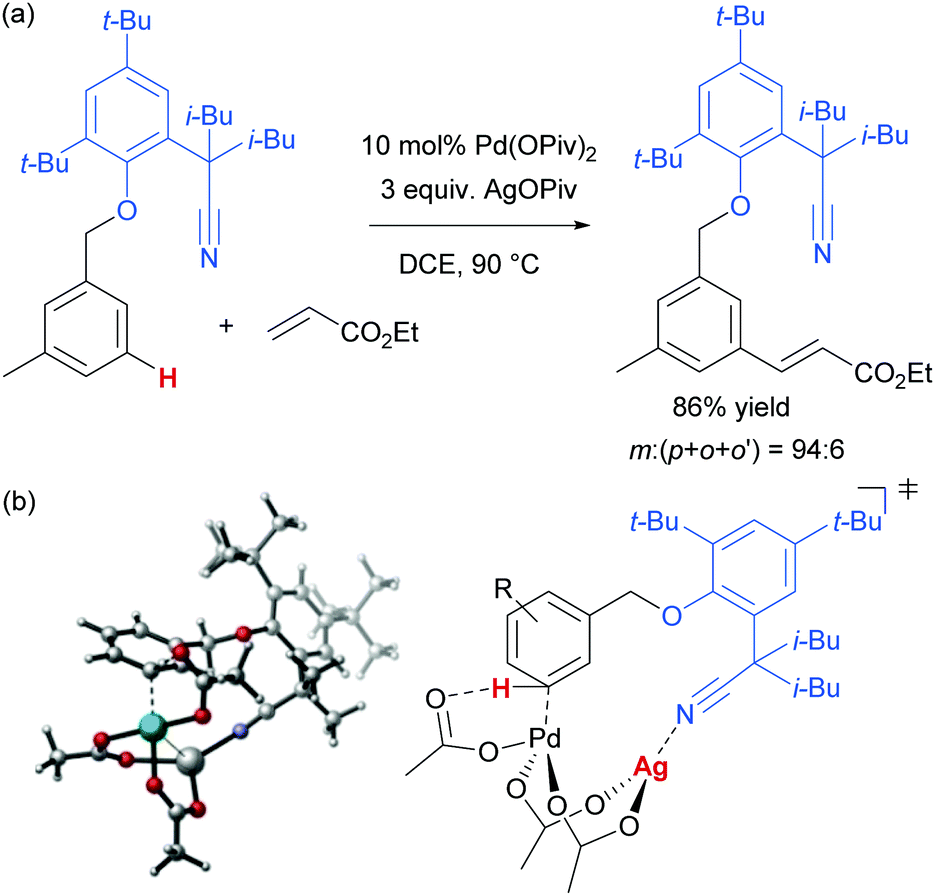 | ||
| Fig. 3 (a) Original report from Yu and co-workers of template-directed meta C–H activation by palladium. (b) Calculated transition state showing rate and regioselectivity-determining C–H activation occurring via a Pd–Ag heterodimeric complex. Reprinted with permission from J. Am. Chem. Soc., 2014, 136, 344–355. Copyright 2014 American Chemical Society. | ||
Since this first example, this template and modified versions have proven to be highly effective for meta-selective C–H activation reactions (for example, alkenylation, arylation, oxygenation)20 of a range of substrates incorporating related templates, including phenols,21,22 aromatic amines,23,24 arylacetic acids,25 benzyl alcohols,26 benzylsulfonic acids,27–29 benzoic acids,30 phenylpropanoic acids,31 biaryl systems,32 benzylphosphonates33 and longer chain alcohols34 (see Fig. 4 for selected examples). From a mechanistic standpoint, Yu, Wu, Houk and co-workers performed a computational investigation of Yu's original template-directed olefination of toluene derivatives. As predicted, the reaction consists of four steps: C–H activation, olefin insertion, β-hydride elimination and reductive elimination. The C–H activation was both rate-determining and regio-determining and they considered four possibilities as the active catalytic species. Interestingly, calculations suggested that the crucial C–H activation event actually occurs via a Pd–Ag heterodimeric transition state in which the nitrile interacts with the silver metal centre which is then complexed with the palladium atom through bridging acetate ligands (Fig. 3b).35 Yu and co-workers have pioneered the use of mono-N-protected amino acid (MPAA) ligands in these transformations, which typically lead to improved yields and shorter reaction times.36 The same authors have also recently disclosed a pyridine-based template which, amongst other reactions, enables meta-selective iodination37 and Maiti and co-workers have developed related pyrimidine and quinoline variants.28,38 The number of different reactions and substrates published using this approach attests to its utility, although the drawback remains incorporation and removal of the bespoke template before and after the desired meta-functionalisation. The robustness and generality of this template approach has also been demonstrated by even longer “D-shaped” templates able to effect para-selective C–H functionalisation.39,40
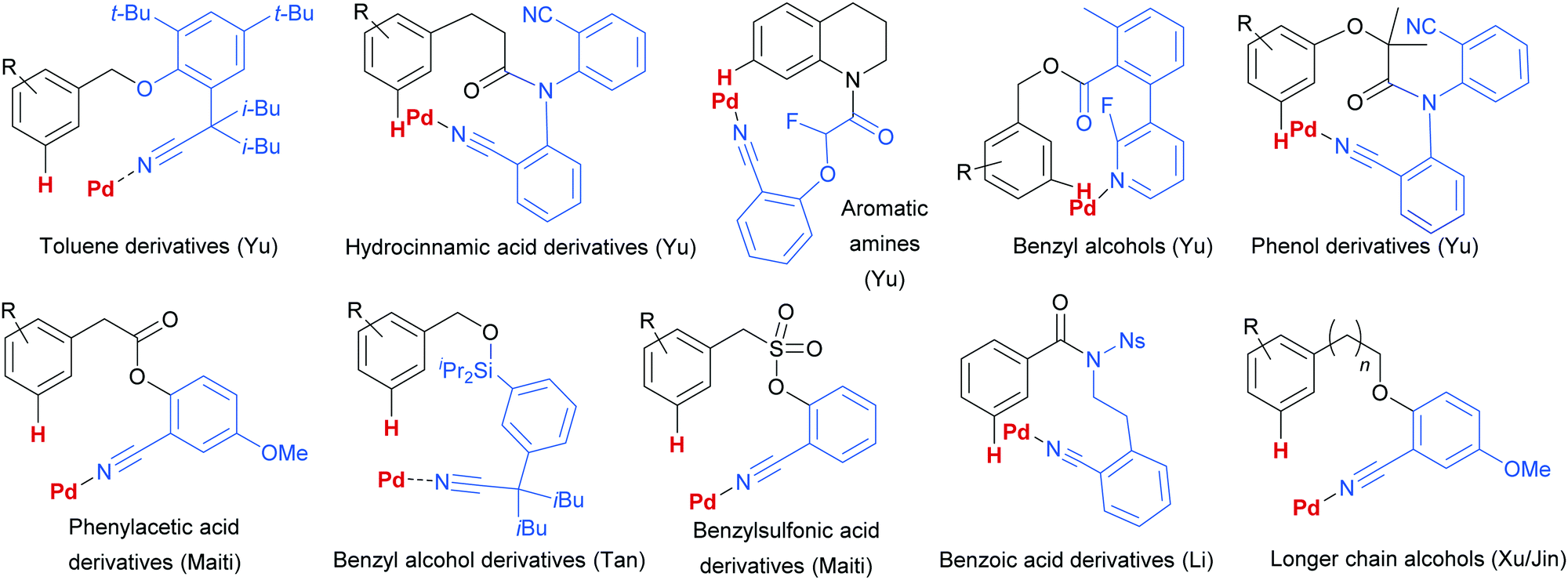 | ||
| Fig. 4 Selected examples of different substrate classes employed with various template directing groups for palladium since 2012. | ||
In another key development in the field, Yu and co-workers disclosed the use of norbornene as a transient mediator to enable meta-selective C–H activation via ortho-palladation.41 This clever strategy is directly inspired by the Catellani reaction and is in effect a relay C–H palladation process (Scheme 1).42–44 Initial ortho-palladation is enabled by a substrate directing group in a relatively conventional way and this is followed by insertion of norbornene and a second C–H palladation by the resulting alkyl palladium(II) complex. This undergoes oxidative addition/reductive elimination with an aryl or alkyl halide (Scheme 1, desired pathway), followed by β-hydride elimination to remove norbornene from the now more sterically congested molecule. If protodemetalation (i.e. the reverse of C–H activation) is viable, then the desired catalytic cycle can be closed. Yu and co-workers were able to achieve this by screening pyridine ligands and identifying one able to prevent the undesired reductive elimination pathway that incorporates norbornene into the product (Scheme 1, undesired pathway). Almost concomitantly, Dong and co-workers reported a similar Catellani-inspired strategy which differed primarily in its use of an amine directing group for the initial directed palladation.45 Since these initial reports, a number of contributions have been made by Yu and co-workers, predominantly concerning arylation and alkenylation of a variety of substrate classes.46–50 Considering more diverse transformations, the recent reports of chlorination,51 amination and alkynylation52 demonstrate the potential generality of this approach.
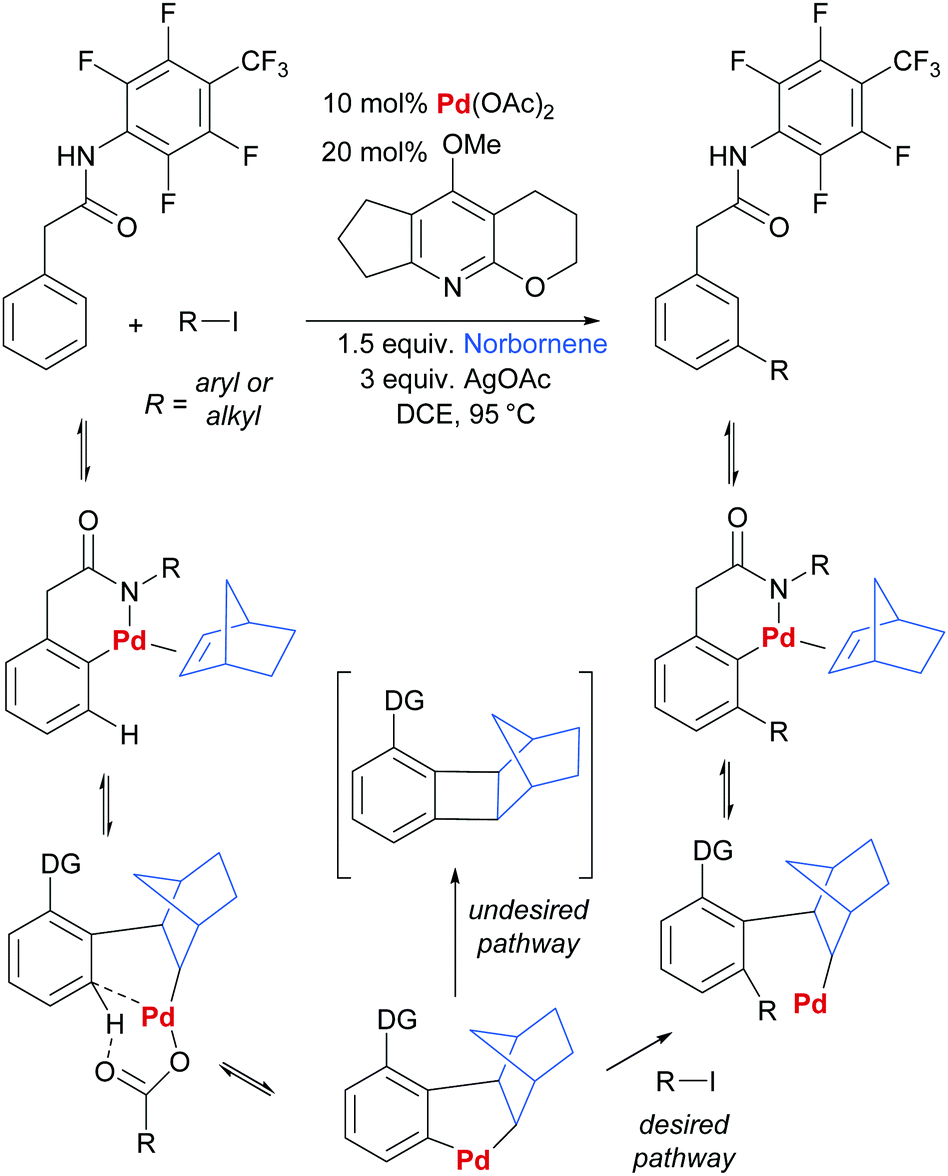 | ||
| Scheme 1 Outline of general reaction and proposed mechanism for Yu and co-workers’ norbornene-mediated meta arylation and alkenylation of aryl acetic acid derivatives. | ||
Very recently, Yu and co-workers reported a meta-functionalisation method which was designed to mimic their template approach but which aimed to exploit reversible coordination between substrate and template via a bridging metal centre (Fig. 5a).53 This was designed to enable the template to be used in a catalytic amount, seeking to address one of the main drawbacks of the prior strategy. They assessed a range of bis-amide and bis-sulfonamide scaffolds bearing two C3 pyridyl groups. They envisaged that metal chelation to the bis-amide would form a reversible binding site for a suitable basic group on the substrate. With the correct substrate structure, the template pyridyl groups should then be suitably positioned to bind to palladium (or potentially a Pd–Ag complex) and hold it over the meta position of the arene in the substrate. Using this approach, meta-olefination of 3-phenylpyridines was achieved using a catalytic amount of template (Fig. 5b). The authors also showed that site-selective olefination of quinolines and other heterocycles could be achieved, although this required stoichiometric template and palladium. This design approach is elegant, although the precise role of each metal in the process is difficult to pinpoint at this early stage in development, as three different metals are employed in the catalytic system.
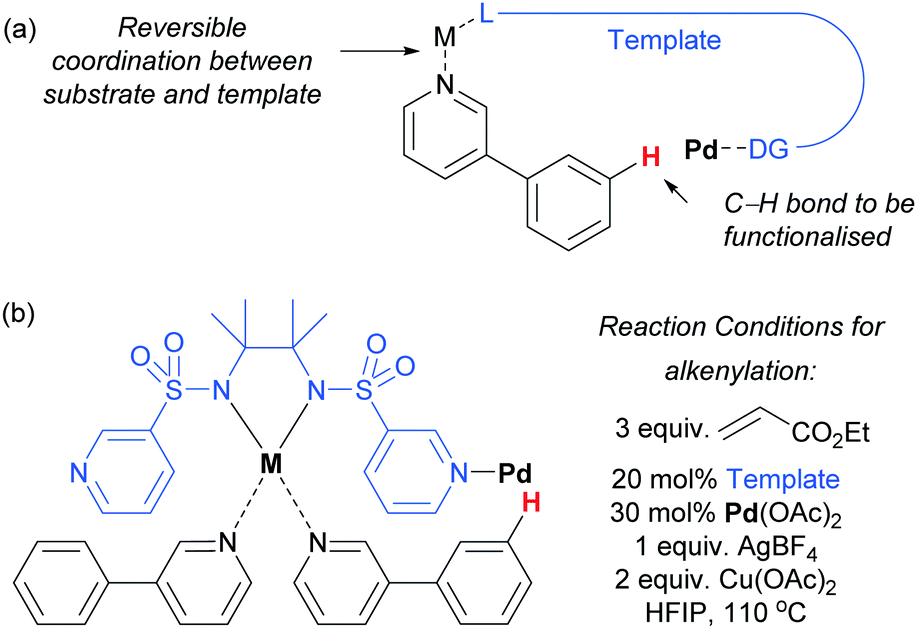 | ||
| Fig. 5 (a) Outline of template approach via reversible metal–ligand coordination. (b) Successful catalytic template design that enabled selective olefination of 3-phenylpyridine and reaction conditions. | ||
Building on their previous work,54 Larrosa and co-workers have developed a clever relay strategy via a series of sequential operations in a single flask, resulting in a formal meta-selective arylation of phenols.55 This proceeds by carboxylation ortho to the phenol, palladium-catalysed arylation ortho to the carboxylate and final decarboxylation, releasing the meta-arylated phenol.
There will undoubtedly be further developments in the future using palladium catalysis to access the arene meta position. The following sections, focussing on other metals, attempt to show that these different metals often function in very different ways. Thus, considering diverse metal catalysts should naturally lead to diversification in mechanistic manifolds that can only have a positive role in the discovery and development of new reactivity and selectivity paradigms in order to tackle this important challenge.
3. Iridium catalysis
Iridium catalysis has been successful in several important and distinct classes of C–H activation56 and amongst these has arguably delivered one of the most broadly applied for the functionalisation of arene C–H bonds: catalytic C–H borylation.57Whilst initial studies on the conversion of C–H bonds to C–B bonds focussed on alkanes, the first iridium catalysed arene borylation was described in 1999 by Iverson and Smith using a Cp*Ir complex and strong electron-donating alkylphosphine ligands to achieve borylation of toluene.58 Subsequently, the efficiency of the process was greatly improved by the use of bidentate phosphine ligands (Smith, Maleczka, and co-workers),59 or bipyridine ligands (Ishiyama, Miyaura, Hartwig and co-workers).60 In detailed mechanistic studies, the latter were able to isolate an Ir(dtbpy)(COE)(BPin)3 complex believed to be the active catalyst precursor in this process. Based on these studies, the now generally accepted mechanism for the iridium catalysed C–H borylation of arenes with bipyridine ligands was proposed (Scheme 2).61 Upon the formation of the active catalyst precursor, COE dissociates to allow for a coordination site to open up, giving I. This is followed by an oxidative addition of an arene C–H arene to give Ir(V) species II and a subsequent reductive elimination of the borylated product to give III. The active catalyst I is then regenerated by the oxidative addition of B2Pin2 and reductive elimination of BHPin.
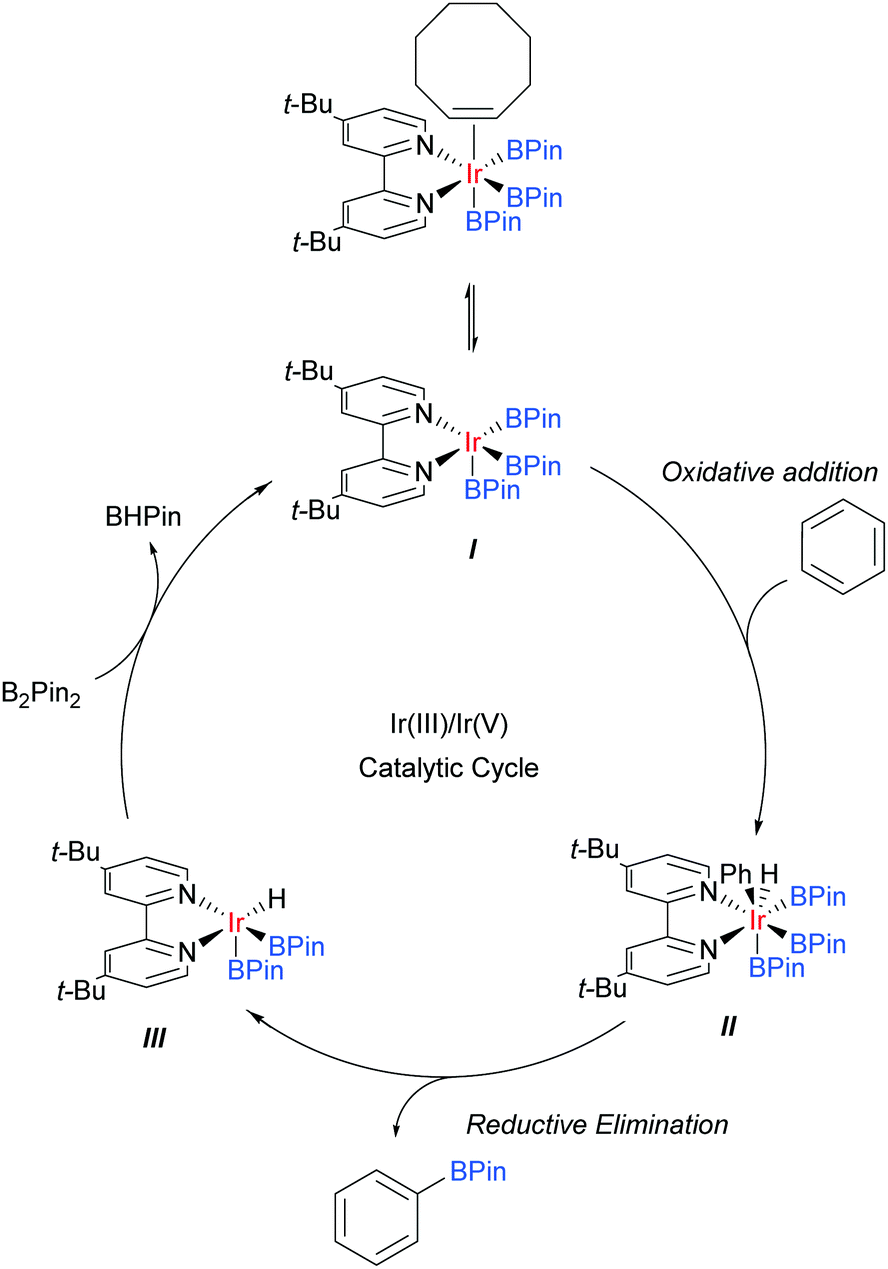 | ||
| Scheme 2 Proposed mechanism for iridium catalysed C–H borylation of arenes. | ||
The regioselectivity of this borylation process is particularly interesting and rather unique. Unlike most classical arene chemistry, in which electronics are key in determining regiochemistry, and many other arene C–H activation reactions, where location relative to a directing group is key, the regioselectivity is almost completely controlled by the steric demand of the substituents.62 As such, this process in its original form can be regarded as being meta selective for most 1,3-disubstituted arenes, which typically afford a single product at the 5-position. However, this selectivity is lost for monosubstituted arenes, 1,2-disubstituted arenes, and those bearing substituents of negligible steric demand, such as fluorines. In these cases, inseparable mixtures of meta- and para-regioisomers typically result, which are often challenging to separate. There are a number of strong drivers for the development of modified borylation methods to overcome these regioselectivity drawbacks. The first is that the process is highly tolerant of existing functionality within substrates, for example basic heteroatoms and carbon–iodine bonds. The second is that the process typically operates under very mild conditions, often as low as room temperature, and this makes it particularly amenable for use on complex molecules and at a late stage in a synthetic sequence.63 These factors can offer considerable advantages over many other C–H activation methods and as a consequence much effort has gone into manipulating borylation regioselectivity beyond simple steric control. The majority of examples relate to proximal directing strategies that enable ortho-selective borylation and much success has been achieved in this area.64 The last few years have seen a surge in developments aimed at marshalling borylation to the more remote meta and para positions. Description of the former will be the topic of the following section. Of the few strategies for para-selective borylation, two are based on incorporating increased steric bulk into the catalytic strategy65,66 and one employs a coordinative non-covalent interaction to direct borylation.67
The first report on meta-selective borylation not to be based on steric control was published in 2015 by Kuninobu, Kanai and co-workers.68 The strategy employed to achieve this was to design a modified bipyridine ligand bearing a distal functionality capable of undergoing a non-covalent interaction with a suitable group on the substrate (Fig. 6). The authors envisioned that placing a hydrogen bond donor urea onto the ligand and thus having it in close proximity to the iridium metal centre would allow for regiocontrolled C–H activation of a substrate bearing a Lewis basic functionality. A range of bipyridine ligands bearing urea functionality were synthesised and one particular ligand (L1) stood out, giving excellent meta:para selectivity in the borylation of a model aryl amide (Fig. 6a). Apart from aryl amides, the substrate scope included arenes bearing other Lewis basic functionalities such as phosphonates and phosphine oxides (Fig. 6b). Control borylations using dtbpy as a ligand were conducted for all substrates to show the poor regioselectivity under typical borylation conditions. The scope also included five- and six-membered heteroarenes bearing amide and ester functionalities, although in these cases only in some examples was the ligand able to perturb the intrinsic borylation regioselectivity.
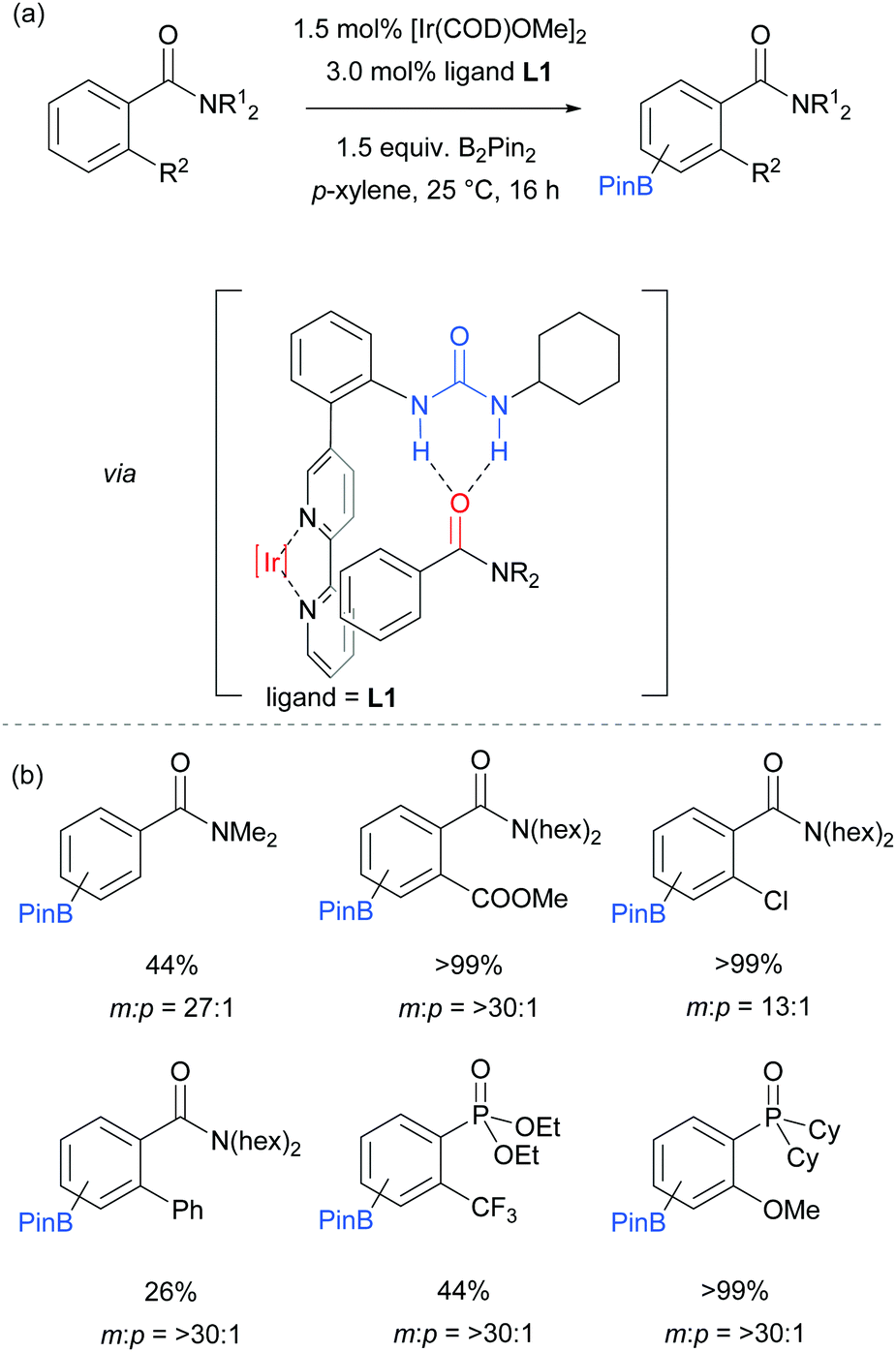 | ||
| Fig. 6 (a) General reaction scheme for the meta-selective borylation of arenes developed by Kuninobu, Kanai and co-workers and the proposed interaction responsible for the selectivity; (b) representative examples from the substrate scope. | ||
In order to support their hypothesis of a hydrogen bond directed borylation, a range of control experiments were conducted in order to show the importance of the urea moiety present on the ligand. Titration experiments between the ligand and an amide substrate showed a downfield shift of the urea N–H protons. In addition, when using a ligand where one or both nitrogens on the urea moiety were methylated, a severe drop in selectivity was observed (Fig. 7a). When changing the relative position of the urea functionality on the bipyridine ligand or varying the linker between the two, a drop in selectivity was observed (e.g.Fig. 7b). Running the reaction with a 2,2′-bipyridine ligand in the presence of cyclohexylphenylurea did not give any selectivity, showing that the two must be covalently linked for meta-selectivity to be achieved (Fig. 7c). Furthermore, the reactions tended to proceed with better yields and selectivity in non-polar solvents, at lower temperatures and under higher concentrations. All these observations were in favour of the proposed mechanism and clearly showed the importance of a properly positioned hydrogen bond donor. A somewhat related ligand design which allowed ortho-selective borylation of aryl sulfides was recently disclosed by the same authors.69
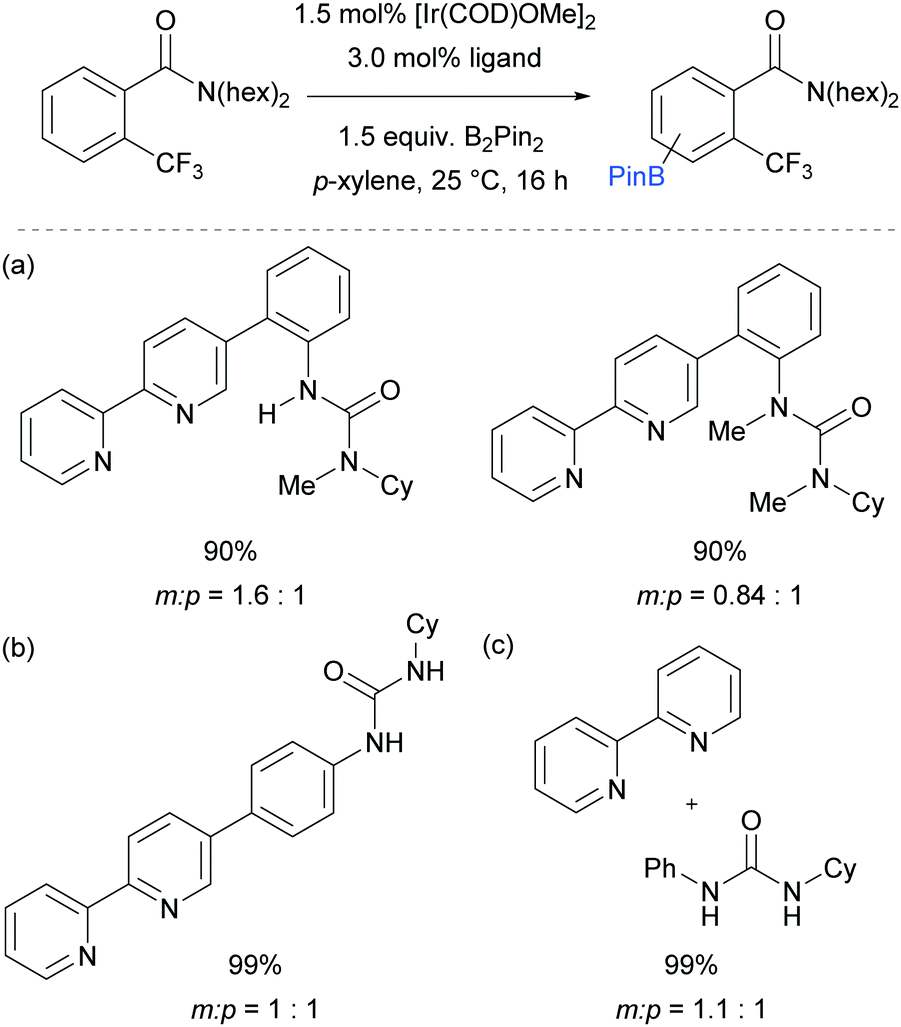 | ||
| Fig. 7 Control experiments undertaken by Kuninobu, Kanai and co-workers to provide support for their hypothesis of hydrogen bond directed borylation. | ||
An effective method for the meta-selective borylation of aromatic aldehydes was recently disclosed by Bisht and Chattopadhyay.70 A key element of their approach was the conversion of the aromatic aldehydes into imines in situ by condensation with an amine. Remarkably, subsequent borylation of this imine using the electron-rich 5,6,7,8-tetramethylphenanthroline (TMP) as ligand resulted in very high selectivity for the meta-position (Fig. 8). The mechanistic origin behind the observed meta-selectivity has not been rigorously determined but the authors advanced a hypothesis based on the following reasoning. The importance of using an electron rich non-labile ligand as well as the observation that smaller steric bulk of the amine additive tends to give better selectivity led the authors to hypothesise the transition state depicted in Fig. 8a. An important feature of the proposed transition state is the formation of a dative bond between the boron of the BPin and the nitrogen of the imine, which allows for ideal positioning of the meta C–H bond over the iridium centre. Although the proposed transition state seems to correlate well with the experimental results, the authors stress that this is still speculative. Interestingly, this work features a switchable catalytic system capable of changing the selectivity between ortho- and meta-depending on the ligand employed. A hemilabile 8-aminoquinoline ligand gave exclusively ortho-borylated product. This can be rationalised by partial dissociation of the hemilabile ligand, opening an extra coordination site on the iridium and thus allowing for coordination to the imine nitrogen, directing the C–H oxidative addition at the ortho position.64 The substrate scope included a range of 2-, 3- and 4-substituted benzaldehydes, all of which gave excellent meta selectivities using methylamine as the amine partner for imine formation (Fig. 8b). In some cases, tert-butylamine could also be used with good selectivity and improved yield.
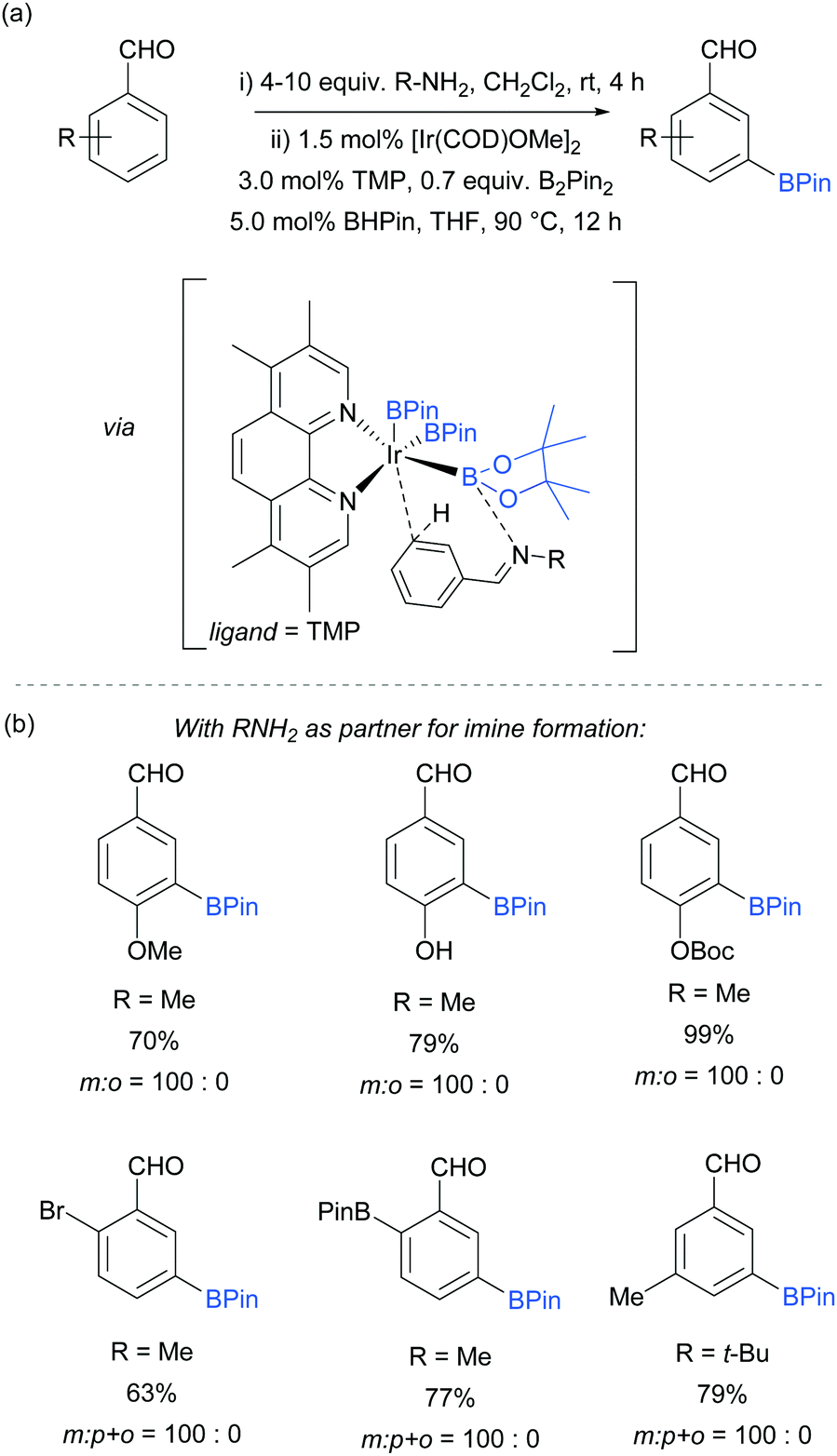 | ||
| Fig. 8 (a) General reaction scheme for the meta-selective borylation of arenes developed by Chattopadhyay and co-workers and the proposed interactions responsible for the observed selectivity; (b) representative examples from the substrate scope. | ||
Our group is particularly interested in the idea of incorporating non-covalent interactions into catalyst scaffolds for control of regioselectivity and site-selectivity.71 In 2016 we reported the borylation of aromatic quaternary ammonium salts directed by ion-pairing interactions with an anionic ligand.72 We synthesised a selection of bipyridine ligands bearing tethered sulfonate groups with the intention that an ion-pairing interaction between ligand and substrate may allow for suitable alignment of the substrate to undergo regioselective oxidative addition at the meta C–H bond of the arene. We tested these ligands on 1,2-disubstituted arenes, which typically give poor regiocontrol in iridium-catalysed borylation. Of the four anionic ligands tested, where the position and the length of the tether of the sulfonate functionality was varied, a single ligand (L2) gave very good levels of meta-selectivity with the test substrate, quaternised 2-chlorobenzylamine (Fig. 9a). In order to assess the importance of the hypothesised ion pairing, negative control experiments were carried out on substrates where the quaternary ammonium was substituted for a tertiary amine and a tert-butyl group; in both cases selectivity was lost. The addition of Bu4NOTs to the reactions led to a decrease in regioselectivity, potentially due to a negative impact on the ion-exchange that leads the substrate to be ion paired with the ligand. A good scope of quaternised benzylamines was demonstrated, with tolerance of halogens and heterocycles, amongst others (Fig. 9b). Furthermore, the same ligand was also highly effective for the meta-selective borylation of aniline-derived quaternary ammonium salts. This was surprising, as even with a substrate methylene removed, modification of the ligand structure was not required in order to achieve high regiocontrol (Fig. 9c). Chemoselective cross coupling of the reaction products was demonstrated.
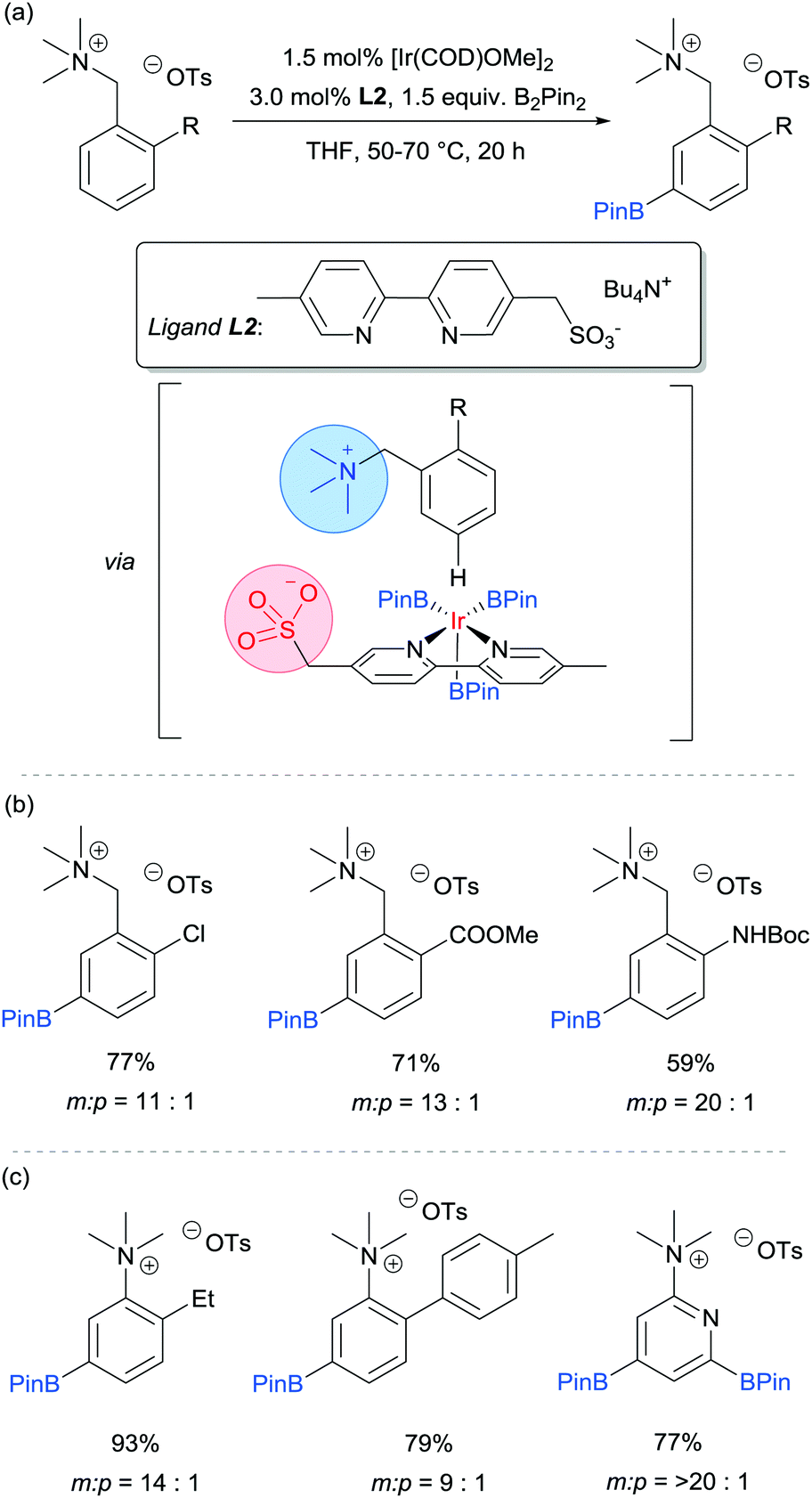 | ||
| Fig. 9 (a) General reaction scheme for the ion pair-directed meta-selective borylation of arenes developed by Phipps and co-workers and the proposed interaction responsible for the observed selectivity; (b) representative examples from scope of quaternised benzylamines; (c) representative examples from scope of quaternised anilines. | ||
We subsequently discovered that the sulfonate ligand L2 is able to function very effectively as a hydrogen bond accepting ligand to direct iridium-catalysed borylation (Fig. 10a).73 Alongside L2, a range of ligands bearing neutral hydrogen bond acceptor groups (amide, sulfoxide and phosphine oxide) were evaluated but the sulfonate-containing L2 was found to be the most effective by a large degree. This enabled aromatic C–H borylation of amides derived from benzylamines with high levels of meta-selectivity (Fig. 10b). Furthermore, the chain length could be extended to encompass phenethylamine (Fig. 10c) and phenylpropylamine-derived (Fig. 10d) amides without any loss of regioselectivity. Several conformationally constrained substrates gave poor selectivity and we hypothesised that some conformational freedom in the substrate is desirable to enable a productive hydrogen bonding interaction with the ligand. The fact that a single ligand design delivers high selectivity with numerous substrate classes could be attributed to the fact that the negative charge on the sulfonate is delocalised across three oxygen atoms, providing a relatively diffuse area of electron density at a specific distance from the catalytically active iridium centre (Fig. 10a). Thus, if a low energy substrate conformation can be achieved that would allow a product hydrogen bond with the ligand then meta-selective borylation may ensue. Control experiments with N-alkylated amide substrates gave poor regiocontrol, in line with the hypothesis that the borylation is controlled by a ligand–substrate hydrogen bonding interaction.
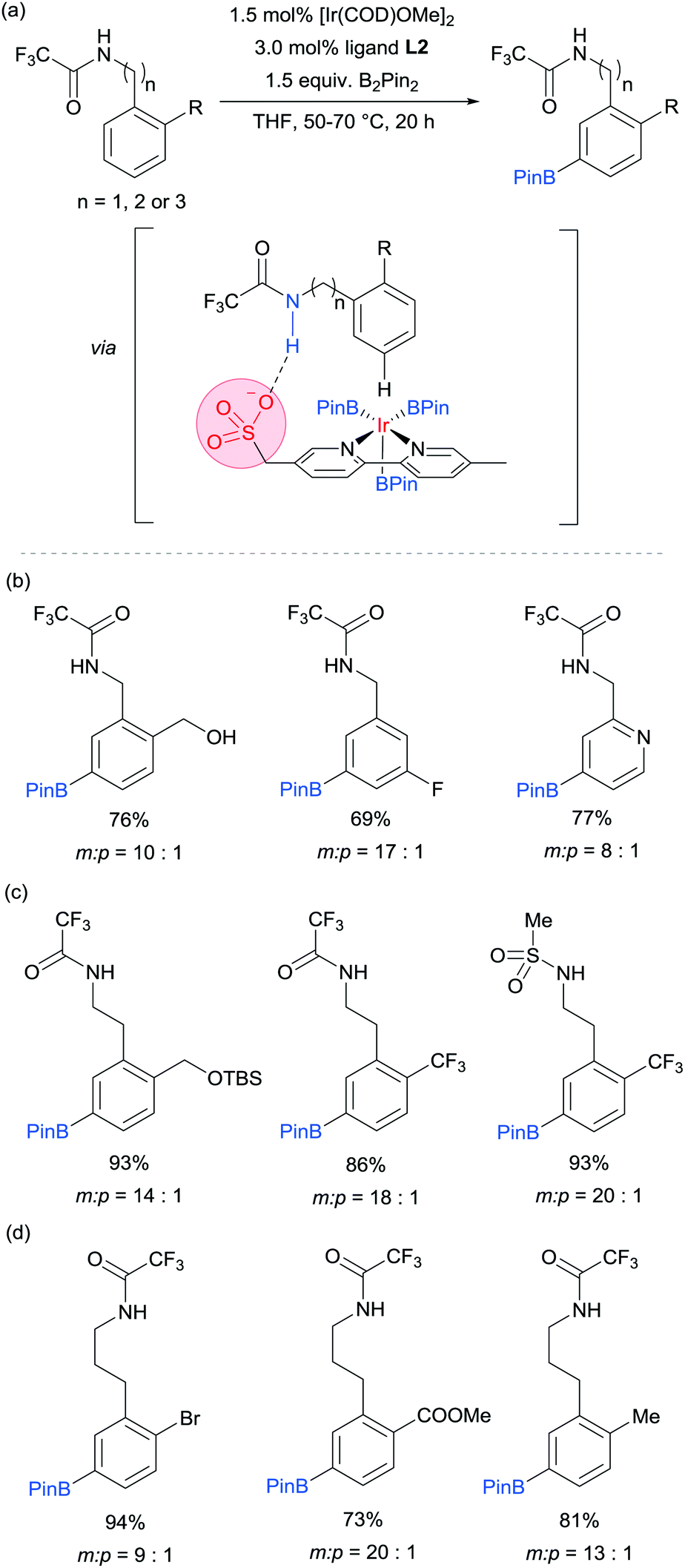 | ||
| Fig. 10 (a) General reaction scheme for the meta-selective borylation of arenes developed by Phipps and co-workers and the proposed hydrogen bonding interaction responsible for the observed selectivity; representative examples from each of the three classes of substrates: (b) benzylamines, (c) phenethylamines, (d) phenylpropylamines. | ||
4. Ruthenium catalysis
The use of ruthenium complexes to effect C–H functionalisation has a rich history, a leading example being the pioneering report of Murai and Chatani regarding the carbonyl-directed C–H hydrometalation of olefins.3 Since this time, a range of diverse C–H functionalisation processes have been developed, utilising both Ru(0) and Ru(II) catalysts.5,74 The vast majority of these proceed by a cyclometalation mechanism and deliver ortho-functionalised arene products.75,76 However, in 2011 Frost and co-workers reported a surprising result in the meta-selective sulfonation of 2-arylpyridines using Ru(II) catalysis.77 Despite almost two decades of research into C–H bond functionalisation using ruthenium catalysis, this was the first example of a meta-selective process. Interestingly, this transformation had been previously reported under palladium catalysis to selectively afford ortho-substituted products via a chelation-assisted C–H bond activation process (Fig. 11a).78 This study demonstrated that the regioselectivity of this reaction can be switched from ortho to meta by simply replacing the Pd(II) with a Ru(II) source (Fig. 11b).79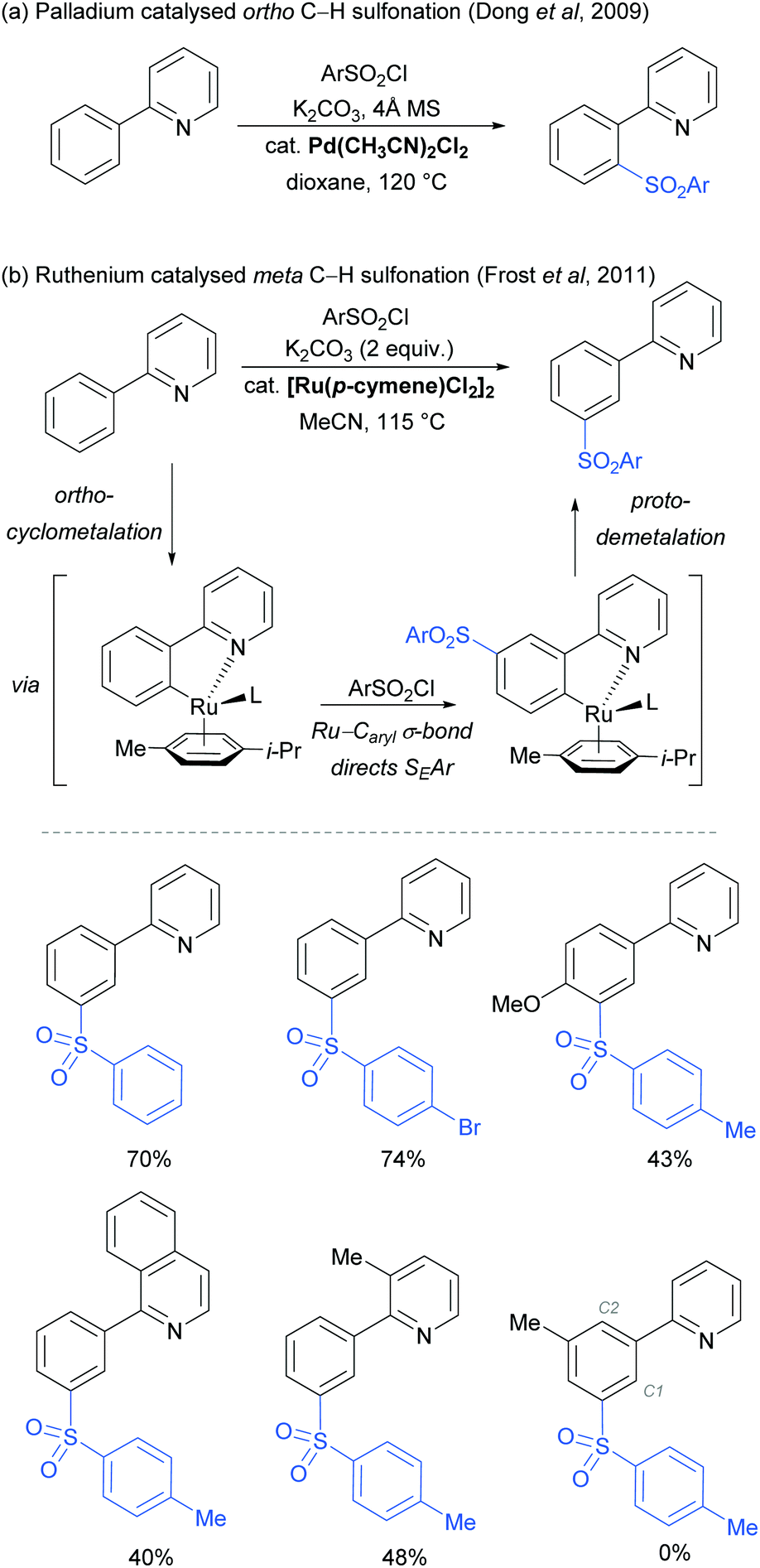 | ||
| Fig. 11 (a) Pd-Catalysed ortho C–H sulfonation; (b) Ru-catalysed meta C–H sulfonation: proposed reaction mechanism and selected examples. | ||
In their initial report, the authors hypothesised that the Ru-catalysed reaction proceeds via an initial chelation-assisted ortho cycloruthenation, followed by SEAr para to the Ru–Caryl σ bond (Fig. 11b). A key factor in obtaining this regioselectivity was thought to be the relative stability of the Ru–Caryl bond along with its activating, ortho-/para-directing effect.80 Presumably, no reactivity was observed at the position ortho- to the Ru–Caryl bond due to steric shielding by the p-cymene ligand. Subsequent proto-demetalation affords meta sulfonation relative to the pyridine directing group. This mechanism was supported by experiments run with premade complex IV, which led to quantitative formation of meta-functionalised 2-aryl pyridine when exposed to the reaction conditions (Scheme 3). The sulfonation reaction tolerates various substituents on the sulfonyl chloride, as well as on the phenyl and pyridine rings (Fig. 11b). Interestingly, no conversion was observed for a substrate bearing a meta-methyl substituent, indicating that the initial Ru–Caryl σ bond forms at the least sterically hindered site (C1), in which case the subsequent sulfonation would be blocked by the presence of the methyl group.
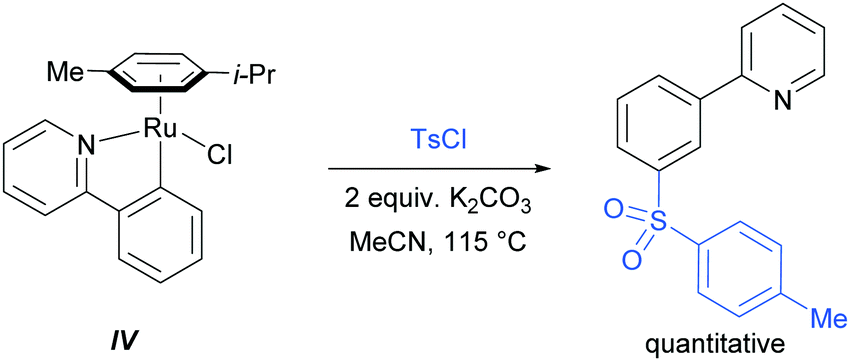 | ||
| Scheme 3 Sulfonation with preformed complex IV; Ts = p-toluenesulfonyl. | ||
The same authors subsequently reported the sequential functionalisation of the meta then ortho positions of 2-phenylpyridine using ruthenium followed by palladium catalysis.81
In 2013, the Ackermann group reported a meta-selective alkylation of arenes using secondary alkyl halides, under the catalysis of [RuCl2(p-cymene)]2/MesCO2H.82 Secondary alkyl halides are known to be challenging coupling partners in transition metal catalysis due to the difficulty of the oxidative addition into a sterically hindered C–H bond and the tendency of the resulting complex to undergo β-hydride elimination. Essential for achieving reactivity in this transformation was the use of carboxylate additives which had previously been reported to aid the cyclometalation step.7 In addition, complexes V and IV were prepared and when exposed to the reaction conditions, V proved catalytically competent, while the chloro-analogue IV did not, indicating the crucial role of the carboxylate ligand in this reaction (Fig. 12).
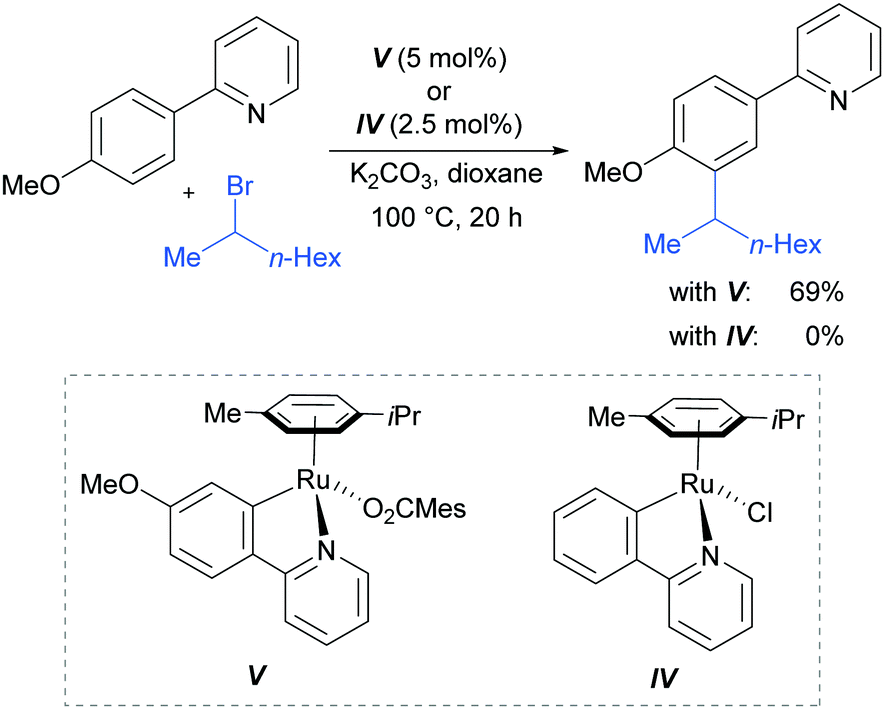 | ||
| Fig. 12 Investigation of complexes IV and V as catalytically relevant species. Mes = mesityl. | ||
This meta-alkylation reaction performed well not only on 2-phenyl pyridines, but also on various other heterocycle-substituted arenes, such as pyrazolyl-, imidazolyl- and benzimidazolyl variants (Fig. 13).
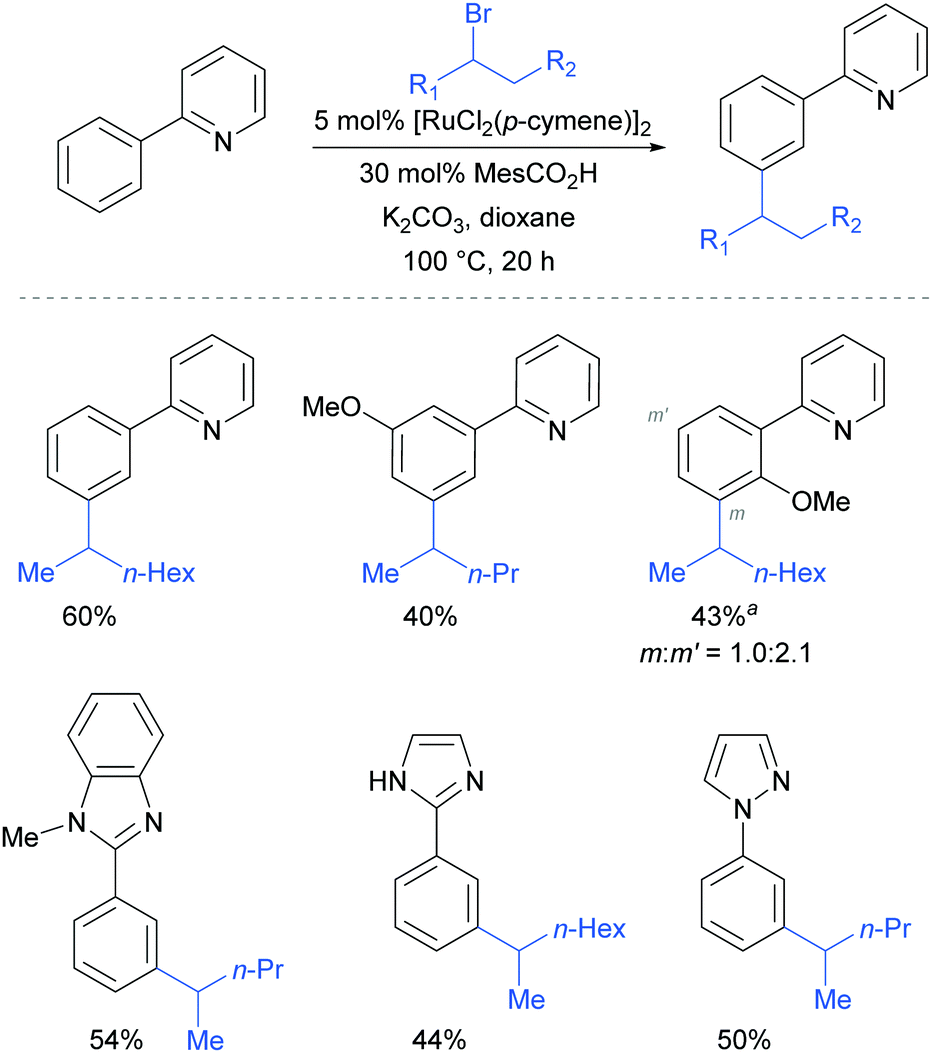 | ||
| Fig. 13 meta-Selective alkylation using secondary alkyl halides; a 2.5 mol% [RuCl2(p-cymene)]2. | ||
On the basis of mechanistic studies, the authors hypothesised that the reaction proceeds via a similar mechanism to the one initially proposed by Frost: a cycloruthenation at the arene ortho position, followed by an electrophilic substitution at the position para to the metal. Also consistent with observations reported by Frost, lower yields were obtained with meta-substituted arenes and in the case of ortho-substituted ones, the more sterically hindered products were obtained due to formation of the Ru–C bond at the less sterically hindered ortho-position (Fig. 14).
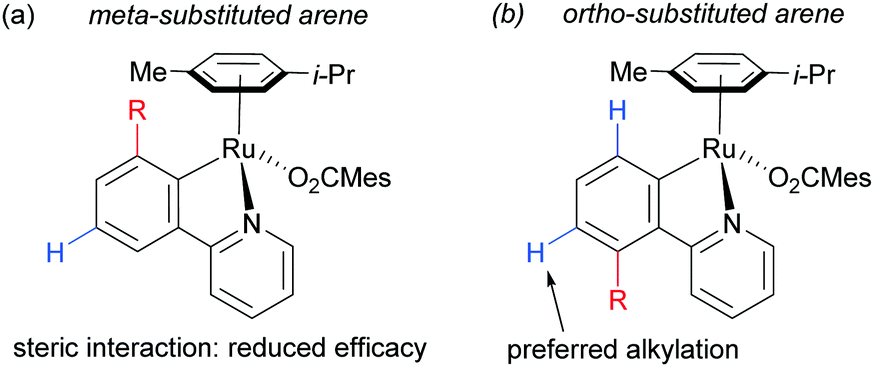 | ||
| Fig. 14 meta-Substituted arenes vs. ortho-substituted arenes. | ||
Competition studies revealed that primary alkyl halides lead exclusively to ortho substituted products whilst secondary alkyl halides give meta, a process described by the authors as chemo-specific (Scheme 4). This was rationalised as a result of the differing steric interactions and electrophilicities of the two types of alkyl halides prompting a change in mechanism. It was also noted that when enantiomerically pure secondary halides were used, the stereochemical information was lost and that inclusion of TEMPO as an additive inhibited the reaction. Experiments with deuterated substrates suggested that, whilst the initial ortho-metalation is reversible, the cleavage of the meta C–H bond is not kinetically relevant, in line with an SEAr mechanism.
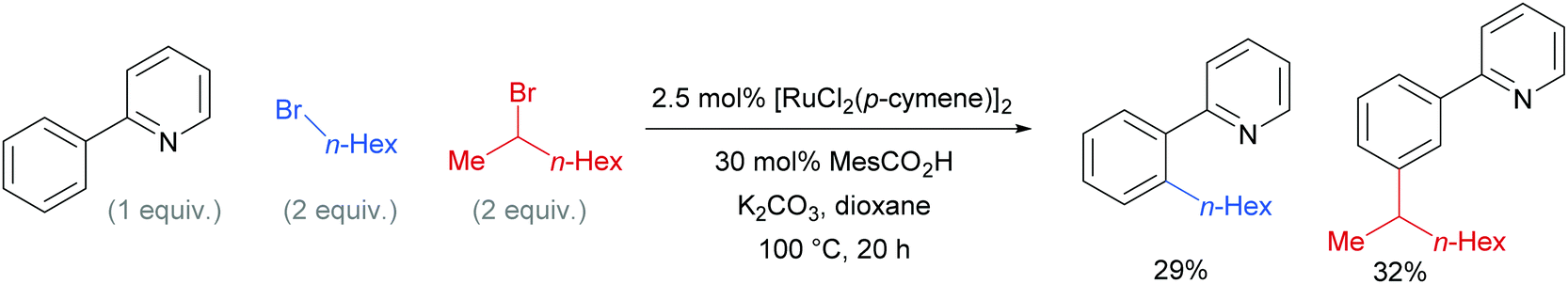 | ||
| Scheme 4 Competition experiments between primary and secondary alkyl bromides for ortho- and meta-alkylation of 2-phenyl pyridine. | ||
In 2015, the Frost group described a modification of their previous sulfonation method to achieve meta-alkylation of 2-aryl pyridines using tertiary alkyl bromides and chlorides.83 The key modification in comparison to their previous conditions was inclusion of an acetate base. Various substituents were tolerated at the para position of the phenyl ring, with the exception of strongly electron withdrawing groups, to form quaternary carbon centres on the electrophile (Fig. 15).
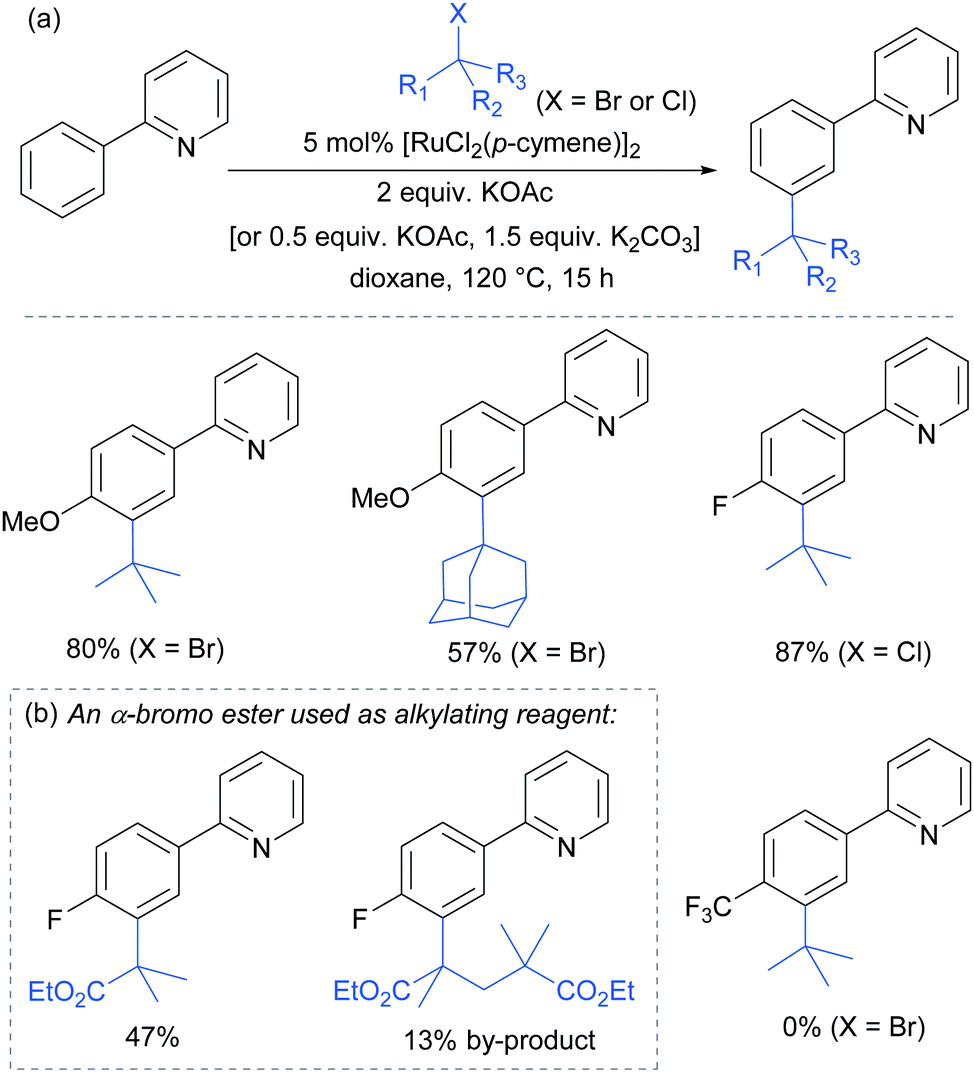 | ||
| Fig. 15 meta-Selective tert-alkylation of 2-arylpyridines – selected examples; KOAc (2 equiv.) was used with alkyl bromides and a mixture of KOAc (0.5 equiv.) and K2CO3 (1.5 equiv.) for alkyl chlorides. | ||
Interestingly, an α-bromo ester could also be used as alkylating agent. This result gave particular clues as to the mechanism because it is unlikely that this substrate could participate in an SN1 pathway as the required carbocation adjacent to an ester would be strongly disfavoured. SN2 type reactivity also seems unlikely given the fact that tertiary alkyl halides are viable. This led to the hypothesis that tertiary alkyl radicals could be intermediates. Evidence for this was seen in the nature of the by-products formed in the alkylation with α-bromo esters (Fig. 15b). These could plausibly derive from elimination of HBr under the reaction conditions, followed by radical conjugate addition. Furthermore, running the reaction in the presence of TEMPO as an additive gave no desired product. The above observations together with other experiments led the authors to propose that a radical mechanism is likely involved in the formation of the C–C bond (Scheme 5). In this proposed mechanism, electron transfer from Ru(II) to the electrophile generates a tertiary alkyl radical which adds to the aromatic ring para to the Ru–C σ bond. Rearomatisation would then occur via electron transfer back to the Ru(III) complex, followed by deprotonation.
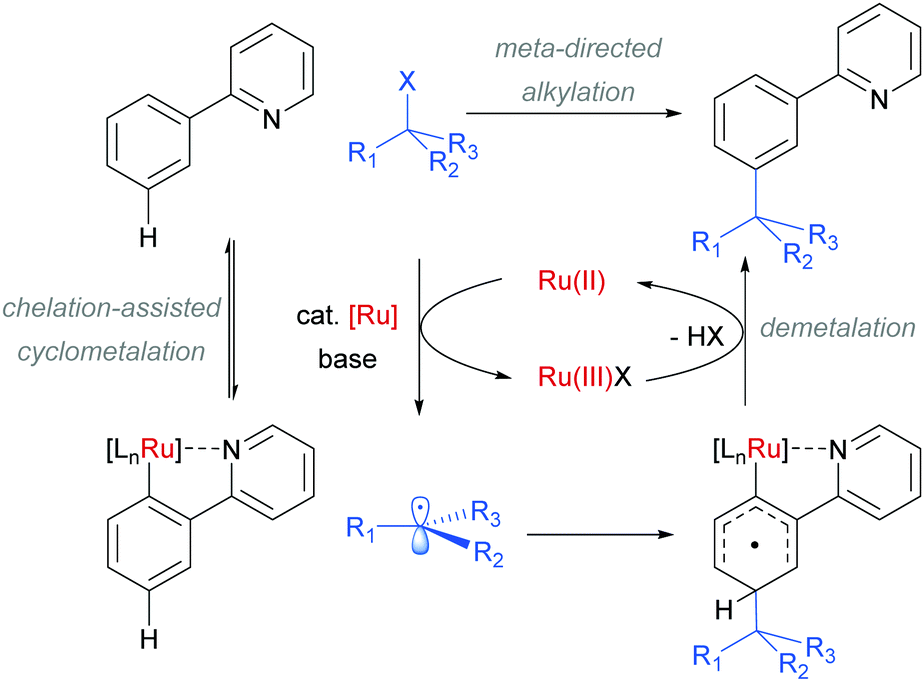 | ||
| Scheme 5 Proposed radical mechanism for the meta-selective tert-alkylation of 2-aryl pyridines. | ||
Very shortly afterwards, the Ackermann group also reported meta selective alkylation using tertiary alkyl bromides.84 Notably, in this report they demonstrated the use of removable directing groups which significantly adds to the synthetic utility of the process. Mono protected amino acids (MPAA), whose use has been pioneered by Yu in Pd catalysis,85 were identified as ideal ligands to accelerate this transformation to give high catalytic activity (Fig. 16). The scope of this tert-alkylation reaction included protected anilines with various substituents in the para position. ortho-Substituents were also tolerated on the phenyl ring when the MPAA ligand was changed to the Ad-Ile-OH. The authors further demonstrated that this methodology is applicable to arenes with a pyridine, pyrazole or pyrimidine directing group.
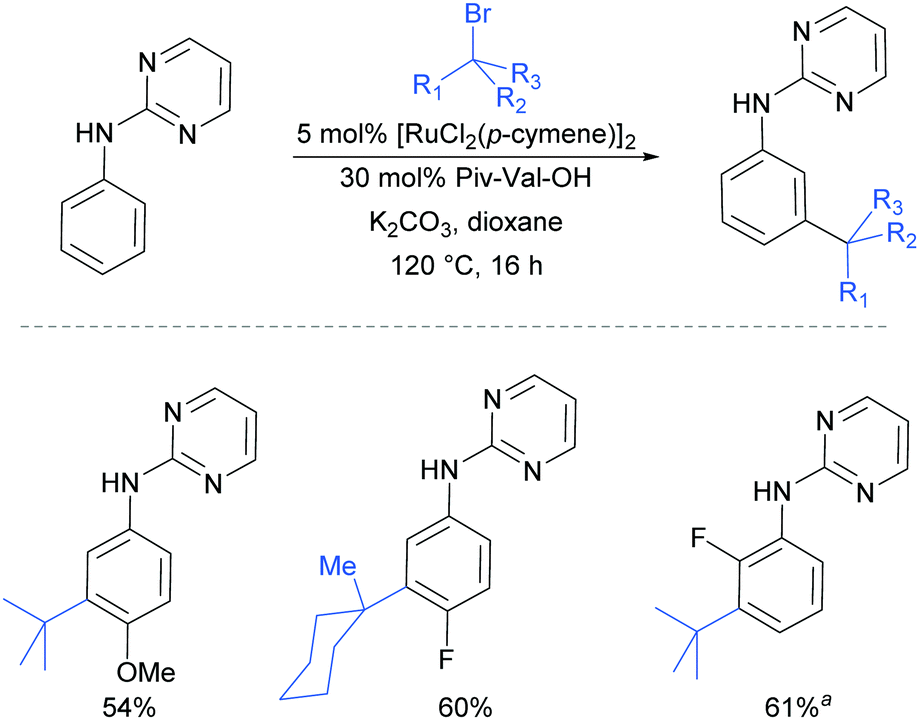 | ||
| Fig. 16 meta-Selective tert-alkylation of N-(pyrimidine-2-yl)anilines. a Ad-Ile-OH ligand used. | ||
The pyrimidine directing group could be easily removed to reveal the aniline (Scheme 6), which is an advantage over the previous methodologies, especially since meta-alkylated anilines may be difficult to access using classical methods.
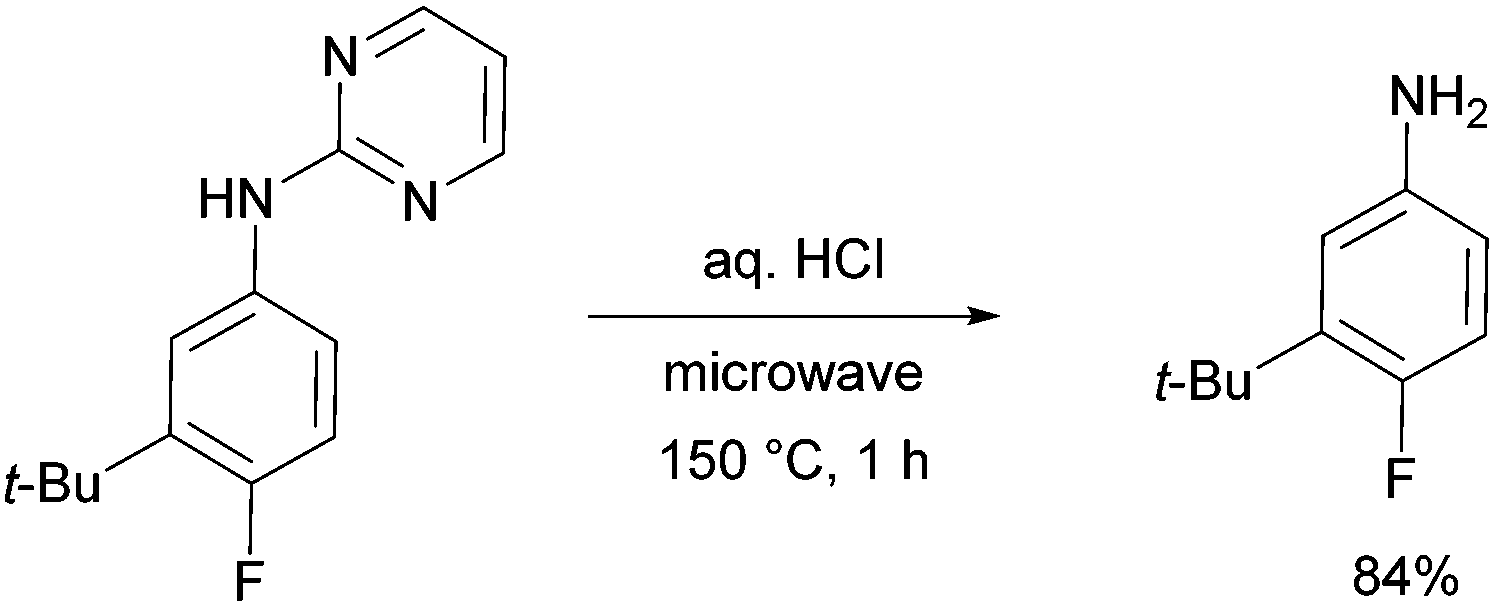 | ||
| Scheme 6 Deprotection to the aniline after meta-alkylation. | ||
Detailed mechanistic studies supported the previously advanced sequence of reversible cycloruthenation, followed by meta C–H cleavage and irreversible C–C bond formation. Careful isotopic labelling implicated a dual role for the electrophile in also providing the proton source for the final protodemetalation step of the catalytic cycle. A series of isolated complexes illustrated the key importance of carboxylate assistance. The scrambling of stereodefined alkyl bromides and ring-opening of α-bromo cyclopropanes pointed strongly to a radical mechanism for C–Br bond cleavage. Crucially, they also determined that the reaction order in [Ru] was second order providing strong evidence that a second ruthenium complex is involved in the fragmentation of the tertiary alkyl halide. These observations are summarised in the below catalytic cycle (Scheme 7).
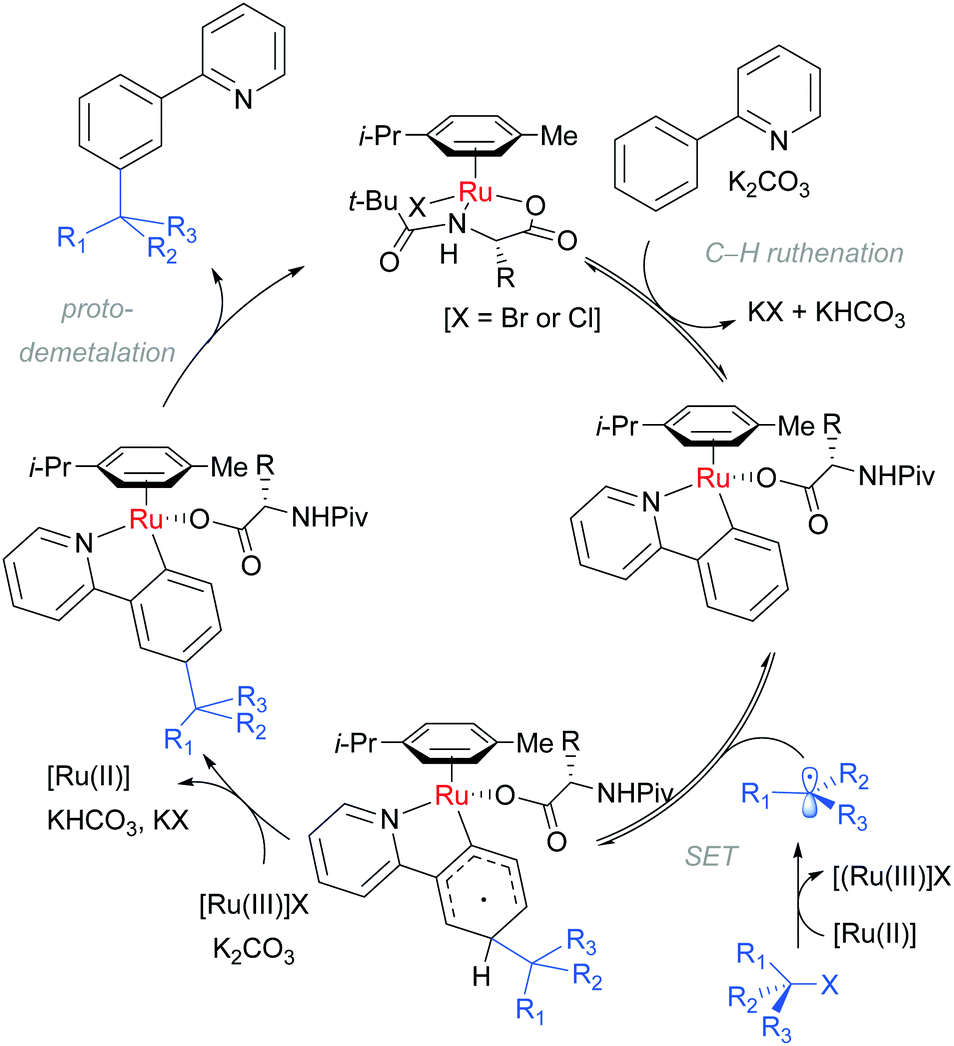 | ||
| Scheme 7 Proposed mechanism for meta tert-alkylation via a radical pathway. | ||
Following these reports which strongly implicated a radical-type mechanism, the Frost group published a detailed mechanistic study of their pioneering meta-selective sulfonation in which they had originally hypothesised an SEAr-type mechanism.86 Importantly, for the first time, they were able to isolate and fully characterise the meta-tosylated ruthenium complex VI (Scheme 8a) and show that it is catalytically active (Scheme 8b), proving the long hypothesised pathway of the reaction via activation para to the ruthenium. Their study also provided strong evidence, in line with other studies, that the C–S bond formation occurs via a radical mechanism as well as further insight into the protodemetalation step of the mechanism.
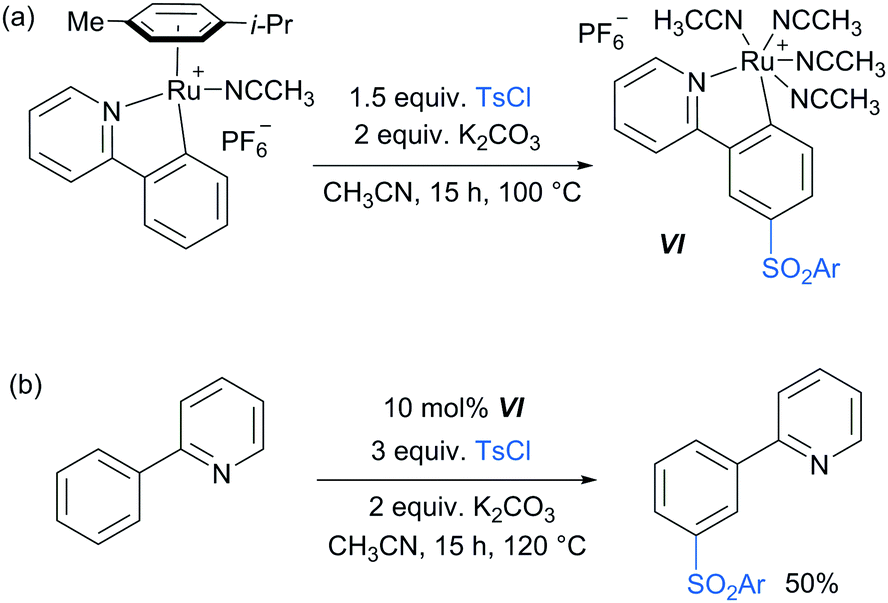 | ||
| Scheme 8 (a) Synthesis of meta-tosylated complex VI; (b) meta-sulfonation of 2-phenyl pyridine using catalytic amounts of complex VI. | ||
The first example of a meta-selective transition metal catalysed C–H bromination was reported by the Greaney group in 2015.87 Tetrabutylammonium tribromide (TBATB) was used as a brominating agent, in the presence of [RuCl2(p-cymene)]2 to achieve meta-bromination of various 2-arylpyridines (Fig. 17). Substituents were tolerated on the pyridine ring as well as at the para position of the phenyl ring. The utility of this methodology was illustrated with one pot procedures: bromination followed by Suzuki coupling or a Heck reaction to yield meta-arylated and meta-alkenylated products respectively.
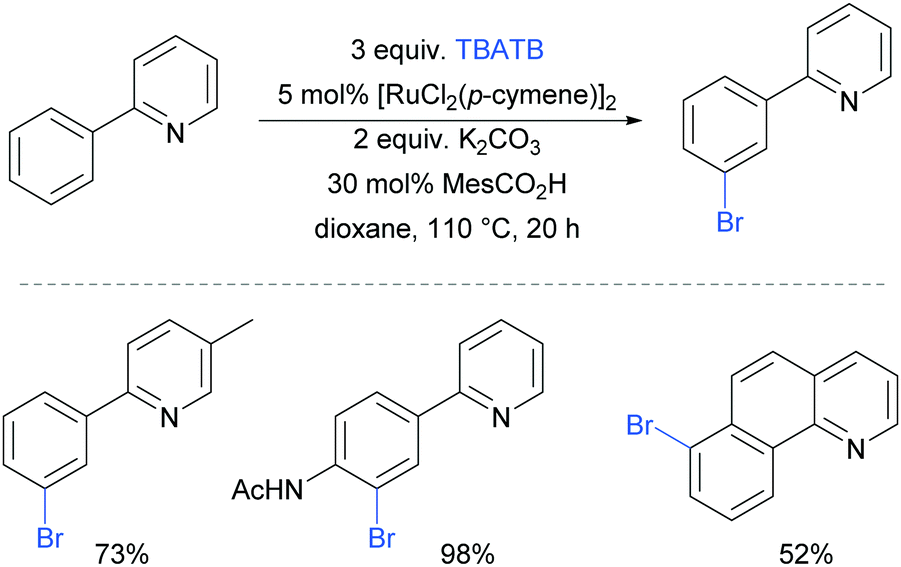 | ||
| Fig. 17 meta-Bromination of 2-aryl pyridines and related substrates. | ||
Very shortly after, Huang et al also reported meta-bromination of 2-arylpyridines, using [RuCl2(p-cymene)]2/NBS.88 The authors demonstrated that this reaction performs well on 2-aryl pyridines with various substitution on both the phenyl and the pyridine ring. Other heterocycles were also used as directing groups (Fig. 18).
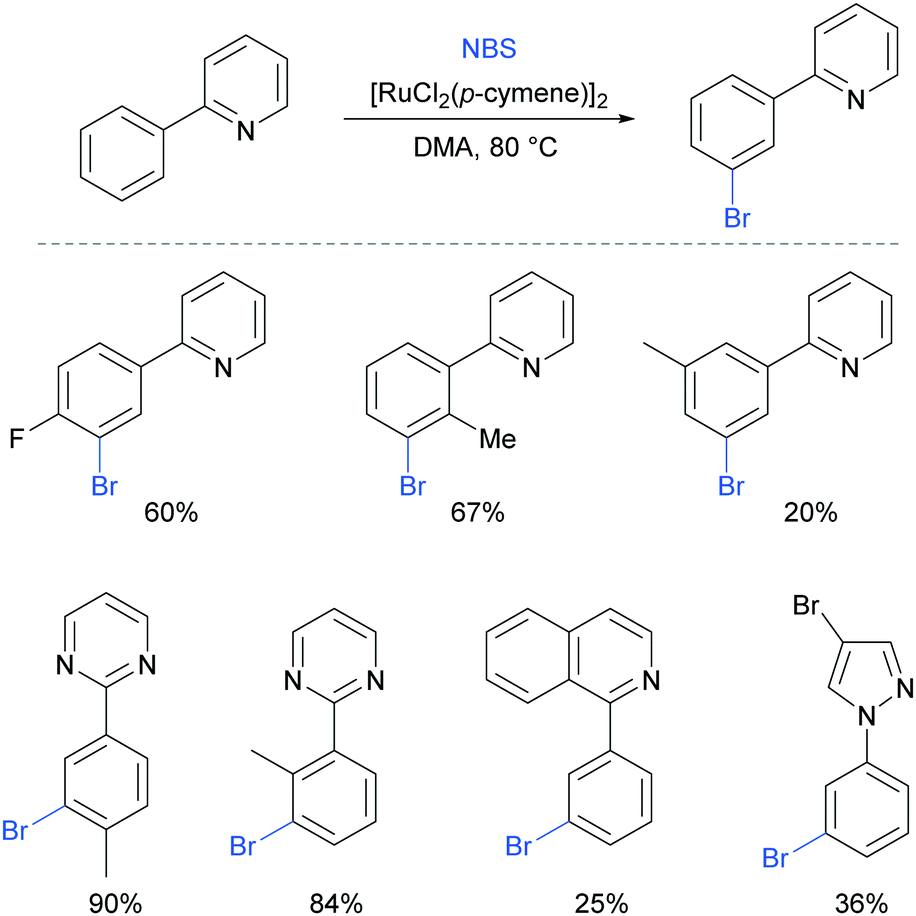 | ||
| Fig. 18 meta-Selective bromination of arenes with heterocyclic directing groups. | ||
The authors proposed that after the ortho-cycloruthenation, a second C–H activation could occur with another molecule of 2-aryl pyridine, to give intermediate VIII (Scheme 9). This could undergo oxidative addition with NBS to generate IX, followed by a SET bromination and ligand exchange to release the product. This mechanism was supported by experiments using isolated IV which did not provide the expected meta-brominated product when exposed to the reaction conditions (Scheme 9b).
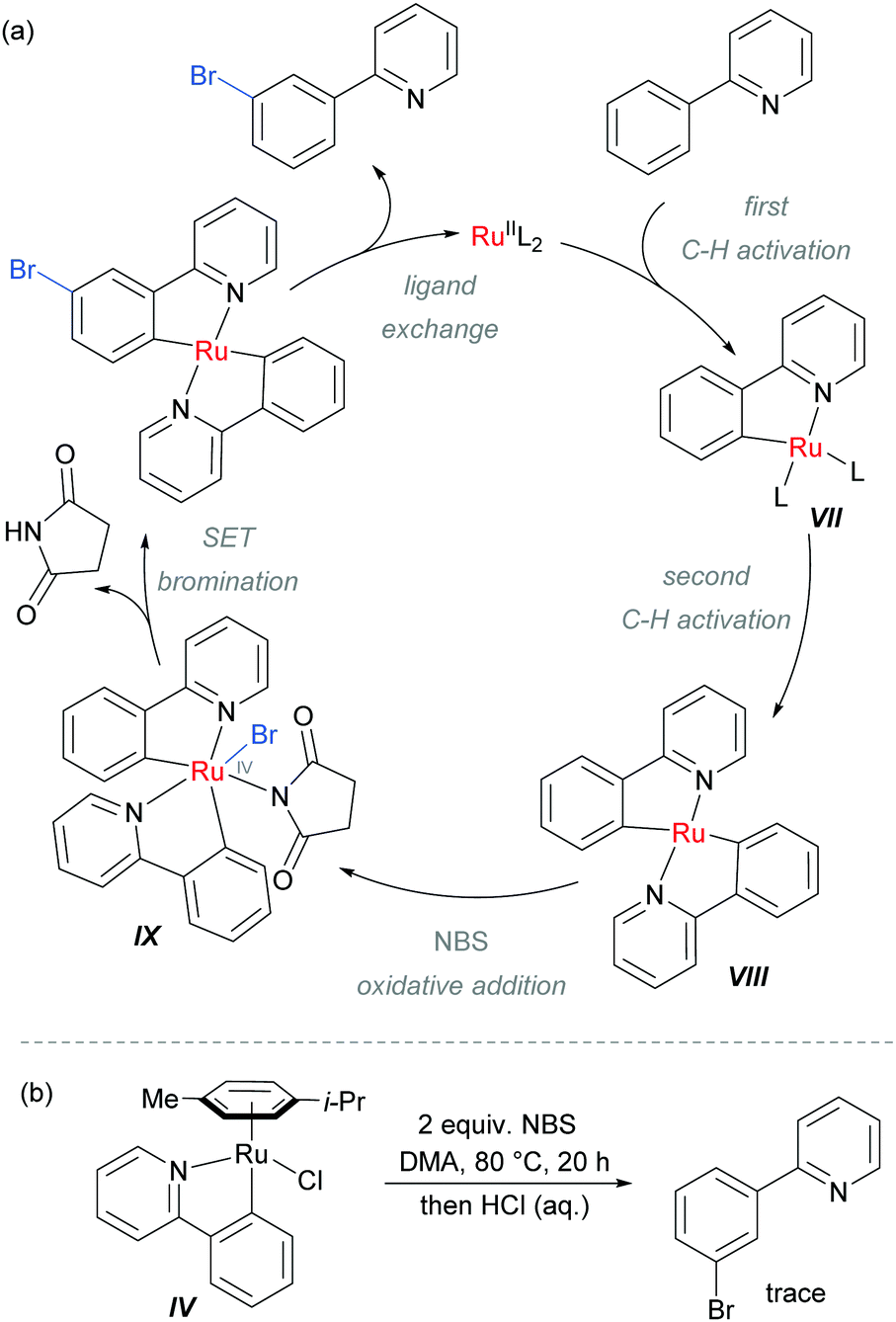 | ||
| Scheme 9 (a) Proposed mechanism of meta-bromination via RuIV complex IX; (b) attempted bromination of isolated ruthenium complex IV. | ||
The meta-selective nitration of arenes bearing heterocycle directing groups was reported by Zhang et al. in 2016, using Ru3(CO)12 as a catalyst and Cu(NO3)·3H2O as nitro source.89 The reaction components also included a silver salt as a proposed radical initiator, Oxone as oxidant and tetrabutylammonium acetate (TBA-OAc) as a phase-transfer catalyst (Fig. 19). With 2-aryl pyridines, substitution is tolerated at all positions on the phenyl ring as well as on the pyridine ring, albeit with a lower yield when a methyl substituent was present adjacent to the nitrogen, presumably due to steric interference with the Ru complex formation. Various other heterocycles were also investigated as directing groups, as well as a single example of an oxime.
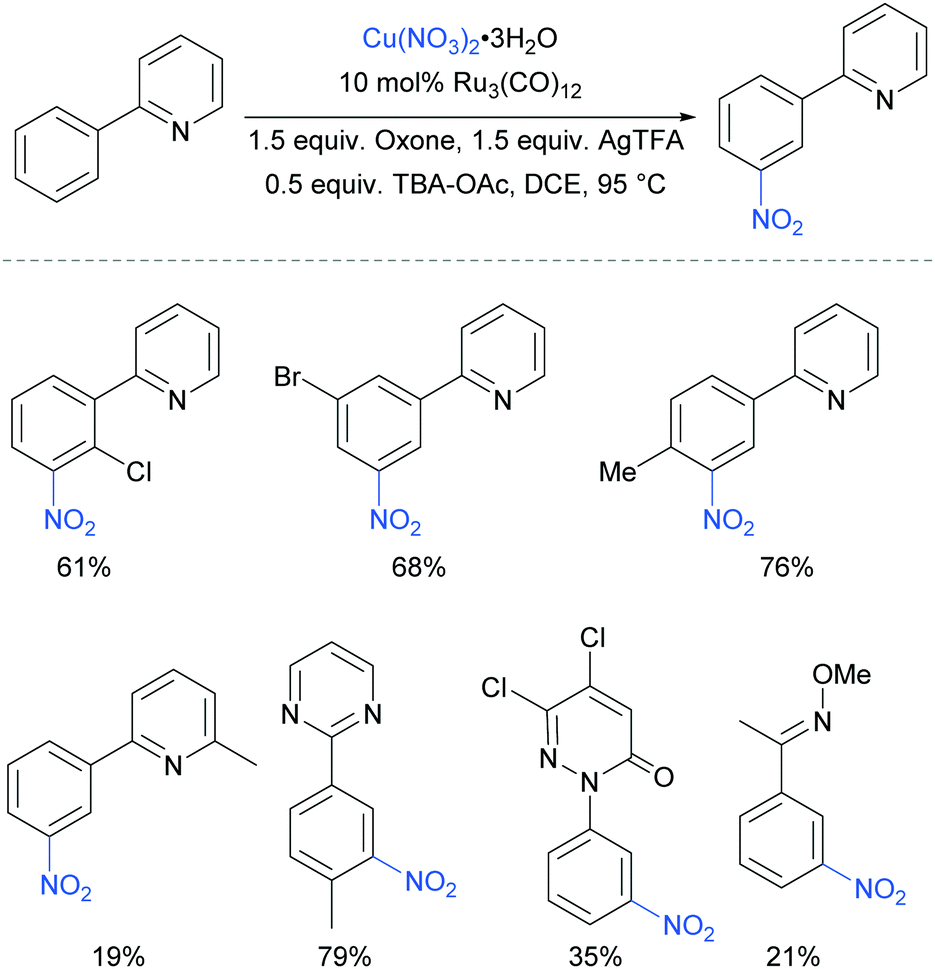 | ||
| Fig. 19 meta-Nitration of arenes with heterocyclic directing groups. | ||
Regarding the nature of the active catalyst, when complex X was isolated and exposed to the reaction conditions, the nitration product was obtained in 70% yield (Scheme 10). Complex X was also found to be a competent catalyst for the nitration of 2-phenylpyridine, further supporting its role as an intermediate in the catalytic cycle. As in previous studies, the authors observed TEMPO inhibition, and carried out a range of isotope labelling experiments. These resulted in a proposed mechanism that closely mirrors the previously proposed mechanisms for analogous transformations, in this case differing in how the nitrogen dioxide radical is formed, which is hypothesised to be via a silver-mediated radical process (Scheme 11).
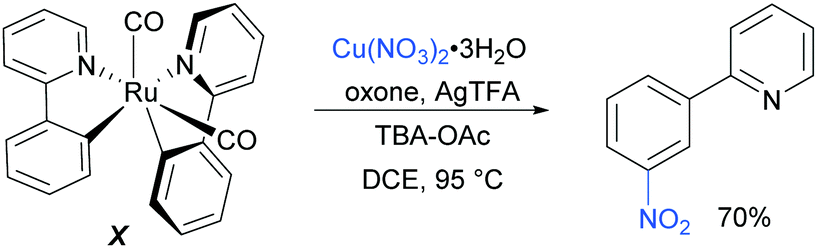 | ||
| Scheme 10 Nitration of preformed ruthenium complex X. | ||
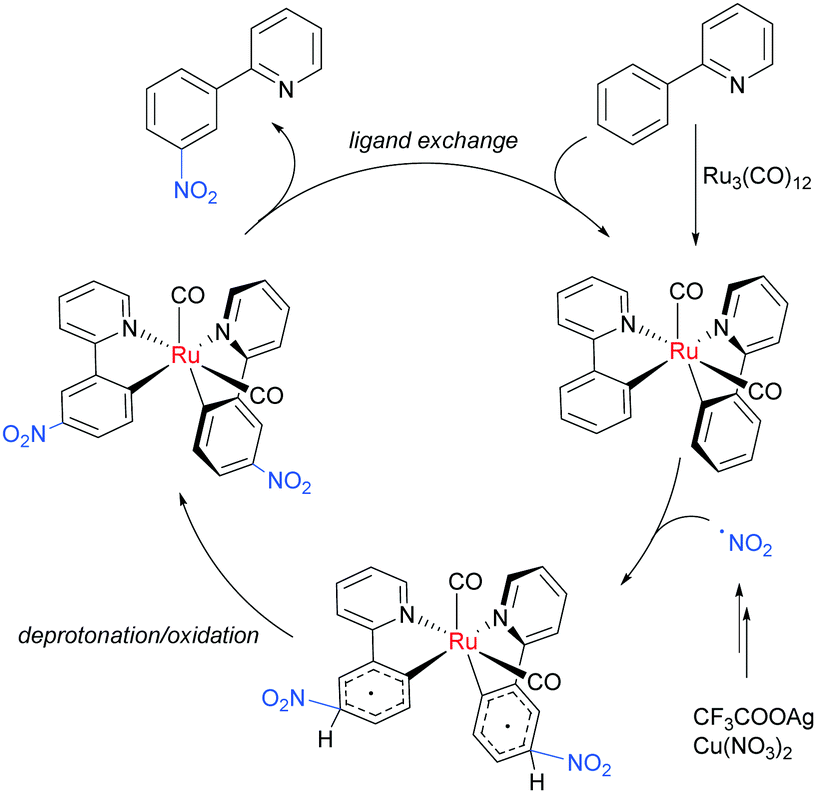 | ||
| Scheme 11 Proposed mechanism for meta-nitration. | ||
The same authors recently published an expansion of this methodology whereby they are able to use removable oximes as directing group for the transformation.90 The high yields obtained are enabled by the use of a combination of PhI(TFA)2 and O2 as oxidants, rather than Oxone, as in the previous report.
In 2017, Li, Yang and co-workers disclosed azoarenes as an additional class of masked aniline substrates for meta-selective functionalisation in the presence of [RuCl2(p-cymene)]2.91 Pivalic acid was used as a key additive to achieve meta-alkylation of various azoarenes with secondary and tertiary alkyl bromides (Scheme 12a). Using this protocol with primary bromides led to selective formation of ortho-substituted products, in line with previous reports. Li, Wang and co-workers92 later reported the meta-sulfonation of azoarenes with arylsulfonyl chlorides under [RuCl2(p-cymene)]2 catalysis. In both papers, the authors demonstrated that the azo group can be readily cleaved to generate meta-alkylated91 or meta-sulfonated anilines,92 respectively.
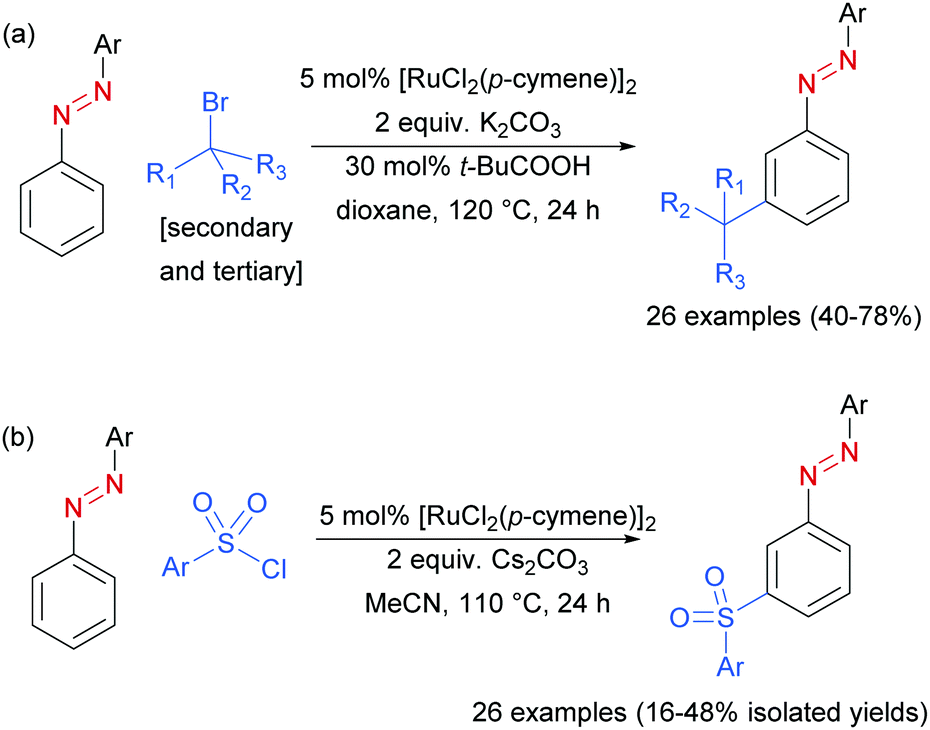 | ||
| Scheme 12 (a) meta-Selective sec- and tert-alkylation of azoarenes; (b) meta-selective sulfonation of azoarenes. | ||
A further example of substrates with removable groups was also published by the Li group in 2017.93 In this report, 2-phenoxypyridines were meta-selectively alkylated in the presence of [RuCl2(p-cymene)]2, using both secondary and tertiary alkyl bromides (Fig. 20). The pyridine directing group was able to be cleaved under relatively harsh conditions to afford meta-alkylated phenols (Scheme 13).
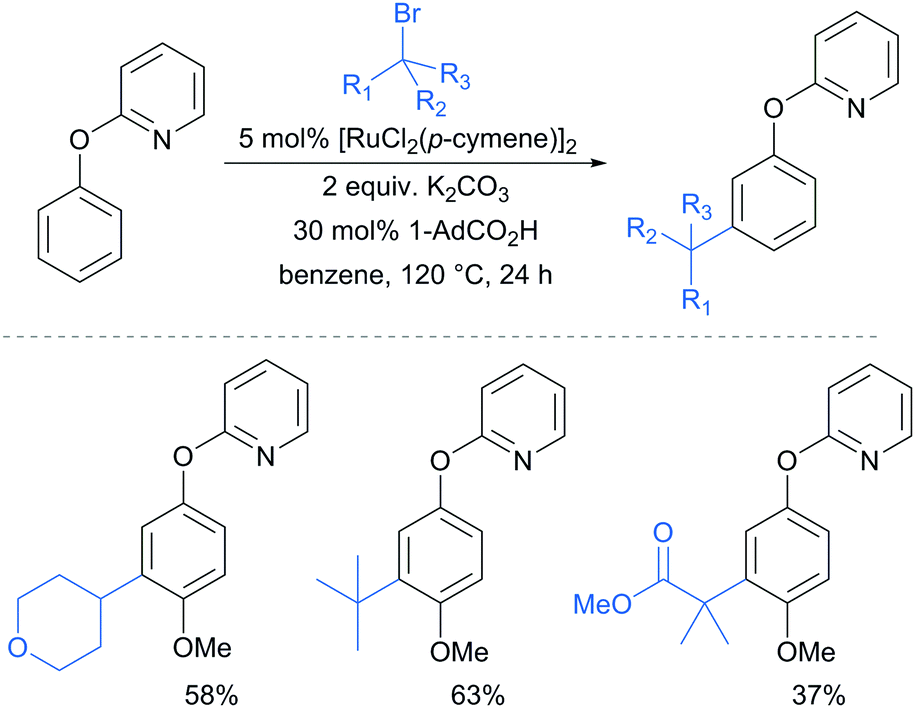 | ||
| Fig. 20 meta-Selective sec- and tert-alkylation of protected phenols. | ||
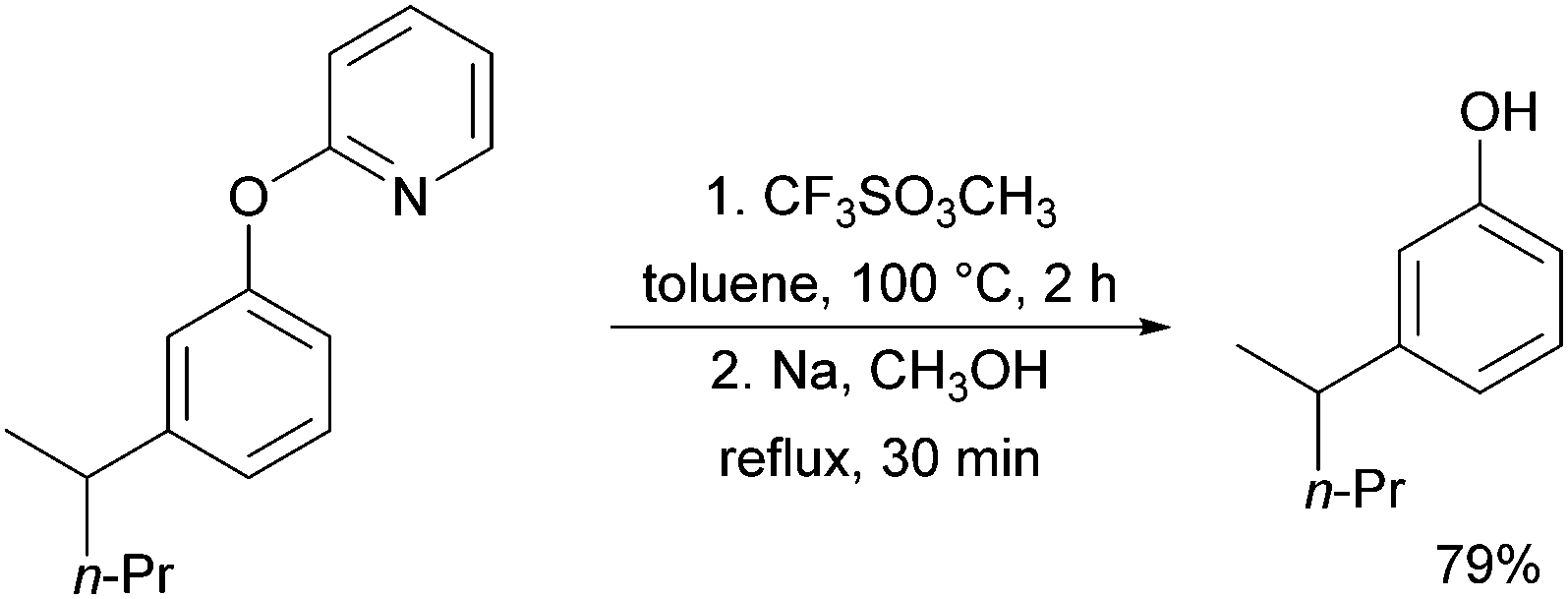 | ||
| Scheme 13 Deprotection of phenols after alkylation. | ||
Later in 2017, the same group reported a dual ortho/meta selective functionalisation of 2-phenoxypyri(mi)dines with sulfonyl chlorides.94 The reaction generates products bearing a meta-sulfonyl group and a chloride at the non-adjacent ortho position. Selected examples are shown in Fig. 21.
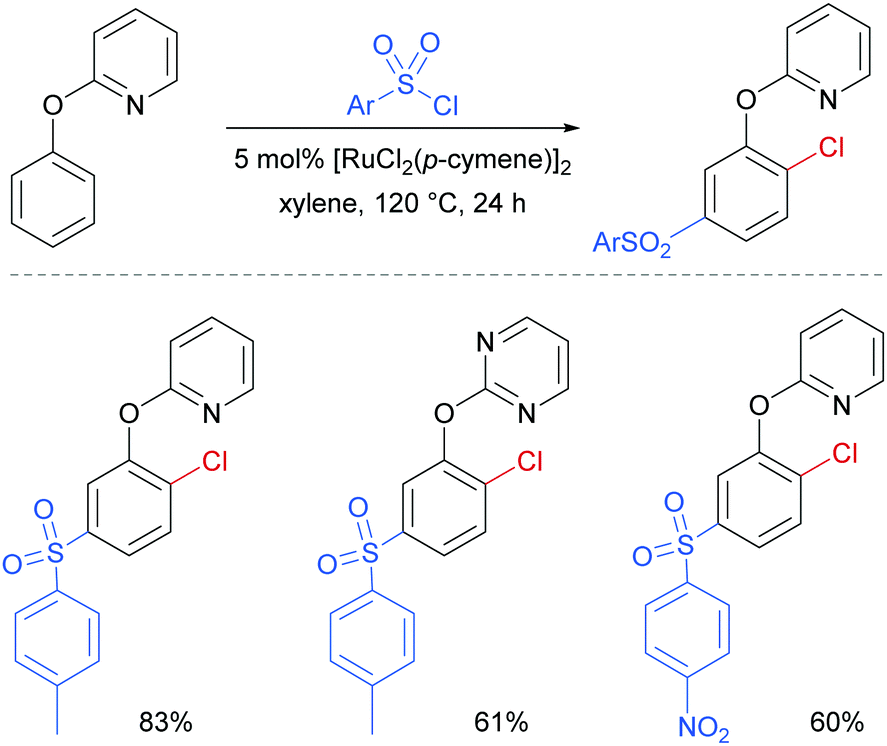 | ||
| Fig. 21 Selected examples for dual ortho/meta selective functionalisation of 2-phenoxypyri(mi)dines. | ||
The authors proposed that this reaction proceeds via an initial ortho-ruthenation, followed by an SEAr with sulfonyl chloride at the C–Ru para position, similarly to examples discussed above. The formation of the ortho C–Cl bond was rationalised by an oxidative addition of intermediate XI into sulfonyl chloride to form XII, which after reductive elimination affords the ortho and meta functionalised product (Scheme 14). This mechanism is supported by experiments run with premade complexes XI and XII which afford the desired ortho/meta functionalised product in nearly quantitative yield when exposed to the reaction conditions. In addition, this reaction is thought to proceed via a radical mechanism as indicated by TEMPO experiments and the presence of 1-(chloromethyl)-methylbenzene by-product. Another by-product, S-p-tolyl-4-methylbenzenesulfonothioate (Scheme 14, inset) was detected by GC-MS and this provides evidence of an additional role that the sulfonyl chloride might play in this reaction, as an oxidant.
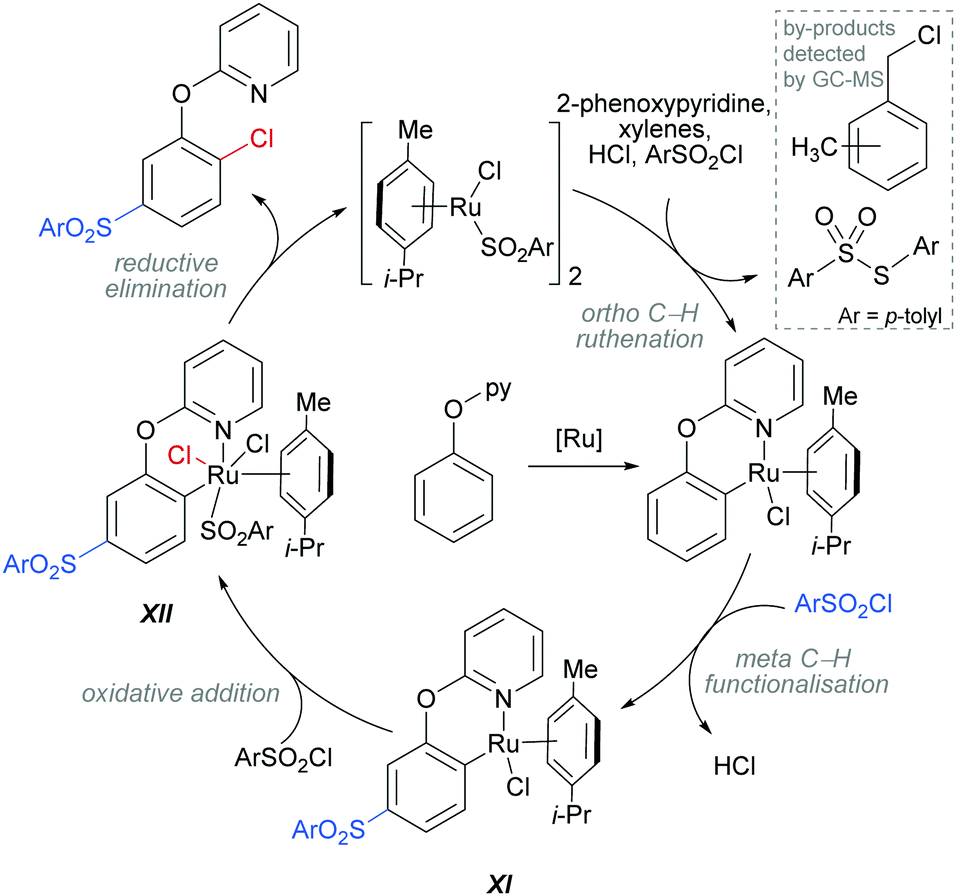 | ||
| Scheme 14 Proposed mechanism for the dual meta-sulfonation/ortho-chlorination of 2-phenoxypyridines. | ||
Earlier this year, the Zhao group published a ligand-enabled benzylation of 2-arylpyridines using toluene derivatives, where both the ortho- and meta-products were selectively accessed by changing the ligand and the Ru source (Scheme 15).95
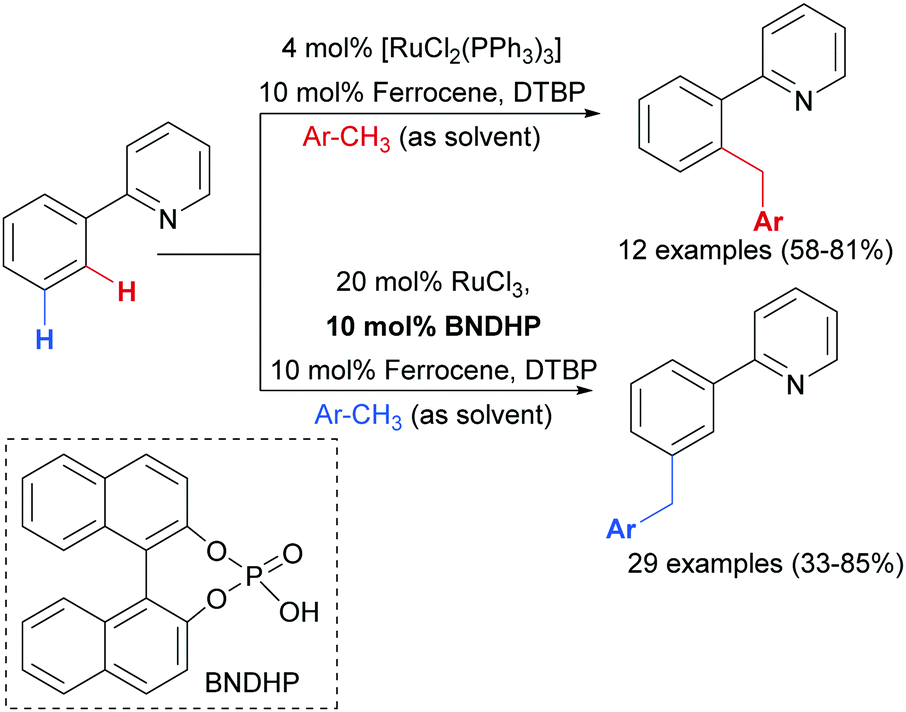 | ||
| Scheme 15 ortho- and meta-Selective benzylation of 2-arylpyridines. | ||
For the meta-selective pathway, the authors proposed a single-electron process in line with previous reports, in this case with ferrocene being involved in the generation of the benzylic radical. A possible structure for the active catalyst was proposed to be XIII (Fig. 22). This was based on experiments with isolated complex X, which when exposed to the benzylation conditions (with BNDHP), afforded significantly more meta than ortho products. A similar experiment with complex IV led to selective ortho benzylation, which is consistent with previous reports from Ackermann and Frost. Therefore, the active catalyst involved in the meta-benzylation pathway was proposed likely to be more similar in structure to X, possibly with L ligands instead of CO, although this postulated complex could not be isolated.
 | ||
| Fig. 22 Complexes isolated and postulated structure of intermediate complex for the meta-benzylation. | ||
Shortly after, the Shi group also reported a meta-selective benzylation of 2-aryl N-heterocycles with toluene derivatives, using heptafluoroisopropyl iodide (iC3F7I) as a radical initiator as well as oxidant (Scheme 16).96
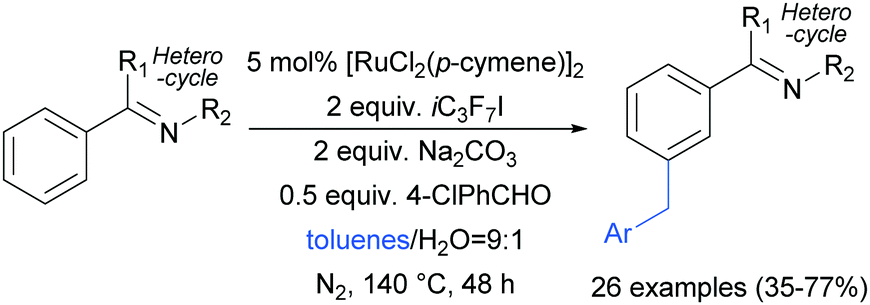 | ||
| Scheme 16 meta-Selective benzylation of arenes with heterocyclic directing groups. | ||
In 2017, the Ackermann group reported a heterogeneous ruthenium catalyst for meta-bromination of purine bases, achieving selectivity at the meta position of a pendant arene, even in the presence of the particularly acidic C8 position of the purine scaffold (Fig. 23a).97 The authors demonstrated that the silica-derived heterogenous catalyst ‘Ru@SiO2’ can be recycled several times without significant decrease in activity and outperformed the related homogenous system. The same protocol was also applied effectively to meta-bromination of a series of 2-aryl pyridine and pyrimidines (Fig. 23b).
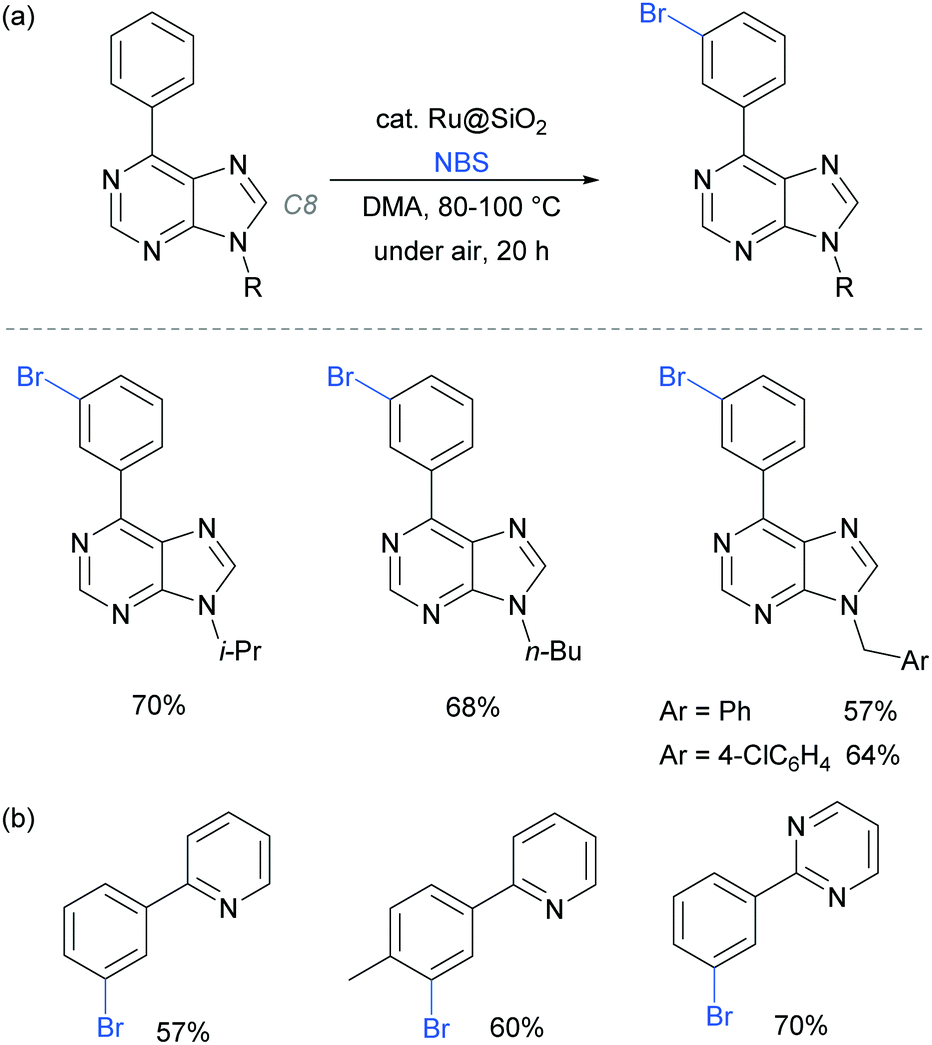 | ||
| Fig. 23 (a) meta-Bromination of purine-base derivatives using a heterogenous catalyst; (b) meta-bromination of 2-aryl pyridines and pyriminides using the same conditions as in (a). | ||
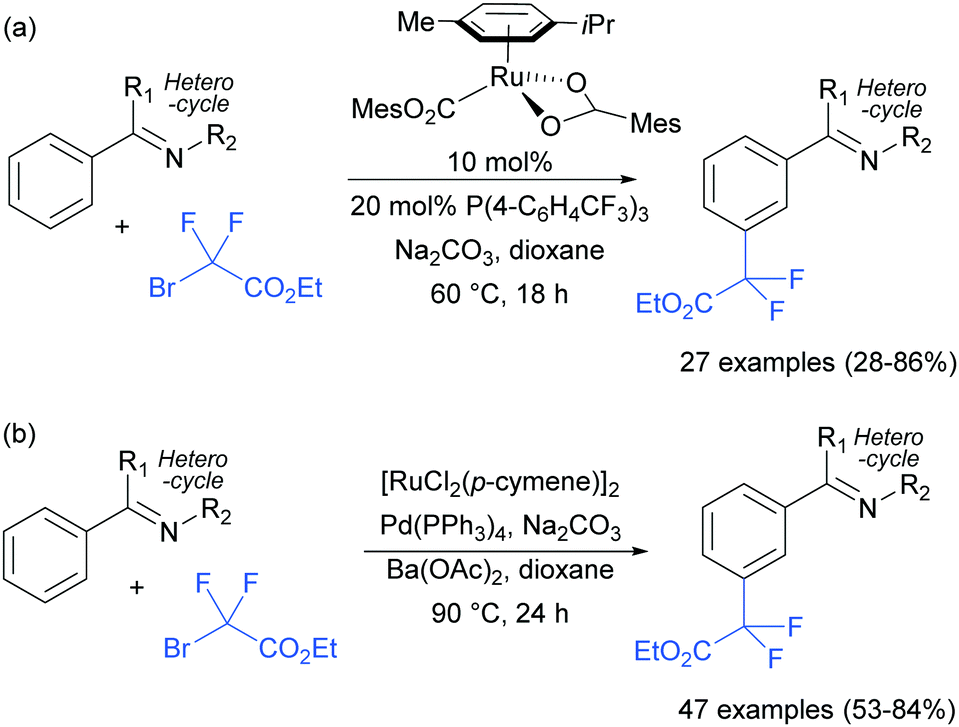
In two papers published almost simultaneously, the Ackermann98 and the Wang99 groups demonstrated that PPh3-containing additives enable the ruthenium-catalysed mono- and di-fluoromethylation of arenes with a heterocyclic directing group (Scheme 17). Control experiments in both publications reveal that no reaction takes place in the absence of the PPh3 source. A subsequent paper on the same topic also appeared soon after.100
In 2017, Frost and co-workers reported an expansion of their previously reported methodology to C6-selective tert-alkylation of pyrimidinyl-indole derivatives (Fig. 24).101 An ancillary ester group at C3 was found to be essential for directing the alkylation at the C6 position. α-Bromo esters and ketones were both compatible as coupling partners.
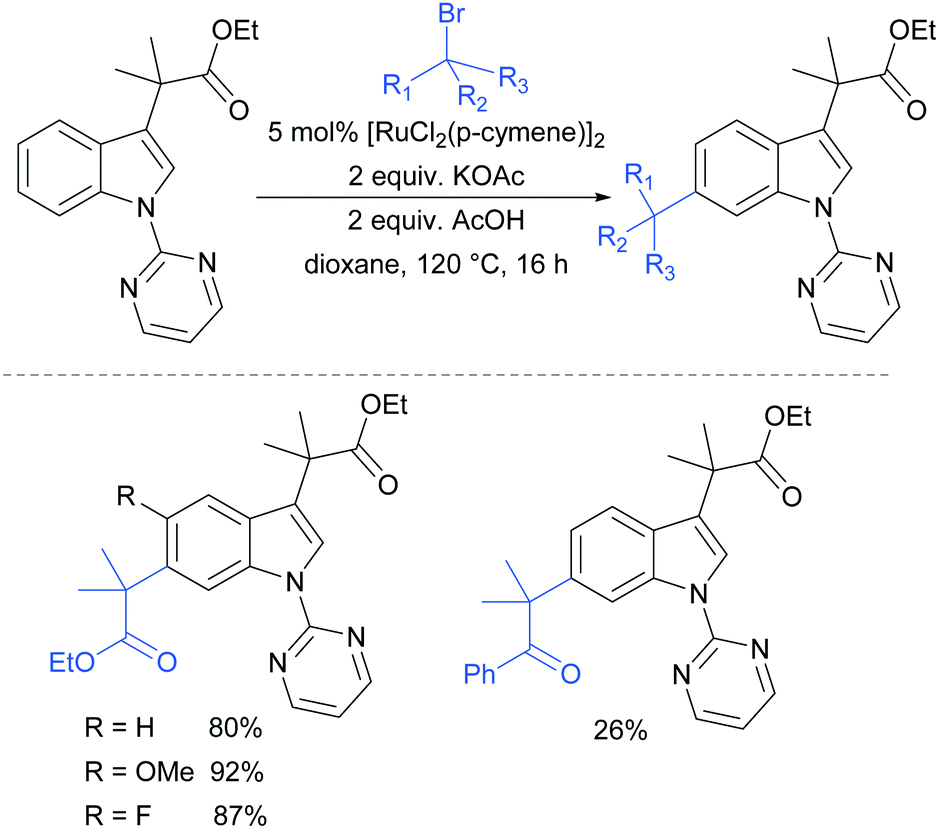 | ||
| Fig. 24 C6-Alkylation of indoles bearing a pyrimidine protecting group on the nitrogen, and an ancillary ester directing group at C3. | ||
The Frost group also recently reported the use of α-halocarbonyls for meta-selective primary, secondary and tertiary alkylations of various arenes bearing heterocyclic directing groups (Fig. 25).102 Notably, the meta-alkylation with primary alkyl bromides was enabled by the use of a triarylphosphine source as an additive, as in the aforementioned mono- and di-fluoromethylation reactions. Indeed, without this phosphine source, poor selectivity between ortho and meta products was obtained. The exact role played by the phosphine ligand remains unknown, however the authors speculate that they might serve to fill coordination sites on the ruthenium which would otherwise lead to ortho-functionalisation. Their preliminary mechanistic studies together with computational work suggested that the secondary and tertiary alkylations with α-halo carbonyls follow a radical mechanism, while with the primary alkylation it is also possible that it follows a more electrophilic rather than radical mechanistic pathway.
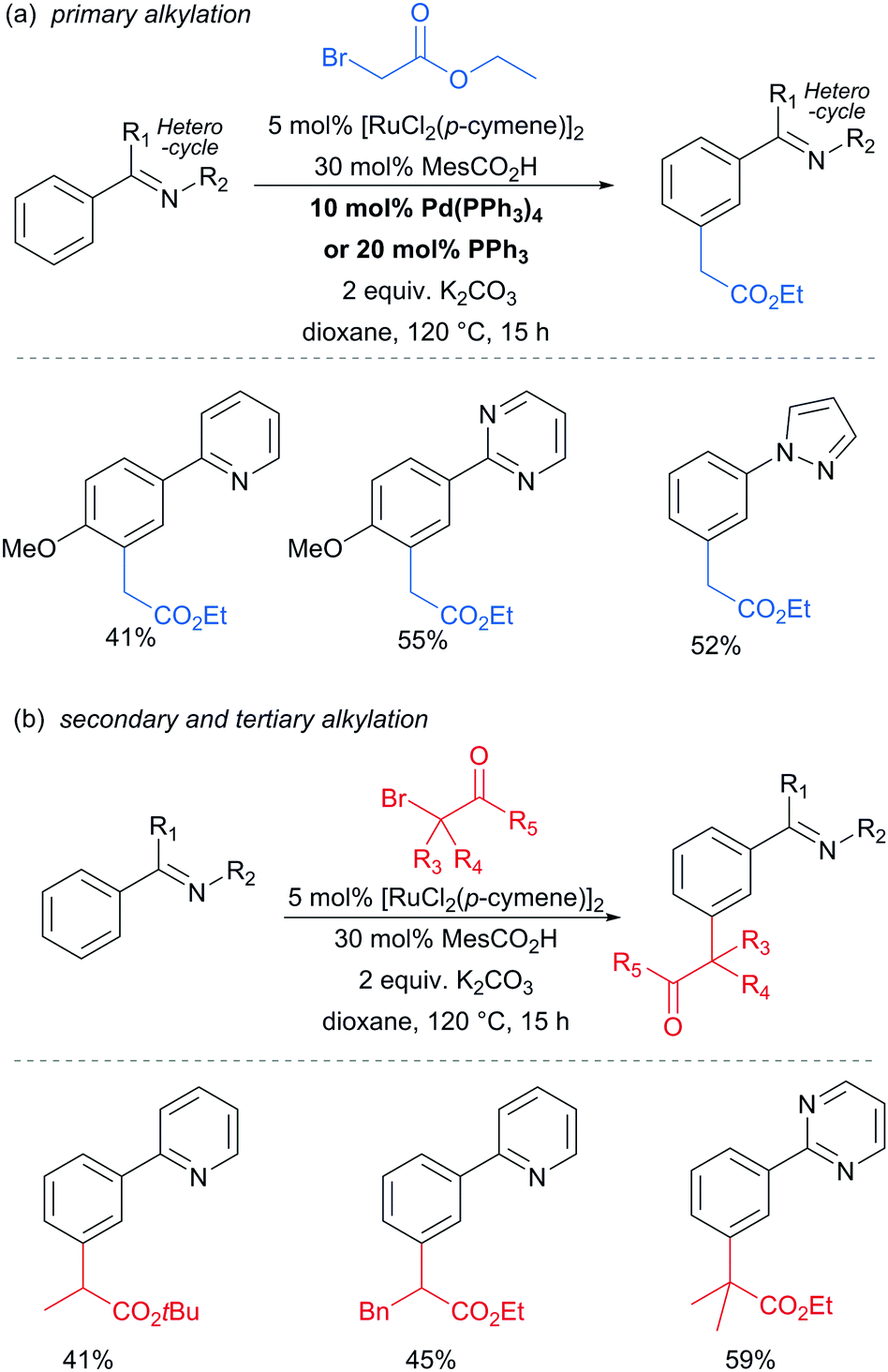 | ||
| Fig. 25 meta-Alkylation with of α-halocarbonyls. | ||
Most of the protocols reported to date on ruthenium-catalysed meta-selective functionalisation employ nitrogen-based heterocyclic directing groups, which constitutes the main limitation for broader application of this methodology, as these directing groups are often difficult to remove or modify. A report by the Ackermann group in 2017 addressed this limitation by employing imines as substrates for a meta-selective alkylation (Fig. 26a).103 Following the alkylation, the imine could be easily converted into a range of other functional groups such as amines or ketones. The authors also showed a one-pot meta C–H alkylation and ortho C–H arylation/alkylation, which did not require further catalyst to be added for the second step (Fig. 26b).
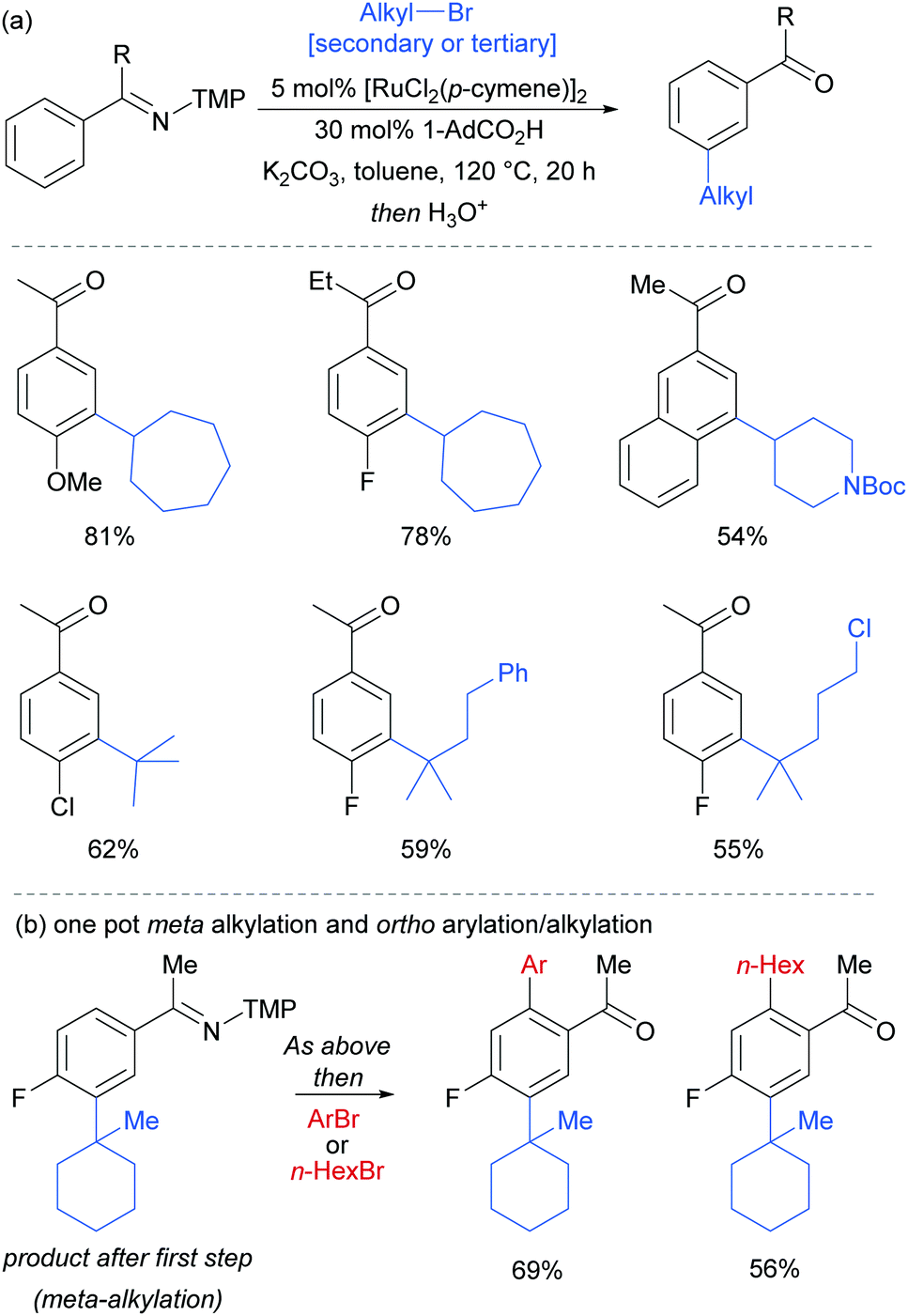 | ||
| Fig. 26 (a) meta-Selective alkylation of ketimines (b) one-pot procedure for meta followed by ortho alkylation of ketimines; TMP = 3,4,5-trimethoxyphenyl. | ||
Very recently, an intriguing report from Frost and co-workers detailed how Ru catalysis could be used to obtain para-selective alkylation by changing the nature of the substrate.104 This was achieved by using anilines bearing a pyrimidine auxiliary which results in Ru–N cyclometalation as opposed to Ru–C. The authors hypothesised that the sigma activation that results in this case leads to para-selective alkylation through a similar SET-type mechanism to the meta-selective examples, a hypothesis that was supported by DFT calculations.
5. Copper catalysis
The use of copper salts to catalyse the decomposition of diaryl iodonium salts has been established for many years.105 But it was not until 2008 that this was investigated in combination with heteroarene nucleophiles to form biaryl bonds when Gaunt and co-workers demonstrated the copper-catalysed arylation of indoles.106 The regioselectivity was found to be controllable – for free NH indoles, C3 arylation was observed whereas with N-acyl indoles, the arylation was directed to the C2 position, potentially through coordination of the active arylating agent to the carbonyl. This led the authors to subsequently investigate the outcome of the reaction when applied to simpler arenes bearing coordinating groups. This resulted in the arylation occurring at the meta position of anilides, a surprising and unprecedented result.107 They found that a range of substituted pivaloyl-protected anilines were compatible and that the arylation occurred under relatively mild conditions (Fig. 27).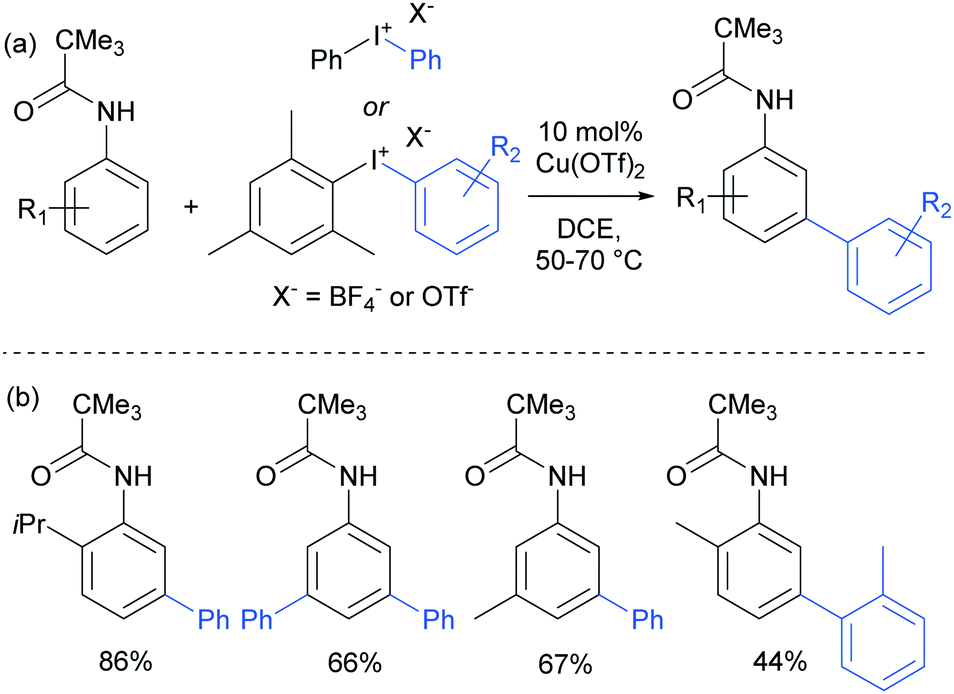 | ||
| Fig. 27 (a) Overview of copper-catalysed anilide arylation with iodonium salts; (b) selected examples of anilide substrates. | ||
The only case in which meta-selectivity was not obtained was in the case of a meta-methoxy group, which directed the arylation to occur in the ortho position (para to the strongly electron donating methoxy group). In addition to the broad anilide scope, a variety of substituted iodonium salts transferred smoothly. The selectivity outcome was particularly surprising given that analogous reactants under palladium catalysis had been previously established to give ortho-arylation via a presumed cyclometalation mechanism.108 This is another example of a switch away from palladium to an alternative metal resulting in meta-selectivity (see Ruthenium section).79 Given this, it seemed likely that the carbonyl group was interacting in a relatively unconventional manner with the postulated electrophilic aryl–Cu(III) complex produced by oxidation of a Cu(I) catalyst by the strongly oxidising iodonium salt.109
The following year, Gaunt and co-workers reported that amides derived from aryl acetic acids also underwent meta-selective arylation under these conditions.110 In this class of substrates the carbonyl group is in approximately the same position relative to the aromatic ring and provided support to the hypothesis that this carbonyl group was playing a key role in enabling the novel meta-selectivity (Fig. 28). An (S)-ibuprofen derivative underwent meta selective arylation without loss of integrity at the benzylic chiral centre. Whilst the Weinreb amide-derived substrates resulted in the highest reactivity, it was interesting to note that ketones were also effective in delivering meta-selective arylation. In this substrate class, α,α-disubstitution was tolerated without erosion of selectivity.
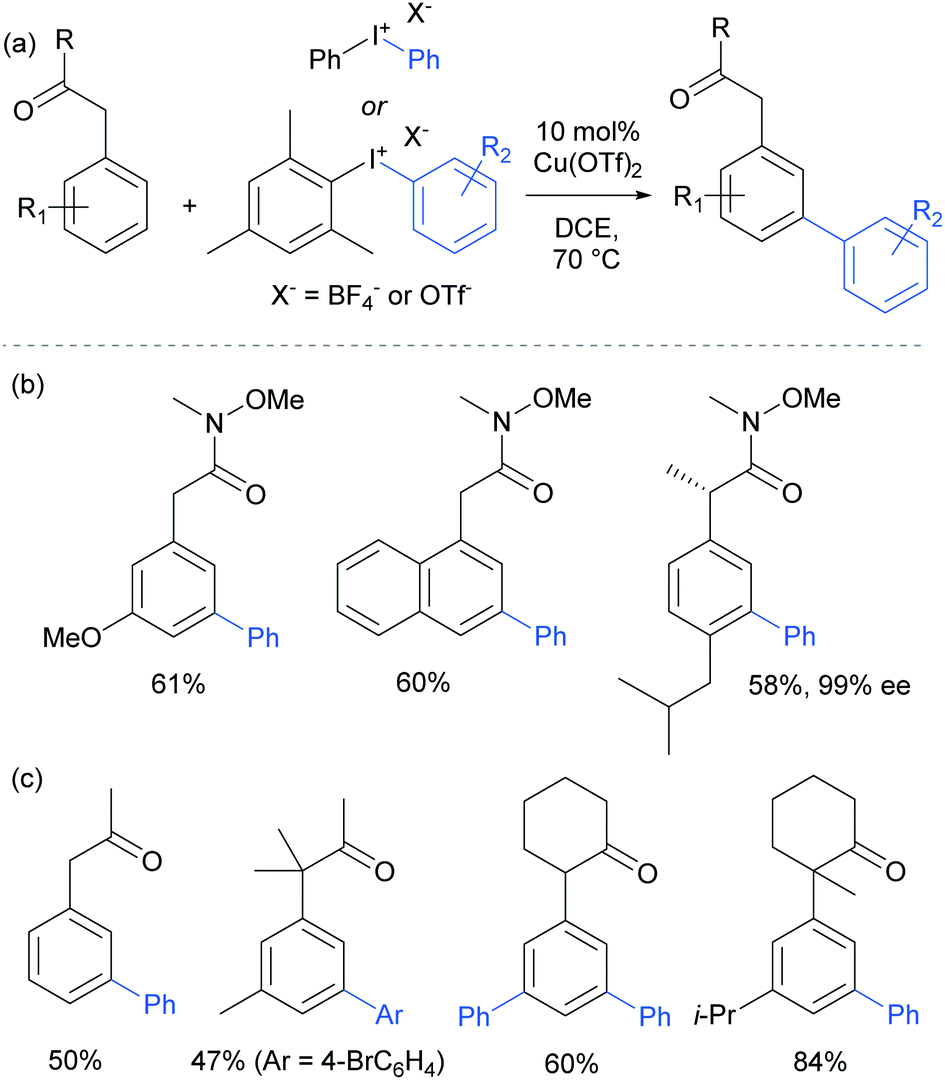 | ||
| Fig. 28 (a) Overview of copper-catalysed arylation of α-aryl carbonyl compounds with iodonium salts; (b) selected examples of substrates with an amide as directing group; (c) selected examples of substrates with a ketone as directing group. | ||
Parallel studies by Gaunt and co-workers showed that electron rich arenes that do not bear potential coordinating groups result in the para-selectivity that would typically be associated with an electrophilic aromatic substitution.111 From a mechanistic standpoint, this finding provided further evidence for the carbonyl group being crucial to the observed meta-selectivity. In 2011, Li, Wu and co-workers published a computational study of the meta-arylation of anilides.112 They investigated three possible reaction pathways for acetanilide. In the first (not shown), direct cupration at the meta position with the amide oxygen attacking concomitantly at the ortho position, as postulated in the original report, was found to be prohibitively high in energy. The second and third both commenced with complexation of the putative electrophilic aryl–Cu(III) species with the amide carbonyl. In the second, leading to the ortho product, cupration occurred at the ortho position by a concerted-metalation–deprotonation (CMD) mechanism prior to reductive elimination (Fig. 29a). The third, leading to the observed meta-product, follows a Heck-like four membered-ring transition state in which aromaticity was proposed not be completely destroyed (Fig. 29b). Following C–Cu bond cleavage to regenerate Cu(I), rearomatisation leads to the meta-arylated product. The transition state for this meta pathway was found to be approximately 2 kcal mol−1 lower than the ortho pathway, which provided a good basis for the observed selectivity of most substrates. With some modification, their model was able to account for the ortho-selectivity observed with the meta-methoxy substrate. Further mechanistic evidence regarding arylation using iodonium salts and copper catalysis remains elusive, but it is generally accepted to proceed via the typically postulated aryl–Cu(III) intermediates. In some processes, including those detailed above, there have been instances wherein reasonable yields of the arylation product could be obtained without the addition of a copper catalyst if the reaction was carried out at higher temperature than normal. Given the very large rate accelerations (up to three orders of magnitude)112 involved upon addition of catalytic amounts of copper to such reactions, it remains a possibility that trace copper contaminants could account for the copper-free reactivity that has been observed.
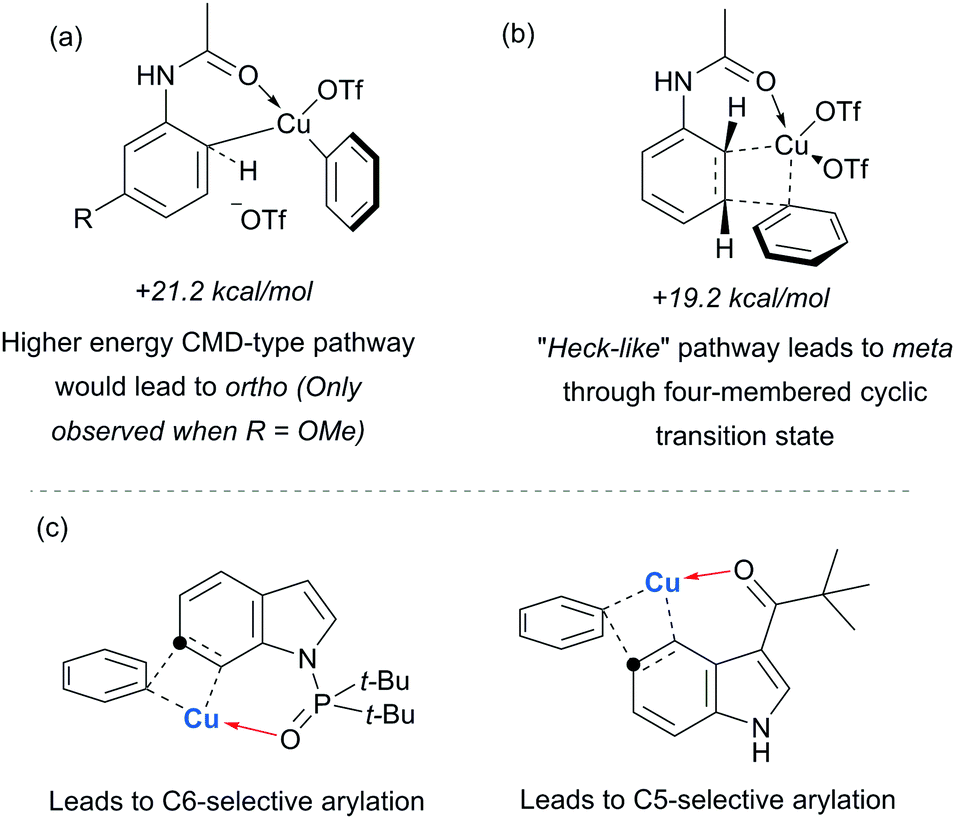 | ||
| Fig. 29 Computational investigation of the reaction pathway disfavoured (a) a CMD-type mechanism which would lead to the ortho-arylated product and suggested (b) a “Heck-like” transition state leading to the meta-arylated product. (c) Subsequent work by Shi and co-workers on directed C6 and C5 arylation of indoles with suitable directing groups. | ||
Gaunt's carbonyl-directed meta-arylation strategy has recently been elegantly applied by Shi and co-workers to selectively access the C5 and C6 positions of indole, which are typically very challenging to access using C–H bond functionalisation. For the C6 position, a bulky phosphinoyl group (N-P(O)tBu2) is installed on the indole nitrogen (Fig. 29c).113 It is thought that the bulky t-Bu groups block the C2 position and force the phosphoryl oxygen to project towards the arene portion of the benzene. In direct analogy with the aforementioned meta-selective processes this now directs the arylation to occur at the indole C6 position and this is proved highly effective on a range of indoles substituted with the N-P(O)tBu2 group. The same group has since reported that if a ketone group (specifically pivaloyl) is installed at the indole C3 position, it is now the C5 position that is functionalised, in the ‘meta’ position with respect to the directing group (Fig. 29c).114 Again this strategy is quite general and the authors demonstrate that the pivaloyl group can be readily removed after the arylation. Additionally, Gaunt's anilide arylation was recently adapted to a modular flow chemistry process where all operations, including iodonium salt synthesis and aniline deprotection were carried out in flow.115
6. Rhodium catalysis
Rhodium has been a key catalyst throughout the development of the field of C–H functionalisation and has been responsible for a variety of important advances.116–118 It is therefore somewhat surprising that it is only in the last year that methods have been disclosed using rhodium catalysis to access the arene meta position.In early 2017, Yu, Sun and co-workers published a Rh(III)-catalysed meta-olefination of hydrocinnamic acid derivatives using one of their U-shaped templates that had been initially very successful with palladium (Fig. 30a).119 In this method, Cu(II) is used in an O2 atmosphere as oxidant, which is an advantage compared to the stoichiometric Ag oxidants commonly used in the related Pd-catalysed processes, although it was not possible to use the Cu in catalytic amounts. A small modification to the previously used template enabled a slightly better ratio of mono:di olefination in the unsubstituted model substrate. In addition to a large scope of hydrocinnamic acid derivatives the authors also demonstrated that a broad variety of electron deficient olefin coupling partners are compatible, including acrolein, enones, acrylamides and a vinyl sulfone (Fig. 30b). Furthermore, they showed that two examples of aniline derivatives incorporating a different template also undergo meta-selective olefination using this catalytic system (Fig. 30c).
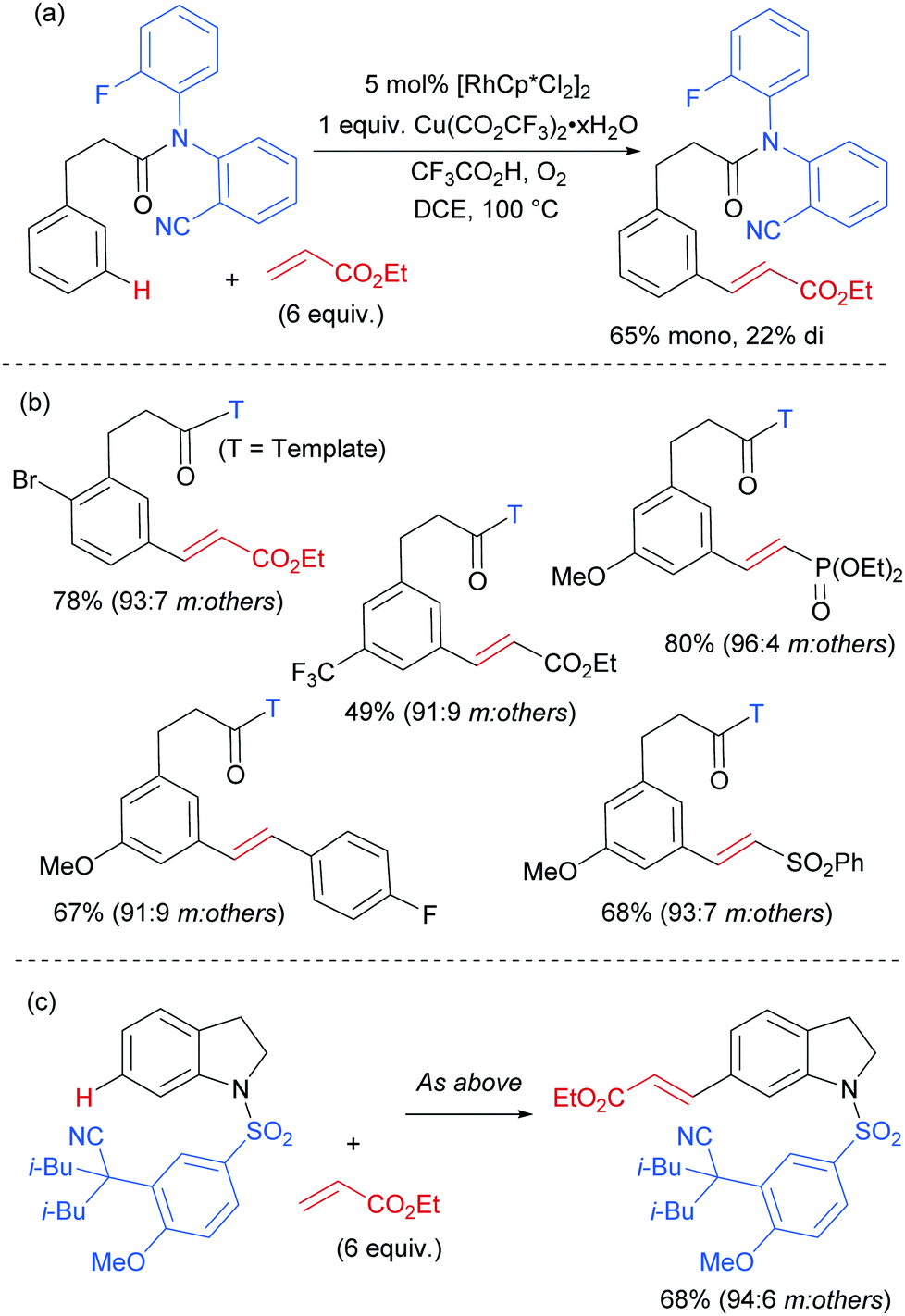 | ||
| Fig. 30 (a) Overview of rhodium-catalysed olefination of hydrocinnamic acid derivatives using a U-shaped template. (b) Examples of substrates effective in the reaction. (c) Example shown using a different template of an indoline derivative being compatible with the rhodium catalytic system. | ||
Several months later, Maiti reported the rhodium-catalysed olefination of benzylsulfonyl ester and phenyl acetic acid ester derivatives, again enabled by the incorporation of a nitrile-containing template into the substrate (Scheme 18).120 The substrates/templates they employed were those that had previously been shown to be successful in Pd-catalysed olefination25 and these translated smoothly to rhodium catalysis. They found it necessary to use a phosphine ligand and that bulky XPhos gave optimal yields and reduced the amount of di-olefination. A stoichiometric copper salt in combination with V2O5 as co-oxidant was found to give the best yields. In comparison to their previous Pd-catalysed olefination, DCE could now be used as solvent rather than hexafluoroisopropanol (HFIP). This constituted an advantage over the previous system as with HFIP as solvent, transesterification to the HFIP ester after olefination was found to be unavoidable. In addition, the meta-selectivity with the Rh system was found to be superior, which could be due to the lack of transesterification in this Rh catalysed system. Although the authors used Rh(I) catalysts, they suggest that the active catalyst is a Rh(III) complex obtained after oxidation of the pre-catalyst and this is supported by UV-vis studies and the fact that Rh(III) complexes are also efficient pre-catalysts. They also showed that it is unlikely that XPhos is oxidised under the reaction conditions.
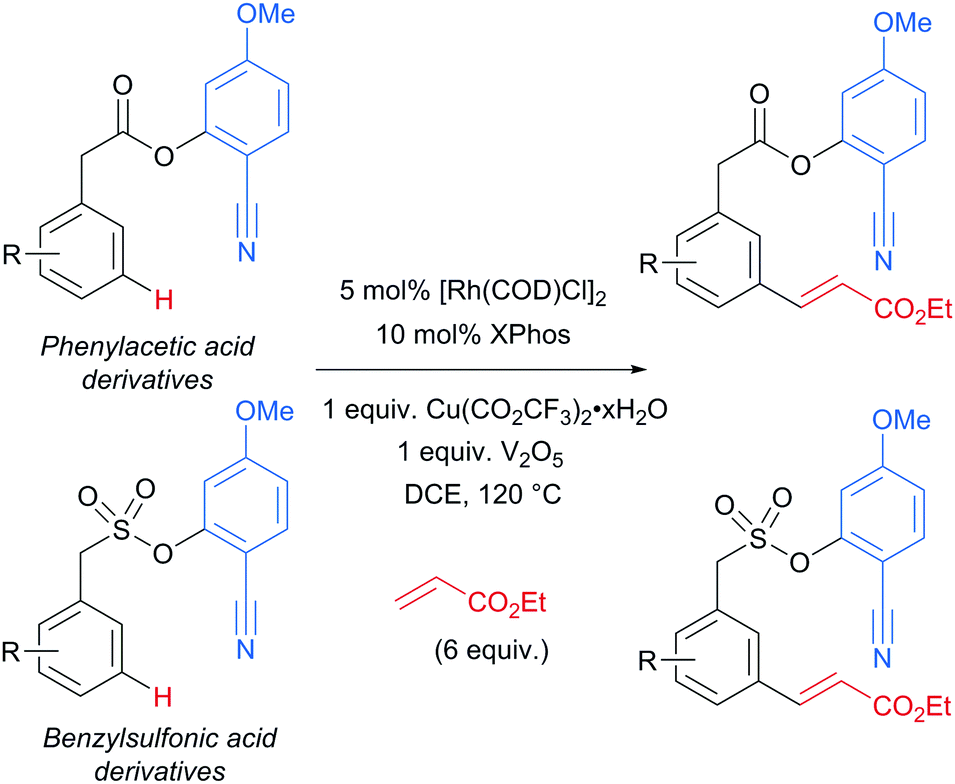 | ||
| Scheme 18 Overview of rhodium-catalysed olefination of phenylacetic acid derivatives and benzenesulfonic acid derivatives using a U-shaped template. | ||
For both reactions, the proposed catalytic cycle is as shown below (Scheme 19, shown for Yu's substrate). In Yu's work, the KIE was determined to be 1.8 in parallel experiments and under Maiti's it was found to 3.4. Maiti also determined the reaction to be first order in arene substrate and zero order in acrylate, which together with the large KIE suggests that the C–H activation is likely rate-determining.
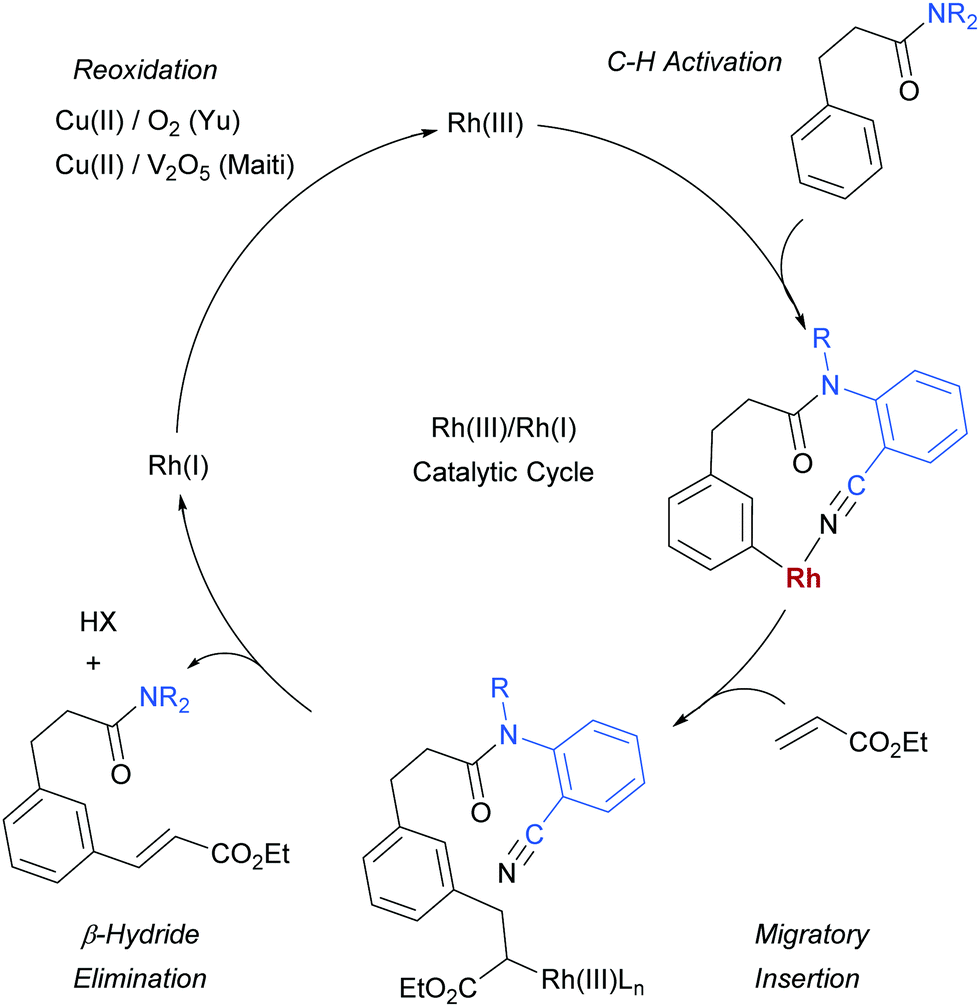 | ||
| Scheme 19 Proposed catalytic cycle for Rh-catalysed olefination using meta-directing templates. | ||
These two very recent disclosures suggest that the application of rhodium catalysis to these template-bearing molecules could be rather general, although as yet a substantial distinction between this and the related Pd catalysed methods remains to be demonstrated. The breath of ortho-selective C–H activation processes which rhodium has enabled certainly suggests that the potential is significant.
7. Conclusions
The notion that transition metals can offer diverse, enabling and sometimes surprising reactivity when used as catalysts is widely appreciated. Although it is sometimes the case that several different metals can undergo analogous catalytic cycles, it is also equally likely that new reaction pathways may be followed by changing from one to another. For example, in both Section 4 (Ruthenium) and Section 5 (Copper), reactions that were known to be ortho-selective under palladium catalysis were rendered meta-selective by switching to an alternative metal, initially surprising results which have been used as springboards for further productive development. In the case of iridium catalysis (Section 3), rational catalyst–substrate directing strategies have played a central role in harnessing the steric-based selectivity of these versatile C–H activation catalysts so that they can be applied to a wider range of molecules than was previously possible.Another notable aspect is that the vast majority of existing methods for meta-functionalisation require precious metal catalysts. Increasing attention in the field of C–H functionalisation is being given to switching to non-precious metal catalysts such as Cu, Ni, Co and Fe.121 This is important not only for the obvious economic and sustainability concerns but also as these largely represent uncharted territory in which novel reactivity and/or selectivity may await.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We are grateful to the Royal Society for a University Research Fellowship (R. J. P.), AstraZeneca for a PhD studentship (M. T. M.) through the AZ-Cambridge PhD Program and the EPSRC (G. R. G., EP/N005422/1). We thank Prof. Steven V. Ley, Prof. Matthew J. Gaunt and Iain Cumming (AstraZeneca) for support and useful discussions.References
- D. Astruc, Modern Arene Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2004, pp. i–xviii DOI:10.1002/3527601767.fmatter.
- C. G. Hartung and V. Snieckus, Modern Arene Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2004, pp. 330–367 DOI:10.1002/3527601767.ch10.
- S. Murai, F. Kakiuchi, S. Sekine, Y. Tanaka, A. Kamatani, M. Sonoda and N. Chatani, Nature, 1993, 366, 529–531 CrossRef CAS.
- T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS PubMed.
- P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879–5918 CrossRef CAS PubMed.
- D. A. Colby, A. S. Tsai, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2012, 45, 814–825 CrossRef CAS PubMed.
- L. Ackermann, Chem. Rev., 2011, 111, 1315–1345 CrossRef CAS PubMed.
- K. M. Engle and J.-Q. Yu, J. Org. Chem., 2013, 78, 8927–8955 CrossRef CAS PubMed.
- M. Gagliardo, D. J. M. Snelders, P. A. Chase, R. J. M. Klein Gebbink, G. P. M. van Klink and G. van Koten, Angew. Chem., Int. Ed., 2007, 46, 8558–8573 CrossRef CAS PubMed.
- A. Dey, S. Maity and D. Maiti, Chem. Commun., 2016, 52, 12398–12414 RSC.
- P. C. Andrikopoulos, D. R. Armstrong, D. V. Graham, E. Hevia, A. R. Kennedy, R. E. Mulvey, C. T. O'Hara and C. Talmard, Angew. Chem., Int. Ed., 2005, 44, 3459–3462 CrossRef CAS PubMed.
- D. R. Armstrong, W. Clegg, S. H. Dale, E. Hevia, L. M. Hogg, G. W. Honeyman and R. E. Mulvey, Angew. Chem., Int. Ed., 2006, 45, 3775–3778 CrossRef CAS PubMed.
- R. E. Mulvey, Acc. Chem. Res., 2009, 42, 743–755 CrossRef CAS PubMed.
- A. J. Martínez-Martínez, A. R. Kennedy, R. E. Mulvey and C. T. O’Hara, Science, 2014, 346, 834–837 CrossRef PubMed.
- A. Dey, S. Agasti and D. Maiti, Org. Biomol. Chem., 2016, 14, 5440–5453 CAS.
- J. Yang, Org. Biomol. Chem., 2015, 13, 1930–1941 CAS.
- J. Li, S. De Sarkar and L. Ackermann, in C–H Bond Activation and Catalytic Functionalization I, ed. P. H. Dixneuf and H. Doucet, Springer International Publishing, Cham, 2016, pp. 217–257 DOI:10.1007/3418_2015_130.
- K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788–802 CrossRef CAS PubMed.
- D. Leow, G. Li, T.-S. Mei and J.-Q. Yu, Nature, 2012, 486, 518–522 CrossRef CAS PubMed.
- S. Bag and D. Maiti, Synthesis, 2016, 804–815 CAS.
- H.-X. Dai, G. Li, X.-G. Zhang, A. F. Stepan and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 7567–7571 CrossRef CAS PubMed.
- L. Wan, N. Dastbaravardeh, G. Li and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 18056–18059 CrossRef CAS PubMed.
- R.-Y. Tang, G. Li and J.-Q. Yu, Nature, 2014, 507, 215–220 CrossRef CAS PubMed.
- G. Yang, P. Lindovska, D. Zhu, J. Kim, P. Wang, R.-Y. Tang, M. Movassaghi and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 10807–10813 CrossRef CAS PubMed.
- M. Bera, A. Modak, T. Patra, A. Maji and D. Maiti, Org. Lett., 2014, 16, 5760–5763 CrossRef CAS PubMed.
- S. Lee, H. Lee and K. L. Tan, J. Am. Chem. Soc., 2013, 135, 18778–18781 CrossRef CAS PubMed.
- M. Bera, A. Maji, S. K. Sahoo and D. Maiti, Angew. Chem., Int. Ed., 2015, 54, 8515–8519 CrossRef CAS PubMed.
- S. Bag, R. Jayarajan, R. Mondal and D. Maiti, Angew. Chem., Int. Ed., 2017, 56, 3182–3186 CrossRef CAS PubMed.
- A. Maji, B. Bhaskararao, S. Singha, R. B. Sunoj and D. Maiti, Chem. Sci., 2016, 7, 3147–3153 RSC.
- S. Li, L. Cai, H. Ji, L. Yang and G. Li, Nat. Commun., 2016, 7, 10443 CrossRef PubMed.
- A. Modak, A. Mondal, R. Watile, S. Mukherjee and D. Maiti, Chem. Commun., 2016, 52, 13916–13919 RSC.
- S. Maity, E. Hoque, U. Dhawa and D. Maiti, Chem. Commun., 2016, 52, 14003–14006 RSC.
- M. Bera, S. K. Sahoo and D. Maiti, ACS Catal., 2016, 6, 3575–3579 CrossRef CAS.
- L. Zhang, C. Zhao, Y. Liu, J. Xu, X. Xu and Z. Jin, Angew. Chem., Int. Ed., 2017, 56, 12245–12249 CrossRef CAS PubMed.
- Y.-F. Yang, G.-J. Cheng, P. Liu, D. Leow, T.-Y. Sun, P. Chen, X. Zhang, J.-Q. Yu, Y.-D. Wu and K. N. Houk, J. Am. Chem. Soc., 2014, 136, 344–355 CrossRef CAS PubMed.
- G.-J. Cheng, Y.-F. Yang, P. Liu, P. Chen, T.-Y. Sun, G. Li, X. Zhang, K. N. Houk, J.-Q. Yu and Y.-D. Wu, J. Am. Chem. Soc., 2014, 136, 894–897 CrossRef CAS PubMed.
- L. Chu, M. Shang, K. Tanaka, Q. Chen, N. Pissarnitski, E. Streckfuss and J.-Q. Yu, ACS Cent. Sci., 2015, 1, 394–399 CrossRef CAS PubMed.
- U. Dutta, A. Modak, B. Bhaskararao, M. Bera, S. Bag, A. Mondal, D. W. Lupton, R. B. Sunoj and D. Maiti, ACS Catal., 2017, 7, 3162–3168 CrossRef CAS.
- S. Bag, T. Patra, A. Modak, A. Deb, S. Maity, U. Dutta, A. Dey, R. Kancherla, A. Maji, A. Hazra, M. Bera and D. Maiti, J. Am. Chem. Soc., 2015, 137, 11888–11891 CrossRef CAS PubMed.
- T. Patra, S. Bag, R. Kancherla, A. Mondal, A. Dey, S. Pimparkar, S. Agasti, A. Modak and D. Maiti, Angew. Chem., Int. Ed., 2016, 55, 7751–7755 CrossRef CAS PubMed.
- X.-C. Wang, W. Gong, L.-Z. Fang, R.-Y. Zhu, S. Li, K. M. Engle and J.-Q. Yu, Nature, 2015, 519, 334–338 CrossRef CAS PubMed.
- F. Faccini, E. Motti and M. Catellani, J. Am. Chem. Soc., 2004, 126, 78–79 CrossRef CAS PubMed.
- J. Ye and M. Lautens, Nat. Chem., 2015, 7, 863–870 CrossRef CAS PubMed.
- N. Della Ca’, M. Fontana, E. Motti and M. Catellani, Acc. Chem. Res., 2016, 49, 1389–1400 CrossRef PubMed.
- Z. Dong, J. Wang and G. Dong, J. Am. Chem. Soc., 2015, 137, 5887–5890 CrossRef CAS PubMed.
- Q. Ding, S. Ye, G. Cheng, P. Wang, M. E. Farmer and J.-Q. Yu, J. Am. Chem. Soc., 2017, 139, 417–425 CrossRef CAS PubMed.
- P.-X. Shen, X.-C. Wang, P. Wang, R.-Y. Zhu and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 11574–11577 CrossRef CAS PubMed.
- P. Wang, M. E. Farmer, X. Huo, P. Jain, P.-X. Shen, M. Ishoey, J. E. Bradner, S. R. Wisniewski, M. D. Eastgate and J.-Q. Yu, J. Am. Chem. Soc., 2016, 138, 9269–9276 CrossRef CAS PubMed.
- P. Wang, M. E. Farmer and J.-Q. Yu, Angew. Chem., Int. Ed., 2017, 56, 5125–5129 CrossRef CAS PubMed.
- Y. Feng, D. Holte, J. Zoller, S. Umemiya, L. R. Simke and P. S. Baran, J. Am. Chem. Soc., 2015, 137, 10160–10163 CrossRef CAS PubMed.
- H. Shi, P. Wang, S. Suzuki, M. E. Farmer and J.-Q. Yu, J. Am. Chem. Soc., 2016, 138, 14876–14879 CrossRef CAS PubMed.
- P. Wang, G.-C. Li, P. Jain, M. E. Farmer, J. He, P.-X. Shen and J.-Q. Yu, J. Am. Chem. Soc., 2016, 138, 14092–14099 CrossRef CAS PubMed.
- Z. Zhang, K. Tanaka and J.-Q. Yu, Nature, 2017, 543, 538–542 CrossRef CAS PubMed.
- J. Cornella, M. Righi and I. Larrosa, Angew. Chem., Int. Ed., 2011, 50, 9429–9432 CrossRef CAS PubMed.
- J. Luo, S. Preciado and I. Larrosa, J. Am. Chem. Soc., 2014, 136, 4109–4112 CrossRef CAS PubMed.
- J. Choi, A. H. R. MacArthur, M. Brookhart and A. S. Goldman, Chem. Rev., 2011, 111, 1761–1779 CrossRef CAS PubMed.
- I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef CAS PubMed.
- C. N. Iverson and M. R. Smith, J. Am. Chem. Soc., 1999, 121, 7696–7697 CrossRef CAS.
- J.-Y. Cho, M. K. Tse, D. Holmes, R. E. Maleczka and M. R. Smith, Science, 2002, 295, 305–308 CrossRef CAS PubMed.
- T. Ishiyama, J. Takagi, K. Ishida, N. Miyaura, N. R. Anastasi and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 390–391 CrossRef CAS PubMed.
- T. M. Boller, J. M. Murphy, M. Hapke, T. Ishiyama, N. Miyaura and J. F. Hartwig, J. Am. Chem. Soc., 2005, 127, 14263–14278 CrossRef CAS PubMed.
- J. F. Hartwig, Chem. Soc. Rev., 2011, 40, 1992–2002 RSC.
- T. Cernak, K. D. Dykstra, S. Tyagarajan, P. Vachal and S. W. Krska, Chem. Soc. Rev., 2016, 45, 546–576 RSC.
- A. Ros, R. Fernandez and J. M. Lassaletta, Chem. Soc. Rev., 2014, 43, 3229–3243 RSC.
- Y. Saito, Y. Segawa and K. Itami, J. Am. Chem. Soc., 2015, 137, 5193–5198 CrossRef CAS PubMed.
- L. Yang, K. Semba and Y. Nakao, Angew. Chem., Int. Ed., 2017, 56, 4853–4857 CrossRef CAS PubMed.
- M. E. Hoque, R. Bisht, C. Haldar and B. Chattopadhyay, J. Am. Chem. Soc., 2017, 139, 7745–7748 CrossRef CAS PubMed.
- Y. Kuninobu, H. Ida, M. Nishi and M. Kanai, Nat. Chem., 2015, 7, 712–717 CrossRef CAS PubMed.
- H. L. Li, Y. Kuninobu and M. Kanai, Angew. Chem., Int. Ed., 2017, 56, 1495–1499 CrossRef CAS PubMed.
- R. Bisht and B. Chattopadhyay, J. Am. Chem. Soc., 2016, 138, 84–87 CrossRef CAS PubMed.
- H. J. Davis and R. J. Phipps, Chem. Sci., 2017, 8, 864–877 RSC.
- H. J. Davis, M. T. Mihai and R. J. Phipps, J. Am. Chem. Soc., 2016, 138, 12759–12762 CrossRef CAS PubMed.
- H. J. Davis, G. R. Genov and R. J. Phipps, Angew. Chem., Int. Ed., 2017, 56, 13351–13355 CrossRef CAS PubMed.
- F. Kakiuchi and S. Murai, Acc. Chem. Res., 2002, 35, 826–834 CrossRef CAS PubMed.
- P. Nareddy, F. Jordan and M. Szostak, ACS Catal., 2017, 7, 5721–5745 CrossRef CAS.
- N. Y. P. Kumar, A. Bechtoldt, K. Raghuvanshi and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 6929–6932 CrossRef CAS PubMed.
- O. Saidi, J. Marafie, A. E. W. Ledger, P. M. Liu, M. F. Mahon, G. Kociok-Köhn, M. K. Whittlesey and C. G. Frost, J. Am. Chem. Soc., 2011, 133, 19298–19301 CrossRef CAS PubMed.
- X. Zhao, E. Dimitrijević and V. M. Dong, J. Am. Chem. Soc., 2009, 131, 3466–3467 CrossRef CAS PubMed.
- L. Ping, D. S. Chung, J. Bouffard and S.-g. Lee, Chem. Soc. Rev., 2017, 46, 4299–4328 RSC.
- A. M. Clark, C. E. F. Rickard, W. R. Roper and L. J. Wright, Organometallics, 1999, 18, 2813–2820 CrossRef CAS.
- W. R. Reynolds, P. M. Liu, G. Kociok-Köhn and C. G. Frost, Synlett, 2013, 2687–2690 CAS.
- N. Hofmann and L. Ackermann, J. Am. Chem. Soc., 2013, 135, 5877–5884 CrossRef CAS PubMed.
- A. J. Paterson, S. St John-Campbell, M. F. Mahon, N. J. Press and C. G. Frost, Chem. Commun., 2015, 51, 12807–12810 RSC.
- J. Li, S. Warratz, D. Zell, S. De Sarkar, E. E. Ishikawa and L. Ackermann, J. Am. Chem. Soc., 2015, 137, 13894–13901 CrossRef CAS PubMed.
- D.-H. Wang, K. M. Engle, B.-F. Shi and J.-Q. Yu, Science, 2010, 327, 315–319 CrossRef CAS PubMed.
- P. Marce, A. J. Paterson, M. F. Mahon and C. G. Frost, Catal. Sci. Technol., 2016, 6, 7068–7076 CAS.
- C. J. Teskey, A. Y. W. Lui and M. F. Greaney, Angew. Chem., Int. Ed., 2015, 54, 11677–11680 CrossRef CAS PubMed.
- Q. Yu, L. a. Hu, Y. Wang, S. Zheng and J. Huang, Angew. Chem., Int. Ed., 2015, 54, 15284–15288 CrossRef CAS PubMed.
- Z. Fan, J. Ni and A. Zhang, J. Am. Chem. Soc., 2016, 138, 8470–8475 CrossRef CAS PubMed.
- Z. Fan, J. Li, H. Lu, D.-Y. Wang, C. Wang, M. Uchiyama and A. Zhang, Org. Lett., 2017, 19, 3199–3202 CrossRef CAS PubMed.
- G. Li, X. Ma, C. Jia, Q. Han, Y. Wang, J. Wang, L. Yu and S. Yang, Chem. Commun., 2017, 53, 1261–1264 RSC.
- G. Li, X. Lv, K. Guo, Y. Wang, S. Yang, L. Yu, Y. Yu and J. Wang, Org. Chem. Front., 2017, 4, 1145–1148 RSC.
- G. Li, P. Gao, X. Lv, C. Qu, Q. Yan, Y. Wang, S. Yang and J. Wang, Org. Lett., 2017, 19, 2682–2685 CrossRef CAS PubMed.
- G. Li, B. Zhu, X. Ma, C. Jia, X. Lv, J. Wang, F. Zhao, Y. Lv and S. Yang, Org. Lett., 2017, 19, 5166–5169 CrossRef CAS PubMed.
- G. Li, D. Li, J. Zhang, D.-Q. Shi and Y. Zhao, ACS Catal., 2017, 7, 4138–4143 CrossRef CAS.
- B. Li, S.-L. Fang, D.-Y. Huang and B.-F. Shi, Org. Lett., 2017, 19, 3950–3953 CrossRef CAS PubMed.
- S. Warratz, D. J. Burns, C. Zhu, K. Korvorapun, T. Rogge, J. Scholz, C. Jooss, D. Gelman and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 1557–1560 CrossRef CAS PubMed.
- Z. Ruan, S.-K. Zhang, C. Zhu, P. N. Ruth, D. Stalke and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 2045–2049 CrossRef CAS PubMed.
- Z.-Y. Li, L. Li, Q.-L. Li, K. Jing, H. Xu and G.-W. Wang, Chem. – Eur. J., 2017, 23, 3285–3290 CrossRef CAS PubMed.
- C. C. Yuan, X. L. Chen, J. Y. Zhang and Y. S. Zhao, Org. Chem. Front., 2017, 4, 1867–1871 RSC.
- J. A. Leitch, C. L. McMullin, M. F. Mahon, Y. Bhonoah and C. G. Frost, ACS Catal., 2017, 7, 2616–2623 CrossRef CAS.
- A. J. Paterson, C. J. Heron, C. L. McMullin, M. F. Mahon, N. J. Press and C. G. Frost, Org. Biomol. Chem., 2017, 15, 5993–6000 CAS.
- J. Li, K. Korvorapun, S. De Sarkar, T. Rogge, D. J. Burns, S. Warratz and L. Ackermann, Nat. Commun., 2017, 8, 15430 CrossRef PubMed.
- J. A. Leitch, C. L. McMullin, A. J. Paterson, M. F. Mahon, Y. Bhonoah and C. G. Frost, Angew. Chem., Int. Ed., 2017 DOI:10.1002/anie.201708961.
- M. Fañanás-Mastral, Synthesis, 2017, 1905–1930 CrossRef.
- R. J. Phipps, N. P. Grimster and M. J. Gaunt, J. Am. Chem. Soc., 2008, 130, 8172–8174 CrossRef CAS PubMed.
- R. J. Phipps and M. J. Gaunt, Science, 2009, 323, 1593–1597 CrossRef CAS PubMed.
- O. Daugulis and V. G. Zaitsev, Angew. Chem., Int. Ed., 2005, 44, 4046–4048 CrossRef CAS PubMed.
- A. J. Hickman and M. S. Sanford, Nature, 2012, 484, 177–185 CrossRef CAS PubMed.
- H. A. Duong, R. E. Gilligan, M. L. Cooke, R. J. Phipps and M. J. Gaunt, Angew. Chem., Int. Ed., 2011, 50, 463–466 CrossRef CAS PubMed.
- C.-L. Ciana, R. J. Phipps, J. R. Brandt, F.-M. Meyer and M. J. Gaunt, Angew. Chem., Int. Ed., 2011, 50, 458–462 CrossRef CAS PubMed.
- B. Chen, X.-L. Hou, Y.-X. Li and Y.-D. Wu, J. Am. Chem. Soc., 2011, 133, 7668–7671 CrossRef CAS PubMed.
- Y. Yang, R. Li, Y. Zhao, D. Zhao and Z. Shi, J. Am. Chem. Soc., 2016, 138, 8734–8737 CrossRef CAS PubMed.
- Y. Yang, P. Gao, Y. Zhao and Z. Shi, Angew. Chem., Int. Ed., 2017, 56, 3966–3971 CrossRef CAS PubMed.
- H. P. L. Gemoets, G. Laudadio, K. Verstraete, V. Hessel and T. Noël, Angew. Chem., Int. Ed., 2017, 56, 7161–7165 CrossRef CAS PubMed.
- V. Ritleng, C. Sirlin and M. Pfeffer, Chem. Rev., 2002, 102, 1731–1770 CrossRef CAS PubMed.
- D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624–655 CrossRef CAS PubMed.
- G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651–3678 RSC.
- H.-J. Xu, Y. Lu, M. E. Farmer, H.-W. Wang, D. Zhao, Y.-S. Kang, W.-Y. Sun and J.-Q. Yu, J. Am. Chem. Soc., 2017, 139, 2200–2203 CrossRef CAS PubMed.
- M. Bera, S. Agasti, R. Chowdhury, R. Mondal, D. Pal and D. Maiti, Angew. Chem., Int. Ed., 2017, 56, 5272 CrossRef CAS PubMed.
- G. Pototschnig, N. Maulide and M. Schnürch, Chem. – Eur. J., 2017, 23, 9206–9232 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |