
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdsorptive removal of gaseous contaminants using biomass-based adsorbents
Kayode Adesina Adegoke *ab,
Omolabake Abiodun Okon-Akan†
acd,
Tosin Adewumi Adebusuyi†e,
Oluwatobi Idowu Adewuyi†f,
Peter Oluwatosin Adu†g,
Abayomi Bamisaye†h,
Oyeladun Rhoda Adegoke†a,
Cecilia Opeyemi Babarinde†i and
Olugbenga Solomon Bello*ab
*ab,
Omolabake Abiodun Okon-Akan†
acd,
Tosin Adewumi Adebusuyi†e,
Oluwatobi Idowu Adewuyi†f,
Peter Oluwatosin Adu†g,
Abayomi Bamisaye†h,
Oyeladun Rhoda Adegoke†a,
Cecilia Opeyemi Babarinde†i and
Olugbenga Solomon Bello*ab
aDepartment of Pure and Applied Chemistry, Ladoke Akintola University, P. M. B. 4000, Ogbomoso, Nigeria. E-mail: kwharyourday@gmail.com; osbello@lautech.edu.ng
bLAUTECH SDG 11 Sustainable Cities and Communities Research Group, Nigeria
cWood and Paper Technology Department, Federal College of Forestry Jericho, Ibadan, Nigeria
dForestry Research Institute of Nigeria, Nigeria
eDepartment of Chemical Sciences, Augustine University, Ilara-Epe, Lagos State, Nigeria
fDepartment of Agricultural and Environmental Engineering, University of Ibadan, Ibadan 200255, Nigeria
gDepartment of Chemistry, Obafemi Awolowo University, Ile-Ife, Osun State, Nigeria
hDepartment of Chemistry, Faculty of Natural and Applied Sciences, Lead City University, Ibadan, Oyo State, Nigeria
iDepartment of Biochemistry and Forensic Science, Abiola Ajumobi Technical University, Ibadan, Nigeria
First published on 30th April 2025
Abstract
Biomass-based adsorbents have emerged as attractive materials for the adsorptive removal of gaseous pollutants due to their abundance in nature, low cost, and environmental friendliness. The modification of the adsorbent surfaces has been regarded as an intriguing technique for improving and enhancing their adsorption capacity for efficient removal of pollutants. The present study investigates the most recent developments and applications of biomass-derived adsorbents for removing various gaseous contaminants from air and gas streams. The use of biomass materials such as agricultural waste and wood residue to synthesize adsorbents provides a long-term solution to environmental pollution. This is due to the fact that biomass-derived adsorbents can be designed to have a large surface area, porosity, and surface functionality, thereby increasing their adsorption capacity and selectivity for target pollutants using a variety of chemical processes such as carbonization, activation, and modification. This study presents a comprehensive report on the use of biomass-based adsorbents for the removal of various gaseous pollutants such as carbon dioxide (CO2), volatile organic compounds (VOCs), nitrogen oxides (NOx), sulfur dioxide (SO2), hydrogen sulphide (H2S) and multi-gas components. The surface chemistry of biomass adsorbents, in addition to their porous nature, is discussed. Multi-gas adsorption properties and the regeneration of biomass adsorbent are also discussed. The challenges and future prospects for developing biomass-based adsorbents for gaseous pollutant removal are also discussed, emphasizing the importance of a thorough understanding of adsorption mechanisms, scalability of manufacturing processes, and integration with existing air purification technologies. The findings of this study present biomass-derived adsorbents as a promising alternative for mitigating the challenges associated with the danger of gaseous pollutants, contributing to sustainable environmental management and public health protection.
1. Introduction
Over the past few years, there has been a significant rise in public concern about air quality and environmental degradation. The combustion of fossil fuels and rising greenhouse gas emissions, particularly carbon dioxide (CO2), have driven global warming. Human activities have increased CO2 levels from 280 ppm to >400 ppm, primarily through electricity generation, transportation, and industrialization.1–8 This increase in anthropogenic atmospheric pollutants is a significant challenge for humanity in the 21st century. The utilization of fossil fuels for diverse activities deteriorates urban air quality at an accelerated pace. Gaseous pollution damages both the physical and emotional health of metropolitan people. Studies reveal that by 2025, environmental contamination will generate irreversible environmental damage.9,10 Air pollution, which is the marker of economic progress, is not a recent problem and is produced by the existence of a diverse collection of gaseous and particulate components in the atmosphere.Statistics on mortality attributable to air pollution originated in London in the 18th, 19th, and early 20th centuries. Since the 1990s, epidemiological research has shown the human health issues linked to air pollution, based on total hospital admissions and death data.11–15 The World Health Organization (WHO) claimed that in 2012, one in nine fatalities was attributable to disorders related to air pollution.16 According to the 2012 Census in India, the urban population of emerging nations rose from 1.5 million to 17 million. Concurrently, there has been an exponential increase of motorized vehicles throughout both the private and public sectors. Consequently, the proportion of gaseous pollutants in the air from traffic emissions significantly escalated. Furthermore, the utilization of coal, diesel, and similar fuels for in situ electrical production by diverse sectors contributes to the elevated concentration of atmospheric pollutants.17 The principal gaseous pollutants comprise carbon monoxide (CO; kinetic diameter (K.D.) = 0.376 nm), nitrogen oxides (NO with K.D. = 0.320 nm and N2O having a K.D. of 0.330 nm), sulfur dioxide (SO2; K.D. = 0.360 nm), ozone (K.D. = 0.58 nm), and volatile organic compounds (VOCs) such as benzene (K.D. = 0.360 nm), ethylbenzene (K.D. = 0.6 nm), formaldehyde (K.D. = 0.4 nm), methyl ethyl ketone (K.D. = 0.52 nm), toluene (K.D. = 0.59 nm), and xylene (K.D. = 0.73 nm), among others. CO2 is a greenhouse gas contributing to global warming and climate change. Exposure to these contaminants can negatively impact both people and other organisms.
The prevalent symptoms of gaseous pollutants encompass persistent headache, dizziness, cognitive impairment, fatigue, muscular spasms, irritation of the eyes, throat, and nasal passages, as well as discomfort in the joints, bones, and spinal column. However, exposure to gases such as carbon monoxide can result in severe symptoms, including disorientation, long-term neurological impairments, unconsciousness, cardiorespiratory failure, coma, and death.
Consequently, there has been substantial interest in the pursuit of scientific investigations on cutting-edge and eco-friendly technologies capable of eliminating gaseous and particulate matter pollutants.18–21 Specialized materials with the inherent ability to selectively attract, collect, and retain gaseous and particulate matter pollutants under different design conditions are known as adsorbents. Adsorbent materials attract impurities onto their surfaces via chemical or physical interactions.22–25 Adsorbent materials are used in various industries and applications for nano-purification purposes because of their capacity to adsorb molecules or particles onto their surfaces.26–28
Research into the discovery of efficient and sustainable adsorbents remains essential, and remarkable efforts in experiments using different materials for the adsorptive removal of contaminants have been reported in the literature.29–34 Consequently, various adsorbents with unique characteristics and suitability for different pollutants have been experimented with and deployed to remove gaseous and particulate contaminants from air samples. Carbon-based materials are widely utilized adsorbents renowned for their porous structure and vast surface area, effectively capturing a diverse range of gaseous and particulate pollutants.35,36 Activated carbon is widely used to remove contaminants and pollutants in multiple applications, including water treatment.37 Also, activated carbon has been used in air purification, CO2 capture, separation of gases, and removal of other gaseous contaminants from the atmosphere by pressure and temperature swing adsorption techniques.38–41
The previous reviews in this field are all incredibly beneficial for comprehending the benefits of biomass in adsorbing common gaseous and organic/inorganic substances that might potentially pose atmospheric risks. Nevertheless, these reviews are insufficient in explaining the precise mechanisms by which biomass functions as an efficient adsorbent for removing gaseous pollutants using adsorption technology. Unlike other reviews, which often include sorption from aqueous media, this review solely examines the adsorptive removal of gaseous pollutants onto biomass-based adsorbents of agricultural waste origin. This distinction is important to prevent confusion in describing the sorption of inorganic or other organic pollutants (e.g. dyes, pharmaceuticals, endocrine descriptor compounds, etc.). In this review, we carefully considered the choice of biomass, the derivative sources of porous carbon, and the surface chemistry of biomass-based adsorbents prior to evaluating the performance of biomass-based sorbents for different categories of gaseous pollutants, namely, CO2, SO2, H2S, NOx, volatile organic compounds (VOCs) and multi-gas components. The corresponding mechanism of adsorption is also discussed in each case. The present review, therefore, advances the technology and theory of gaseous adsorption onto biomass waste, which is not only green but sustainable. In addition, this review seeks to give comprehensive knowledge of the relationship between the biomass properties, adsorptive removal conditions, and the corresponding performance of various biomass-based adsorbents towards different types of gaseous pollutants. Regeneration studies of the spent biomass adsorbents are also discussed, which are essential for a successful adsorption–desorption process. Lastly, the knowledge gaps to foster the development of biomass-based sorbents and their applications for real-world implementation for gaseous pollutants removal are discussed.
2. Why biomass?
In the face of rising environmental challenges such as poor waste management, researchers are devising a multidisciplinary approach that leverages the circular economy concept toward identifying specific byproducts/waste materials with applicable adsorbent properties.42,43 This innovative strategy addresses the increasing demand for effective pollution control while underscoring the importance of sustainable and environmentally friendly solutions, particularly by exploring biomass-derived materials as a basis for these adsorbents. In this regard, agricultural waste products such as rice husks and coconut shells have gained attention as sustainable biomass-derived adsorbents.44–46 These biomass-based adsorbents exhibit favourable adsorption characteristics and contribute to the utilization of agricultural byproducts and waste reduction. Thus, biomass-based adsorbents have emerged as a viable option because of their unique eco-friendliness, renewability, and outstanding adsorption characteristics.47 Thus, investigating biomass-based adsorbents for the adsorptive removal of contaminants is a critical step toward developing environmentally sensitive technologies that solve the multiple issues linked with environmental pollution control and agricultural waste handling.According to the waste data provided by the European Commission,48 activities within the coal industry (including mining and energy activities), agricultural industry, water treatment, and home sector are responsible for producing and accumulating significant quantities of solid waste. In 2008, the EU-27 produced a total of 2.62 billion tonnes of waste. The construction sector contributed 32.9% of the total waste, the mining industry contributed 27.8%, the manufacturing industry contributed 13.1%, waste and water management contributed 7.3%, households contributed 8.5%, agriculture and forestry contributed 1.7%, and the energy sector contributed 3.5%. Current waste management techniques, namely the use of landfills, contribute to climate change and have the potential to contaminate water and soil, as well as cause local air pollution. To address the escalating environmental issues caused by the growing volume of waste being disposed of in landfills, numerous research initiatives are concentrating on creating novel waste management approaches. These efforts involve producing valuable products derived from readily available waste materials.48
According to the United Nations Environment Programme, the agricultural sector produces a total of 140 billion metric tons of biomass annually on a worldwide scale.48 Prior studies have documented the use of biomass waste, namely agricultural leftovers, in the production of carbonaceous adsorbents.49 Numerous studies have demonstrated the use of lignocellulosic materials such as soybean, bagasse, olive stones, shells, almond shells, rice husk, and coconut as precursors for carbon-based sorbents in the adsorption of various gaseous pollutants at low-medium temperatures.50–55 For example, various by-products derived from biomass, such as rice husk ash, have been utilized for manufacturing solid adsorbents for CO2 capture at low temperatures.56 A typical example is the annual global output of rice husk ash which is now around 20 million tons, accounting for approximately 20% of the total dry rice husk.57
3. Biomass-derived porous carbons
The process of converting biomass into porous carbons requires the use of carbonization (pyrolysis) and activation, which can be achieved by physical and/or chemical means. Physical activation is a two-step procedure. First, the raw material is heated to temperatures ranging from 400 to 850 °C in the absence of oxygen, causing it to undergo carbonization. Then, the resulting char is subjected to activation by exposing it to gases such as steam, air, nitrogen, oxygen, ammonia, carbon dioxide, or a combination of these gases. This activation process occurs at temperatures around 600 to 1000 °C.58,59 The utilization of CO2 in the activation process results in the formation of porous carbons with a narrow distribution of micropores, which is ideal for achieving optimal pore size for separating CO2 and CH4. The steam activation produces carbons with a wider range of pore sizes and a smaller volume of micropores.58 However, there is a less precise regulation of the porosity in the physical activation process. The reactions that occur during physical activation can form oxygen functional groups on the surface. On the other hand, activation with NH3 introduces nitrogen-containing groups onto the carbon surface. This process is, however, typically combined with another gas to increase the porosity of the material.60 The elevated temperatures often employed in physical activation pose an energy disadvantage.Chemical activation can be performed in either one step or two steps. This involves impregnating the biomass or the resultant char from the initial carbonization stage with activating chemicals such as dehydrating agents and/or oxidants. Following impregnation, the combination of precursor and activation agent is subjected to elevated temperatures ranging from 400 to 800 °C under an inert environment.58,59 Chemical activation, like physical activation, leads to the formation of porosity and functional groups on the surface of carbon. Chemical activations of biomass precursors often involve the use of acids, alkalis, and salts such as H3PO4, H2SO4, ZnCl2, K2CO3, NaOH, and KOH.59,61 Chemical activation is commonly used to create biomass-derived porous carbons with large surface areas and precise control over the porosity. However, the process becomes time-consuming and energy-intensive when the produced carbon needs to be washed to remove the residual activating agent in the carbon matrix. Additionally, this washing process is not environmentally friendly.
4. Surface chemistry of biomass-based adsorbents
The surface chemistry of biomass-based adsorbents is influenced by the presence of heteroatoms, such as oxygen, hydrogen, nitrogen, and sulfur, on its surface. These components can be supplied by the precursor, added during activation, or delivered by posttreatment. The presence of these heteroatoms on the surface of the carbon materials determines whether it is neutral, acidic, or basic and greatly affects its ability to adsorb substances. Fig. 1 provides an overview of the many surface functional complexes on the carbon surfaces, determining their basic or acidic properties. | ||
| Fig. 1 (a) Nitrogen and oxygen surface functional groups on carbons.62 Different surface functional groups on the surface biomass: (b) oxygenated functional groups (c) nitrogen functional groups (d) sulfur functional groups. | ||
Carboxylic acids, phenols, lactones, anhydrides, and lactols possess acidic properties, but ether and carbonyl groups are considered neutral. Quinone, pyrone, nitrogen, and chromene groups exhibit fundamental functions.63,64 Different techniques are utilized to alter the surface chemistry of AC and generate acid or basic sites. These post-treatments impact both the characteristics and the amount of the surface functional groups. In general, oxidation processes are used to impart an acidic nature to activated carbon.65 Particularly, oxygen surface groups are introduced onto carbon materials by reactions conducted either in the gas phase (using reagents like oxygen or ozone) or in the liquid phase (utilizing nitric acid or hydrogen peroxide). Typically, the oxidation of carbon in a liquid phase is employed under mild reaction conditions (low temperature and short reaction time) to generate surface oxygen functionalities, such as carboxylic groups, without causing major changes to the textural qualities.66
In contrast, gas phase oxidation necessitates elevated temperatures and extended reaction durations resulting in a comparatively limited production of hydroxyl and carbonyl groups. This method is employed when there is a need to alter the porosity structure.67,68
The fundamental nature of activated carbon is determined by two types of structures: (i) the existence of basic surface functionalities such as chromene, pyrone, ketone, and basic amines. (ii) Oxygen-free Lewis basic sites on graphitic layers. The first method involves subjecting the material to high-temperature heat treatment in the presence of inert gases such as H2, He, and N2. This process selectively removes the acidic surface groups. The second technique involves nitrogenation, which refers to the addition of nitrogen to the surface of the activated carbon. This type of treatment can enhance the basicity of the activated carbon by promoting the development of amine bases. Elevated temperatures and prolonged treatments cause the conversion of nitrogen groups from amide and amine to imide and imine and ultimately to pyridine and nitrile.69–71
5. Biomass-based adsorbent for CO2 removal
In recent times, the worldwide issue of global warming which is caused by the release of greenhouse gases, has generated significant alarm and apprehension.72–74 The CO2 capture and sequestration is a very efficient method for extracting and storing CO2.75–77 The utilization of solid materials as an adsorbent offers a more environmentally friendly and economically efficient alternative to the traditional CO2 capture technique which involves aqueous amine adsorption methods. This is mostly owing to the energy-intensive regeneration phase included in the current method.78 Various adsorbents have been employed for the purpose of CO2 adsorption. These include porous carbons,79–82 carbon nanotubes,83,84 zeolitic imidazole frameworks (ZIFs),85 metal–organic frameworks (MOFs),86,87 zeolites,88,89 and mesoporous silica.90 Porous carbon is considered a very promising material for adsorbing CO2 due to its affordability, lightweight, extensive surface area, strong chemical and thermal stability, and rapid adsorption kinetics.91,92Recently, biomass-derived carbon has gained attention as a cost-effective and ecologically benign substitute for synthetic adsorbents.93,94 These carbon compounds may be synthesized from abundant sources such as foods, animals, and agricultural wastes.95 Several studies have shown a significant focus on biomass-derived porous carbons for capturing CO2 in order to mitigate global warming (Table 1). This attention is primarily due to their well-developed textural properties, relatively easy synthesis methods, and outstanding ability to adsorb CO2.96–100 When compared to other earliest substances, biomass has various advantageous qualities for creating CO2 adsorbents, including pores, inexpensive nature, sustainability, renewable, and readily available.101 Additionally, the utilization of biomass-derived porous carbon for capturing CO2 is advantageous in addressing climate change and environmental pollution resulting from insufficient biomass management concurrently. Due to its carbon-neutral properties, biomass-derived porous carbon may be used to trap CO2 and help emission-intensive companies and sectors reduce their carbon footprint. Additionally, it can contribute to negative emissions technologies for mitigating climate change.102
| Adsorbents | Initial conditions | Isotherm models | Kinetic models | qmax (mg g−1) | Adsorption mechanism | Findings | Ref. |
|---|---|---|---|---|---|---|---|
| Microporous carbons obtained from hydrothermally treated biomass waste tobacco stem by KOH activation with analogous textural properties and oxygen functional groups | 0.5 g of pretreated sample/hydrochar and 1.0 g of KOH dissolved in 50 mL water | BET (Brunauer–Emmett–Teller) | Non-local density functional theory | 8.0 mmol g−1 (at 0 °C, 1 bar) | Electrostatic interactions | Oxygen-containing functional groups on porous carbons are critically sensitive to CO2 uptake, especially with carboxyl and hydroxyl groups | 137 |
| Initial PH: 7 | 4.8 mmol g−1 (at 25 °C, 1 bar) | Physical sorption, intraparticle diffusion | |||||
| Temperature: 500–800 °C at 3 °C min−1 for 1 h | |||||||
| Nitrogen-doped porous carbons are obtained from biomass lotus stalks | Powdery lotus stalks with sizes of 74–150 nm blended with melamine; the mixture pyrolysis at 500 °C for 2 h to yield sample/LSM | BET (Brunauer–Emmett–Teller) at 25 °C | 4.25 mmol g−1 (at 25 °C, 1 bar) | Physical sorption, intraparticle diffusion | The narrow microporosity of the N-doped carbonaceous adsorbent influences the CO2 adsorption positively. Specific features of the Lotus stalk-derived adsorbents are high CO2/N2 selectivity, recyclability, adequate heat of adsorption, quick kinetics, excellent stability, and good dynamic adsorption capacity, which make them suitable for supercapacitor electrodes | 154 | |
| KOH/LSM mass ratio of 1, 2, and 3 at 500, 550, and 600 °C, respectively | D-R (Dubinin-Radushkevich) at 0 °C | 6.59 mmol g−1 (at 0 °C, 1 bar) | |||||
| Activated carbon from corn cobs (CC), date seeds (DS), peanut shells (PS), pomegranate peels (PP), and rice husk (RH), in powder form – with KOH and H3PO4 activating agents | 150 g CC + 85% KOH at 800 °C and 35 min, | ||||||
| 100 g DS + 85% H3PO4 at 800 °C and 45 min, | |||||||
| 200 g PS + 85% KOH at 680 °C and 30 min, | |||||||
| 300 g PP + 85% KOH at 700 °C and 35 min, | |||||||
| 250 g RH + 90% H3PO4 at 800 °C and 45 min |
Although the initial CO2 adsorption capacity of pyrolyzed biomass is rather poor (<0.5 mmol g−1 at 298 K and 1 bar), several changes are recognized to improve its capacity and selectivity, specifically for CO2.95 For example, the addition of basic groups or heteroatoms, such as nitrogen, increases the alkalinity and ability of carbon to adsorb CO2.103 Nitrogen doping is mostly accomplished by subjecting carbon to a nitrogen-rich component or gas with basic properties, which results in the introduction of nitrogen atoms onto the surface of the carbon.104 Additionally, biomass precursors that are rich in nitrogen provide an easy and consistent approach for introducing nitrogen heteroatoms.
Furthermore, augmenting the microporosity of carbon using several physical and chemical activation methods proves to be a successful approach in expanding its ability to adsorb CO2. Specifically, micropores with a diameter of less than 1 nm are extremely efficient in enhancing the CO2 capacity. This is because they are similar in size to the kinetic diameter of CO2 and may interact with it through overlapping adsorption forces and potential fields generated by nearby pore walls.105 Previous studies have employed a post-synthesis activation method to enhance the microporosity of carbon derived from biomass. This process entailed using CO2 or steam as activating agents at temperatures ranging from 500 to 900 °C to increase the development of micropores and enhance the ability of CO2 to be adsorbed.94 Nevertheless, the processes of carbon synthesis and activation are time-consuming and energy-intensive due to their two-step nature. Furthermore, the ultimate porosity can be influenced by factors such as temperature, flow rate, and the type of synthesis equipment used. Thus, a more streamlined and effective method is necessary.96 The process of transforming biomass to porous carbon materials for CO2 capture commonly involves carbonization (pyrolysis, gasification, and hydrothermal),106–112 activation (physical and chemical)113–124 and surface modification (N, S, Mg, etc.)125–135 as illustrated in Fig. 2.
 | ||
| Fig. 2 (a–d) Schematic diagram of practical functional biomass-derived porous carbon for CO2 capture. | ||
The majority of activated carbon has a CO2 uptake that is less than 3–4 mmol g−1 at 25 °C and 1 bar pressure.136,137 The biomass-derived porous carbon composites nevertheless have a value higher than 4 mmol g−1 at 25 °C and 1 bar, as reported in the literature.136,138,139 Particularly, several studies have proposed that a limited micropore volume in porous carbons improves their ability to adsorb CO2. For example, the biomass-derived porous carbon produced by Coromina et al.118 demonstrated outstanding CO2 adsorption capacity of 5.0 mmol g−1 at a pressure of 1 bar and a temperature of 25 °C. In their study, Haffner-Staton et al.139 reported that pre-carbonized wood, which was used to produce microporous carbon via KOH activation, gave a maximum CO2 capture of 5.0 mmol g−1 under 1 bar at 25 °C. Similarly, Sevilla et al.138 produced an effective microporous surface area of activated carbon by using KOH activation on biomass via hydrothermal method. This activated carbon had a CO2 adsorption capacity of 4.8 mmol g−1 at 25 °C under 1 bar of pressure. Nevertheless, these investigations just ascribed the exceptional absorption of CO2 by porous carbon to the presence of many small micropores that were less than 0.7 nm. However, the information about the impact of oxygen-containing functional groups on biomass-based porous carbons has been rarely reported.
Parshetti investigated carbon-based adsorbents with microporous architecture and high specific surface area (up to 2511 m2 g−1), which were produced from lignocellulosic feedstock using hydrothermal carbonization and chemical activation enabling CO2 adsorption.81 These adsorbents underwent extensive microscopic and spectroscopic investigations to determine their morphological, textural, and structural properties. Microporous carbons have a remarkable CO2 absorption capacity of 3.71 mmol g−1 at 25 °C and 1 atm pressure. These materials exhibit high selectivity for CO2 over NO gas and a hydrophobic core, making them ideal for CO2 capture. The Freundlich equilibrium model correlated well with the experimental adsorption isotherm data, while the traditional micropore diffusion model well characterized the adsorption kinetics. The Virial model was used to compute Henry's law constant and indicated substantial electrostatic interactions and dispersion forces between CO2 and porous carbons. These carbon-based adsorbents maintained their adsorption capacity for up to 10 reuse cycles for CO2 capture. The study found that microporous adsorbents are a cost-effective and environmentally friendly method for trapping CO2 from post-fossil fuel combustion processes.
Singh et al. found that the most effective adsorbent materials for CO2 adsorption have outstanding textural features, tunable porosity, and low cost.140 These materials are efficient adsorbents for CO2 capture due to their shape and physicochemical features, which are created through various activation procedures, including solid-state activation. Another group provided a full overview of the pyrolysis mechanisms for cellulose, hemicellulose, and lignin.141
Sher et al. investigated the comparative screening of three groups of biomasses: soft or non-woody (peanut shell), intermediate woody (walnut shell), and hard woody (pine wood) for the development of adsorbents/activated carbons for post-combustion CO2 capture (over N2 balance).142 Three different groups of biomass residues are selected to study the role and nature of the material in adsorption and selection of the raw material for CO2 adsorbent synthesis for future research, because of the hot issue of anthropogenic CO2 emissions. The adsorption isotherms studied by the thermal gravimetric analyzer (TGA) revealed that CO2 adsorption capabilities are in the range of 2.53–3.92 mmol g−1 (over N2 balance) at 25 °C. The newly synthesized activated carbons (ACs) exhibited a fast adsorption rate at 41–94% in the initial 2 min. Porous surface development with catalytic KOH activation is seen clearly through SEM surface morphological analyses and mathematically confirmed from SBET ranges from 146.86 to 944.05 m2 g−1. FTIR and XRD peaks verify the generation of basic or inorganic O2-rich moieties that help in acidic CO2 capture. It has also been observed from adsorption isotherms that the order of higher adsorption groups is; peanut shell > pine wood > walnut shell, while the best activation mass ratio (sample/KOH) is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3. The synthesized low-cost ACs with an amount of 1.93 US$ per kg production could help to overcome the environmental hazards and problems caused by CO2 and biomass waste.
:3. The synthesized low-cost ACs with an amount of 1.93 US$ per kg production could help to overcome the environmental hazards and problems caused by CO2 and biomass waste.
According to Bade et al.,143 large-scale applications of activated carbon from a single substrate of agricultural waste have been found inefficient in the adsorption of CO2. Thus, a composite activated carbon (CAC) was prepared using the chemical activation properties of four different agricultural waste substrates. The developed CAC exhibited the highest CO2 removal efficiency compared to other activated carbon-based single substrate-derived adsorbents at a capacity of 8.86 wt%. The study conducted by Lui et al.125 showed that beyond the inherent potential of biomass-based porous carbon materials for efficient CO2 adsorption due to the abundance of narrow pores of less than 0.7 nm, the oxygen-containing functional groups on the porous carbons are established to be critically crucial for CO2 uptake. Based on the density functional theory, the proliferation of oxygen-functional groups within the porous carbon material firmly grasps CO2 through electrostatic interactions. The study reported by Valdebenito et al.144 considered modifying a cellulose nanofibril (CNF) film to produce CO2-adsorbent materials. The produced absorbent materials were assessed and found to be thermally stable with remarkable CO2 adsorption when subjected to 99.9% CO2 flow at 25 °C.
In the study reported by Yuan et al.,101 a machine-learning approach was deployed to systematically map out gathered datasets on the CO2 adsorption properties of biomass waste-derived porous carbon adsorbent materials as a function of the textural and compositional properties, as well as the adsorption parameters. From the results, the machine learning model revealed the significance of the adsorption parameter, textural properties, and compositional properties (in that order of superiority) for the considered biomass waste-derived porous carbon-based CO2 adsorption materials. The study conducted by Khosrowshahi et al.145 investigated the effects of external adsorption parameters such as time, temperature, and pressure on CO2 adsorption properties of carbonaceous wastes from celery biomass. The effects of the presence of carboxyl, hydroxyl, and nitrogenous functional groups on CO2 adsorption were studied using molecular dynamics. It was observed that pyridinic nitrogen absorbs more CO2 than graphitic nitrogen, and the study also revealed that increasing the simulation increases the optimality of the CO2 adsorption rates.
The research by Hanif et al.93 studied the use of nitrogen-rich Albizia procera leaves as the substrate for activated carbon adsorbent materials utilizing single-stage pyrolysis at high temperatures in the presence of NaHCO3 as the activating agent. The study shows the subtle relationship between the surface characteristics and the resulting nitrogen content, which affects the CO2 adsorption performance of the resultant adsorbent.
The study by Nguyen et al.146 revealed a revolutionary way to generate energy and develop CO2-adsorbent materials from agricultural byproducts. Macadamia nut shells, bagasse, and rice straw were gasified for energy, and the leftovers were collected and analyzed for CO2 adsorption. The study found residual char from Macadamia nut shells absorbed CO2 the best, followed by bagasse and rice straw. It also shows better CO2 and N2 recyclability than advanced CO2 adsorbents reported in prior studies. The authors posited that the ultra-microporous volumes and aromatic functional groups on the char surface of Macadamia nut shells accounted for the excellent performances.
Fig. 3 illustrates the potential mechanisms of CO2 sequestration onto the surface of a sorbent by physical and chemical adsorption. Creamer et al. reported the sorption of CO2 on non-activated porous carbons derived from sugarcane bagasse and hickory wood. It was noted that the primary sorption mechanism is physisorption, which is driven by weak van der Waals forces.147 The CO2 molecule's quadrupole nature was proposed as a valuable characteristic for establishing the surface interactions with adsorbent through the induction and dispersion processes. The author further suggested that the surface areas remain the primary determinant of the physisorption process of CO2 onto the adsorbent. Martín-Martínez et al. synthesized a range of physically activated carbons from anthracite and investigated the process of CO2 sorption, focusing on the morphology and dimensions of the pores.140,148 Based on their observations, tiny micropores (1σ) were filled through physical sorption and exhibited a curved sorption isotherm, whereas larger micropores (2σ) were filled through a surface sorption and displayed rectilinear isotherm.
 | ||
| Fig. 3 Physisorption and chemisorption mechanistic process of CO2 onto the surface of the biomass.140 | ||
The process of impregnating porous carbon with metal oxide has been observed to enhance surface chemistry and increase the capacity for pollutant adsorption.149 ACs derived from biomass can be impregnated with metal compounds such as silver, aluminum, copper, and iron, owing to their markedly higher adsorption capabilities. The impact of the impregnation ratio must be understood. This is the weight ratio of the activation agents to the precursor. The ratio is paramount in the activation process via chemical methods, as an increasing ratio is anticipated to enhance the biomass sorbent surface area.
In contrast to N- and/or S-doping treatments, the incorporation of metal oxides onto biomass-derived porous carbon has garnered relatively little attention.114,150 In this context, further research has been conducted on metal oxide-doped CO2 adsorbents derived from biomass. Lahijani et al. formulated a magnesium-doped CO2 sorbent exhibiting a capacity of 1.86 mmol g−1 at 25 °C and 1 bar, achieving adsorption equilibrium within 30 minutes.134 MgO-doped porous carbon sourced from biomass has been formulated by employing MgCl2 (4500 ppm) present in seawater.151 The qmax of 5.45 mmol g−1 was recorded for CO2 adsorption and attained equilibrium within a span of 30 minutes. Another study detailed the development of biocarbon-supported MgO nanoparticles via the MgCl2 decomposition method,152 which demonstrated effective CO2 capture through physisorption and the formation of magnesium carbonate, achieving a maximum CO2 uptake of 5.34 mmol g−1. Nonetheless, achieving adsorption equilibrium requires over 90 minutes for this adsorbent, indicating that the sorption kinetic rate diminishes as the MgO/biomass ratio increases.96,134,151–153
6. Biomass-based adsorbents for the removal of sulfur dioxide (SO2)
The use of fossil fuels as energy sources is increasing due to their high energy production capability despite ongoing efforts of green and renewable technology.155 Sulfur dioxide (SO2) and other obnoxious gases such as hydrocarbons, CO2, NOx, and CO generate VOCs and particulate matter during the combustion of fossil fuels.156 These emitted gases are transported over long distances for 3–5 days, depending on the meteorological conditions of the environment causing environmental pollution effects such as photochemical smog, ozone depletion, greenhouse effect, and global warming.157 Sulfur dioxide, which is an established precursor of acid rain, causes acidification of water bodies, corrosion of buildings, damage of agricultural crops, etc.158 The adverse effects of SO2 on the health of animals and humans such as pulmonary, cardiovascular, and respiratory diseases due to its high toxicity.159 Air pollution caused by SO2 is becoming a serious environmental issue driven by an increase in urban migration, energy consumption, industrialization, transportation, and urbanization.160 The release of SO2 into the atmosphere can be via natural and anthropogenic activities. The major contributor to the release of SO2 into the environment is the burning of fossil fuels such as coal, natural gas, and oil.161 According to a report by the World Energy Council (2013), 82% of the primary energy supply in 2011 was from the burning of fossil fuel.161 Urban air quality monitoring database obtained from low- and middle-income earning countries revealed that 98% of cities with an average of one hundred thousand inhabitants do not meet the World Health Organization (WHO) air quality guidelines, whereas only 56% of cities in high-income countries meet with the guidelines.161 Fossil fuel consumption per capita in the twenty-largest world population is shown in Fig. 4. | ||
| Fig. 4 Fossil fuel usage per capita in the top 20 countries with the largest populations.162 | ||
A viable means of decontaminating the environment of SO2 is by adsorption because of its simplicity, efficiency, economics, and possible regeneration of adsorbent.163 Biomass-based activated carbon (AC) is commonly utilized for the adsorption of SO2 to simultaneously solve two environmental problems: the adsorption of released SO2 in the environment and the disposal of plant and animal organic wastes. Activated carbon has been widely utilized for adsorption of pollutants from air, liquids, and soil.99 They are porous substances with high mechanical strength, large surface area, and high adsorption capacity.164 Adsorption of pollutants on them does occur mainly on the pores on the surface of the particles. ACs used for commercial purposes are generally produced from coal, which is a non-renewable source of carbon, but it is relatively expensive. As a viable alternative, ACs are now derived from biomass such as palm, bamboo, and coconut shell.165 Carbonization is the first step used in producing AC which leads to char production. This first step removes moisture and all volatile compounds present in the AC before subjecting the char to physical or chemical activation. Activating agents such as air, CO2, steam, or a combination of these gases at temperatures of 800–1250 K are employed for physical activation, while acids and alkali metal hydroxides are used for chemical activation. AC is a commonly used adsorbent for water and gas treatments.99 However, the adsorption capacity of AC decreases with an increase in temperature, making AC suitable for low-temperature applications.166 The industrial application of AC as an acid gas adsorbent is restricted due to its low selectivity at high temperatures, high regeneration cost, and poor adsorption in the presence of water vapor. The regeneration of AC usually occurs at a temperature range of 400–500 °C.167
6.1. Adsorption of SO2 using activated carbon and carbonized biomass-based adsorbents
Lignocellulosic biomass is a widely recognized source of low-cost biomass materials for the effective production of activated carbons used in the adsorption of SO2.168 Various carbonaceous materials have been utilized in the production of ACs and applied for various purposes such as water purification, medical application, sewage treatment, gold purification, filter masks, gas purification, and filters in compressed air among other similar uses. They have also been widely used in removing gaseous pollutants such as sulphur dioxide, hydrogen sulphide, carbon dioxide, volatile organic compounds, and nitrogen oxides.169 ACs of commercial value are produced from various carbonaceous precursors such as wood (∼33%), lignite and coal (∼42%), peat (∼10%), and coconut shells. However, agricultural wastes are preferred in ACs production because of their commercial availability, low cost, abundance, low inorganic and high carbon contents. A report by Daud and Ali170 showed that at any level of burn-off, palm shell biomass-based AC had higher microspore volume than coconut-based AC due to properties of palm shell biomass-based AC such as lignocellulosic composition, botanical family, and texture. Palm shell activated carbon (PSAC) obtained from oil palm biomass wastes has been successfully used in the removal of SO2, as reported by Guo and Lua171 and the results obtained show that the AC gave better adsorptive capacity than commercial AC. Furthermore, the textural characterization of prepared PSAC gave higher values of solid density, total porosity, BET surface area, and micropore surface area of 2.03 g cm−3, 66%, 1366 m2 g−1, and 985 m2 g−1, respectively, compared with raw palm shell which gave values of 1.53 g cm−3, 3.9%, 1.6 m2 g−1, and 0.2 m2 g−1 respectively while char gave values of 1.63 g cm−3, 17.2%, 176 m2 g−1, 108 m2 g−1, respectively for the measured parameters. Oil palm empty fruit bunch (EFB) was used in making AC for adsorptive removal of SO2. At optimized parameters of 70 °C, 1 g of prepared AC, and 2000 ppm of SO2, using Box-Behnken Design (BBD); approximately 1101 mg SO2 per g activated carbon was observed. The results obtained show significant agreement between predicted and experimental values obtained. Inlet SO2 concentration and adsorbent dosage were found to be the most significant factors which affected SO2 adsorption with adsorbent dosage being the most significant.155 Shukor et al.172 studied the adsorption rate of SO2 using ACs prepared from different biomass, namely coconut shell (CS-AC), rubber seed pericarp (RSP-AC), and their blends (CSRSP-AC). The prepared activated carbons were tested for their adsorption capacity for SO2 using an evolved gas analyzer. The results obtained show that the single CS-AC and RSP-AC samples gave breakthrough time for SO2 adsorption at 14 min and 23 min respectively. Meanwhile, the longest breakthrough time of 15 min was observed for the blended AC (CSRSP-AC) with a 20:80. It was observed that the more the amount of microporous RSP in the blend, the better the SO2 adsorption capacity of the blend. This is an indication that a high ratio of RSP over CS in the blend (CSRSP-AC) increased adsorption capacity for SO2. Furthermore, the presence of fly ash/Ca(OH)2 catalyst in the 20:80 ratio of the blended CSRSP-AC improved its SO2 adsorption capacity and gave a breakthrough time of 36 min at 35 °C adsorption temperature. The use of adsorbent prepared from banana peels for the removal of SO2 from motorcycle emissions was studied by Viena et al..157 The banana peel AC was activated thermally and then used for the SO2 adsorption experiment. The results obtained show that SO2 emission of 24523 ppm from motorcycle emission was totally removed to 0 ppm. This indicates 100% efficiency of banana peels AC in the removal of SO2 from motorcycle emission. Sumathi et al.156 investigated the effectiveness of using palm shell activated carbon (PSAC) for adsorptive removal of SO2. The results obtained show that the breakthrough time for the removal of SO2 was approximately 120 min for PSAC, which was higher than the reported for PSAC impregnated with Fe, Ni, and V but less than what was reported for Ce. A walnut shell was used in the preparation of AC for the removal of SO2 from a simulated gaseous mixture obtained from coal combustion by Guo et al.173 The results obtained show that prepared AC from walnut had a surface area of 401.2 m2 g−1 which was less than what was reported for the AC prepared by impregnation with TiO, TiO2, and Fe2O3 with values 937.1, 661.8, and 791.0 m2 g−1 respectively. Furthermore, the breakthrough time and breakthrough sulphur capacity of raw AC were reported to have the lowest values when compared with AC impregnated with metals. Guo and Lua,174 reported that the adsorption of SO2 using AC prepared from oil palm shell and activated using CO2 gave a better breakthrough time of 181.4 min when compared with the ones prepared via impregnation with 40% KOH (147.3 min) and 40% H3PO4 (155.2 min) but gave a lesser breakthrough time compared with 10% KOH impregnation (190.5 min). However, it gave a lower adsorption capacity (14.55 mg g−1) for the adsorption of SO2 when compared with other prepared adsorbents except for 40% KOH, which gave an adsorption capacity of 11.37 mg g−1. The adsorption of SO2 onto activated carbon prepared from pistachio nut shells by physical activation with CO2 was studied theoretically and experimentally in a fixed-bed column. The experimental results showed that 89.6 mg g−1 was the adsorption capacity of the AC towards SO2 while the breakthrough time was 380 min. Furthermore, there was a very good level of agreement between the results obtained experimentally and theoretically.175 The summary of the adsorption of SO2 by physical activation is summarized in Table 2.
| Carbon source | Activation condition | Inlet SO2 concentration (ppm) | Breakthrough time (min) | Adsorption capacity (mg g−1) | Reference |
|---|---|---|---|---|---|
| Palm shell | Gas inlet flow rate of 30–90 cm3 min−1 at 298 K, 1 atm, on 20 mg AC | 1000 | 203.8 | 35.2 | 171 |
| Oil palm empty fruit | Temperature of 70 °C on 1 g AC | 2000 | — | 1101 | 155 |
| Pistachio nut shell | Gas inlet flow rate of 345 cm3 min−1 at 25 °C, and 1 atm | 1000 | 380 | 89.6 | 175 |
| Oil palm shell | Gas inlet flow rate of 30–90 cm3 min−1 at 25 °C, and 1 bar | 2000 | 181.4 | 14.55 | 174 |
| Coconut shell | Gas inlet flow rate of 500 mL min−1 at 35 °C, 1 atm, on 5 g AC | 1000 | 14 | — | 172 |
| Rubber seed pericarp | Gas inlet flow rate of 500 mL min−1 at 35 °C, 1 atm, on 5 g AC | 1000 | 23 | — | |
| Coconut and rubber seed blend | Gas inlet flow rate of 500 mL min−1 at 5 °C, 1 atm, on 5 g AC | 1000 | 29 | — | |
| Without fly ash and Ca(OH)2 | Gas inlet flow rate of 500 mL min−1 at 25 °C, 1 atm, on 5 g AC | 1000 | 36 | — | |
| With fly ash and Ca(OH)2 | |||||
| Banana peels | Motorcycle emission | 24523 |
— | — | 157 |
| Palm shell activated carbon | Gas inlet flow rate of 0.15 L min−1 at 150 °C, 1 atm, on 1 g AC | 2000 | ∼120 | — | 156 |
| Walnut shell activated carbon | Gas inlet flow rate of 0.12 L min−1 at 90 °C, space velocity of 600 h−1, on fixed mass of AC | — | 5 | 63 | 173 |
6.2. Adsorption of SO2 using chemically modified biomass-based adsorbents
The surface chemistry of an adsorbent determines its efficiency. Surface modification of adsorbents can be achieved through two major means which are: physical and chemical methods.176 Surface modification by chemical means is highly preferred to physical methods because it has a direct influence on the surface of the adsorbent.177 It is a viable means of transforming inexpensive materials from different precursors into highly valuable products with high adsorptive capacities. Chemical surface modifications give the material new surface properties that are not dependent on those of the bulk polymer.178 Acid, alkaline, and neutral solutions have been employed as chemical agents in modifying the surface of adsorbent to achieve better adsorption capacity.179 Acid modification can be done using mineral acids and oxidants such as H2SO4, HCl, HNO3, HClO, H3PO4, and H2O2.180 Organic acids are seldom used for surface chemical modification because they have low strength and, therefore, produce a weaker effect.181 Similarly, the surface functional groups of adsorbents can be influenced by using reducing agents. Alkali modification of the adsorbent's surface can improve the non-polar surface and hence enhance the adsorption capacity of the adsorbent for non-polar substances.180 Alkali treatment of an adsorbent leads to the adsorption of a positive charge on the surface and hence leads to the adsorption of negatively charged species.182 Alkali surface modification can be achieved using chemical agents such as NaOH, Na2CO3, KOH, Na2SiO3, LiOH, and oxides. Other chemical agents, which are oxidizing agents such as H2O2, and KMnO4; metal impregnation such as cerium, zirconium, ferric chloride, hydroxides, carbonates, nitrates, or chromates; and organic agents such as ethanol have also been employed for chemical modification. Surface modification of AC-based adsorbents has been reported to improve their adsorption capacities for the removal of various contaminants, as reported.176 Adsorption of SO2 on oil palm-activated carbon in a fixed bed was carried out with and without pre-impregnation. SO2 adsorption capacity on AC from oil palm shells pre-impregnated with KOH and H3PO4 of various concentrations was carried out. The results obtained show that AC pre-impregnated with 10% KOH gave a better adsorption capacity for the adsorption of SO2 (Breakthrough time) (BTT) of 190.5 min and 15.34 mg g−1 adsorption capacity (W) compared with AC activated with CO2 (BTT = 181.4 min and W = 14.55 mg g−1) and 40% H3PO4 (BTT = 155.2 min and W = 16.08) pre-impregnation. However, the AC with 40% KOH (BTT = 147.3 min and W = 11.37 mg g−1) pre-impregnation did not give a better adsorption capacity than 10% KOH sample, as likely expected. Over-gasification during preparation might be responsible for this, resulting in a reduction in pore surface area. Furthermore, pre-impregnation with 40% H3PO4 gave a better adsorption capacity but lesser breakthrough time when compared with AC without impregnation and the one pre-impregnated with 10% KOH.174 Kraft lignin from Eucalyptus was used to make AC for the adsorption of SO2 by impregnating it with ZnCl2, acting as an activating agent. The results obtained show that the presence of oxygen and water vapour in the inlet stream increased the adsorption of SO2 by the prepared AC due to the oxidation of SO2 to SO3 on the surface of the AC and subsequent hydration of the produced SO3 to H2SO4.183 Lee et al.184 studied the adsorption of NOx and SO2 on coconut shell-prepared granulated activated carbon individually and simultaneously impregnated with KOH (KOH-IAC). The results obtained for individual adsorption experiments show that KOH-IAC gave a higher adsorption capacity for NOx (0.734 mmol g−1) than for SO2 (0.6243 mmol g−1). Meanwhile, simultaneous adsorption of NOx and SO2 using KOH-IAC showed that SO2 adsorption was higher than NOx. The report confirmed that the impregnation of the AC with KOH is a determining factor in the adsorption of NOx and SO2, thus establishing the surface chemical behaviour. Adsorbents prepared from wood and activated with phosphoric acid and impregnated with various calcium compounds (CaCO3, CaO, Ca(CH3COO)2, Ca(OH)2, and Ca(C2H5COO)2 were used as calcium precursors for the adsorption of SO2 at low temperature. The CaO/AC adsorbents were prepared by various methods such as wet impregnation, rotary evaporator impregnation, physical mixing, ion exchange, and complex formation. The results showed that SO2 adsorption on prepared adsorbents depended on calcium loading and dispersion. The adsorption capacity obtained for AC without impregnation was 26 mg g−1, while the highest adsorption capacity recorded for impregnated AC (calcium acetate impregnation) was 120 mg g−1. Other impregnated adsorbents gave adsorption capacities within the range of 30 mg g−1–80 mg g−1 for SO2 adsorption. Furthermore, it was observed that the physical mixing method gave the best calcium loading but low calcium dispersion. However, high calcium dispersion was obtained with samples prepared by complex formation method.185The interaction between carbon on the surface of AC and metallic oxides increases SO2 adsorption through the formation of basic oxygen-containing functional groups such as pyridine oxide, pyrones, quinones, or the deposition of metals on the pores of the AC surface. Oxides of various metals such as iron,186 nickel,187 magnesium,188 cerium,189 etc. have been doped on AC. Palm kernel shell doped with cerium oxide gave a maximum breakthrough time of 455 min and a minimum breakthrough time of 245 min for SO2 adsorption;156 rice husk ash doped with a blend of calcium and cerium oxides gave a breakthrough time of 52 min;190 walnut shell doped with iron(III) oxide, titanium(IV) oxide, and a mixture of iron(III) oxide and titanium(IV) oxide gave breakthrough times of 1080 min, 1429.8 min, and 1321.8 min, respectively;173 while coconut shell doped with copper gave a breakthrough time of 42 min for SO2 adsorption.191 The summary of the adsorptive removal of SO2 using impregnated activated carbons is presented in Table 3.
| Carbon source | Activation condition | Inlet SO2 concentration (ppm) | Breakthrough time (min) | Adsorption capacity (mg g−1) | Reference |
|---|---|---|---|---|---|
| Oil palm shell impregnated with KOH | Gas inlet flow rate of 30–90 cm3 min−1 at 25 °C, and 1 bar | 2000 | 190.5 | 13.34 | 174 |
| Oil palm shell impregnated with H3PO4 | Gas inlet flow rate of 30–90 cm3 min−1 at 25 °C, and 1 bar | 2000 | 155.2 | 16.08 | 174 |
| Kraft lignin from eucalyptus impregnated with ZnCl2 | Gas inlet flow rate of 200 cm3 min−1 at 25 °C, 1 atm, on 0.2 g AC | 2500 | 12 | 94.8 | 183 |
| Coconut shell impregnated with KOH | Adsorption temperature of 403 K and pressure of 1 atm on 10.67 g AC | 1006 | 69 | 40 | 184 |
| Wood impregnated with H3PO4 | Gas inlet flow rate of 60 mL min−1 at 573 K on 0.2 g AC | — | — | 120 | 185 |
| Rice husk ash AC modified with calcium and cerium oxides | Gas inlet flow rate of 0.15 L min−1 at 150 °C, 1 atm, on 0.7 g AC | 2000 | 52 | 46.33 | 190 |
| Walnut shell AC modified with iron(III) oxides | Gas inlet space velocity of 600 h−1 at 90 °C, 1 atm, on 0.5 g AC | 2000 | 1080 | — | 173 |
| Walnut shell AC modified with iron(III) oxides | Gas inlet space velocity of 600 h−1 at 90 °C, 1 atm, on 0.5 g AC | 2000 | ∼1430 | — | 173 |
| Mixture of iron(III) and titanium(IV) oxides | 2000 | ∼1322 | — | 173 | |
| Coconut shell and copper nitrate modified AC | Gas inlet flow rate of 0.40 L min−1 at 50 °C, 1 atm | 2000 | 42 | — | 191 |
To demonstrate the influence of impregnating various materials onto biomass/biochar sorbent on the adsorption of SO2, CO2 activation and impregnation with methyl diethanolamine (MDEA) were simultaneously utilized to enhance the SO2 adsorption capacity of biochar and increase its physicochemical properties.159 The influence of pore shape and nitrogen functionality on SO2 adsorption was also investigated. The results indicate that following CO2 activation, the specific surface area of biochar rose from 56.91 m2 g−1 to 755.35 m2 g−1, but this value decreased to 25.54 m2 g−1 or lower after MDEA impregnation. Despite the degradation of the pore structure in activated biochar, the surface nitrogen content of the nitrogen-enriched biochar (with 10% MDEA impregnation) rose from 1.46% to a peak of 7.20%. Following 10% MDEA impregnation, the highest SO2 sorption capacity rose from 57.8 mg g−1 to 156.2 mg g−1.159 Zhang et al. also reported the methyl diethanolamine-methanol impregnation method to prepare precursors for nitrogen-enriched biochar from peanut shells, corn stalks, and corncobs under different activation conditions to study the influence of precursors (raw biochar) on SO2 sorption,192 demonstrating an improved sorption capacity of 216.19 mg g−1 at 120 °C with O2 and 500 ppm NO.
Wang et al.193 discovered that the biochar impregnated with NH3 has an enhanced specific surface area of 453 m2 g−1, following steam activation but diminished to a minimum of 4.81 m2 g−1 subsequent to ammonia impregnation. Prior research also validated that the impregnation of amines might obstruct the surface pores of biochar, particularly the micropores.192 The co-pyrolysis/activation modification approach utilizing nitrogen-containing compounds can streamline production and yield nitrogen-doped biochar with superior pore structure characteristics. The thermal reactivity of nitrogen-containing compounds and activators complicates nitrogen stabilization in the solid phase, leading to a low nitrogen concentration in nitrogen-doped biochar. When carbon precursors were co-pyrolyzed with urea (a nitrogen source) and KOH (an activator), the highest nitrogen content of nitrogen-doped porous carbon was found to be just 1.52%.194 Consequently, the one-step modification of biochar, premised on a well-developed pore structure and efficient nitrogen doping, is essential for creating a cost-effective material with superior SO2 adsorption capabilities.195 In another research,196 CO2 activation and high-temperature ammonia (nitrogen doping) have demonstrated efficacy in altering biochar, with the SO2 adsorption capabilities of the changed biochar achieving 191.1 and 199.8 mg g−1, respectively.
7. Biomass-based adsorbents for the removal of hydrogen sulfide (H2S)
Gaseous pollutants are not easy to control because of their high mobility and difficulty in visualizing them. Some gaseous pollutants such as H2S, which are contained in ambient air or biogas, are said to have high levels of toxicity and, therefore, pose health risks.197 The production of biogas, which is a renewable source of energy, has been growing in recent decades.198 However, H2S, which is one of the constituents of biogas with a concentration range of 100–10000 ppm, is a poisonous acidic gas.198,199 Thus, burning of H2S-containing biogas would cause serious corrosion of the combustion engine, boiler, and various parts of the system.197,200 Furthermore, SO2 generated during the combustion of H2S-containing biogas could pose serious human health challenges.197 In order to overcome this problem, several means have been used to mitigate air pollution caused by H2S emissions. Ambient temperature catalytic oxidation of H2S into sulfur in a solid state is one of the most promising methods used for removing H2S air pollutants with low concentration, i.e., <50000 ppm, due to being secondary pollution-free and having moderate operating conditions.201,202 Various adsorbent materials, such as porous natural and synthetic zeolites, metal oxides, and activated carbons, have been used to remove H2S. The materials can either be amorphous or crystalline, which can be porous or non-porous with the ability to be modified to improve their H2S affinity. Interfering compounds present in feedstock can be a determining factor in choosing the material to be used as an adsorbent for H2S removal. A polar adsorbent surface helps to separate a polar H2S from a non-polar molecule like CH4 in a fuel gas mixture. However, separation based on polarity alone can be difficult in the presence of other polar compounds, such as CO2 and water molecules. Water molecules present in the gas mixture may improve or hinder H2S adsorption depending on the amount of water in the mixture and the strength of interaction between the adsorbent and the water molecules. The presence of CO2 in the feed mixture poses further problems since both are acidic.203 Several categories of adsorbents have been synthesized in the last two decades, which possess the capability to remove acid gases such as H2S, and they are: carbon-based adsorbents, microporous and mesoporous silica, and MOFs. A good adsorbing material must possess high adsorption capacity, low rate of friction, fast adsorption kinetics, stability in a redox environment, regenerability, and ability to withstand high temperature.204 Adsorption using carbon-based materials such as activated carbon205 has been widely utilized for H2S removal at room temperature due to their large pore size, high surface area and good surface chemistry.206 The pore size of activated carbon is sufficient for gaseous adsorption to take place, and it has a large enough surface area that enables rapid reactions to occur, thus controlling air pollution.207 The average size of H2S pollutants is <2 nm, and this makes the filtration method alone not sufficient to remove them completely.135 One of the major factors that affect the desulfurization performance of carbon-based materials is their porous structure.208 Microporous and mesoporous carbon-based materials are quite suitable for the removal of H2S. Micropores provide the adsorption site for the removal of H2S,209 while mesopores are suitable for deposition of desulfurization products i.e., sulfur, along with rapid mass transfer of H2S gas molecules.208 Chemical activation is the most common activating method for the pore structure development of carbon materials.114 However, chemical activation develops micropores into carbon-based materials with the generation of fewer mesopores.114 The traditional means of preparing mesoporous carbon is by using a sacrificial template method such as the use of SiO2. Another major factor that affects the efficiency of H2S removal by carbon-based materials is surface composition.208,210,211 A means of improving the surface composition of carbon materials is the use of N-doped carbon which increases catalytic activity towards H2S at room temperature. The introduction of nitrogen could lead to an increase in redox chemistry and chemical reactivity for acid–base reaction through improved basic catalytic sites.135,212 Furthermore, it can increase the rate of dissociation and oxidation of H2S in the presence of water molecules.
7.1. Adsorption of H2S using activated carbon and carbonized biomass-based adsorbents
The use of ACs and other carbon-based materials as absorbents, as presented in Table 4, is very common for low-temperature desulfurization material due to their properties such as large surface area, which is usually greater than 1000 m2 g−1, high thermal stability, controllable surface chemistry, and high pore volume. Production of AC is generally cheaper than other types of adsorbents because they are obtained from easily available carbon materials such as wood, agricultural wastes, peat, coal, etc. The surface chemistry of ACs is highly controlled by modification of the functional groups on their surfaces.209,219 The functionalization determines the extent of adsorbate–adsorbent interactions and consequently affects the chemical and physical adsorption rate. This has led to a variety of functionalized carbon-based sorbents produced by impregnation, deposition–precipitation, or doping with heteroatoms.202,220,221 The direct integration method was recently proposed as a better alternative to the impregnation method.222| Carbon source | Activation condition | Inlet SO2 concentration (ppm) | Breakthrough time (min) | Adsorption capacity (mg g−1) | Reference |
|---|---|---|---|---|---|
| a Key: superscript w = wet, mw = moisture and wet. | |||||
| Granulated activated carbon | Gas inlet flow rate of approximately 22.3 m3 h−1 at 20–25 °C | 1350 | 124 | 1293 | 213 |
| Cypress sawdust | Gas inlet flow rate of 150 mL min−1 at 25 °C on 0.2 g AC | 1000 | 12 | 12.5 | 214 |
| Coffee | Gas flow rate of 450 mL min−1 at 22 °C on 1.0 g AC | — | 212 | ||
| CP7PA | 149.6w | ||||
| CDA | 215.6mw | ||||
| Tobacco | |||||
| TP7PA | 21mw | ||||
| TDA | 178.4mw | ||||
| Corn cobs | |||||
| CCP7PA | 119.1w | ||||
| CCDA | 159.4w | ||||
| Cherry stones | |||||
| CSP7PA | 1.3mw | ||||
| CSDA | 19.5mw | ||||
| Hay | Gas flow rate of 450 mL min−1 on 1.0 g AC | 39.0w | 215 | ||
| HPA7-15 | 216 | ||||
| Hay | Gas flow rate of 450 mL min−1 on 1.0 g AC | 2000 | 217 | ||
| H5A | 1.1w | 218 | |||
| H6A | 10.8w | ||||
| H7A | 40.5w | ||||
| Used wood pallets | Gas flow rate of 180 mL min−1 at 22 °C on 1.0 g AC | 12.92 | |||
| Rice hull | Gas flow rate of 40 mL min−1 | — | 620 | 382.7 | |
| Bamboo | 360 | 109.3 | |||
| Shell-derived AC | 120 | 35.6 | |||
| Wood based AC | Gas flow rate of 150 mL min−1 on 1.0 g AC | 1000 | — | 6.6w | 210 |
Ou et al.213 investigated using granular activated carbon (GAC) for the adsorption of H2S from the biogas generated by small to medium-sized cattle or pig farms. The GAC sorbent performance was conducted on a long-term basis for biogas generated from treating wastewater. Various adsorption parameters, such as adsorbent masses, volumes, and inlet H2S concentrations, were analyzed to ascertain the impact of the different factors. The results obtained show that at H2S inlet concentrations of 932–1560 ppm and 100 ppm breakthrough concentration, GAC gave breakthrough capacities of 745–1293 mg g−1 at 20–25 °C. The comparison of these values to other types of adsorbents shows that it is high. The choice of GAC as an acceptable adsorbent for the removal of H2S is due to its low cost and high adsorption capacities. Further studies have shown that cellulose and lignin contents of the waste biomass have positive impacts on the yield and microporous surface area of the resulting biochar.223 Yang et al.221 carried out an adsorption experiment of H2S using wood-based AC, and the results obtained show that at an H2S concentration of 1000 ppm and inlet flow rate of 500 mL min−1 on a 2 cm3 of sample packed into a column with particle size of 30–40 mesh, the H2S breakthrough capacity was 5.6 mg g−1. The breakthrough capacity obtained is higher than the value obtained when ZnFe2O4 was used alone as a sorbent for H2S (1.6 mg g−1) but lower than the values obtained when wood-based AC was impregnated with various masses of ZnFe2O4. In a related study carried out by Chen et al.214 using cypress Sawdust to produce activated carbon, which was used for the adsorption of H2S at an initial concentration of 1000 ppm, gave a breakthrough capacity of 12.5 mg g−1, which was less than the value obtained for N-doped cypress sawdust without K2CO3 activation. Nowicki et al.212 carried out a series of adsorption studies using various biomass-based materials, which are coffee, tobacco, corn cobs, and cherry stones. The sample materials were crushed, sieved, and subjected to pyrolysis at 700 °C before physical activation using CO2 at 800 °C. The products obtained were code-named CP7PA, TP7PA, CCP7PA, and CSP7PA for coffee, tobacco, corn cobs, and cherry stones, respectively. They were then used for the adsorption of H2S under dry and wet conditions with/without moisture. The results obtained show that CP7PA gave the highest H2S breakthrough capacity of 149.6 mg g−1 under wet conditions without moisture. In comparison, CSP7PA gave the lowest H2S breakthrough capacity of 1.3 mg g−1 under wet conditions with 70% moisture. Furthermore, the adsorbent obtained by directly activating coffee using CO2 at 800C gave the highest H2S breakthrough capacity of 215.6 mg g−1 under moist wet conditions. In contrast, direct activation of coffee (CDA) adsorbent for adsorption under dry conditions gave the lowest value of 6.0 mg g−1 among adsorbents prepared by direct activation. Adsorption of H2S under wet conditions gave better adsorption breakthrough capacities of H2S. A study carried out by Kazmierczak-Razna and Pietrzak,216 used low-quality hay that was subjected to pyrolysis at different temperatures and times in a muffle chamber in a microwave furnace. The physical activation was done using CO2, which was done after pyrolysis or by direct activation. The two temperatures used for pyrolysis were 700C and 800C in a muffle furnace for either 15 min or 30 min. The adsorbents obtained after pyrolysis and physical activation were code-named HPA7-15, HPA7-30, HPA8-15, and HPA8-30, while those obtained by direct activation were code-named HDA7-15, HDA7-30, HDA8-15, and HDA-30. The prepared adsorbents were used for the adsorption of H2S under dry and wet conditions and the results obtained show that HPA7-15 gave the highest H2S breakthrough capacity of 39.0 mg g−1 under wet conditions. The HDA7-30 gave the lowest H2S breakthrough capacity of 5.2 mg g−1 under dry conditions. Furthermore, the lowest H2S breakthrough capacity of 12.3 mg g−1 was the lowest under wet conditions. This is an indication that the adsorption of H2S under wet conditions gave a better result than under dry conditions. Another similar study by Kazmierczak-Razna et al.215 used low-quality hay for the synthesis of activated carbon using a microwave oven for the pyrolysis of the material at three different temperatures of 500 °C, 600 °C, and 700 °C. The prepared char was then subjected to physical activation in a microwave oven at 500 °C under a stream of CO2. The synthesized adsorbents prepared at 500 °C, 600 °C, and 700 °C with CO2 activation were code-named H5A, H6A, and H7A, respectively. They were then used for the adsorption of H2S under dry and wet conditions, and the results obtained show that H7A under wet conditions gave the highest H2S breakthrough capacity of 40.5 mg g−1 while H5A under dry conditions gave the lowest H2S breakthrough capacity of 1.0 mg g−1. The highest H2S breakthrough capacity of 40.5 mg g−1 obtained for H7A is comparable to the value of 39.0 mg g−1 obtained under similar conditions in an earlier study by Kazmierczak-Razna and Pietrzak.216 Hervey et al.217 carried out a study on the adsorption of H2S with activated carbon prepared from used wood pallets (UWP). The results from the study show that AC obtained from the pyrolysis of UWP via steam activation gave H2S a breakthrough capacity of 12.92 mg g−1. Meanwhile, c.UWP prepared by pyrolysis of UWP without steam activation gave H2S breakthrough capacity of 0.04 mg g−1 while ox.UWP, which was produced from c.UWP by oxygenation gave H2S a breakthrough capacity of 1.81 mg g−1. Shang et al.218 carried out a study on the adsorption of H2S using adsorbent prepared from the pyrolysis of rice hull and bamboo and compared their adsorption capacity towards H2S with shell-derived commercial AC. The results obtained show that SR gave the highest H2S breakthrough capacity of 382.7 mg g−1 while bamboo and AC gave H2S breakthrough capacity of 109.3 mg g−1 and 35.6 mg g−1, respectively. Activated carbon of wood origin without thermal treatment was used for the adsorption of H2S with the application of a custom-designed dynamic tester. The tests were carried out under dry and wet conditions, and the results obtained show that adsorption under dry conditions gave H2S breakthrough capacity of 3.1 mg g−1 and 5.7 mg g−1 under dry and wet conditions respectively. Furthermore, AC with thermal treatment at 450 °C and 950 °C were also used for the adsorption of H2S under dry and wet conditions. The results obtained show that the H2S breakthrough capacities at 450 °C under dry and wet conditions are 0.6 mg g−1 and 2.6 mg g−1, respectively, while the H2S breakthrough capacities under wet conditions at 950 °C are 2.0 mg g−1 and 6.6 mg g−1 respectively. This further confirms that wet condition favours the adsorption of H2S.210
7.2. Adsorption of H2S using chemically modified biomass-based adsorbents
Modification of the surface of an adsorbent involves adjusting the surface of a material by biological, chemical, and physical means to form new material with different forms that can be used for the desired purpose. It involves physical and chemical changes to remove surface impurities on the adsorbent. Surface modification of an adsorbent usually leads to alteration in the functional groups on the surface. Surface modification of adsorbents can be done through thermal, mechanical, or chemical means.224 Chemical modification involves the use of alkali, acid, or salt to improve the functional group on the surface of the adsorbent (Table 5). Meanwhile, the physical process improves the physical properties of the adsorbent such as surface area and pore sizes. Furthermore, surface modification can be achieved through the combination of methods, such as thermochemical and mechanochemical processes.227| Carbon source | Activation condition | Inlet SO2 concentration (ppm) | Breakthrough time (min) | Adsorption capacity (mg g−1) | Reference |
|---|---|---|---|---|---|
| a Key: superscript w = wet, mw = moisture and wet. | |||||
| Almond impregnated with KOH | Gas flow rate of 1.5 L min−1 on 2 cm bed height at ambient temperature | 970 | 10 | 230 | 225 |
| Coffee impregnated with KOH | 130 | 22 | |||
| Eucalyptus impregnated with KOH | 180 | 490 | |||
| Coffee impregnated with ZnCl2 | Gas flow rate of 250 mL min−1 on 1 g AC | 1000 | ∼44 | 81.3 | 208 |
| COFAC-0.5 | ∼65 | 127.0 | |||
| COFAC-1 | ∼1.0 | 18.3 | |||
| COFAC-2 | |||||
| Wood impregnated with ZnFe2O4 | Gas flow rate of 500 mL min−1 on 2 g AC at room temperature | 1000 | ∼108 | 122.5 | 221 |
| Rice impregnated with ZnFe2O4 | Gas flow rate of 100 mL min−1 on 0.2 g AC at 25 °C | 300 | ∼380 | 54.29 | 226 |
| RZF-500-3:1 |
∼960 | 117.06 | |||
| RZF-500-2:1 |
∼1370 | 228.29 | |||
| RZF-500-1:1 |
∼1100 | 153.8 | |||
| Coffee impregnated with KOH | Gas flow rate of 450 mL min−1 at room temperature on 1.0 g AC | ∼5 | 11.1mw | 212 | |
| CP7CA | ∼87 | 76.3w | |||
| Tobacco impregnated with KOH | ∼12 | 17.4w | |||
| TP7CA | ∼19 | 10.6md | |||
| Corn cobs impregnated with KOH | |||||
| CC7CA | |||||
| Cherry stones | |||||
| CS7CA | |||||
| Wood impregnated with melamine or urea | Gas flow rate of 150 mL min−1 on 1.0 g AC | 1000 | ∼370 | 51.6w | 210 |
Sawalha et al.225 carried out an adsorption study using activated carbons synthesized from spent almond shells, coffee grains (COF), and eucalyptus barks, which were impregnated with potassium hydroxide and zinc chloride, respectively, to capture H2S from biogas containing an average concentration of 970 ppm H2S. The adsorption performance of eucalyptus barks impregnated with KOH for the removal of H2S was the best among the three for a bed height of 2 cm and gas flow rate of 1.5 L min−1 at ambient conditions. It gave an adsorption capacity of 490 mg g−1 and a saturation time of 180 min, while COF gave a very poor adsorption capacity of 22 mg g−1 and a saturation time of 10 min. The results obtained further revealed that the more the lignin and cellulose contents of the prepared adsorbents, the better their adsorption capacity. Furthermore, they did a comparative study of the effect of an impregnation agent on the adsorption capacity of eucalyptus for the adsorption of H2S. The results obtained show that eucalyptus barks impregnated with KOH gave H2S a breakthrough time of 180 min, whereas the one impregnated with ZnCl2 gave a breakthrough time of 70 min. This indicates that KOH is a better impregnation agent than ZnCl2 for the adsorption of H2S.
A similar study using spent coffee and zinc chloride as an impregnation agent at various ratios was carried out by Kante et al.,208 The ratio of dry coffee to ZnCl2 was 1:0.5 (COFAC-0.5), 1:1 (COFAC-1), and 1:2 (COFAC-2). These prepared adsorbents were used for the adsorption of H2S, and the results obtained show that COFAC-1 gave the highest H2S breakthrough capacity of 127.0 mg g−1 while COFAC-2 gave the lowest H2S breakthrough capacity of 18.3 mg g−1. Another zinc compound (ZnFe2O4) was used as an impregnation agent in a study by Yang et al.221 Wood-based carbon, which was activated with phosphoric acid, was used in the study. Various ratios of combinations between wood-based carbon and ZnFe2O4 were used. The synthesized adsorbents were referred to as ZFOB-x, where ZFO represents a combination of ZnFe2O4 and wood-based carbon, and x represents the amount of ZnFe2O4 in the composite. The synthesized adsorbents, along with wood-based carbon only, were used for the adsorption of H2S, and the results obtained show that ZFOB-10 had the highest H2S breakthrough capacity of 122.5 mg g−1 while ZFO had the lowest H2S breakthrough capacity of 1.6 mg g−1, a value which is lower than 5.6 mg g−1 which was obtained when wood-based carbon alone was used for the adsorption of H2S.
In another study by Yuan et al.,226 using ZnFe2O4 as an impregnation agent with leftover rice as biomass was carried out. The prepared adsorbents (ZnFe2O4)-loaded porous biochar (RZF) were code-named according to different activation temperatures and activation ratios as RZF-T-X:Y, where T is the activation temperature and X:Y is the mass ratio of leftover rice to ZnFe2O4. Among the various adsorbents prepared and used for the adsorption of H2S, RZF-500-1:1 gave the highest H2S breakthrough capacity of 228.29 mg g−1, while the material prepared directly at the carbonization temperature of 500 °C without activator (RBC) that is adsorbent prepared from leftover rice alone without impregnation with ZnFe2O4 gave the lowest H2S breakthrough capacity of 12.11 mg g−1.
Alkaline chemical activation of coffee, tobacco, corn cobs, and cherry stones using KOH was carried out by Nowicki et al.,212 The prepared adsorbents were coded CP7CA, TP7CA, CCP7CA, and CSP7CA for adsorbents prepared from coffee, tobacco, corn cobs, and cherry stones. The adsorbents were used for the adsorption of H2S under dry and wet conditions with or without moisture. The results obtained show that TP7CA had the highest H2S breakthrough capacity of 76.3 mg g−1 under wet conditions without moisture, while TP7CA under dry conditions without moisture had the lowest H2S breakthrough capacity of 3.7 mg g−1. Chen et al.,214 used another potassium compound (K2CO3) to activate cypress sawdust doped with carbon nitride (CN) at various ratios. The produced adsorbents were coded as NPC-n, where n is the ratio of CN to CS. The H2S breakthrough testing results showed that NPC-1 gave the highest breakthrough value of 426.2 mg g−1 while NPC-0.5 gave the lowest value of 119.1 mg g−1 among the NPC-n-prepared adsorbents. However, the H2S breakthrough value obtained for NPC-0.5 is far higher than what was obtained for porous carbon without CN loading (PC), which gave a value of 12.5 mg g−1 and N-doped carbon without K2CO3 activation, which gave a value of 19.5 mg g−1.
The effect of nitrogen doping of wood-based commercial activated carbon using urea or melamine at 450 °C or 950 °C was carried out by Seredych and Bandosz.210 In all, a total of eleven different adsorbents were prepared with code-named BAX (raw activated carbon), CBAX-A (activated carbon heated at 450 °C), CBAX-AM (activated carbon heated at 450 °C and doped with melamine), CBAX-AMO (activated carbon heated at 450 °C, doped with melamine and preoxidized with HNO3), CBAX-B (activated carbon heated at 950 °C), CBAX-BM (activated carbon heated at 950 °C and doped with melamine), CBAX-BMO (activated carbon heated at 950 °C, doped with melamine and preoxidized with HNO3), CBAX-AU (activated carbon heated at 450 °C and doped with urea), CBAX-AUO (activated carbon heated at 450 °C, doped with urea and preoxidized with HNO3), CBAX-BU (activated carbon heated at 950 °C and doped with urea), and CBAX-BUO (activated carbon heated at 950 °C, doped with urea and preoxidized with HNO3). All the prepared adsorbents were used for the adsorption of H2S under dry and wet conditions. The results obtained show that CBAX-BM under wet conditions gave the highest breakthrough capacity of 64.1 mg g−1, while CBAX-BU gave the highest breakthrough capacity under dry conditions with a value of 21.6 mg g−1. The lowest H2S breakthrough capacity under dry and wet conditions was observed with CBAX-A (0.6 mg g−1) and CBAX-AM (2.5 mg g−1), respectively. The value for CBAX-A under dry conditions is less than that obtained for BAX alone (3.1 mg g−1) under dry conditions. Similarly, BAX alone, used as an H2S adsorbent, gave a higher breakthrough capacity of 5.7 mg g−1 under wet conditions. The results showed that N-doping and wet adsorption conditions of H2S gave better results.
Prior studies228,229 on H2S sorption utilizing copper sorbent-impregnated rice husk and cocoa AC. with NaOH as a substitute material for copper to assess its impact on the surface chemistry of AC. It was observed that NaOH significantly enhances the adsorption efficacy of AC by generating reactive functional groups, particularly the –OH functional group, following the modification of AC with NaOH. Impregnating of AC with KOH and KI to enhance the chemisorption of H2S has been reported.230 The H2S sorption efficacy was evaluated within a temperature range of 30–550 °C employing the temperature-programmed sorption approach to ascertain the influence of sorption temperature on the material's sorption properties. At ambient temperature, the impregnation of AC with KOH enhances its H2S sorption capacity, but impregnation with KI yields no noticeable advantage. At elevated adsorption temperatures (up to 550 °C), the impregnation of AC with KOH and KI significantly enhances its H2S sorption performance, specifically regarding adsorption capacity and breakthrough time. N2 adsorption, SEM, and EDS measurements indicated that the chemical interactions between H2S and alkaline substances (KOH and KI) are enhanced at elevated temperatures. Utilizing all experimental data, the equilibrium adsorption model employing the linear isotherm was formulated to forecast the sorption behavior of these sorbents concerning the equilibrium isotherm constant and mass transfer coefficient for further scaling-up processes.
8. Biomass-based adsorbent for nitrogen dioxide removal
Nitrogen dioxide (NO2) is an inorganic air pollutant that poses detrimental impacts on animal life, human health, and the environment. NOx gas is emitted by natural occurrences including, volcanic eruptions, lightning strikes, and forest fires. In recent times, nanostructured metal oxides such as ZnO, TiO2, and CuO have gained significant popularity as gas sensor components due to their distinct structure and surface-to-volume ratio, which sets them apart from layered materials.231–234 Additionally, composites that incorporate MOFs in their structures are utilized for the purpose of adsorbing NO2.235,236Several studies have conducted comparisons of the adsorption capabilities of NO2 at low temperatures using various activated carbons. The activated carbons for NO2 removal were obtained either commercially or by chemical/physical activation of different precursors such as lignocellulosic biomasses or mineral coals. Many techniques were employed in the literature to alter the texture and surface chemistry of the material and enhance the efficacy of activated carbon for removing NO2. Table 6 summarizes the performance of biomass-based materials for NO2 removal under different operating environments.
| Precursor | Activation conditions | SBET (m2 g−1) | Vt (cm3 g−1) | Vmic (cm3 g−1) | Total content of surface oxides (mmol g−1) | Adsorption test conditions | NO2 sorption capacity (mg g−1) | Ref. |
|---|---|---|---|---|---|---|---|---|
| Bituminous coal | Carbonisation at 700 °C | 1856 | 0.875 | 0.863 | 1.32 | 0.450 L min−1 | 43.5 | 237 |
| KOH at 700 °C | 0.1% NO2, moist air (70% humidity) | |||||||
| Walnut shell | Pyrolyzed at 400 °C | 2305 | 1.15 | 1.12 | 1.64 | 0.450 L min−1 | 66.3 | 238 |
| KOH activation at 800 °C | ||||||||
| Direct CO2 activation at 800 °C | 697 | 0.37 | 0.34 | 1.09 | 0.1% NO2, dry air | 58.1 | ||
| Plum stone | Pyrolyzed at 400 °C | 2570 | 1.35 | 1.30 | 1.17 | 0.450 L min−1 | 67 | 239 |
| KOH activation at 800 °C | 0.1% NO2, dry air | |||||||
| Pine sawdust pellets | CO2 activation at 800 °C for 60 min | 275 | 0.15 | 0.14 | 1.92 | 0.450 L min−1 | 45.3 | 240 |
| 0.1% NO2, moist air (70% humidity) | ||||||||
| Coffee industry waste | Pyrolyzed at 800 °C | 1553 | 1.06 | 0.76 | 3.25 | 0.450 L min | 44.5 | 241 |
| KOH activation at 700 °C | 0.1% NO2, moist air (70% humidity) | |||||||
| Wood-based activated carbon BAX-1500 | BAX150, impregnation with 5% Ag | 1772 | 1.09 | 0.41 | — | 0.450 L min−1 | 66.0 | 242 |
| 0.1% NO2, dry air |
Although previous studies were conducted on the interaction between NO2 and carbonaceous materials in the early 1900s, the first study on the use of AC for NO2 removal was published in 2007 by Pietrzak and Bandosz.243 The efficacy of BAX-1500, a commercially available wood-based activated carbon, was assessed for adsorbing NO2 using a custom-designed dynamic test. This experiment involved injecting either dry or wet air (with 70% humidity) that contained 0.1% volume/volume of NO2. The air was injected at a flow rate of 0.45 L min−1 into a glass column that contained a packed activated carbon. The residence time was 0.4 s, which is consistent with the value commonly employed in industrial air filtering systems. It was observed that the adsorption and reduction of NO2 took place on the surface of the activated carbon.
Similarly, Jeguirim et al. previously discovered this behavior when studying the interaction between NO2 and black carbon.244 The BAX-1500 exhibited adsorption capabilities of 42.7 and 63.8 mg g−1 under dry and wet conditions, respectively. The analysis of the AC's characteristics before and after adsorption testing using different analytical techniques revealed an enhancement in the acidity properties of the AC following NO2 adsorption which was also evident in the colour change. The rise in acidity was ascribed to the surface oxidation and the generation of carboxylic groups during the reduction of NO2, as well as the production of nitric acid during the interaction of NO2 with hydroxyl groups. In addition, the application of NO2 treatment resulted in a small reduction (10–15%) in the textural characteristics, such as the specific surface area and volume of micropores, due to surface oxidation. This suggests that both the physical characteristics and chemical composition of the adsorbent's surface are significant factors in the adsorption and interaction of NO2 with activated carbon.
Nowicki et al. conducted a study to examine the impact of surface oxygen groups by comparing the effectiveness of activated carbons produced by chemical (KOH) and physical (CO2) activations of coffee residues.241 The adsorption capacity of 44.5 mg g−1 was achieved using activated carbon with a surface oxygen basic group concentration of 0.9 mmol g−1. However, the physically activated carbon showed a reduced adsorption capacity for CO2, despite the presence of a greater quantity of basic surface groups of 11.32 mmol g−1. This behavior might be ascribed to the lack of micropores. In addition, both the existence of micropores and the presence of surface oxygen basic groups are crucial for facilitating effective interactions between the NO2 molecules and the carbon surface.
Similarly, Belhachemi et al. conducted a study to assess the effectiveness of several activated carbons. These carbons were made using chemical (CZn) and physical (CCO2) activation of date pits, as well as using commercial activated carbon (CAC). Modified activated carbons were also created through chemical oxidation (CAC-O) and thermal treatment (CAC-O-T). The experiments to measure the adsorption of NO2 were conducted in a fixed-bed reactor, as seen in Fig. 5. The tests were carried out at normal room temperature and under dry circumstances.245 For each test, a concentration of 500 parts per million by volume of NO2 in nitrogen gas was introduced into the reactor. The flow rate of the gas was set at 20 NL h−1, and the reactor included 100 mg of activated carbon. The experiment revealed NO2 adsorption capabilities ranging from 78 to 136 mg g−1, as seen in Fig. 5b. Furthermore, a substantial emission of nitrogen monoxide (NO) resulting from the reduction of NO2 on the carbon surface was observed. The quantity of released NO exhibited a pattern that closely resembled the adsorption of NO2. This behavior suggests that the conversion of NO2 to NO is an essential process for the adsorption of NO2, as previously proposed.244 Moreover, examining the textural and surface characteristics revealed that the volume of micropores and the existence of stable oxygen groups were the primary factors that influenced the interaction between NO2 and activated carbons. The quantity of strong acidic groups exhibits an inverse correlation with the quantity of remaining oxygen subsequent to the reduction of NO2 to NO. thus, the presence of highly acidic groups hinders the reduction of NO2 to NO and thus prevents the adsorption of NO2 on the surface of adsorbent.
 | ||
| Fig. 5 (a) Scheme of the set-up for the NO2 adsorption experiments. (b) NO2 adsorption capacities at 25 °C for the different activated carbons.245 | ||
Ghouma et al.246 discovered that the adsorption capacity of 131 mg g−1 which closely aligns with the findings of Belhachemi et al.245 Nevertheless, the activated carbon derived from olive stone exhibited a smaller volume of micropores when compared to the activated carbon produced from date stone.245 This increased capability was ascribed to the greater quantity of basic groups (1.86 mmol g−1). The examination of the gases generated during Temperature Programmed Desorption (TPD) revealed that NO2 can undergo either physisorption directly onto the activated carbon or chemisorption by interacting with the surface oxygen groups. Ghouma et al. conducted a study to determine the efficiency of activated carbon produced by water vapor activation. The performances of three distinct activated carbons made from the same precursor (olive stone) were examined.246 The authors developed two physically activated carbons, one activated using CO2 and the other using H2O, as well as a chemically activated carbon using H3PO4. Their performance in breakthrough experiments was evaluated using a fixed-bed flow reactor setup.
Copper salt-impregnated carbon was subjected to a reductive atmosphere utilizing hydrazine hydrate or nitrogen heat treatment at 925 °C.247 Impregnating copper increased NO2 sorption and NO retention on carbon after NO2 reduction. That enhancement is due to copper metal's strong surface dispersion. Both reduction techniques reduced copper, but the carbon surface reacted differently. Heat treatment increases metallic copper and reduces carbon matrix oxygen functional groups, whereas hydrazine reduces copper and incorporates nitrogen. The data indicates that NO2 is mostly transformed into copper nitrates, while N2 reduction is possible. Hydrazine-treated samples have high capacity due to metallic copper dispersion on carbon. Similarly, the impregnation of urea and heat-treated wood-based AC has been reported to positively impact NO2 adsorption. In dry-air experiments, positively charged nitrogen centers may be involved in electron transfer for NO2 chemisorption and superoxide ion oxidation to single-bond NO3.242
9. Biomass-based adsorbent for volatile organic compounds removal activated carbon
Volatile organic compounds (VOCs) are a specific category of air contaminants that have garnered significant environmental concern. They are prominent aromatic compounds that are commonly found in industrial processes, including those in the paint and petroleum sectors.248,249 They have significant biological toxicity resulting in respiratory tract infections, lung cancer, brain damage, and several other disorders.250,251 Studies have demonstrated that adsorption is a cost-effective and efficient method for treating low levels of VOCs.248,252,253 Currently, the development of adsorbent from biomass is considered a green strategy for obtaining carbon with functional properties (Table 7). For example, Shen et al.262 synthesized rice husk-based porous carbon via ball milling and KOH activation. The resulting carbon material exhibited a toluene adsorption capacity of 250.6 mg g−1 at 20 °C, significantly higher than the original biochar's adsorption capacity of just 0.72 mg g−1. Jin et al.267 discovered that utilizing the air pre-oxidation technique in producing nitrogen-rich porous biochar enhanced the efficiency and augmented the toluene sorption capability from 296.4 mg g−1 to 437.8 mg g−1 at 30 °C.| Biomass precursors | Activating conditions | Biochar preparation | Adsorption conditions | Adsorption capacity | Ref. |
|---|---|---|---|---|---|
| Bamboo-based biochar | Cu-BTC solvothermal synthesis with biochar of 0.3, 0.6, 0.9 g | FBR. Toluene 500, 1000, 1500, 2000 ppm, N2; 60, 90, 120, 150 °C | BiocharCu-BTC:toluene: 501.8 mg g−1, 60 °C, 1000 ppm |
254 | |
| 88.8 mg g−1, 150 °C, 1000 ppm | |||||
| Pistachio | Biomass + melamine, N2: 700 °C | FBR. Toluene 110 ± 5 mg m−3, 0, 40, 80% humidiy; N2; 40 °C | Biocharmelamine:toluene: 233 mg g−1, 40 °C |
248 | |
| Shells | |||||
| Gasification biochar | KOH 1:3, 850 °C |
— | Gas chromatograph with a flame ionization detector. Benzene 50 ppm; 80 °C | BiocharKOH:benzene: 144 mg g−1, 80 °C |
255 |
| Hickory wood, peanut hull | H3PO4 or KOH:C = 1:1, 600 °C |
HTC: 200 °C | TGA. VOC (50 mL min−1), N2; 20 °C | BiocharH3PO4:acetone: 147.77 mg g−1, 20 °C |
256 |
| Cyclohexane: 159.66 mg g−1, 20 °C | |||||
| Milled hickory, wood chips, peanut shell | CO2: 600, 700, 800, 900 °C | HTC: 200 °C | TGA. VOC (50 mL min−1), N2; 20, 40, 60 °C | BiocharCO2:acetone 121.74 mg g−1, 20 °C |
257 |
| Cyclohexane: 155.41 mg g−1, 40 °C | |||||
| Oxford Hardwood, wheat straw, corn straw, rice straw, rape straw | — | N2: 300, 400, 500 °C | Bespoke testing system. Xylene isomer (100–400 ppm), N2; 25, 35, 45 °C | P-Xylene: 50.88 mg g−1, 25 °C | 258 |
| M-Xylene: 36.82 mg g−1, 25 °C | |||||
| O-Xylene: 45.37 mg g−1, 25 °C | |||||
| Bamboo, pepper wood, sugarcane | — | N2: 300, 450, 600 °C | Mass = 5 mg. T = 150 °C flow rate = 10 °C min−1 under 50 mL min−1 N2 atmosphere | Toluene: 62.91 mg g−1, 20 °C | 259 |
| Acetone: 91.16 mg g−1, 20 °C | |||||
| Bagasse, hickory wood, sugar beet tailings | Cyclohexane: 69.33 mg g−1, 20 °C | ||||
| Hickory wood | Ball milling, 1:100 |
N2: 300, 450, 600 °C | TGA. VOC (50 mL min−1), N2; 20 °C | Toluene: ∼70 mg g−1, 20 °C. Acetone: 103.4 mg g−1, 20 °C | 260 |
| Cyclohexane: 72.5 mg g−1, 20 °C | |||||
| Ethanol: ∼65 mg g−1, 20 °C | |||||
| Chloroform: 87.0 mg g−1, 20 °C | |||||
| Neem, sugarcane, bamboo | — | N2: 350, 450, 550 °C | TGA. Benzene, toluene, methyl chloride, xylene, chloroform, carbon tetrachloride; 120 °C | Benzene: 54.6 mg g−1, 120 °C | 261 |
| Toluene: 65.5 mg g−1, 120 °C | |||||
| Methyl chloride: 39.61 mg g−1, 120 °C | |||||
| Xylene: 60.2 mg g−1, 120 °C | |||||
| Chloroform: 30.81 mg g−1, 120 °C | |||||
| Carbon tetrachloride: 40.99 mg g−1, 120 °C | |||||
| Rice husk | Ball-milling, 30 min | N2: 450 °C | FBR. Toluene 300 ppm, phenol 60 ppm; 20 °C | BiocharKOH:toluene: 264 mg g−1, 20 °C |
262 |
| KOH:C = 1, 3; 750 °C |
|||||
| Phenol: 6.53 mg g−1, 20 °C | |||||
| Oil palm | — | N2: 500, 600, 700, 800 °C | Batch equilibrium tests. Formaldehyde (0.5, 0.75, 0.9, 2.1 ppm), H2O 50%; 25 °C | 410 mg g−1, 2.1 ppm, 25 °C | 263 |
| Wheat straw, bagasse | — | N2: 500 °C | Bespoke testing system. Acetone, hexane, toluene, p-xylene (200–220 ppm), N2; 25 °C | Acetone; 110.9 mg g−1, 25 °C | 264 |
| Toluene: 45.2 mg g−1, 25 °C. p-Xylene: 51.1 mg g−1, 25 °C | |||||
| Hexane: 36.8 mg g−1, 25 °C | |||||
| Multi-component: 109.1 mg g−1, 25 °C | |||||
| Corn stalk | Ball milling, 3:200; H2O2/NH4OH |
N2: 600 °C | TGA. VOC (50 mL min−1), N2; 25 °C | BiocharH2O2/NH4OH:benzene: ∼118 mg g−1, 25 °C |
265 |
| M-Xylene: ∼125 mg g−1, 25 °C | |||||
| O-Xylene: ∼120 mg g−1, 25 °C | |||||
| P-Xylene: 130.21 mg g−1, 25 °C | |||||
| Coconut shell | Steam: 900 °C | N2: 900 °C | FBR. Toluene 80 ppm, chlorobenzene, 80 ppm; 25, 30, 40 °C | BiocharH2O:toluene: 255 mg g−1, 25 °C |
266 |
| Chlorobenzene: 272 mg g−1, 25 °C |
Guo et al.268 studied the impact of activated carbon characteristics on removing chlorobenzene. They found that the pore structure of the activated carbon was the primary influencing factor. It has been discovered that benzene and toluene prefer to be adsorbed in the confined micropores of activated carbon.269 Regarding surface properties, several researchers have posited that certain carbonyl surface groups establish a bond with the aromatic ring of phenol based on a donor–acceptor hypothesis.270 However, some individuals hypothesized that graphene layers interact with the π electrons of the aromatic ring of phenols, resulting in the presence of electron-rich areas (π–π argument).271
The surface acidity of biochar and its enhanced sorption of polar and hydrophilic VOCs are primarily attributed to the presence of oxygenic functional groups.261 These functional groups, however, have a detrimental effect on the sorption of hydrophobic VOCs by inhibiting the interactions between the hydrophobic VOCs and electron-rich regions of biochar.256 Chemical functional groups impact dispersive and electrostatic interactions between the biochar and VOCs, mostly through van der Waals interaction.272,273 The attractive forces that affect the sorption uptake include π–π dispersion interactions and hydrogen bonding. Hence, it is necessary to take into account the interplay between the pore property, chemical functional group of biomass-based materials, and characteristics of VOCs in order to assess the sorption efficiency of common VOCs.257 Fig. 6 depicts the primary adsorption mechanism of VOC onto biomass-based materials. Therefore, understanding the sorption process and structural–functional relationship requires a thorough understanding of the in-depth knowledge of adsorption equilibrium.
 | ||
| Fig. 6 Adsorption mechanism of VOCs onto biomass/biochar. | ||
This involves physical sorption, which includes surface sorption, pore filling, and partitioning to noncarbonized organic matter. It also involves intermolecular forces such as π–π interaction, CH–π bonding and electrostatic attraction. The sorption characteristics of VOCs are influenced by their molecular sizes, molecular weights, and boiling points.
Li et al.274 investigated five common straws as potential materials for preparing straw-based activated carbon (SAC) and characterized them using a scanning electron microscope, thermo-gravimetric analysis, and the Brunauer–Emmett–Teller method. Millet straw-derived activated carbon shows superior properties in SBET, Smic and adsorption capacities of both toluene and ethyl acetate. The preparation process of millet straw activated carbon was optimized via response surface methodology, using carbonization temperature, carbonization time, and impregnation ratio as variables and toluene adsorption capacity, ethyl acetate adsorption capacity, and activated carbon yield as responses. The optimal preparation conditions include a carbonization temperature of 572 °C, carbonization time of 1.56 h and impregnation ratio (ZnCl2/PM, w/w) of 1.60, which was verified experimentally, resulting in millet straw activated carbon with a toluene adsorption capacity of 321.9 mg g−1 and ethyl acetate adsorption capacity of 240.4 mg g−1. Meanwhile, the adsorption isothermals and regeneration performance of millet straw-activated carbon prepared under the optimized conditions were evaluated. The descriptive ability of the isothermals via the Redlich–Peterson equation suggests a heterogeneous surface on millet straw-activated carbon. Recyclability testing has shown that millet straw-activated carbon maintained a stable adsorption capacity throughout the second to fifth cycles. The results of this work indicate that millet straw-activated carbon is a potential volatile organic compound adsorbent for industrial application.
Hierarchical porous carbons (HPCs) derived from biomass were produced using a cost-effective method that integrates pyrolysis and KOH impregnation.275 The maximum surface area of HPCs reaches 3936 m2 g−1, demonstrating unprecedented acetone (26.1 mmol g−1 at 18 kPa) and methanol (46.9 mmol g−1 at 15 kPa) sorption capacities at 25 °C. Experimental and molecular modeling results indicate that the overall pore volume predominantly influences the sorption capacity of acetone and methanol at elevated pressures. Due to the electrostatic interactions between the gaseous molecules and carbon frameworks, the oxygen groups offer sorption sites for acetone and methanol at low pressure. The small micropore influences acetone/methanol selectivity at low pressure, but the oxygen groups do not enhance this selectivity. Kim et al. enhanced activated carbon by impregnating it with diverse acids or bases. The findings indicated that activated carbon modified with 1 wt% H3PO4 exhibited the highest adsorption capacity for toluene and benzene, representing a potential hybrid system sorbent for mitigating low-concentration VOC emissions.276
Shen et al. reported an exceptional carbon adsorbent developed from corncobs using KOH as an impregnating agent for the selective sorptive removal of VOCs in humid environments.277 The carbon sorbent derived from corncobs demonstrated superior sorption capacities for volatile benzene (851.3 mg g−1) and toluene (854.9 mg g−1), significantly surpassing those obtained from KOH grinding methods, which are three times greater than commercial activated carbon (benzene 275 mg g−1, toluene 310.5 mg g−1). It was then established that the optimized structural characteristics of the synthesized adsorbent exhibited a strong association with the sorption effectiveness of VOCs.
Coconut shell-based carbons were impregnated with phosphoric acid, ammonia, sulfuric acid, sodium hydroxide, and nitric acid to identify optimal modifications for enhancing the sorption capacity of hydrophobic VOCs on granular activated carbons (GAC).278 The results indicated that alkali-impregnated GAC exhibited superior o-xylene adsorption ability. The uptake quantity increased by 26.5% and decreased by 21.6% following treatment with NH3H2O and H2SO4. In comparison to the original, acid-impregnated GAC exhibited reduced adsorption capability. Other authors have reported that the impregnation method modified and enhanced the sorption ability of adsorbent for removing VOCs.279–283
10. Biomass-based adsorbents for multi-gas removal
To date, there are limited studies on the adsorptive removal of multi-gas components using biomass-based adsorbents. Table 8 provides an overview of the preparation conditions of biomass sorbents and their corresponding performance for multigas removal. The palm shell-activated carbon coated with metal oxides was used to simultaneously remove SO2 and NOx. It demonstrated an increased in sorption capacity in the order Fe2O3 < NiO < V2O5 < CeO2.156 The metal oxides with strong oxidizing and oxygen retention capabilities added new oxygenic functional groups, such as C–O, COOH, and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, to the obtained adsorbent. This enhances the oxidation reaction of NO to NO2, and SO2 to SO3, resulting in a highly effective removal of NOx and SO2.156 It has been discovered that the biochar amine obtained from the feedstock of peanut shells, corn stalks, and corncobs had a significantly higher capacity for removing SO2 in the presence of both NO and SO2 compared to when NO was absent.192 The SO2 removal capacity initially increased and then decreased as the NO concentration increased. The maximum sorptive removal capacity was observed at 120 °C and 500 ppm NO, reaching 216.19 mg g−1. The interaction between SO2 and NO takes place, particularly at the N-containing sorption sites of biochar during the sorption process (Fig. 7). This interaction could potentially lead to creating (SO3)(NO2) intermediates.192
O, to the obtained adsorbent. This enhances the oxidation reaction of NO to NO2, and SO2 to SO3, resulting in a highly effective removal of NOx and SO2.156 It has been discovered that the biochar amine obtained from the feedstock of peanut shells, corn stalks, and corncobs had a significantly higher capacity for removing SO2 in the presence of both NO and SO2 compared to when NO was absent.192 The SO2 removal capacity initially increased and then decreased as the NO concentration increased. The maximum sorptive removal capacity was observed at 120 °C and 500 ppm NO, reaching 216.19 mg g−1. The interaction between SO2 and NO takes place, particularly at the N-containing sorption sites of biochar during the sorption process (Fig. 7). This interaction could potentially lead to creating (SO3)(NO2) intermediates.192
| Biomass precursors | Biochar preparation | Activating conditions | Adsorption conditions | Typical adsorption capacity | Ref. |
|---|---|---|---|---|---|
| Palm shell | N2: ∼700 °C | CO2: 1100 °C | FBR. SO2 2000 ppm, NO 500 ppm, O2 10%, N2; 100, 200, 300 °C | SO2: biocharCe, ∼180 min (breakthrough time), 200 °C | 284 |
| Ni, Ce: 5% | NOx: biocharCe, ∼170 min, 200 °C | ||||
| Palm shell | N2: ∼700 °C | CO2: 1100 °C | FBR. SO2 2000 ppm, NO 500 ppm, O2 10%, N2; 100 °C | SO2: biocharCe, 155 min, 100 °C | 156 |
| Ni, V, Ce, Fe: 10% | NOx: biocharCe, 65 min, 100 °C | ||||
| Palm shell | N2: ∼700 °C | CO2: 1100 °C | FBR. SO2 (1000, 1500, 2000, 2500 ppm), NO (100, 300, 500, 700 ppm), O2 10%, N2; 150–300 °C | BiocharCe: 2500 ppm SO2/700 ppm NO: SO2, 120.25 mg g−1, 150 °C; NO, 3.52 mg g−1, 150 °C | 189 |
| Ce: 10% | GHSV: 23527 h−1 |
||||
| Palm shell | N2: ∼700 °C | CO2: 1100 °C | FBR. SO2 (500–2500 ppm), NO (100–700 ppm), H2O (15, 30, 45, 60%), O2 10%, N2; 100–300 °C | BiocharCe: 2000 ppm SO2/500 ppm NO: SO2, 121.7 mg g−1, 150 °C; NO, 3.46 mg g−1, 150 °C | 285 |
| Ce: 10% | GHSV: 14000–36000 h−1 |
||||
| Rice husk | N2: 600 °C | NH4Cl, NH4Br: 1%, 2% | FBR. Hg (33 μg m−3), SO2 1000 ppm, NO 200 ppm, O2 10%, N2; 150 °C | BiocharNH4Br:FBR. Hg 30 μg g−1 |
286 |
| GHSV: 23527 h−1 |
|||||
| Coal-fired flue gas, 150 °C | Flue gas (SO2 1177 ppm, NO 254 ppm): Hg 80%, SO2 ∼35%, NO ∼35%, 150 °C | ||||
| Food waste | N2: 500, 700 °C | CO2: 900 °C | SVA. CO2, 25 °C, 0–101 kPa | Biocharmelamine:CO2: 4.41 mmol g−1, 25 °C |
287 |
| HNO3: 1:10; melamine:N2 450 °C |
Gravimetric sorption. Benzene; 25, 35, 45 °C | ||||
| Benzene: 380.7 mg g−1, 25 °C | |||||
| Maize straw | ZnCl2: 1:1 |
Co/Ce:C = 5, 15, 25% (Co:Ce = 4:6, 5:5, 6:4); N2 550 °C |
FBR. Hg (100 μg m−3), SO2 1000 ppm, NO 600 ppm, NH3 600 ppm, O2 6%, N2; 80–320 °C | BiocharCo/Ce:NO: 84.7%, 230 °C |
288 |
| N2: 750 °C | GHSV: 100000 h−1 |
||||
| Hg0: 96.8%, 230 °C | |||||
| Pinecone | N2: 500 °C | H2O2: 10:1; Cu/Mn:C = 8, 12, 14% (Cu/Mn = 1–3:1, 1:2–5); N2 500 °C |
FBR. Hg (700 μg m−3), HCHO 100 ppm, SO2 (400, 800 ppm), NOx (300, 600 ppm), NH3 300 ppm, O2 5%, N2; 100–300 °C | BiocharCu1Mn1:HCHO: 89%, 175 °C |
289 |
| GHSV: 13000 h−1 |
|||||
| Hg: 83%, 175 °C | |||||
| Walnut shell | N2: 500–900 °C | KOH: 2:1, N2: 600 °C |
FBR. Hg (100 μg m−3), SO2 200 ppm, NOx 300 ppm, NH3 300 ppm, O2 5%, N2; 150 °C.GHSV: 10000 h−1 |
BiocharFe:SO2 100%, 250 °C |
290 |
| Al, Cu, Fe, Mn: 5% | |||||
| NOx ∼90%, 300–400 °C | |||||
| Hg ∼90%, 250 °C | |||||
| Rice straw | Ball milling; 1 (Fe/Mn = 1.1–2.0); N2 800 °C | — | FBR:Hg (35 μg m−3), o-Xylene 20 ppm, O2 5%, N2; 100–350 °C |
BiocharFe/Mn:O-Xylene: ∼88%, 100 °C; ∼75%, 350 °C |
291 |
| GHSV: Xylene 30000 h−1, Hg0 90000 h−1 |
|||||
| Hg: ∼96%, 100 °C; ∼72%, 350 °C |
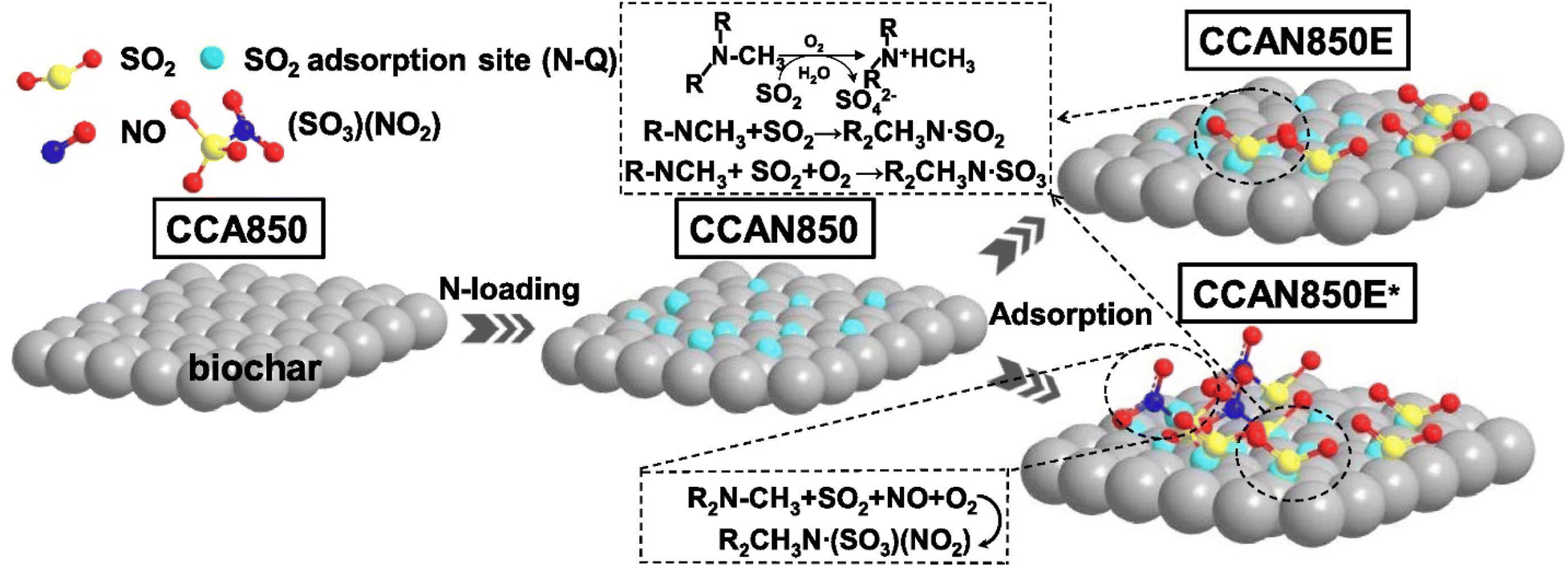 | ||
| Fig. 7 Schematic representation of the adsorption of SO2 onto nitrogen content of biochars.192 | ||
Another study reported the competitive adsorptive removal of NO and SO2 onto the active sites of biochar produced from palm shell activated carbon supported with cerium. It was demonstrated that molecules with a high boiling point have stronger intermolecular interactions and van der Waals attractive forces. Therefore, biochar efficiently adsorbs SO2 relative to NOx because SO2 has a high boiling point than NOx.156,292 Elevating the concentration of SO2 (up to 2500 ppm) could effectively displace and remove the NO from the active sites on the biochar thereby limiting the sorption of NO.189 In addition, the flue gas exhibited a much greater concentration of NO (up to 700 ppm) compared to that of SO2, resulting in a more pronounced catalytic bonding of NO on the active sites of biochar supported with cerium. Consequently, it decreased the number of active sites available for the removal of SO2.189
The simultaneous adsorption of NO and SO2 onto cerium-impregnated palm shell-activated carbon (Ce/PSAC) sorbent has been reported.285 In the presence of 15% H2O, the water layer contained a higher concentration of NO2 molecules due to NO2 being more soluble in water and having stronger oxidizing properties than O2 and NO. As a result, the reactions between NO2 and HSO3− and SO32− to form SO42− were enhanced. Consequently, more SO2 molecules were captured into the water layer, leading to an overall increase in the removal of SO2 compared to the scenario where the simultaneous removal of NO was not considered.285
Recently, Qin et al. prepared a defective walnut shell-based carbon that was modified with transition metal (Fig. 8). The removal efficiencies of SO2, NOx, and Hg0 onto the prepared adsorbent varied as the carbonization temperature increased. This change was due to the impact of the carbonization temperature on the porous characteristics and surface defects of the adsorbent (Fig. 8a–d). Carbonizing at 700 °C significantly increased the degree of graphitization, the presence of a rich pore structure, and the contents of surface defects in the adsorbent compared to other temperatures. This enhancement was advantageous for the loading and dispersion of the active constituents of Fe species, thereby affecting the adsorbent activity for the simultaneous removal process.290 The DFT simulation demonstrated a significant sorption interaction between Mn and Fe species and NOx, SO2, and Hg0 (Fig. 8f–i). This indicates that biochar Fe/Mn has a higher simultaneous removal effectiveness of about 80% at temperatures ranging from 300 to 350 °C.290 When biochar sorbents were introduced into coal-fired flue gas at a temperature of 150 °C in an entrained-flow reactor, almost 80% of gaseous mercury was eliminated. Additionally, the SO2 concentration was reduced by 35.6% while that of NO decreased by 36.0%.290
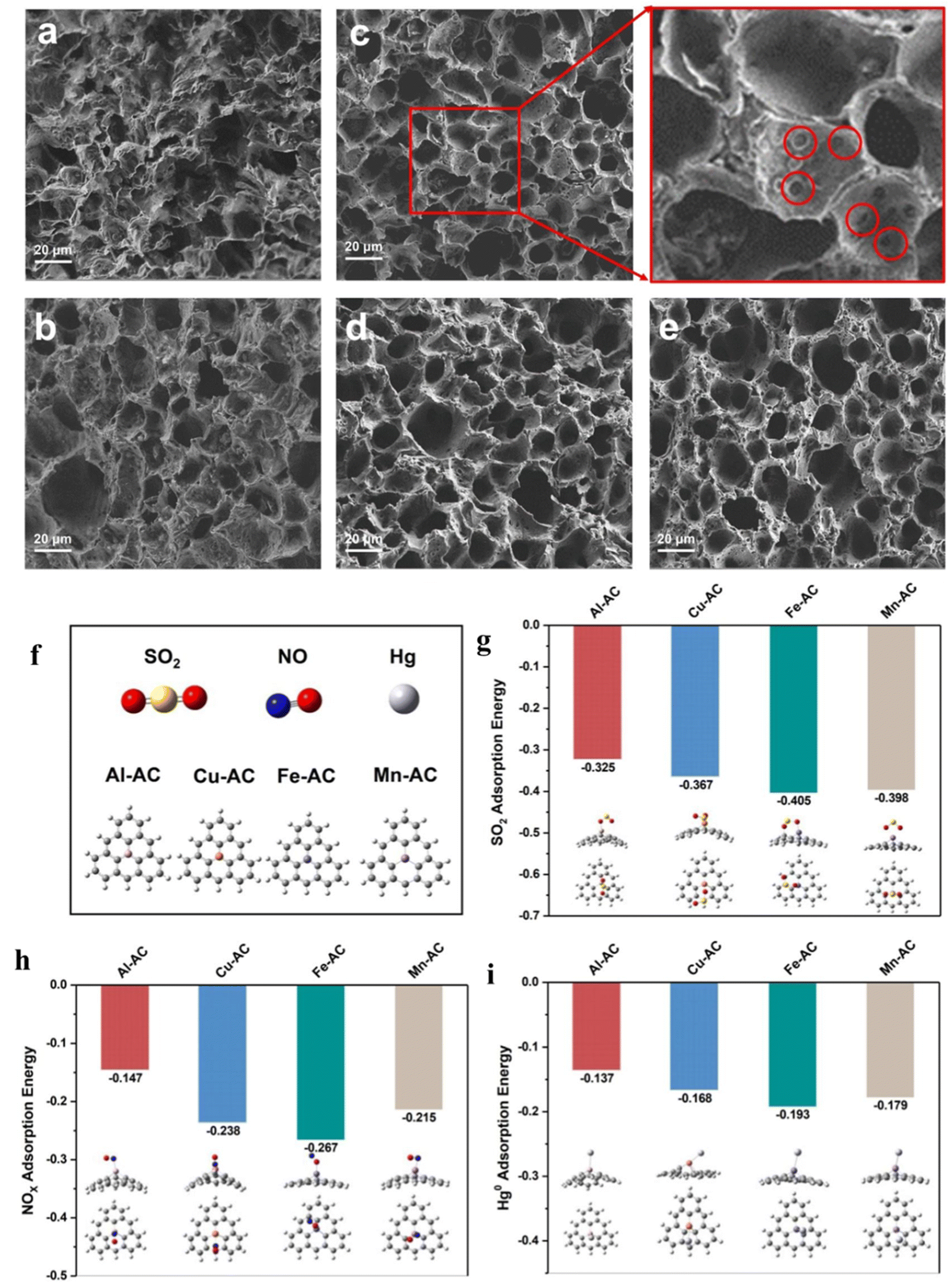 | ||
| Fig. 8 SEM images at (a) 500 °C (b) 600 °C (c) 700 °C (d) 800 °C (e) 900 °C. (f–i) Molecular structures and energy states of SO2, NOx and Hg0 adsorbed on walnut shell-based carbon.290 | ||
Similarly, the presence of 6% O2 and 10% H2O has been reported to have a beneficial effect on the oxidation reactions of SO2 to SO3 and H2SO4, as well as the oxidation of NO to NO2.286 An inconsistent impact of SO2 was seen in the elimination of Hg0. Specifically, low levels of SO2 appeared to enhance its removal, whereas high levels of SO2 (above about 700 ppm) had an inhibitory effect.293 When both SO2 and NO are present, SO2 consumes the active oxygen and produces sulfate, which causes permanent damage to the active sites on the adsorbent. As a result, the interaction between Hg0 and NO2, which was generated from NO, becomes impeded by SO2.294 Gao et al. found that the presence of adsorbed SO2 can hinder the adsorption of NO, NH3, and Hg0 through competitive inhibition.288 The combined impact of SO2 and H2O had a more detrimental effect as their competing adsorption and the formation of ammonium sulfates led to the degradation of the porous structure and the obstruction of the active sites on the adsorbent.
Sumathi et al. simultaneously removed both NOx and SO2 by impregnating coconut shell-AC with KOH.295 Moreover, the activated carbon may penetrate the pore structure with the addition of potassium, markedly improving the efficacy of NOx and SO2 reductions due to its surface's heightened reactivity. The simultaneous elimination of SO2 and NO from simulated flue gas with cerium oxide supported on palm shell AC (Ce/PSAC) was investigated in a fixed bed adsorber.285 The influences of adsorber temperature, humidity present, feed gas concentration, and space velocity were examined as process parameters. The experimental results indicated that increased space velocity decreased the sorption capacity of SO2 and NO. Humidity increased the SO2 sorption capacity but inhibited NO sorption at levels exceeding 15%. Temperature significantly influenced the simultaneous elimination of SO2 and NO by cerium supported on PSAC. The qmax of SO2 and NO were attained at a temperature of 150 °C, measuring 121.7 mg g−1 and 3.5 mg g−1, respectively. This demonstrates that inexpensive biomass-derived AC may serve as an effective sorbent for the simultaneous removal of SO2 and NO from flue gas.
A highly efficient porous carbon filter for indoor air pollutant removal was created utilizing NaOH-impregnated AC (NaOH/AC) for the sorption of H2S and CH3COOH.296 The NaOH/AC filter was studied using several methods, demonstrating favorable physical and chemical features, particularly the presence of –OH functional groups, for the sorption of air contaminants. The NaOH/AC filter was subjected to various curing temperatures and dwell periods to analyze the impact of curing conditions on sorption efficacy. The optimal performances were achieved with the NaOH/AC filter cured at 100 °C for 20 min, effectively removing the starting concentration of 400 ppm of CH3COOH within 15 min and H2S within 30 min at 20 °C and 60% relative humidity. Isotherm and kinetic models were employed to examine the sorption process. Both the Langmuir isotherm and pseudo-second-order kinetic models exhibited the most accurate fit for the sorption of CH3COOH and H2S on the NaOH/AC filter. The sorption process was governed by intraparticle diffusion in conjunction with film diffusion. The NaOH/AC filter exhibited a qmax of 473 mg g−1 for H2S and 550 mg g−1 for CH3COOH. The wasted NaOH/AC filter was regenerated for subsequent usage.
11. Regeneration studies
In an industrial application, regenerating used biomass adsorbent is essential for a successful adsorption–desorption process and it is important to strive for a lower regeneration temperature and a short cycle duration. The quick kinetics process is advantageous for reducing the duration of the adsorption cycle. Increased chemisorption strength hinders the regeneration of biomass adsorbent, necessitating higher regeneration or desorption temperatures and resulting in greater energy consumption.297Atanes and colleagues298 discovered that the SO2 removal onto acidic biomass adsorbents by physisorption may be easily regenerated for several sorption cycles. It has been observed that heat treatment mostly impacted the physical structure of the used adsorbent.299 During this process, H2SO4 that had been adsorbed onto the biomass was eliminated, resulting in a reduction in carbon content. A comparison of the two different treatments has been experimented.300 It was found that approximately half of the adsorbed SO2 was regenerated after the first cycle. Additionally, low treatment temperatures resulted in greater levels of residual sulfur which led to a drop in SO2 sorption. The efficacy of SO2 sorption decreased as the number of regeneration cycles increases to the third time, due to the degradation of pore texture and active adsorption sites.301
The effectiveness of biomass H2O regeneration during 6 cycles has been reported.299 It was discovered that the pore structure of biomass changed after the regeneration cycles leading to a moderate increase in both micropore volumes and surface areas starting with the fourth cycle. In addition, the acid surface that resulted from SO2 adsorption was broken down during the desorption test at 600 °C. This process creates new basic surfaces which may enhance the ability of the biomass to adsorb more SO2 compared to its initial state. Surprisingly, these two factors resulted in increased SO2 adsorption capabilities during the fourth–sixth cycles compared to the first-thirdcycles.
Two primary types of adsorption technology considered suitable for capturing CO2 in post-combustion processes and can be used to effectively regenerate the adsorbents involve temperature swing adsorption (TSA) and pressure swing adsorption (PSA), or in certain cases, vacuum swing adsorption (VSA).50 The biomass may be easily regenerated by either raising the temperature in the TSA process or lowering the system pressure in the PSA process.302 Nevertheless, implementing a traditional PSA method to compress the flue gases with a higher flow rate would be impractical and challenging due to its lack of cost-effectiveness. A pressure level of 5 kPa was found to be a suitable lower pressure during the desorption stage of CO2 to keep operation expenses low.302 In contrast, the regeneration of adsorbent with a high concentration of CO2 by increasing the temperature of the reactor in the TSA process has been reported.50 This may be readily accomplished using the heat generated by an operating industrial facility through the addition of a heating exchanger.50 Due to a weak interaction between the biomass and CO2, the energy requirement for its regeneration is low. This process has been widely observed in various studies.50,134,138
Most reported biomass can be easily and quickly regenerated over multiple cycles without a significant decrease in their sorption capacity and these are achieved through the TSA process81,134,303 or the PSA process.151,303–305 The sorbent's excellent reusability indicates that the physisorption process was the main mechanism. The decrease in CO2 capture efficiency via cyclic processes may be attributed to diminished surface activity, agglomeration, and insufficient regeneration of the adsorbent.306 It is important to note that the CO2 uptake in the spent biomass adsorbent may be used immediately for soil amendment and for carbon storage. Under normal soil and atmospheric temperature, the CO2 will not escape (leak). Therefore, there is no need to regenerate the biomass for an extended period of time.147
A study by Zhang et al.256 showed that the temperature of desorption of acetone on biomass was less than that of cyclohexane due to the higher porosity, numerous functional groups on the biomass, and the characteristics of VOCs. The VOCs that are absorbed in the small micropores typically necessitate a high temperature for desorption. Also, the hydrochar H3PO4 which has an excess of oxygenic functional groups could enhance the adhesion force with hydrophilic acetone. In addition, the adsorbate with a higher boiling point possesses a stronger attraction to the sorbent thereby explaining that cyclohexane has high boiling point than acetone and is more challenging to desorb. After the 5 cycles, about 90% of the acetone's adsorption capacity was retained, whereas cyclohexane had 83.30% of its capacity retained.257 In another findings, there was a decrease of just 2.5% in cyclohexane and 0.9% in acetone throughout the past four cycles.256 The significant reduction in the adsorption capacity of VOCs during the initial cycle might be ascribed to either the partial release of VOCs due to their strong affinity for biomass,259 or the formation of persistent bonds that require higher temperatures to break them.265,273
Literature studies demonstrate that impregnation is not only an efficient option for viable adsorbents in gaseous adsorption but also cost-effective due to its superior regeneration capacity. Therefore, evaluating their influence on adsorbent regeneration cannot be overemphazied. For example, the research conducted by Creamer demonstrated that aluminium-impregnated cottonwood AC has a 99% regeneration capacity, in contrast to magnesium at 96% and iron at 90%.307 In another study,151 X-ray Photoelectron Spectroscopy (XPS) was employed to determine the stability of the regenerated sorbent, and it was ascertained that the elemental and chemical composition of the regenerated sorbent remained mostly unaltered, having qmax of 5.45 mol kg−1. It may, therefore, be inferred that after more than 20 cycles, the elevated desorption temperature (673 K) diminishes the number of active sites and compromises the adsorbent's surface integrity. On the other hand, a thin layer shape using SEM analysis on the surface of Mg-based biomass AC indicates surface degradation during several cyclic sorption processes.308
The use of dual adsorption sites on metal-impregnated biomass AC could further enhance CO2 selectivity.309 For instance, metal ions with hydroxyl groups were used to impregnate biomass,310 demonstrating that the resultant sorption exceeded that of a singular site. Here, Metal–Organic Frameworks (MOFs) containing a hydroxyl group derived from Mg, Zn, and Cu were reported to exhibit remarkably rapid adsorption kinetics, with gravimetric CO2 adsorption capacities of 0.08, 0.13, and 1.24 mmol g−1, and CO2/N2 selectivity of 182, 1700, and 2000, respectively. The efficacy of CO2 sorption was significantly enhanced by the integration of these two adsorption sites. The adsorption capacity of a nickel-based biomass sorbent has been developed311 to exhibit 5.22 mmol g−1 at 25 °C, surpassing the capacity of commercial AC (3.37 mmol g−1) at ambient temperature. Mg-based MBAC has been used for post-combustion CO2 collection in binary mixed conditions, achieving a CO2 adsorption capacity of 5.87 mmol g−1 at 25 °C.312 It has superior CO2 selectivity compared to N2 gas. Metal oxides exhibit significant sensitivity to certain gases and are more stable than other precursors.313 However, investigations indicate that metal oxides alone exhibit poor selectivity for certain gases while possessing a greater surface area.314 Nonetheless, the drawback of metal oxide can be mitigated by impregnating it with carbonaceous biomass materials.
12. Density function theory calculation and molecular dynamics studies
Density functional theory (DFT) is a renowned computational chemistry technique that can be used to investigate micromechanisms at the molecular level. This is useful for understanding the interaction between small molecules and the adsorbent surface in the process of gas adsorption.315,316 For instance, Rafizah Rahamathullah et al.317 conducted an integrated study combining DFT simulations and CO2 adsorption experiments using biochar derived from desiccated coconut waste (amine-biochar@DCW). The study introduced a new class of amine-functionalized biochar as potential CO2 adsorbents. Among the tested materials, triethylenetetramine (TETA)-biochar@DCW exhibited the highest CO2 adsorption capacity at 61.78 mg g−1. Before the experiments, DFT simulations were performed using the B3LYP/6-31G (d,p) level of theory to analyze the energy band gap, global chemical reactivity descriptors (GCRD), and molecular electrostatic potentials (MEP). The simulated data showed that TETA-biochar@DCW had the lowest HOMO–LUMO gap of 2.7890 eV before adsorption, which increased after CO2 was adsorbed. The 3D MEP plots also highlighted TETA-biochar@DCW as a highly reactive adsorbent for CO2. The combined theoretical and experimental results suggest that amine-biochar@DCW, particularly the TETA-functionalized variant, is a cost-effective and efficient material for CO2 capture, demonstrating great potential as a sustainable adsorbent.Marziyeh Ahmadi et al.318 studied the improvement of CO2 adsorption using activated carbon modified with lithium hydroxide (LiOH). Their research focused on how operating conditions and the properties of the adsorbent affect performance. Experimental findings suggest that the structure of activated carbon can be approximated as a fullerene made up of heptagonal and pentagonal rings.319 A model fullerene containing 28 carbon atoms (C28) was designed and analyzed to represent this structure in simulations, as shown in Fig. 9a. Using DFT calculations, the study revealed that in its optimal configuration, the C28 structure adsorbs CO2 molecules at a distance of 3.3 Å, with a binding energy of approximately 0.06 eV. The adsorption occurs mainly at the heptagonal faces on the top and bottom of the structure. In contrast, the pentagonal faces showed weaker CO2 adsorption. The C28 model is considered the smallest simulated structure for activated carbon, with a diameter of about 5.33 Å, and bond lengths ranging between 1.39 Å and 1.56 Å. Fig. 9b illustrates the distribution of electrical charges in the C28 structure using the Mulliken scale.320 Further modeling indicated that applying pressure reduces the distance between CO2 molecules and the C28 structure, thus enhancing adsorption. LiOH nanostructures were also introduced to improve CO2 adsorption performance. Fig. 9c shows the periodic arrangement of LiOH crystals. To compare the effects of LiOH with the AC model, smaller LiOH structures of similar dimensions were designed. This approach avoided periodic boundary condition calculations as LiOH particles were modeled as free-standing structures, allowing for high-accuracy Gaussian functions.
 | ||
| Fig. 9 (a): Modeled structure of AC with chemical formula C28, which fullerene includes pentagonal and heptagonal rings; (b): the electric charge distribution of the C28 structure on the Mulliken scale; (c): LiOH crystal structure showing the top and side view.318 | ||
The study concluded that combining AC and LiOH nanostructures significantly improves CO2 adsorption. DFT simulations modeled the adsorption behaviour based on binding energy and evaluated the performance of hybrid systems composed of AC and LiOH structures. The findings highlight that LiOH nanoclusters enhance the surface interactions of materials, optimizing the adsorption process and increasing the overall efficiency of the hybrid system (C28 + Li4(OH)5 + CO2) recorded 0.13 eV and 2.4 Å for binding energy (eV), distance of CO2 from the adsorber (Å), respectively, exhibited superior CO2 adsorption capabilities while C28 + CO2 recorded 0.06 eV and 3.3 Å corroborating the experimental findings and highlighting the synergistic effects of combining AC with LiOH.
Furthermore, Lihua Deng et al.321 investigated the CO2 adsorption properties of straw-based biochar prepared through multi-step KOH activation, focusing on the structure-effect relationship under atmospheric and pressurized conditions. The study analyzed the CO2 adsorption capacity of KOH-activated biochar, and the adsorption performance of different sites at atmospheric pressure is illustrated in Fig. 10c. At 0 °C, larger nanopores (0.7 nm) and nitrogen-containing functional groups were the primary contributors to CO2 adsorption (Fig. 10(c1)). At 25 °C, smaller nanopores (0.5 nm) and oxygen-containing groups dominated adsorption (Fig. 10(c2)), though the oxygen-containing groups showed no significant difference in adsorption capacity at different temperatures. These findings are critical for designing high-performance CO2 adsorbents with targeted properties.
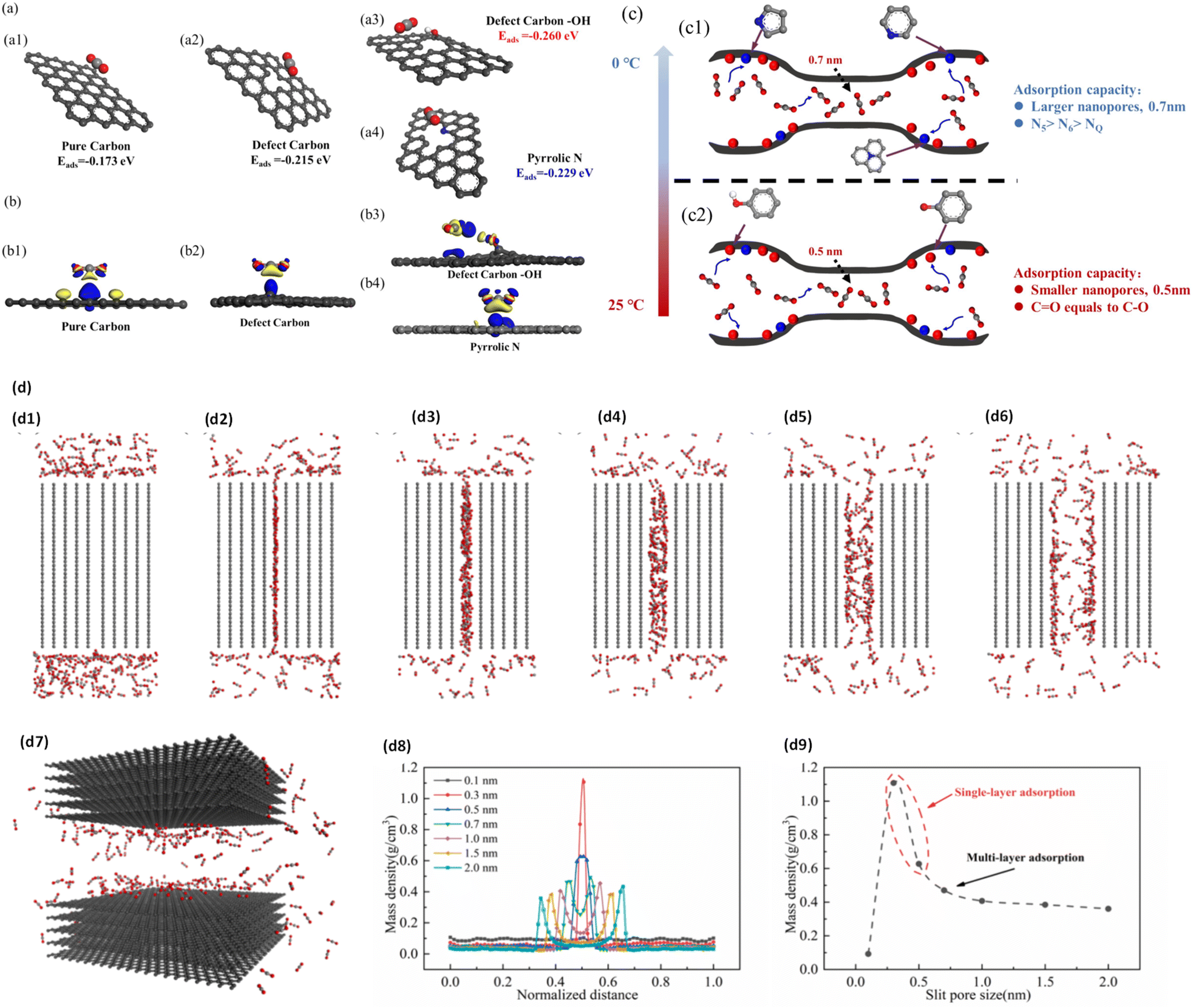 | ||
| Fig. 10 Adsorption energies of CO2 on (a1) pure carbon, (a2) defect carbon, (a3) defect carbon –OH, (a4) defect carbon + pyrrolic N; (b) differential charge density of CO2 capture on (b1) pure carbon, (b2) defect carbon, (b3) defect carbon –OH, (b4) defect carbon + pyrrolic N (c): adsorption of CO2 by adsorption sites at different temperatures: (d) diffusion of CO2 in slit pore models with different effective pore sizes (d1) 0.1 nm, (d2) 0.3 nm, (d3) 0.5 nm, (d4) 0.7 nm, (d5) 1.0 nm, (d6) 1.5 nm, (d7) 2.0 nm, (d8) mass density distribution of CO2 molecules (d9) the relationship between the mass density distribution and the effective pore size.321 | ||
A slit pore model was constructed with pore sizes ranging from 0.1 to 2.0 nm, and the equilibrium results of the system are shown in Fig. 10(d1–d9). At a pore size of Deff = 0.1 nm, CO2 diffusion into the pores was restricted, causing molecules to cluster around the pore openings. As pore size increased, CO2 molecules showed a stronger tendency to adsorb on the carbon wall's surface, transitioning from monolayer adsorption to bilayer adsorption. At 0.5 nm, the transition between monolayer and multilayer adsorption was observed (Fig. 10(d9)). Additionally, as pore size increased, the mass density of CO2 in the pores decreased (Fig. 10(d8)), which explains why ultramicro pores are the primary storage sites for CO2 under atmospheric conditions.
To evaluate the adsorption energy of various carbon structures, the study created models of intrinsic carbon, defective carbon, and structures incorporating hydroxyl and N-5 groups, as determined from earlier analyses. Simulation results revealed that CO2 molecules tend to align either parallel or perpendicular to carbon surfaces for adsorption, as shown in Fig. 10a. Differential charge calculations (Fig. 10b) indicated that modifying the carbon structure with heteroatoms, such as nitrogen and oxygen, enhances CO2 capture. Adsorption energy increased from322 (pure carbon) to −0.215 eV, −0.260 eV, and −0.229 eV for carbon structures with heteroatoms. Adding heteroatoms improved the carbon surface's electronegativity, increasing CO2 adsorption. Nitrogen-containing groups acted as Lewis bases, modifying the charge distribution to enhance adsorption, while oxygen-containing groups strengthened hydrogen bonding between the carbon surface and CO2 molecules. Hydroxyl and N-5 groups showed greater electron transfer than pure carbon, further improving CO2 capture efficiency, as shown in Fig. 10b. The doping of nitrogen (N) and oxygen (O) atoms into the carbon substrate modifies the charge distribution, increases the polarity of carbon atoms, and enhances the local electron density in regions rich in N, O, and C. This creates more effective adsorption sites for CO2. These findings provide valuable insights into optimizing biochar's pore structure and tailoring the carbon surface's chemical properties to enhance CO2 adsorption capacity. Under pressurized conditions, the CO2 adsorption capacity of biochar is positively correlated with its total pore volume. However, at atmospheric pressure, the optimal adsorption sites differ based on temperature. At 25 °C, ultramicro pores (0.5 nm) and oxygen-containing functional groups dominate adsorption, whereas, at 0 °C, ultramicro pores (0.7 nm) and N-5 functional groups play a key role. Simulation results further indicate that the transition from monolayer to bilayer adsorption occurs at a pore size of 0.5 nm. Both oxygen- and nitrogen-containing functional groups effectively enhance the CO2 adsorption capacity of biochar, making them critical for improving adsorption performance.
Yang et al. studied the adsorption properties of seaweed-based biochar for greenhouse gases (CO2, CH4, N2O) using DFT.323 Different models of seaweed-based biochar were constructed and the interactions with greenhouse gases were analyzed through structural parameters, adsorption energy, charge transfer, and surface electronic properties. The study found that biochar doped with nitrogen (N) and oxygen (O) heteroatoms exhibited improved adsorption performance. By calculating the lowest energy configurations for various adsorption sites (e.g., top site and side site) and molecular orientations of greenhouse gases, the most stable adsorption configuration of greenhouse gas molecules on biochar (BC) is presented in Fig. 11a. The results of multiple linear regression analysis are shown in Fig. 11b, where the red dots represent adsorption energy data obtained from the analysis above. In contrast, the black dots represent adsorption energy data from a different N-doped biochar system calculated separately to validate the accuracy of the regression model. These findings suggest that parameters such as Vs,max, lowest occupied molecular orbital (ELOMO), and ΔEgap can serve as reliable descriptors for the preliminary screening of biochar material models with high greenhouse gas adsorption energy.
 | ||
| Fig. 11 (a) Optimized structures of the most stable configurations for the (i) CO2-top, (ii) CH4-top, (iii) N2O-top, (iv) CO2-side, (v) CH4-side, and (vi) N2O-side adsorbed on the BC and (b) Scatter plot showing Eads (i) CO2, (ii) CH4, (iii) N2O.323 | ||
The results also showed that biochar was more sensitive to CO2 and N2O than CH4. Specifically, the adsorption energies for CO2 and N2O on N-doped biochar increased by 58.1% and 21.4%, respectively. Additionally, quantitative structure–activity relationships were developed, linking the adsorption energy of greenhouse gases to key electronic properties of the biochar surface. The electrostatic potential, the energy of the ELOMO, and the energy gap (ΔEgap) of α orbitals showed a strong linear correlation with the adsorption energy. These properties can be used as predictors for the preliminary screening of greenhouse gas adsorbents. This research provides molecular-level insights into the greenhouse gas adsorption mechanism and offers guidance for designing more effective materials for environmental remediation.
13. Concluding remarks
This review has presented significant advancements in the adsorption processes of various gaseous pollutants, highlighting biomass materials as attractive candidates for a green and sustainable ecosystem. The study has noted that while intensified research and awareness regarding the hazards of gaseous compounds are crucial, the substantial enhancement of global security and the quest for environmental sustainability should further inspire scientists and engineers to create innovative and unconventional sorbents derived from agricultural waste. Despite significant advancements, there are still challenges that must be addressed for the effective deployment of these materials. This would enable them to exhibit superior adsorption capabilities relative to conventional benchmark materials.The swift advancement of the social economy has increasingly brought environmental issues to light. The major focus of contemporary researchers involves the selection of clean and effective technologies for pollution removal in order to achieve a balance between development and environmental protection. The cost-effective and reusable properties of sorption materials derived from biomass should perfectly align with the sustainable development criteria. The conventional techniques employed in biochar production exhibit low efficiency, necessitating a substantial quantity of raw ingredients. When considering the application of biochar materials in engineering practices, it is important to consider the quality relationship between these materials and the raw materials. Therefore, continuous investigation into techniques for modifying and regenerating is still a research hotspot.
Currently, there have been few endeavours conducted on adsorbents for typical gaseous pollutants. For example, the current studies on CO2 adsorption primarily depend on simulated gas mixtures, including only a few key gas components (such as N2, CO2, and H2O) or even pure CO2. Further research is required to investigate the competitive adsorption of biomass adsorbent in industrial flue gas that contains contaminants. The adsorption of VOCs onto solid sorbents often decreases as the adsorption temperature increases. This is mostly due to the physical exothermic interaction. Significantly, elevated temperatures enhance molecular diffusion and chemisorption, resulting in intricate impacts on the adsorption of VOCs, particularly when metal oxide-modified biochar is involved. In addition, humidity can impact the adsorption of VOCs onto carbon materials because the H2O molecules can compete to occupy the pore sites. Nevertheless, there has been limited research on the impact of temperature and humidity on the sorption of VOCs onto biomass-based sorbents.
The use of advanced analytical techniques should be encouraged. Various characterization techniques have been utilized to quantify the pore textural characteristics and the presence of surface functional groups of the adsorbent before and after the adsorption process. For example, Abdulrasheed et al.292 and Shafeeyan et al.64 provided a comprehensive description of the common methods used, such as chemical titration, FTIR, X-ray photoelectron spectroscopy (XPS), temperature-programmed reduction (TPR), and temperature-programmed desorption (TPD). Typically, NH3-TPD may be utilized to assess the surface acidity of biochar, whereas H2-TPR measurement was employed to analyze the redox properties of metal oxides. Nevertheless, the assessment of biochar's functional groups was conducted using an offline method, which may undergo alterations throughout the cooling process of the used sorbent. Hence, it is imperative to develop in situ sophisticated techniques for quantitatively analyzing the adsorbents in order to accurately identify the sorption mechanism of the gaseous pollutants.
Current studies on multi-gaseous adsorption onto biomass adsorbent is quite limited. The previous studies did not attempt to simultaneously test more than three gaseous pollutants. Several suggestions may be considered to achieve an enhanced sorption of several gaseous components through synergistic adsorption. The oxygen-containing functional groups present in biomass materials are often acidic and positively impact the sorption of polar and hydrophilic VOCs. Moreover, these groups offer many active sites where NH3 may be adsorbed, resulting in better efficiency in removing NO. Meanwhile, the oxygenation anchoring sites serve as a useful intermediary stage for introducing nitrogen functional groups. These functional groups are typically more successful than the pore characteristics in adsorbing acidic CO2 and SO2, particularly at temperatures over 100 °C. Also, developing biochar that can efficiently remove several gas pollutants in a single-step procedure is currently challenging due to the unavoidable issue of competing adsorptions.
Furthermore, the majority of investigations on the adsorption of gaseous pollutants onto biomass sorbent have been conducted using a laboratory scale. However, it may be challenging to achieve the same level of adsorption capacity under complex industrial-relevant conditions as those observed in laboratory-scale platforms, primarily due to the unique particle properties of adsorbents and the necessity to consider reactor bed layouts. The potential configurations for integrating these sorbents with combustion flue gas streams include injection systems, fixed-bed, moving-bed, and fluidized-bed setups.324 The multiphase reactor enhances the mass transfer and diffusion of gases, which are the primary rate-controlling stages in gaseous adsorption, thereby resulting in an improved sorption efficiency.325
The effectiveness of using biomass adsorbent in large-scale industrial applications will determine the practicality and cost-effectiveness of employing biomass-based adsorption technology for removing gaseous contaminants. This study recommends that an ideal adsorbent for gaseous pollutants decontamination should meet the following industrial criteria (Fig. 12): (i) produced using cost-effective and energy-efficient methods such as ultrasonic, microwave, template, and plasma techniques. (ii) Exhibit a higher adsorption capacity and selectivity for N2, while also being able to tolerate other flue gas components such as SOx, NOx, H2O, and light hydrocarbons, as it is preferable for them to be able to simultaneously remove multiple gaseous pollutants. (iii) Ensure long-term adsorption–desorption operation cycles, such adsorbent needs to have fast adsorption and desorption kinetics, lower energy requirements for desorption, and good mechanical regeneration stability.
 | ||
| Fig. 12 Expected characteristics of an ideal sorbent for gaseous pollutant removal. | ||
The disposal approach for spent biomass-based sorbents needs to be revisited. The major focus of the industry is on regenerating and renewing the used adsorbents until their ability to remove gas pollutants becomes ineffective. This is mostly achieved by thermal regeneration at temperatures of about 200–300 °C, which can be facilitated by transferring heat with a flowing flue gas in industrial facilities. The desorbed gas pollutants, such as NOx and SO2, can be efficiently collected and used as chemical feedstock. Before returning the waste biomass to the boiler, it is essential to note that it has a higher heating value and energy density compared to its original biomass form, making it a more efficient fuel. Due to its higher concentration, CO2 is in high demand for disposal compared to other types of gases. Biochar, which has the special ability to adsorb CO2, could be buried as a form of carbon sequestration before it loses its effective adsorption capability. In this scenario, biomass functions as a soil supplement that sequesters carbon.
Data availability
All data and materials used in this study are available within this article.Author contributions
Kayode Adesina Adegoke: conceptualization, methodology, validation, visualization, writing – original draft, and writing – review, and editing and supervision. Omolabake Abiodun Okon-Akan: validation, visualization, writing – original draft, and writing – review and editing. Tosin Adewumi Adebusuyi: validation, visualization, writing-original draft, and writing – review and editing. Oluwatobi Idowu Adewuyi: validation, visualization, writing – original draft, and writing – review and editing. Peter Oluwatosin Adu: validation, visualization, writing – original draft, and writing – review, and editing. Abayomi Bamisaye: validation, visualization, writing – original draft, and writing – review, and editing. Oyeladun Rhoda Adegoke: validation, visualization, writing – original draft, and writing – review and editing. Cecilia Opeyemi Babarinde: validation, visualization, writing – original draft, and writing – review and editing. Olugbenga Solomon Bello: writing – review, and editing and supervision.Conflicts of interest
No conflicts of interest.Acknowledgements
Authors acknowledge their respective universities for the platform given to carry out this work.References
- K. A. Adegoke and N. W. Maxakato, J. CO2 Util., 2023, 69, 102412 CrossRef CAS.
- IEA, Key World Energy Statistics 2009, OECD Publishing, 2009, DOI:10.1787/9789264039537-en.
- BP Energy, BP Energy Outlook 2019 Edition, 2019 Search PubMed.
- S. Solomon, G. K. Plattner, R. Knutti and P. Friedlingstein, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 1704–1709 CrossRef CAS PubMed.
- D. L. Hartmann, A. M. G. Klein Tank, M. Rusticucci, L. V. Alexander, S. Brönnimann, Y. A. R. Charabi, F. J. Dentener, E. J. Dlugokencky, D. R. Easterling, A. Kaplan, B. J. Soden, P. W. Thorne, M. Wild and P. Zhai, in Climate Change 2013 the Physical Science Basis: Working Group I Contribution to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, 2013, vol. 9781107057, pp. 159–254 Search PubMed.
- R. Lindsey, Climate Change: Atmospheric Carbon Dioxide, https://www.climate.gov/news-features/understanding-climate/climate-change-atmospheric-carbon-dioxide, accessed 20 February 2025.
- K. A. Adegoke and N. W. Maxakato, Coord. Chem. Rev., 2022, 457, 214389 CrossRef CAS.
- K. A. Adegoke and N. W. Maxakato, Mater. Today Chem., 2022, 24, 100838 CrossRef CAS.
- A. Palliyarayil, H. Saini, K. Vinayakumar, P. Selvarajan, A. Vinu, N. S. Kumar and S. Sil, Emerg. Mater., 2021, 4, 607–643 CrossRef CAS.
- G. McGranahan and F. Murray, Air Pollution and Health in Rapidly Developing Countries, Routledge, 2012 Search PubMed.
- L. Filleul, A. Zeghnoun, C. Declercq, C. Le Goaster, A. Le Tertre, D. Eilstein, S. Medina, P. Saviuc, H. Prouvost and S. Cassadou, Rev. Mal. Respir., 2001, 18, 387–395 CAS.
- F. Dominici, R. D. Peng, M. L. Bell, L. Pham, A. McDermott, S. L. Zeger and J. M. Samet, JAMA, 2006, 295, 1127–1134 CrossRef CAS PubMed.
- R. D. Peng, H. H. Chang, M. L. Bell, A. McDermott, S. L. Zeger, J. M. Samet and F. Dominici, JAMA, 2008, 299, 2172–2179 CrossRef CAS PubMed.
- K. Bhaskaran, S. Hajat, A. Haines, E. Herrett, P. Wilkinson and L. Smeeth, Heart, 2009, 95, 1746–1759 CrossRef CAS PubMed.
- H. Mustafić, P. Jabre, C. Caussin, M. H. Murad, S. Escolano, M. Tafflet, M.-C. Périer, E. Marijon, D. Vernerey and J.-P. Empana, Jama, 2012, 307, 713–721 CrossRef PubMed.
- World Health Organization, Ambient air pollution: A global assessment of exposure and burden of disease, 2016 Search PubMed.
- S. K. Guttikunda, R. Goel and P. Pant, Atmos. Environ., 2014, 95, 501–510 CrossRef CAS.
- K. Sheoran, S. S. Siwal, D. Kapoor, N. Singh, A. K. Saini, W. F. Alsanie and V. K. Thakur, ACS Eng. Au., 2022, 2, 378–396 CrossRef CAS PubMed.
- T. Pettit, P. J. Irga and F. R. Torpy, J. Hazard. Mater., 2018, 360, 594–603 CrossRef CAS PubMed.
- K. Wróblewska and B. R. Jeong, Environ. Sci. Eur., 2021, 33, 110 CrossRef PubMed.
- İ. A. Reşitoğlu, K. Altinişik and A. Keskin, Clean Technol. Environ. Policy, 2015, 17, 15–27 CrossRef.
- G. Crini, E. Lichtfouse, L. D. Wilson and N. Morin-Crini, Environ. Chem. Lett., 2019, 17, 195–213 CrossRef CAS.
- L. Pellenz, C. R. S. de Oliveira, A. H. da Silva Júnior, L. J. S. da Silva, L. da Silva, A. A. Ulson de Souza, S. M. de, A. G. U. de Souza, F. H. Borba and A. da Silva, Sep. Purif. Technol., 2023, 305, 122435 CrossRef CAS.
- K. A. Adegoke, O. O. Adesina, O. A. Okon-Akan, O. R. Adegoke, A. B. Olabintan, O. A. Ajala, H. Olagoke, N. W. Maxakato and O. S. Bello, Curr. Res. Green Sustain. Chem., 2022, 5, 100274 CrossRef CAS.
- K. A. Adegoke and O. S. Bello, Water Resour. Ind., 2015, 8–24, DOI:10.1016/j.wri.2015.09.002.
- M. Hemalatha, G. S. Lakshmi, V. Megha and B. C. Kotibagar, Applications of Nanomaterials for Water Treatment: Current Trends and Future Scope, in Modern Nanotechnology, ed. J. A. Malik and M. J. Sadiq Mohamed, Springer, Cham, DOI:10.1007/978-3-031-31111-6_7.
- V. F. Gold, A. Bamisaye, M. O. Adesina, K. A. Adegoke, A. R. Ige, O. Adeleke, M. O. Bamidele, Y. A. Alli, A. K. Oyebamiji and O. O. Ogunlaja, J. Dispers. Sci. Technol., 2024, 1–17 CrossRef.
- A. A. Adaramaja, A. Bamisaye, S. M. Abati, K. A. Adegoke, M. O. Adesina, A. R. Ige, O. Adeleke, M. A. Idowu, A. K. Oyebamiji and O. S. Bello, RSC Adv., 2024, 14, 12703–12719 RSC.
- Y. Dai, Q. Sun, W. Wang, L. Lu, M. Liu, J. Li, S. Yang, Y. Sun, K. Zhang, J. Xu, W. Zheng, Z. Hu, Y. Yang, Y. Gao, Y. Chen, X. Zhang, F. Gao and Y. Zhang, Chemosphere, 2018, 211, 235–253 CrossRef CAS PubMed.
- Z. Ajmal, M. ul Haq, Y. Naciri, R. Djellabi, N. Hassan, S. Zaman, A. Murtaza, A. Kumar, A. G. Al-Sehemi, H. Algarni, O. A. Al-Hartomy, R. Dong, A. Hayat and A. Qadeer, Chemosphere, 2022, 308, 136358 CrossRef CAS PubMed.
- A. Othmani, A. Kadier, R. Singh, C. A. Igwegbe, M. Bouzid, M. O. Aquatar, W. A. Khanday, M. E. Bote, F. Damiri, Ö. Gökkuş and F. Sher, Environ. Res., 2022, 215, 114294 CrossRef CAS PubMed.
- S. F. Ahmed, M. Mofijur, S. Nuzhat, A. T. Chowdhury, N. Rafa, M. A. Uddin, A. Inayat, T. M. I. Mahlia, H. C. Ong, W. Y. Chia and P. L. Show, J. Hazard. Mater., 2021, 416, 125912 CrossRef CAS PubMed.
- Z. Bano, N. Z. Ali, M. A. Khan, S. Mutahir, S. Zhu, F. Wang and M. Xia, Ceram. Int., 2022, 48, 8409–8416 CrossRef CAS.
- X. Wang, M. A. Khan, M. Xia, S. Zhu, W. Lei and F. Wang, J. Mater. Sci. Mater. Electron., 2019, 30, 5503–5515 CrossRef CAS.
- M. M. Sabzehmeidani, S. Mahnaee, M. Ghaedi, H. Heidari and V. A. L. Roy, Adv. Mater., 2021, 2, 598–627 RSC.
- J. Zhou, X. Wang and W. Xing, in Post-Combustion Carbon Dioxide Capture Materials, ed. Q. Wang, The Royal Society of Chemistry, 2018, p. 0 Search PubMed.
- J. Jjagwe, P. W. Olupot, E. Menya and H. M. Kalibbala, J. Bioresour. Bioprod., 2021, 6, 292–322 CAS.
- R. Ahmed, G. Liu, B. Yousaf, Q. Abbas, H. Ullah and M. U. Ali, J. Clean. Prod., 2020, 242, 118409 CrossRef CAS.
- Z. Zhang, Z. P. Cano, D. Luo, H. Dou, A. Yu and Z. Chen, J. Mater. Chem. A, 2019, 7, 20985–21003 RSC.
- L. Chen, S. Deng, R. Zhao, L. Zhao, S. Li, Z. Guo, Y. Lu, J. Zhao and K. Wu, Energy Technol., 2021, 9, 2000756 CrossRef CAS.
- K. A. Adegoke, K. G. Akpomie, E. S. Okeke, C. Olisah, A. Malloum, N. W. Maxakato, J. O. Ighalo, J. Conradie, C. R. Ohoro, J. F. Amaku and K. O. Oyedotun, Sep. Purif. Technol., 2024, 331, 125456 CrossRef CAS.
- F. O. Ochedi, Y. Liu and Y. G. Adewuyi, Process Saf. Environ. Prot., 2020, 139, 1–25 CrossRef CAS.
- V. Acevedo-García, E. Rosales, A. Puga, M. Pazos and M. A. Sanromán, Sep. Purif. Technol., 2020, 242, 116796 CrossRef.
- H. Mumtaz, M. Farhan, M. Amjad, F. Riaz, A. H. Kazim, M. Sultan, M. Farooq, M. A. Mujtaba, I. Hussain, M. Imran, S. Anwar, A. M. El-Sherbeeny, F. A. Siddique, S. Armaković, Q. Ali, I. A. Chaudhry and A. Pettinau, Sustain. Energy Technol. Assessments, 2021, 46, 101288 CrossRef.
- D. Mallesh, J. Anbarasan, P. Mahesh Kumar, K. Upendar, P. Chandrashekar, B. V. S. K. Rao and N. Lingaiah, Appl. Surf. Sci., 2020, 530, 147226 CrossRef CAS.
- B. Aghel, S. Behaein and F. Alobaid, Fuel, 2022, 328, 125276 CrossRef CAS.
- M. Karimi, M. Shirzad, J. A. C. Silva and A. E. Rodrigues, J. CO2 Util., 2022, 57, 101890 CrossRef CAS.
- M. Olivares-Marín and M. M. Maroto-Valer, Greenh. Gases: Sci. Technol., 2012, 2, 20–35 CrossRef.
- O. Ioannidou and A. Zabaniotou, Renew. Sustain. Energy Rev., 2007, 11, 1966–2005 CrossRef CAS.
- M. G. Plaza, S. García, F. Rubiera, J. J. Pis and C. Pevida, Sep. Purif. Technol., 2011, 80, 96–104 CrossRef CAS.
- M. G. Plaza, C. Pevida, C. F. Martín, J. Fermoso, J. J. Pis and F. Rubiera, Sep. Purif. Technol., 2010, 71, 102–106 CrossRef CAS.
- M. G. Plaza, C. Pevida, B. Arias, J. Fermoso, M. D. Casal, C. F. Martín, F. Rubiera and J. J. Pis, Fuel, 2009, 88, 2442–2447 CrossRef CAS.
- A. Boonpoke, S. Chiarakorn, N. Laosiripojana, S. Towprayoon and A. Chidthaisong, J. Environ. Sustain., 2013, 2, 77–81 Search PubMed.
- S.-J. Son, J.-S. Choi, K.-Y. Choo, S.-D. Song, S. Vijayalakshmi and T.-H. Kim, Korean J. Chem. Eng., 2005, 22, 291–297 CrossRef CAS.
- J. A. Thote, K. S. Iyer, R. Chatti, N. K. Labhsetwar, R. B. Biniwale and S. S. Rayalu, Carbon, 2010, 48, 396–402 CrossRef CAS.
- H. T. Jang, Y. Park, Y. S. Ko, J. Y. Lee and B. Margandan, Int. J. Greenh. Gas Control, 2009, 3, 545–549 CrossRef CAS.
- S. A. Miller, P. R. Cunningham and J. T. Harvey, Resour. Conserv. Recycl., 2019, 146, 416–430 CrossRef.
- H. Marsh and F. Rodríguez-Reinoso, Activated Carbon, 2006 Search PubMed.
- P. González-García, Renew. Sustain. Energy Rev., 2018, 82, 1393–1414 CrossRef.
- X. Tan, S. Liu, Y. Liu, Y. Gu, G. Zeng, X. Hu, X. Wang, S. Liu and L. Jiang, Bioresour. Technol., 2017, 227, 359–372 CrossRef CAS PubMed.
- M. A. Yahya, Z. Al-Qodah and C. W. Z. Ngah, Renew. Sustain. Energy Rev., 2015, 46, 218–235 CrossRef CAS.
- J. L. Figueiredo and M. F. R. Pereira, Catal. Today, 2010, 150, 2–7 CrossRef CAS.
- J. Rivera-Utrilla, M. Sánchez-Polo, V. Gómez-Serrano, P. M. Álvarez, M. C. M. Alvim-Ferraz and J. M. Dias, J. Hazard. Mater., 2011, 187, 1–23 CrossRef CAS PubMed.
- M. S. Shafeeyan, W. M. A. W. Daud, A. Houshmand and A. Shamiri, J. Anal. Appl. Pyrolysis, 2010, 89, 143–151 CrossRef CAS.
- M. Jeguirim, M. Belhachemi, L. Limousy and S. Bennici, Chem. Eng. J., 2018, 347, 493–504 CrossRef CAS.
- M. I. S. Haider, G. Liu, B. Yousaf, M. Arif, K. Aziz, A. Ashraf, R. Safeer, S. Ijaz and K. Pikon, Environ. Pollut., 2024, 124365 CrossRef CAS PubMed.
- J. L. Figueiredo, J. Mater. Chem. A, 2013, 1, 9351–9364 RSC.
- P. V Samant, F. Gonçalves, M. M. A. Freitas, M. F. R. Pereira and J. L. Figueiredo, Carbon, 2004, 42, 1321–1325 CrossRef.
- F. Kapteijn, J. A. Moulijn, S. Matzner and H.-P. Boehm, Carbon, 1999, 37, 1143–1150 CrossRef CAS.
- C. L. Mangun, K. R. Benak, J. Economy and K. L. Foster, Carbon, 2001, 39, 1809–1820 CrossRef CAS.
- R. Pietrzak, Fuel, 2009, 88, 1871–1877 CrossRef CAS.
- J. Wang, L. Huang, R. Yang, Z. Zhang, J. Wu, Y. Gao, Q. Wang, D. O'Hare and Z. Zhong, Energy Environ. Sci., 2014, 7, 3478–3518 RSC.
- R. Balasubramanian and S. Chowdhury, J. Mater. Chem. A, 2015, 3, 21968–21989 RSC.
- X. Ma, L. Li, R. Chen, C. Wang, K. Zhou and H. Li, Fuel, 2019, 236, 942–948 CrossRef CAS.
- A. Ö. Yazaydın, A. I. Benin, S. A. Faheem, P. Jakubczak, J. J. Low, R. R. Willis and R. Q. Snurr, Chem. Mater., 2009, 21, 1425–1430 CrossRef.
- Y. Xia, R. Mokaya, G. S. Walker and Y. Zhu, Adv. Energy Mater., 2011, 1, 678–683 CrossRef CAS.
- X. Ma, L. Li, R. Chen, C. Wang, H. Li and S. Wang, Appl. Surf. Sci., 2018, 435, 494–502 CrossRef CAS.
- K. Kim, D. Kim, Y.-K. Park and K. S. Lee, Int. J. Greenh. Gas Control, 2014, 26, 135–146 CrossRef CAS.
- Z. Yang, Y. Xia and R. Mokaya, J. Am. Chem. Soc., 2007, 129, 1673–1679 CrossRef CAS PubMed.
- A. Aijaz, N. Fujiwara and Q. Xu, J. Am. Chem. Soc., 2014, 136, 6790–6793 CrossRef CAS PubMed.
- G. K. Parshetti, S. Chowdhury and R. Balasubramanian, Fuel, 2015, 148, 246–254 CrossRef CAS.
- X. Ma, L. Li, Z. Zeng, R. Chen, C. Wang, K. Zhou and H. Li, Appl. Surf. Sci., 2019, 481, 1139–1147 CrossRef CAS.
- Q. Ye, J. Jiang, C. Wang, Y. Liu, H. Pan and Y. Shi, Energy Fuels, 2012, 26, 2497–2504 CrossRef CAS.
- M. M. Gui, Y. X. Yap, S.-P. Chai and A. R. Mohamed, Int. J. Greenh. Gas Control, 2013, 14, 65–73 CrossRef CAS.
- Z. Zhang, S. Xian, Q. Xia, H. Wang, Z. Li and J. Li, AIChE J., 2013, 59, 2195–2206 CrossRef CAS.
- B. Arstad, H. Fjellvåg, K. O. Kongshaug, O. Swang and R. Blom, Adsorption, 2008, 14, 755–762 CrossRef CAS.
- F. Salles, A. Ghoufi, G. Maurin, R. G. Bell, C. Mellot-Draznieks and G. Férey, Angew. Chem. Int. Ed., 2008, 47, 8487–8491 CrossRef CAS PubMed.
- Y. Lee, D. Liu, D. Seoung, Z. Liu, C.-C. Kao and T. Vogt, J. Am. Chem. Soc., 2011, 133, 1674–1677 CrossRef CAS PubMed.
- F. Su, C. Lu, S.-C. Kuo and W. Zeng, Energy Fuels, 2010, 24, 1441–1448 CrossRef CAS.
- C. Chen, S.-T. Yang, W.-S. Ahn and R. Ryoo, Chem. Commun., 2009, 3627–3629 RSC.
- M. Sevilla, J. B. Parra and A. B. Fuertes, ACS Appl. Mater. Interfaces, 2013, 5, 6360–6368 CrossRef CAS PubMed.
- R.-L. Liu, W.-J. Ji, T. He, Z.-Q. Zhang, J. Zhang and F.-Q. Dang, Carbon, 2014, 76, 84–95 CrossRef CAS.
- A. Hanif, M. A. Aziz, A. Helal, M. M. Abdelnaby, A. Khan, R. Theravalappil and M. Y. Khan, ACS Omega, 2023, 8, 36228–36236 CrossRef CAS PubMed.
- A. D. Igalavithana, S. W. Choi, J. Shang, A. Hanif, P. D. Dissanayake, D. C. W. Tsang, J.-H. Kwon, K. B. Lee and Y. S. Ok, Sci. Total Environ., 2020, 739, 139845 CrossRef CAS PubMed.
- P. D. Dissanayake, S. You, A. D. Igalavithana, Y. Xia, A. Bhatnagar, S. Gupta, H. W. Kua, S. Kim, J.-H. Kwon, D. C. W. Tsang and Y. S. Ok, Renew. Sustain. Energy Rev., 2020, 119, 109582 CrossRef CAS.
- S. Li, X. Yuan, S. Deng, L. Zhao and K. B. Lee, Renew. Sustain. Energy Rev., 2021, 152, 111708 CrossRef CAS.
- M. Bui, C. S. Adjiman, A. Bardow, E. J. Anthony, A. Boston, S. Brown, P. S. Fennell, S. Fuss, A. Galindo, L. A. Hackett, J. P. Hallett, H. J. Herzog, G. Jackson, J. Kemper, S. Krevor, G. C. Maitland, M. Matuszewski, I. S. Metcalfe, C. Petit, G. Puxty, J. Reimer, D. M. Reiner, E. S. Rubin, S. A. Scott, N. Shah, B. Smit, J. P. M. Trusler, P. Webley, J. Wilcox and N. Mac Dowell, Energy Environ. Sci., 2018, 11, 1062–1176 RSC.
- M. Fajardy and N. Mac Dowell, Energy Environ. Sci., 2018, 11, 1581–1594 RSC.
- H. Bamdad, K. Hawboldt and S. Macquarrie, Renew. Sustain. Energy Rev., 2017, 1–16 Search PubMed.
- O. A. A. A. Bamisaye, C. O. Eromosele and E. O. Dare, J. Chem. Soc. Niger., 2020, 45, 804–811 Search PubMed.
- X. Yuan, M. Suvarna, S. Low, P. D. Dissanayake, K. B. Lee, J. Li, X. Wang and Y. S. Ok, Environ. Sci. Technol., 2021, 55, 11925–11936 CrossRef CAS PubMed.
- A. I. Osman, M. Hefny, M. I. A. Abdel Maksoud, A. M. Elgarahy and D. W. Rooney, Environ. Chem. Lett., 2021, 19, 797–849 CrossRef CAS.
- M. Sevilla, P. Valle-Vigón and A. B. Fuertes, Adv. Funct. Mater., 2011, 21, 2781–2787 CrossRef CAS.
- J. Xiao, X. Yuan, T. C. Zhang, L. Ouyang and S. Yuan, Sep. Purif. Technol., 2022, 298, 121602 CrossRef CAS.
- Y. Li, L. Liu, H. Yu, Y. Zhao, J. Dai, Y. Zhong, Z. Pan and H. Yu, Sci. Total Environ., 2022, 811, 151384 CrossRef CAS PubMed.
- K. Wiedner, C. Rumpel, C. Steiner, A. Pozzi, R. Maas and B. Glaser, Biomass Bioenergy, 2013, 59, 264–278 CrossRef CAS.
- Z. Zhang, Z. Zhu, B. Shen and L. Liu, Energy, 2019, 171, 581–598 CrossRef CAS.
- M. Tripathi, J. N. Sahu and P. Ganesan, Renew. Sustain. Energy Rev., 2016, 55, 467–481 CrossRef CAS.
- E. Antunes, M. V Jacob, G. Brodie and P. A. Schneider, J. Anal. Appl. Pyrolysis, 2018, 129, 93–100 CrossRef CAS.
- T. Kan, V. Strezov and T. J. Evans, Renew. Sustain. Energy Rev., 2016, 57, 1126–1140 CrossRef CAS.
- A. Zubrik, M. Matik, S. Hredzák, M. Lovás, Z. Danková, M. Kováčová and J. Briančin, J. Clean. Prod., 2017, 143, 643–653 CrossRef CAS.
- H. S. Kambo and A. Dutta, Renew. Sustain. Energy Rev., 2015, 45, 359–378 CrossRef CAS.
- M. Rana, K. Subramani, M. Sathish and U. K. Gautam, Carbon, 2017, 114, 679–689 CrossRef CAS.
- Y. Chen, X. Zhang, W. Chen, H. Yang and H. Chen, Bioresour. Technol., 2017, 246, 101–109 CrossRef CAS PubMed.
- O. Boujibar, A. Souikny, F. Ghamouss, O. Achak, M. Dahbi and T. Chafik, J. Environ. Chem. Eng., 2018, 6, 1995–2002 CrossRef CAS.
- M. Zhu, W. Cai, F. Verpoort and J. Zhou, Chem. Eng. Res. Des., 2019, 146, 130–140 CrossRef CAS.
- S. Li, K. Han, J. Li, M. Li and C. Lu, Microporous Mesoporous Mater., 2017, 243, 291–300 CrossRef CAS.
- H. M. Coromina, D. A. Walsh and R. Mokaya, J. Mater. Chem. A, 2016, 4, 280–289 RSC.
- M. B. Ahmed, M. A. Hasan Johir, J. L. Zhou, H. H. Ngo, L. D. Nghiem, C. Richardson, M. A. Moni and M. R. Bryant, J. Clean. Prod., 2019, 225, 405–413 CrossRef CAS.
- J. Serafin, M. Baca, M. Biegun, E. Mijowska, R. J. Kaleńczuk, J. Sreńscek-Nazzal and B. Michalkiewicz, Appl. Surf. Sci., 2019, 497, 143722 CrossRef CAS.
- R. A. Fiuza-Jr, R. C. Andrade and H. M. C. Andrade, J. Environ. Chem. Eng., 2016, 4, 4229–4236 CrossRef CAS.
- V. Benedetti, E. Cordioli, F. Patuzzi and M. Baratieri, J. CO2 Util., 2019, 33, 46–54 CrossRef CAS.
- L. Yue, L. Rao, L. Wang, L. Wang, J. Wu, X. Hu, H. DaCosta, J. Yang and M. Fan, Ind. Eng. Chem. Res., 2017, 56, 14115–14122 CrossRef CAS.
- J. Serafin, U. Narkiewicz, A. W. Morawski, R. J. Wróbel and B. Michalkiewicz, J. CO2 Util., 2017, 18, 73–79 CrossRef CAS.
- S. Liu, R. Ma, X. Hu, L. Wang, X. Wang, M. Radosz and M. Fan, Ind. Eng. Chem. Res., 2020, 59, 7046–7053 CrossRef CAS.
- H. Wei, J. Chen, N. Fu, H. Chen, H. Lin and S. Han, Electrochim. Acta, 2018, 266, 161–169 CrossRef CAS.
- Z. Geng, Q. Xiao, H. Lv, B. Li, H. Wu, Y. Lu and C. Zhang, Sci. Rep., 2016, 6, 30049 CrossRef CAS PubMed.
- D. Saha, S. E. Van Bramer, G. Orkoulas, H.-C. Ho, J. Chen and D. K. Henley, Carbon, 2017, 121, 257–266 CrossRef CAS.
- H. Wei, H. Chen, N. Fu, J. Chen, G. Lan, W. Qian, Y. Liu, H. Lin and S. Han, Electrochim. Acta, 2017, 231, 403–411 CrossRef CAS.
- B. Chang, W. Shi, H. Yin, S. Zhang and B. Yang, Chem. Eng. J., 2019, 358, 1507–1518 CrossRef CAS.
- Y.-J. Heo and S.-J. Park, Green Chem., 2018, 20, 5224–5234 RSC.
- C. Zhang, W. Song, Q. Ma, L. Xie, X. Zhang and H. Guo, Energy Fuels, 2016, 30, 4181–4190 CrossRef CAS.
- Z. Rouzitalab, D. Mohammady Maklavany, A. Rashidi and S. Jafarinejad, J. Environ. Chem. Eng., 2018, 6, 6653–6663 CrossRef CAS.
- P. Lahijani, M. Mohammadi and A. R. Mohamed, J. CO2 Util., 2018, 26, 281–293 CrossRef CAS.
- X. Kan, X. Chen, W. Chen, J. Mi, J.-Y. Zhang, F. Liu, A. Zheng, K. Huang, L. Shen, C. Au and L. Jiang, ACS Sustain. Chem. Eng., 2019, 7, 7609–7618 CrossRef CAS.
- S. Deng, H. Wei, T. Chen, B. Wang, J. Huang and G. Yu, Chem. Eng. J., 2014, 253, 46–54 CrossRef CAS.
- X. Ma, Y. Yang, Q. Wu, B. Liu, D. Li, R. Chen, C. Wang, H. Li, Z. Zeng and L. Li, Fuel, 2020, 282, 118727 CrossRef CAS.
- M. Sevilla and A. B. Fuertes, Energy Environ. Sci., 2011, 4, 1765–1771 RSC.
- E. Haffner-Staton, N. Balahmar and R. Mokaya, J. Mater. Chem. A, 2016, 4, 13324–13335 RSC.
- G. Singh, K. S. Lakhi, S. Sil, S. V. Bhosale, I. Y. Kim, K. Albahily and A. Vinu, Carbon, 2019, 148, 164–186 CrossRef CAS.
- O. A. Okon-Akan, O. Abiodun, O. Oluwaseun, O. K. Oladayo, O. Abayomi, A. A. George, E. Opatola, R. F. Orah, E. J. Isukuru, I. C. Ede, O. T. Oluwayomi, J. A. Okolie and I. A. Omotayo, Clean Technol., 2023, 5, 934–961 CrossRef.
- F. Sher, S. Z. Iqbal, S. Albazzaz, U. Ali, D. A. Mortari and T. Rashid, Fuel, 2020, 282, 118506 CrossRef CAS.
- M. M. Bade, A. A. Dubale, D. F. Bebizuh and M. Atlabachew, ACS Omega, 2022, 7, 18770–18779 CrossRef CAS PubMed.
- F. Valdebenito, R. García, K. Cruces, G. Ciudad, G. Chinga-Carrasco and Y. Habibi, ACS Sustain. Chem. Eng., 2018, 6, 12603–12612 CrossRef CAS.
- M. S. Khosrowshahi, M. A. Abdol, H. Mashhadimoslem, E. Khakpour, H. B. M. Emrooz, S. Sadeghzadeh and A. Ghaemi, Sci. Rep., 2022, 12, 8917 CrossRef CAS PubMed.
- H. N. Nguyen, D. A. Khuong and T. Tsubota, Therm. Sci. Eng. Prog., 2024, 49, 102446 CrossRef CAS.
- A. E. Creamer, B. Gao and M. Zhang, Chem. Eng. J., 2014, 249, 174–179 CrossRef CAS.
- J. M. Martín-Martínez, R. Torregrosa-Maciá and M. C. Mittelmeijer-Hazeleger, Fuel, 1995, 74, 111–114 CrossRef.
- K. A. Adegoke, S. O. Akinnawo, T. A. Adebusuyi, O. A. Ajala, R. O. Adegoke, N. W. Maxakato and O. S. Bello, Int. J. Sci. Environ. Technol., 2023, 20, 11615–11644 CrossRef CAS.
- N. A. Rashidi and S. Yusup, J. CO2 Util., 2016, 13, 1–16 CrossRef CAS.
- W.-J. Liu, H. Jiang, K. Tian, Y.-W. Ding and H.-Q. Yu, Environ. Sci. Technol., 2013, 47, 9397–9403 CrossRef CAS PubMed.
- A. E. Creamer, B. Gao, A. Zimmerman and W. Harris, Chem. Eng. J., 2018, 334, 81–88 CrossRef CAS.
- J. Gopalan, A. Buthiyappan and A. A. Abdul Raman, J. Ind. Eng. Chem., 2022, 113, 72–95 CrossRef CAS.
- Q. Li, T. Lu, L. Wang, R. Pang, J. Shao, L. Liu and X. Hu, Sep. Purif. Technol., 2021, 275, 119204 CrossRef CAS.
- S. K. Sooriyan and N. Ibrahim, Biointerface Res. Appl. Chem., 2022, 12, 7972–7982 Search PubMed.
- S. Sumathi, S. Bhatia, K. T. Lee and A. R. Mohamed, Chem. Eng. J., 2010, 1093–1096 CAS.
- V. Viena, et al., IOP Conf. Ser. Mater. Sci. Eng., 2018, 012037, DOI:10.1088/1757-899X/334/1/012037.
- M. M. Meimand and N. Javid, J. Health Scope, 2019, 8, DOI:10.5812/jhealthscope.69158.
- J. Shao, J. Zhang, X. Zhang, Y. Feng, H. Zhang and S. Zhang, Fuel, 2018, 224, 138–146 CrossRef CAS.
- B. Chen and H. Kan, Environ. Health Prev. Med., 2008, 13, 94–101 CrossRef CAS PubMed.
- F. P. Perera, Environ. Health Perspect., 2017, 125, 141–148 CrossRef CAS PubMed.
- K. S. Rani, J. Chem. Pharm. Sci., 2014, 2, 63–67 Search PubMed.
- R. Davarnejad and P. Panahi, Sep. Purif. Technol., 2016, 158, 286–292 CrossRef CAS.
- M. B. Yue, B. Sun, Y. Cao, Y. Wang and J. Wang, Chem.–Eur. J., 2008, 3442–3451 CrossRef CAS PubMed.
- D. N. K. Putra Negara, T. G. Tirta Nindhia, I. W. Surata and M. Sucipta, IOP Conf. Ser.:Mater. Sci. Eng., 2017, 201, 012033 CrossRef.
- B.-K. Na, K.-K. Koo, H.-M. Eum, H. Lee and H. K. Song, Korean J. Chem. Eng., 2001, 18, 220–227 CrossRef CAS.
- S. Hao, J. Zhang, Y. Zhong and W. Zhu, Adsorption, 2012, 18, 423–430 CrossRef CAS.
- S. Sethupathi, M. J. K. Bashir, Z. A. Akbar and A. R. Mohamed, Waste Manag. Res., 2015, 303–312, DOI:10.1177/0734242X15576026.
- S. Lu, Y. Ji, A. Buekens, Z. Ma, Y. Jin, X. Li and J. Yan, Waste Manag. Res., 2012, 31, 169–177 CrossRef PubMed.
- W. M. A. W. Daud and W. S. W. Ali, Bioresour. Technol., 2004, 93, 63–69 CrossRef CAS PubMed.
- J. Guo and A. C. Lua, J. Colloid Interface Sci., 2002, 251, 242–247 CrossRef CAS PubMed.
- N. S. A. Shukor, Int. J. Technol., 2018, 1121–1131 CrossRef.
- J. Guo, L. Fan, J. Peng, J. Chen, H. Yin and W. Jiang, J. Chem. Technol. Biotechnol., 2013, 1565–1575, DOI:10.1002/jctb.4240.
- J. Guo and A. C. Lua, Sep. Purif. Technol., 2003, 30, 265–273 CrossRef CAS.
- A. C. Lua and T. Yang, Chem. Eng. J., 2009, 155, 175–183 CrossRef CAS.
- S. M. Abegunde, K. S. Idowu, O. M. Adejuwon and T. Adeyemi-Adejolu, Resour. Environ. Sustain., 2020, 1, 100001 Search PubMed.
- R. R. Manory, Mater. Manuf. Process., 1990, 5, 445–470 CrossRef CAS.
- V. K. Thakur, D. Vennerberg and M. R. Kessler, ACS Appl. Mater. Interfaces, 2014, 6, 9349–9356 CrossRef CAS PubMed.
- M. G. A. Vieira, A. F. A. Neto, M. L. Gimenes and M. G. C. da Silva, J. Hazard. Mater., 2010, 176, 109–118 CrossRef CAS PubMed.
- M. Liu and C. Xiao, E3S Web Conf., 2018, 38, 2–5 Search PubMed.
- M. Kong, L. Huang, L. Li, Z. Zhang, S. Zheng and M. K. Wang, Appl. Clay Sci., 2014, 99, 207–214 CrossRef CAS.
- A. Rehman, M. Park and S. Park, Coatings, 2019, 9, 1–22 CrossRef.
- J. M. Rosas, R. Ruiz-rosas, J. Rodríguez-mirasol and T. Cordero, Chem. Eng. J., 2017, 707–721, DOI:10.1016/j.cej.2016.08.111.
- Y. W. Lee, H. J. Kim, J. W. Park, B. U. Choi, D. K. Choi and J. W. Park, Carbon, 2003, 41, 1881–1888 CrossRef CAS.
- M. C. Macías-Pérez, Fuel, 2007, 86, 677–683 CrossRef.
- J. A. Arcibar-Orozco and J. R. Rangel-Mendez, Chem. Eng. J., 2013, 230, 439–446 CrossRef CAS.
- Y. Wu, Y. Fan, M. Zhang, Z. Ming, S. Yang, A. Arkin and P. Fang, Biochem. Eng. J., 2016, 105, 27–35 CrossRef CAS.
- J. Przepiórski, A. Czyżewski, M. Toyoda, T. Tsumura, R. Pietrzak and A. W. Morawski, Int. J. Greenh. Gas Control, 2012, 10, 164–168 CrossRef.
- S. Sumathi, S. Bhatia, K. T. Lee and A. R. Mohamed, Chem. Eng. J., 2010, 162, 194–200 CrossRef CAS.
- I. Dahlan, K. Teong, A. Harun and A. Rahman, J. Hazard. Mater., 2011, 185, 1609–1613 CrossRef CAS PubMed.
- H. Yi, Y. Zuo and H. Liu, Water, Air, Soil Pollut., 2014, 1–7, DOI:10.1007/s11270-014-1965-2.
- J. Zhang, J. Shao, D. Huang, Y. Feng, X. Zhang, S. Zhang and H. Chen, Chem. Eng. J., 2020, 385, 123932 CrossRef CAS.
- L. Wang, L. Sha, S. Zhang, F. Cao, X. Ren and Y. A. Levendis, Fuel Process. Technol., 2022, 107233, DOI:10.1016/j.fuproc.2022.107233.
- Q. Wang, L. Han, Y. Wang, Z. He, Q. Meng, S. Wang, P. Xiao and X. Jia, RSC Adv., 2022, 12, 20640–20648 RSC.
- X. Zhang, S. Zhang, J. Zhang, G. Li, H. Zheng, J. Shao, S. Zhang, H. Yang and H. Chen, Fuel, 2023, 354, 129266 CrossRef CAS.
- X. Zhang, H. Zheng, G. Li, J. Gu, J. Shao, S. Zhang, H. Yang and H. Chen, J. Anal. Appl. Pyrolysis, 2021, 105119, DOI:10.1016/j.jaap.2021.105119.
- R. J. Lewis, G. B. Copley, R. J. Lewis and G. B. Copley, Crit. Rev. Toxicol., 2014, 93–123, DOI:10.3109/10408444.2014.971943.
- A. Peluso, N. Gargiulo, P. Aprea, F. Pepe and D. Caputo, Sep. Purif. Rev., 2019, 48, 78–89 CrossRef CAS.
- M. C. Castrillon, K. O. Moura, C. A. Alves, M. Bastos-neto, C. S. Azevedo, J. Hofmann, J. Möllmer, W. Einicke and R. Glaser, Energy Fuel., 2016, 9596–9604, DOI:10.1021/acs.energyfuels.6b01667.
- D. Long, Q. Chen, W. Qiao, L. Zhan, X. Liang and L. Ling, Chem. Commun., 2009, 3898–3900 RSC.
- J. Mi, F. Liu, W. Chen, X. Chen, L. Shen, Y. Cao, C. Au, K. Huang, A. Zheng and L. Jiang, ACS Appl. Mater. Interfaces, 2019, 11, 29950–29959 CrossRef CAS PubMed.
- Y. Pan, M. Chen, M. Hu, M. Tian, Y. Zhang and D. Long, Appl. Catal., B, 2019, 118266 Search PubMed.
- A. Pudi, M. Rezaei, V. Signorini, M. Peter, M. Giacinti and S. Soheil, Sep. Purif. Technol., 2022, 298, 121448 CrossRef CAS.
- D. Somashekara and L. Mulky, ChemBioEng Rev., 2023, 10, 491–509 CrossRef CAS.
- Z. Zhang, J. Wang, W. Li, M. Wang, W. Qiao and D. Long, Carbon, 2016, 96, 608–615 CrossRef CAS.
- P. Zhang, H. Zhu and S. Dai, ChemCatChem, 2015, 7, 2788–2805 CrossRef CAS.
- M. Khabazipour and M. Anbia, Ind. Eng. Chem. Res., 2019, 22133–22164, DOI:10.1021/acs.iecr.9b03800.
- K. Kante, C. Nieto-delgado, J. R. Rangel-mendez and T. J. Bandosz, J. Hazard. Mater., 2012, 201–202, 141–147 CrossRef CAS PubMed.
- T. J. Bandosz, J. Colloid Interface Sci., 2002, 20, 1–20 CrossRef PubMed.
- M. Seredych and T. J. Bandosz, Energy Fuel., 2008, 850–859 CrossRef CAS.
- X. Zhang, Y. Tang, S. Qu, J. Da and Z. Hao, ACS Catal., 2015, 5, 1053–1067 CrossRef CAS.
- P. Nowicki, P. Skibiszewska and M. Wis, Adsorption, 2015, 20–31 Search PubMed.
- H. W. Ou, M. L. Fang, M. S. Chou, H. Y. Chang, T. F. Shiao, M. L. Fang, M. S. Chou, H. Y. Chang and T. F. Shiao, Long-term, J. Air Waste Manage. Assoc., 2020, 641–648, DOI:10.1080/10962247.2020.1754305.
- L. Chen, J. Yuan, T. Li, X. Jiang, S. Ma, W. Cen and W. Jiang, Sci. Total Environ., 2021, 768, 144452 CrossRef CAS PubMed.
- J. Kazmierczak-Razna, P. Nowicki and R. Pietrzak, Chem. Eng. Res. Des., 2016, 346–353, DOI:10.1016/j.cherd.2016.02.018.
- J. Kazmierczak-Razna and R. Pietrzak, Adsorption, 2016, 22, 473–480 CrossRef CAS.
- H. W. Ou, M. L. Fang, M. S. Chou and H. Y. Chang, J. Air Waste Manage. Assoc., 2020, 70(6), 641–648 CrossRef CAS.
- G. Shang, G. Shen, L. Liu, Q. Chen and Z. Xu, Bioresour. Technol., 2013, 133, 495–499 CAS.
- W. Shen, Z. Li and Y. Liu, Recent Patents Chem. Eng., 2010, 1, 27–40 CrossRef.
- A. B. Bajaj, H. Jo, S. M. Jo, J. H. Park, K. B. Yi and S. Lee, Appl. Surf. Sci., 2018, 253–257, DOI:10.1016/j.apsusc.2017.06.280.
- C. Yang, M. Florent, G. De Falco, H. Fan and T. J. Bandosz, Chem. Eng. J., 2020, 394, 124906 CAS.
- B. Liu and S. Zuo, Environ. Prog. Sustain. Energy, 2022, 41, e13781 CAS.
- N. M. Nor, L. L. Chung, L. K. Teong and A. R. Mohamed, Biochem. Pharmacol., 2013, 1, 658–666 Search PubMed.
- A. P. Alekhin, G. M. Boleiko, S. A. Gudkova, A. M. Markeev, A. A. Sigarev, V. F. Toknova, A. G. Kirilenko, R. V Lapshin, E. N. Kozlov and D. V. Tetyukhin, Nanotechnol. Russ., 2010, 5, 696–708 CrossRef.
- H. Sawalha, M. Maghalseh, J. Qutaina, K. Junaidi and R. Rene, Bioengineered, 2020, 11, 607–618 CrossRef CAS PubMed.
- Y. Yuan, L. Huang, T. C. Zhang, L. Ouyang and S. Yuan, Sep. Purif. Technol., 2021, 279, 119686 CrossRef CAS.
- I. Tole, K. Habermehl-Cwirzen and A. Cwirzen, Mineral. Petrol., 2019, 113, 449–462 CrossRef CAS.
- H. Nam, S. Wang and H.-R. Jeong, Fuel, 2018, 213, 186–194 CrossRef CAS.
- S. Wang, H. Nam and H. Nam, Environ. Prog. Sustain. Energy, 2019, 38, e13241 CrossRef CAS.
- R. Sitthikhankaew, C. David, A. Suttichai and N. and Laosiripojana, Sep. Sci. Technol., 2014, 49, 354–366 CrossRef CAS.
- D. Langhammer, J. Kullgren and L. Österlund, ACS Catal., 2022, 12, 10472–10481 CrossRef CAS.
- S. Gyawali, R. T. A. Tirumala, M. Andiappan and A. D. Bristow, Proc. SPIE, 2024, 12884, 128840F Search PubMed.
- H. J. Choi, S. H. Kwon, W. S. Lee, K. G. Im, T. H. Kim, B. R. Noh, S. Park, S. Oh and K. K. Kim, Nanomaterials, 2020, 462, DOI:10.3390/nano10030462.
- T. Li, Y. Wu, G. Zhang, C. Guo, X. Zhang, B. Li, X. Zhou, M. Zheng, Y. Xu, S. Gao and L. Huo, Sens. Actuators, B, 2024, 399, 134820 CrossRef CAS.
- B. Levasseur, C. Petit and T. J. Bandosz, ACS Appl. Mater. Interfaces, 2010, 2, 3606–3613 CrossRef CAS.
- M. Sun, A. Hanif, T. Wang, Q. Gu and J. Shang, Sep. Purif. Technol., 2023, 314, 123563 CrossRef CAS.
- R. Pietrzak, Energy Fuels, 2009, 23, 3617–3624 CrossRef CAS.
- P. Nowicki, H. Wachowska and R. Pietrzak, J. Hazard. Mater., 2010, 181, 1088–1094 CrossRef CAS PubMed.
- P. Nowicki, R. Pietrzak and H. Wachowska, Catal. Today, 2010, 150, 107–114 CrossRef CAS.
- R. Pietrzak, Bioresour. Technol., 2010, 101, 907–913 CrossRef CAS PubMed.
- P. Nowicki, P. Skibiszewska and R. Pietrzak, Adsorption, 2013, 19, 521–528 CrossRef CAS.
- S. Bashkova and T. J. Bandosz, J. Colloid Interface Sci., 2009, 333, 97–103 CrossRef CAS PubMed.
- R. Pietrzak and T. J. Bandosz, Carbon, 2007, 45, 2537–2546 CAS.
- M. Jeguirim, V. Tschamber, J. F. Brilhac and P. Ehrburger, J. Anal. Appl. Pyrolysis, 2004, 72, 171–181 CAS.
- M. Belhachemi, M. Jeguirim, L. Limousy and F. Addoun, Chem. Eng. J., 2014, 253, 121–129 CrossRef CAS.
- I. Ghouma, M. Jeguirim, U. Sager, L. Limousy, S. Bennici, E. Däuber, C. Asbach, R. Ligotski, F. Schmidt and A. Ouederni, Energies, 2017, 1508, DOI:10.3390/en10101508.
- B. Levasseur, E. Gonzalez-Lopez, J. A. Rossin and T. J. Bandosz, Langmuir, 2011, 27, 5354–5365 CrossRef CAS PubMed.
- T. Cheng, Y. Bian, J. Li, X. Ma, L. Yang, L. Zhou and H. Wu, Fuel, 2023, 334, 126452 CrossRef CAS.
- C. He, J. Cheng, X. Zhang, M. Douthwaite, S. Pattisson and Z. Hao, Chem. Rev., 2019, 119, 4471–4568 CrossRef CAS PubMed.
- H. Gao, X. Lv, M. Zhang, Q. Li, J. Chen, Z. Hu and H. Jia, Chem. Eng. J., 2022, 434, 134618 CrossRef CAS.
- H. Wang, J. Gao, X. Xu, B. Liu, L. Yu, Y. Ren, R. Shi, Z. Zeng and L. Li, Chem.–Asian J., 2021, 16, 1118–1129 CrossRef CAS PubMed.
- S. A. Halawy, A. I. Osman, N. Mehta, A. Abdelkader, D.-V. N. Vo and D. W. Rooney, Chemosphere, 2022, 295, 133795 CrossRef CAS PubMed.
- X. Ma, H. Lv, L. Yang, Z. Zhang, Z. Sun and H. Wu, J. Hazard. Mater., 2021, 405, 124193 CrossRef CAS PubMed.
- J. Zhang, J. Shao, X. Zhang, G. Rao, G. Li, H. Yang, S. Zhang and H. Chen, Chem. Eng. J., 2023, 452, 139003 CrossRef CAS.
- A. Khan, J. E. Szulejko, P. Samaddar, K.-H. Kim, B. Liu, H. A. Maitlo, X. Yang and Y. S. Ok, Chem. Eng. J., 2019, 361, 1576–1585 CrossRef CAS.
- X. Zhang, B. Gao, J. Fang, W. Zou, L. Dong, C. Cao, J. Zhang, Y. Li and H. Wang, Chemosphere, 2019, 218, 680–686 CrossRef CAS PubMed.
- X. Zhang, W. Xiang, B. Wang, J. Fang, W. Zou, F. He, Y. Li, D. C. W. Tsang, Y. S. Ok and B. Gao, Chemosphere, 2020, 245, 125664 CrossRef CAS PubMed.
- H. Rajabi, M. H. Mosleh, P. Mandal, A. Lea-Langton and M. Sedighi, Chemosphere, 2021, 268, 129310 CrossRef CAS PubMed.
- X. Zhang, B. Gao, Y. Zheng, X. Hu, A. E. Creamer, M. D. Annable and Y. Li, Bioresour. Technol., 2017, 245, 606–614 CrossRef CAS PubMed.
- W. Xiang, X. Zhang, K. Chen, J. Fang, F. He, X. Hu, D. C. W. Tsang, Y. S. Ok and B. Gao, Chem. Eng. J., 2020, 385, 123842 CrossRef CAS.
- A. Kumar, E. Singh, A. Khapre, N. Bordoloi and S. Kumar, Bioresour. Technol., 2020, 297, 122469 CrossRef CAS PubMed.
- Y. Shen and N. Zhang, Bioresour. Technol., 2019, 282, 294–300 CrossRef CAS PubMed.
- N. R. Abdul Manap, R. Shamsudin, M. N. Maghpor, M. A. Abdul Hamid and A. Jalar, J. Environ. Chem. Eng., 2018, 6, 970–983 CrossRef CAS.
- H. Rajabi, M. Hadi Mosleh, T. Prakoso, N. Ghaemi, P. Mandal, A. Lea-Langton and M. Sedighi, Chemosphere, 2021, 283, 131288 CrossRef CAS PubMed.
- X. Zhang, X. Miao, W. Xiang, J. Zhang, C. Cao, H. Wang, X. Hu and B. Gao, J. Hazard. Mater., 2021, 403, 123540 CrossRef CAS PubMed.
- X. Zhao, X. Zeng, Y. Qin, X. Li, T. Zhu and X. Tang, Chemosphere, 2018, 206, 285–292 CrossRef CAS PubMed.
- Z. Jin, B. Wang, L. Ma, P. Fu, L. Xie, X. Jiang and W. Jiang, Chem. Eng. J., 2020, 385, 123843 CrossRef CAS.
- Y. Guo, Y. Li, J. Wang, T. Zhu and M. Ye, Chem. Eng. J., 2014, 236, 506–512 CrossRef CAS.
- M. A. Lillo-Ródenas, D. Cazorla-Amorós and A. Linares-Solano, Carbon, 2005, 43, 1758–1767 Search PubMed.
- T. Garcıa, R. Murillo, D. Cazorla-Amoros, A. M. Mastral and A. Linares-Solano, Carbon, 2004, 42, 1683–1689 Search PubMed.
- S. Haydar, M. A. Ferro-Garcıa, J. Rivera-Utrilla and J. P. Joly, Carbon, 2003, 41, 387–395 CrossRef CAS.
- eR. R. Gil, B. Ruiz, M. S. Lozano, M. J. Martín and E. Fuente, Chem. Eng. J., 2014, 245, 80–88 CrossRef.
- C. Wen, T. Liu, D. Wang, Y. Wang, H. Chen, G. Luo, Z. Zhou, C. Li and M. Xu, Prog. Energy Combust. Sci., 2023, 99, 101098 CrossRef.
- Z. Li, Y. Li, J. Zhu, Z. Li, Y. Li and J. Zhu, Materials, 2021, 3284, DOI:10.3390/ma14123284.
- X. Ma, B. Liu, M. Che, Q. Wu, R. Chen, C. Su, X. Xu, Z. Zeng and L. Li, Sep. Purif. Technol., 2021, 269, 118690 CrossRef CAS.
- K.-J. Kim, C.-S. Kang, Y.-J. You, M.-C. Chung, M.-W. Woo, W.-J. Jeong, N.-C. Park and H.-G. Ahn, Catal. Today, 2006, 111, 223–228 CrossRef CAS.
- X. Shen, R. Ou, Y. Lu, A. Yuan, J. Liu, X. Hu, Z. Yang and F. Yang, ChemistrySelect, 2020, 5, 9162–9169 CrossRef CAS.
- L. Li, S. Liu and J. Liu, J. Hazard. Mater., 2011, 192, 683–690 CrossRef CAS PubMed.
- K. Zhou, L. Li, X. Ma, Y. Mo, R. Chen, H. Li and H. Li, RSC Adv., 2018, 8, 2922–2932 RSC.
- J. L. Figueiredo, M. F. R. Pereira, M. M. A. Freitas and J. J. M. Órfão, Carbon, 1999, 37, 1379–1389 CrossRef CAS.
- W. Shen, Z. Li and Y. Liu, Recent Patents Chem. Eng., 2008, 1, 27–40 CrossRef CAS.
- X. Yu, S. Liu, G. Lin, X. Zhu, S. Zhang, R. Qu, C. Zheng and X. Gao, RSC Adv., 2018, 8, 21541–21550 RSC.
- J. Choma, B. Szczęśniak, A. Kapusta and M. Jaroniec, Molecules, 2024, 29, 5677 CrossRef CAS PubMed.
- S. Sumathi, S. Bhatia, K. T. Lee and A. R. Mohamed, Sci. China, Ser. E: Technol. Sci., 2009, 198–203, DOI:10.1007/s11431-009-0031-6.
- S. Sumathi, S. Bhatia, K. T. Lee and A. R. Mohamed, Chem. Eng. J., 2010, 162, 51–57 CrossRef CAS.
- C. Zhu, Y. Duan, C.-Y. Wu, Q. Zhou, M. She, T. Yao and J. Zhang, Fuel, 2016, 172, 160–169 CrossRef CAS.
- S. A. Opatokun, A. Prabhu, A. Al Shoaibi, C. Srinivasakannan and V. Strezov, Chemosphere, 2017, 168, 326–332 CrossRef CAS PubMed.
- L. Gao, C. Li, J. Zhang, X. Du, S. Li, J. Zeng, Y. Yi and G. Zeng, Chem. Eng. J., 2018, 342, 339–349 CrossRef CAS.
- Y. Yi, C. Li, L. Zhao, X. Du, L. Gao, J. Chen, Y. Zhai and G. Zeng, Environ. Sci. Pollut. Res., 2018, 25, 4761–4775 CrossRef CAS PubMed.
- Y. Qin, C. Wang, X. Sun, Y. Ma, X. Song, F. Wang, K. Li and P. Ning, Fuel, 2021, 293, 120391 CrossRef CAS.
- X. Zhang, K. Ren, Y. Wang, B. Shen, F. Shen and Y. Shang, Energy Fuels, 2021, 35, 15969–15977 CrossRef CAS.
- A. A. Abdulrasheed, A. A. Jalil, S. Triwahyono, M. A. A. Zaini, Y. Gambo and M. Ibrahim, Renew. Sustain. Energy Rev., 2018, 94, 1067–1085 CrossRef CAS.
- G. Li, B. Shen, Y. Li, B. Zhao, F. Wang, C. He, Y. Wang and M. Zhang, J. Hazard. Mater., 2015, 298, 162–169 CrossRef CAS PubMed.
- H. Li, J. Zhang, Y. Cao, C. Liu, F. Li, Y. Song, J. Hu and Y. Wang, Fuel Process. Technol., 2020, 207, 106478 CrossRef CAS.
- S. Sumathi, S. Bhatia, K. T. Lee and A. R. Mohamed, Energy Fuel., 2010, 24, 427–431 CrossRef CAS.
- S. Wang, H. Nam, T. B. Gebreegziabher and H. Nam, Eng. Rep., 2020, 2, e12083 CrossRef CAS.
- A. S. González, M. G. Plaza, F. Rubiera and C. Pevida, Chem. Eng. J., 2013, 230, 456–465 CrossRef.
- E. Atanes, A. Nieto-Márquez, A. Cambra, M. C. Ruiz-Pérez and F. Fernández-Martínez, Chem. Eng. J., 2012, 211, 60–67 Search PubMed.
- F. L. Braghiroli, H. Bouafif and A. Koubaa, Ind. Crops Prod., 2019, 138, 111456 CAS.
- N. Iberahim, S. Sethupathi, C. L. Goh, M. J. K. Bashir and W. Ahmad, J. Environ. Manage., 2019, 248, 109302 CrossRef CAS PubMed.
- N. Iberahim, S. Sethupathi, M. J. K. Bashir, R. Kanthasamy and T. Ahmad, Sci. Total Environ., 2022, 805, 150421 CAS.
- M. G. Plaza, A. S. González, J. J. Pis, F. Rubiera and C. Pevida, Appl. Energy, 2014, 114, 551–562 CAS.
- M.-V. Nguyen and B.-K. Lee, Process Saf. Environ. Prot., 2016, 104, 490–498 CrossRef CAS.
- W. Hao, E. Björkman, M. Lilliestråle and N. Hedin, Appl. Energy, 2013, 112, 526–532 CAS.
- H. Wei, S. Deng, B. Hu, Z. Chen, B. Wang, J. Huang and G. Yu, ChemSusChem, 2012, 5, 2354–2360 CAS.
- P. Murge, S. Dinda and S. Roy, Energy Fuel., 2018, 32, 10786–10795 CAS.
- A. E. Creamer, B. Gao and S. Wang, Chem. Eng. J., 2016, 283, 826–832 CAS.
- W. Gao, T. Zhou and Q. Wang, Chem. Eng. J., 2018, 336, 710–720 CAS.
- C. Chen, J. Kim, D.-A. Yang and W.-S. Ahn, Chem. Eng. J., 2011, 168, 1134–1139 CAS.
- M. Kandiah, M. H. Nilsen, S. Usseglio, S. Jakobsen, U. Olsbye, M. Tilset, C. Larabi, E. A. Quadrelli, F. Bonino and K. P. Lillerud, Chem. Mater., 2010, 22, 6632–6640 CrossRef CAS.
- S. Xiang, Y. He, Z. Zhang, H. Wu, W. Zhou, R. Krishna and B. Chen, Nat. Commun., 2012, 954, DOI:10.1038/ncomms1956.
- H.-J. Kim and J.-H. Lee, Sens. Actuators, B, 2014, 192, 607–627 CrossRef CAS.
- A. Mirzaei, S. G. Leonardi and G. Neri, Ceram. Int., 2016, 42, 15119–15141 CrossRef CAS.
- D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem. Int. Ed., 2010, 49, 6058–6082 CrossRef PubMed.
- D. Jiang, H. Li, S. Wang, X. Cheng, P. Bartocci and F. Fantozzi, Fuel, 2023, 125948, DOI:10.1016/j.fuel.2022.125948.
- M. Rahimi, M. H. Abbaspour-Fard, A. Rohani, O. Yuksel Orhan and X. Li, Ind. Eng. Chem. Res., 2022, 61, 10670–10688 CrossRef CAS.
- R. Rahamathullah, D. S. Zakaria, S. K. M. Rozi, H. N. A. Halim, F. I. A. Razak and S. Sapari, Biomass Convers. Biorefin., 2024, 1–13, DOI:10.1007/s13399-024-05343-5.
- M. Ahmadi, F. Bahmanzadegan, M. Qasemnazhand, A. Ghaemi and H. Ramezanipour Penchah, Sci. Rep., 2024, 13595, DOI:10.1038/s41598-024-64503-9.
- C. S. Allen, F. Ghamouss, O. Boujibar and P. J. F. Harris, Proc. R. Soc. A Math. Phys. Eng. Sci., 2022, 478, 20210580 Search PubMed.
- M. Qasemnazhand, F. Khoeini and M. Badakhshan, Mater. Today Chem., 2023, 28, 101383 CrossRef CAS.
- L. Deng, J. Shi, Y. Zhao, D. Feng, W. Zhang, Y. Yu and S. Sun, Chem. Eng. J., 2024, 153403, DOI:10.1016/j.cej.2024.153403.
- M. Su, W. Peng, Z. Ding, Y. Zhou, H. Gao, Q. Jiang and C. Yu, Bioelectrochemistry, 2024, 160, 108776 CrossRef CAS PubMed.
- X. Yang, D. Jiang, X. Cheng, C. Yuan, S. Wang, Z. He and S. Esakkimuthu, Biomass Bioenergy, 2022, 106519, DOI:10.1016/j.biombioe.2022.106519.
- F. Raganati, F. Miccio and P. Ammendola, Energy Fuel., 2021, 35, 12845–12868 CrossRef CAS.
- W. Yang, Z. Wang and Y. Liu, Energy Fuels, 2020, 34, 13473–13490 CrossRef CAS.
Footnote |
| † Shared the second authorship. |
| This journal is © The Royal Society of Chemistry 2025 |