
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStereo-electronic contributions in yttrium-mediated stereoselective ring-opening polymerization of functional racemic β-lactones: ROP of 4-alkoxymethylene-β-propiolactones with bulky exocyclic chains†
Rama M.
Shakaroun
a,
Ali
Dhaini
a,
Romain
Ligny
a,
Ali
Alaaeddine
b,
Sophie M.
Guillaume
 *a and
Jean-François
Carpentier
*a
*a and
Jean-François
Carpentier
*a
aUniv. Rennes, CNRS, Institut des Sciences Chimiques de Rennes, UMR 6226, F-35042 Rennes, France. E-mail: sophie.guillaume@univ-rennes.fr; jean-francois.carpentier@univ-rennes.fr
bUniv. Libanaise, Campus Universitaire Rafic Hariri Hadath, Faculté des Sciences, Laboratoire de Chimie Médicinale et des Produits Naturels, Beirut, Lebanon
First published on 6th January 2023
Abstract
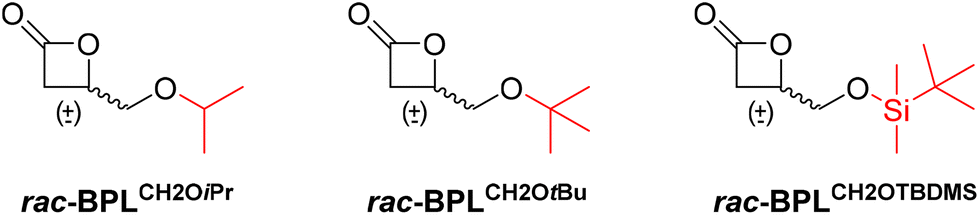
Stereoselective ring-opening polymerization (ROP) of cyclic esters is the privileged strategy to access stereoregular polyesters that are widely applied in various domains, such as in particular the biomedical and packaging fields. The production of synthetic stereo-enriched polyhydroxyalkanoates (PHAs) derived from racemic β-lactones by ROP is still a challenge. In this context, linear, high molar mass, narrowly dispersed PHAs, namely PBPLCH2OiPr, PBPLCH2OtBu and PBPLCH2OTBDMS (Mn,SEC up to 94![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 g mol−1; ĐM = 1.06–1.18; TBDMS = SitBuMe2), with syndiotactic enrichment (Pr = 0.76–0.87) were successfully synthesized by stereoselective ROP of the corresponding functional racemic β-propiolactones, rac-BPLCH2OiPr, rac-BPLCH2OtBu and rac-BPLCH2OTBDMS, respectively, which are promoted by diverse achiral diamino-bis(phenolate) yttrium complexes featuring various R′/R′′ substituents (Y{ONNOR′,R′′}, 2a–d). The influence of the steric hindrance of the BPLFG side-functionality, with FG = CH2OiPr, CH2OtBu, and CH2OTBDMS, on the ROP kinetics, stereoselectivity and thermal properties of the resulting PHAs, as a function of 2a–d catalysts, was compared to that of the previously reported similar but less hindered BPLFG monomers, with FG = CH2OMe, CH2OAllyl, CH2OBn, and CH2OPh. Overall, this study evidenced that, for the newly prepared rac-BPLCH2OiPr, rac-BPLCH2OtBu and rac-BPLCH2OTBDMS monomers, due to steric constraints induced by the monomer alkoxy/silyloxy side-functionality, all ROPs afforded syndio-enriched polyesters, regardless of the catalyst used. Conversely, only combinations of a BPLFG monomer containing two sets of methylene hydrogens within the side-functionality, i.e. with FG = CH2OCH2X with X = H, CH = CH2 and C6H5 as in BPLCH2OMe, BPLCH2OAllyl, and BPLCH2OBn, with a yttrium catalyst bearing ortho/para-chloro substituents (2a), gave isotactic functional PHAs. With the latter three monomers, a catalyst with highly sterically crowded substituents on the ligand platform (2a,b) was necessary to recover syndio-enriched PBPLCH2OMe,OAll,OBn.
300 g mol−1; ĐM = 1.06–1.18; TBDMS = SitBuMe2), with syndiotactic enrichment (Pr = 0.76–0.87) were successfully synthesized by stereoselective ROP of the corresponding functional racemic β-propiolactones, rac-BPLCH2OiPr, rac-BPLCH2OtBu and rac-BPLCH2OTBDMS, respectively, which are promoted by diverse achiral diamino-bis(phenolate) yttrium complexes featuring various R′/R′′ substituents (Y{ONNOR′,R′′}, 2a–d). The influence of the steric hindrance of the BPLFG side-functionality, with FG = CH2OiPr, CH2OtBu, and CH2OTBDMS, on the ROP kinetics, stereoselectivity and thermal properties of the resulting PHAs, as a function of 2a–d catalysts, was compared to that of the previously reported similar but less hindered BPLFG monomers, with FG = CH2OMe, CH2OAllyl, CH2OBn, and CH2OPh. Overall, this study evidenced that, for the newly prepared rac-BPLCH2OiPr, rac-BPLCH2OtBu and rac-BPLCH2OTBDMS monomers, due to steric constraints induced by the monomer alkoxy/silyloxy side-functionality, all ROPs afforded syndio-enriched polyesters, regardless of the catalyst used. Conversely, only combinations of a BPLFG monomer containing two sets of methylene hydrogens within the side-functionality, i.e. with FG = CH2OCH2X with X = H, CH = CH2 and C6H5 as in BPLCH2OMe, BPLCH2OAllyl, and BPLCH2OBn, with a yttrium catalyst bearing ortho/para-chloro substituents (2a), gave isotactic functional PHAs. With the latter three monomers, a catalyst with highly sterically crowded substituents on the ligand platform (2a,b) was necessary to recover syndio-enriched PBPLCH2OMe,OAll,OBn.
Introduction
Stereoselective ring-opening polymerization (ROP) of chiral racemic lactones is a field of topical interest as it allows accessing polymers with variable microstructures (i.e., tacticities), and hence polymer materials with differentiated and controlled properties.1 One of the most ubiquitous examples in this area is probably the ROP of racemic lactide (rac-LA) which has opened large avenues, in particular toward the formation of isotactic PLA stereocomplexes with significantly enhanced thermal characteristics.1a–e The formation of isotactic polyesters from the ROP of racemic lactones is clearly much less common than that of their syndiotactic (heterotactic) counterparts; indeed, the latter syndio/hetero tacticities are often the result of a chain-end stereocontrol1a,b where the minimization of steric tilting in the transition state induces the regular, alternated enchainment of consecutive monomer units with the opposite configuration. On the other hand, the formation of isotactic polyesters from racemic cyclic esters usually requires the use of chiral catalysts, most often metal-based ones,2 to proceed via a so-called enantiomorphic site control; isoselective ROP of racemic lactones with achiral catalysts is even less frequent and largely focused on rac-LA.1,3 A recent, remarkable addition is the stereoselective ROP of eight-membered racemic cyclic diolides mediated by discrete rare earth-based catalysts developed by the group of Chen, which affords a variety of perfectly isotactic (Pm up to 0.99)‡ polyhydroxyalkanoates (PHAs).4 In our longstanding research on the stereoselective ROP of racemic β-lactones5 with achiral alkoxyamino- or diamino-bisphenolate-yttrium catalysts ({Y{ON(X)OR′,R′′}),6 we have reported that only the use of catalysts bearing halogeno ortho/para-substituents (R′,R′′ = Cl, F or Br) on the phenolate ligand platform allowed the highly isoselective ROP of the specific racemic 4-alkoxymethylene-β-propiolactones (rac-BPLCH2OR, Scheme 1);7 actually, this was proved to be effective (i.e., isoselective) only for R = 4-methoxy (OMe), -allyloxy (OAll) or benzyloxy (OBn) derivatives, that is, the monomers having two methylene groups apart from the oxygen in the side-functionality (i.e., with a CH2(referred to as “inner”)-O–CH2(referred to as “outer”)X group).8 As supported by DFT computations, such isoselectivity apparently relies on attractive non-covalent interactions (NCIs)9 between the halogen ortho-substituents on the yttrium ligand with the “inner” and/or “outer” methylene hydrogens within the side-functional group of the last inserted monomer unit within the growing polymer chain (Scheme 1).7 As a matter of fact, the ROP of rac-BPLCH2OR monomers without such an “outer” methylene group, e.g. with R = OPh, or with two “inner” methylene groups, i.e. rac-BPLCH2CH2OCH2Ph, with an isoselective Y{ONNOCl,Cl} catalytic system, all recovered syndio-enriched polymers (Pr = 0.75–0.77).10;§ The contribution of the “outer” methylene group in the pendant arm of BPLCH2OCH2X is thus still an open question. | ||
| Scheme 1 Previously reported stereoselective ROP of 4-alkoxymethylene-β-propiolactones by Y{ON(X)OR′,R′′} catalytic systems, with stereoselectivity outcomes depending on the stereo-electronic characteristics of the catalyst. The bottom-right structure depicts the transition state with attractive non-covalent Cl⋯H interactions allegedly driving the isoselectivity.7 | ||
To gain a better insight into the factors that govern the isoselectivity observed in the ROP of some rac-BPLCH2OR monomers mediated by the Y{ONNOCl,Cl} catalytic system and to probe further the possible decisive influence of “outer” methylene hydrogens (–CH2OCH2X), we have herein explored the yttrium-mediated ROP of three β-propiolactones which do not feature an “outer” methylene group within the alkoxide moiety and which display an alkoxy tertiary or quaternary carbon/silicon, namely rac-BPLCH2OiPr, rac-BPLCH2OtBu, and rac-BPLCH2OTBDMS (TBDMS = SitBuMe2; Scheme 2),11 respectively, in comparison with the related rac-BPLCH2OPh which is depleted of the outer methylene (-OCH2X) moiety.10 These three rac-BPLCH2OiPr/OtBu/OTBDMS monomers, two of which are new and all of which are readily prepared by carbonylation of the parent glycidyl ethers (see the ESI†),11,12 have been chosen so as to replace the “outer” methylene hydrogens (as in R = OCH2H, OCH2CH = CH2 and OCH2Ph) with one or two methyl groups (as in R = OiPr and OtBu). This should allow one to assess whether the “outer” alkoxide methylene may contribute to the iso-stereocontrol of the ROP of such functional BPLCH2ORs. In addition, rac-BPLCH2OTBDMS was selected to probe, besides the impact of a missing “outer” methylene in the side function of the monomer, the impact of the steric bulkiness on the alkoxy functionality (OiPr and OtBu vs. OSiMe2tBu). Further comparison with the β-propiolactone featuring a somewhat bulky aryloxy moiety, namely BPLCH2OPh, which was shown to give a syndiotactic polyester,10 could then be made. In addition, PHAs derived from rac-BPLCH2OTBDMS are of further interest as they could subsequently provide access to hydrophilic polyesters upon deprotection of –OTBDMS into pendant hydroxyl groups also available for post-polymerization modification, as for instance to chemically bound a biological moiety for theranostic outcomes. Four catalysts {Y{ONNOR′,R′′}, 2a–d, having different R′,R′′ ortho/para-substituents installed on the bisphenolate platform and which have previously revealed quite distinctive and effective stereocontrol abilities in the ROP of racemic functional β-lactones due to their different stereo-electronic characteristics,6,7 have been selected for the present study.
 | ||
| Scheme 2 4-Alkoxymethylene-β-propiolactones with bulky alkoxy side-functionalities depleted of an “outer” methylene group, investigated in the present study. | ||
Experimental section
See the ESI† for additional details.Synthesis and characterization of BPLCH2OR monomers
BPLCH2OR monomers were synthesized by carbonylation of the corresponding racemic or enantiopure glycidyl ethers (rac-/(S)-GlycCH2OR) using a previously reported procedure.11,12 All rac-BPLCH2OR and (S)-BPLCH2OR monomers were characterized (refer to the ESI) and stored under argon at −27 °C.Typical BPLCH2OR polymerization procedure
In a typical experiment,13 in a glovebox, a Schlenk flask was charged with [Y(N(SiHMe2)2)3](THF)2 (8.8 mg, 14 μmol) and {ONNOtBu2} (1d, 7.4 mg, 14 μmol), and toluene (0.25 mL) was next added. To this solution, iPrOH (107 μL of a 1% (v/v) solution in toluene, 1 equiv. vs. Y) was added under stirring at room temperature (ca. 20 °C). After 5 min of stirring, a solution of rac-BPLCH2OR (0.84 mmol, 60 equiv.) in toluene (0.5 mL) was added rapidly and the mixture was stirred at 20 °C for 1 h. The reaction was quenched by the addition of acetic acid (ca. 0.5 mL of a 1.6 mol L−1 solution in toluene). The resulting mixture was concentrated to dryness under vacuum and the conversion was determined by 1H NMR analysis of the residue in CDCl3. The crude polymer was then dissolved in CH2Cl2 (ca. 1 mL), precipitated in cold pentane (ca. 5 mL), filtered and dried. PBPLCH2OiPr, PBPLCH2OtBu and PBPLCH2OTBDMS were recovered as a colorless oil, yellowish oil, and colorless solid, respectively. All recovered polymers were then analyzed by NMR spectroscopy, mass spectrometry, SEC, and DSC analyses.Results and discussion
ROP of rac-BPLCH2OiPr
The ROP of rac-BPLCH2OiPr was explored under the same general operating conditions as those used for the above-mentioned reference ROPs mediated by similar Y{ONNOR′,R′′} catalytic systems, that is, in toluene solution at room temperature, using in situ combinations of 2a–d/iPrOH (1:1) (Table 1).10 The reactivity trend observed for the different yttrium catalytic systems followed the one established with other similar β-lactones:6b,7 the Cl substituted catalyst 2d was the least active, with only partial conversion of 30 monomer equiv. even after prolonged reaction time (TOF2d = ca. 0.2 h−1, entry 1); the Me-substituted catalyst 2c achieved nearly complete consumption of ca. 60 and 100 monomer units within 12 and 24 h, respectively (TOF2c = ca. 5 h−1, entry 2); a much higher reactivity was observed with the catalytic systems bearing bulky substituted ligands (cumyl, tBu, 2a–b), and almost quantitative conversion of 30–250 equiv. of rac-BPLCH2OiPr was typically achieved within 5–20 min (TOF2a–b > 750 h−1, entries 4–10).
| Entry | Cat. | [BPLCH2OiPr]0/[2]0/[iPrOH]0 | Timeb (min) | BPLCH2OiPr Conv.c (%) |
M
n,theod (g mol−1) |
M
n,NMRe (g mol−1) |
M
n,SECf (g mol−1) |
Đ
Mf |
P
rg |
|---|---|---|---|---|---|---|---|---|---|
| a Reactions performed with [BPLCH2OiPr]0 = 1.0 M in toluene at room temperature. b Reaction times were not necessarily optimized. c Conversion of BPLCH2OiPr as determined by 1H NMR analysis of the crude reaction mixture. d Molar mass calculated according to Mn,theo = ([BPLCH2OiPr]0/[2]0 × conv.BPL(CH2OiPr) × MBPL(CH2OiPr)) + MiPrOH with MBPL(CH2OiPr) = 144 g mol−1 and MiPrOH = 60 g mol−1. e Molar mass determined by 1H NMR analysis of the isolated polymer, from the resonances of the terminal OiPr group. f Number-average molar mass (uncorrected values) and dispersity (Mw/Mn) determined by SEC analysis in THF at 30 °C vs. polystyrene standards. g P r is the probability of racemic linkages between BPLCH2OiPr units as determined by 13C{1H} NMR analysis of the isolated PBPLCH2OiPrs. h ROP of enantiopure (S)-BPLCH2OiPr. | |||||||||
| 1 | 2d | 30:1:1 |
24 × 60 | 19 | 850 | 1100 | 1400 | 1.15 | 0.70 |
| 2 | 2c | 60:1:1 |
12 × 60 | 96 | 8350 | 7550 | 9500 | 1.09 | 0.71 |
| 3 | 2c | 100:1:1 |
24 × 60 | 90 | 13000 |
15000 |
15900 |
1.18 | 0.72 |
| 4 | 2b | 30:1:1 |
5 | 100 | 4400 | 3700 | 5400 | 1.09 | 0.84 |
| 5 | 2b | 60:1:1 |
5 | 100 | 8700 | 9400 | 10700 |
1.11 | 0.84 |
| 6 | 2b | 100:1:1 |
15 | 100 | 14450 |
14400 |
15300 |
1.13 | 0.85 |
| 7 | 2b | 250:1:1 |
20 | 100 | 36050 |
36500 |
38300 |
1.08 | 0.85 |
| 8 | 2b | 500:1:1 |
210 | 100 | 72100 |
68700 |
48900 |
1.06 | 0.86 |
| 9 | 2a | 60:1:1 |
5 | 100 | 8700 | 9000 | 10050 |
1.10 | 0.82 |
| 10 | 2a | 100:1:1 |
15 | 100 | 14450 |
17000 |
18600 |
1.13 | 0.82 |
| 11h | 2b | 30:1:1 |
60 | 94 | 4100 | 4600 | 5600 | 1.10 | <0.05 |
The ROP of rac-BPLCH2OiPr with the 2a–d/iPrOH systems proceeded with quite good control in terms of macromolecular parameters. All the polymers showed a linear topology with α-isopropoxycarbonyl and ω-hydroxy chain-end groups, as unambiguously established by 1H and J-MOD NMR spectroscopy and MALDI-ToF mass spectrometry analyses (see the ESI; Fig. S1–S6†). Also, alongside narrow dispersities (ĐM = 1.06–1.18), the calculated (Mn,theo) and experimental (Mn,NMR, Mn,SEC) molar mass values were in quite good agreement. A linear relationship between the experimental molar mass values and the rac-BPLCH2OiPr monomer loading/conversion up to 250 equiv. was observed with the 2b/iPrOH (1:1) catalytic system (Fig. 1). Altogether, these results confirm the limited extent or the absence of irreversible transfer/side-reactions (typical inter- and intra-molecular undesirable transesterification reactions, i.e. reshuffling and backbiting reactions, respectively) and suggest essentially active polymerization features.
 | ||
| Fig. 1 Illustration of the variation of Mn,NMR ■, Mn,SEC ○, and Mn,theo (the solid line) molar mass values of PBPLCH2OiPr synthesized from the ROP of rac-BPLCH2OiPr mediated by the 2b/iPrOH (1:1) catalytic system as a function of the BPLCH2OiPr monomer loading/conversion (Table 1, entries 4–8). | ||
A close examination of the carbonyl (δ r = ca. 169.65, δ m = ca. 169.55 ppm), methine (δ r = ca. 68.12, δ m = ca. 68.03 ppm) and methylene (δ r = ca. 35.94, δ m = ca. 36.05 ppm) main-chain carbons’ signals in the 13C NMR spectra allowed establishing the polymers’ tacticity (Fig. 2). For the sake of comparison, a pure isotactic PBPLCH2OiPr system was prepared from the ROP of enantiopure (S)-BPLCH2OiPr (Table 1, entry 11). Regardless of the catalyst used in the 2a–d series, the ROP of rac-BPLCH2OiPr gave syndio-enriched polymers. It is noteworthy that the 2c system (Me substituents) exhibited approximately the same syndiotacticity (Pr = 0.71–0.72) as the one obtained from 2d (Cl substituents, Pr = 0.69–0.70). This suggests the absence of any electronic effect from 2d but, instead, the preponderance of a pure, yet limited steric control, in tuning the tacticity of PBPLCH2OiPr. Along the same line, catalytic systems 2a–b that bear bulkier cumyl and tert-butyl substituents resulted in better syndio-enrichments (Pr = 0.82–0.85), which are close to those obtained for PBPLCH2OMe, PBPLCH2OAll and PBPLCH2OBn (Pr = 0.78–0.90).7
 | ||
| Fig. 2 Zoomed regions of the 13C{1H} NMR spectra (125 MHz, CDCl3, 23 °C) of PBPLCH2OiPr prepared by ROP of rac-BPLCH2OiPr, except for the top spectrum of enantiopure (S)-BPLCH2OiPr (Table 1, entry 11), mediated by the 2a, 2b, 2c, or 2d/iPrOH (1:1) catalytic systems (Table 1, entries 9, 7, 3 and 1, respectively). | ||
ROP of rac-BPLCH2OtBu
The stereoselective ROP of rac-BPLCH2OtBu was similarly examined (Table 2). The activity trend of catalysts 2a–d (ca. 75 monomer equiv.) was very comparable to the aforementioned ROP of rac-BPLCH2OiPr: TOF2d = ca. 0.7 h−1 (entry 1) vs. TOF2c = ca. 10 h−1 (entry 4) vs. TOF2a–b > 900 h−1 (entries 6 and 11). Also, the corresponding characteristic data of PBPLCH2OtBu (NMR spectra, Mn and dispersity values, and linear molar mass increase correspondingly to larger monomer loadings) revealed well-defined α-isopropoxycarbonyl, ω-hydroxy telechelic PHAs with chain-end fidelity and an overall quite good control of the polymerization (see the ESI, Fig. S7–S11†).| Entry | Cat. | [BPLCH2OtBu]0/[2]0/[iPrOH]0 | Timeb (min) |
BPLCH2OtBu Conv. c (%) |
M
n,theo
(g mol−1) |
M
n,NMRe (g mol−1) |
M
n,SECf (g mol−1) |
Đ
Mf |
P
rg |
|---|---|---|---|---|---|---|---|---|---|
| a Reactions performed with [BPLCH2OtBu]0 = 1.0 M in toluene at room temperature. b Reaction times were not necessarily optimized. c Conversion of BPLCH2OtBu as determined by 1H NMR analysis of the crude reaction mixture. d Molar mass calculated according to Mn,theo = ([BPLCH2OtBu]0/[2]0 × conv.BPL(CH2OtBu) × MBPL(CH2OtBu)) + MiPrOH with MBPL(CH2OtBu) = 158 g mol−1and MiPrOH = 60 g mol−1. e Molar mass determined by 1H NMR analysis of the isolated polymer, from the resonances of the terminal OiPr group. f Number-average molar mass (uncorrected values) and dispersity (Mw/Mn) determined by SEC analysis in THF at 30 °C vs. polystyrene standards. g P r is the probability of racemic linkages between BPLCH2OtBu units as determined by 13C{1H} NMR analysis of the isolated PBPLCH2OtBus. h ROP of enantiopure (S)-BPLCH2OtBu. | |||||||||
| 1 | 2d | 25:1:1 |
24 × 60 | 67 | 2600 | 2500 | 2400 | 1.12 | 0.70 |
| 2 | 2d | 75:1:1 |
27 × 60 | 28 | 3300 | 3400 | 3000 | 1.06 | 0.71 |
| 3 | 2c | 25:1:1 |
60 | 100 | 3600 | 3100 | 3200 | 1.09 | 0.74 |
| 4 | 2c | 75:1:1 |
7 h | 90 | 10700 |
10900 |
13600 |
1.16 | 0.75 |
| 5 | 2b | 30:1:1 |
30 | 100 | 4300 | 3900 | 4300 | 1.12 | 0.83 |
| 6 | 2b | 73:1:1 |
5 | 100 | 11600 |
11300 |
14800 |
1.10 | 0.84 |
| 7 | 2b | 120:1:1 |
10 | 100 | 18800 |
18800 |
24000 |
1.14 | 0.83 |
| 8 | 2b | 250:1:1 |
15 | 99 | 39100 |
40000 |
49900 |
1.15 | 0.84 |
| 9 | 2b | 500:1:1 |
15 | 99 | 78300 |
80000 |
94300 |
1.18 | 0.84 |
| 10 | 2a | 30:1:1 |
30 | 100 | 4300 | 4300 | 4000 | 1.12 | 0.78 |
| 11 | 2a | 75:1:1 |
5 | 100 | 11900 |
10900 |
15200 |
1.14 | 0.78 |
| 12h | 2b | 70:1:1 |
30 | 100 | 11100 |
10300 |
14300 |
1.09 | <0.05 |
The stereochemistry of PBPLCH2OtBu prepared by ROP of rac-BPLCH2OtBu mediated by the 2a–d/iPrOH systems closely resembles that of PBPLCH2OiPr. All the isolated PBPLCH2OtBu samples revealed a syndio-enrichment regardless of the catalyst used (2a–d) (Fig. 3) (δ r(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) = ca. 169.77, δ m(CO) = ca. 169.70 ppm; δ r(CH) = ca. 62.18, δ m(CH) = ca. 62.11 ppm; δ r(CH2) = ca. 35.97, δ m(CH2) = ca. 36.06 ppm). While 2d (Cl substituents) afforded almost the same enrichment of PBPLCH2OtBu as that of PBPLCH2OiPr (Pr = ca. 0.70), catalyst 2c (Me substituents) contributed to a slightly higher syndiotacticity (Pr = 0.75 vs. 0.71), while catalyst 2a (cumyl substituents) exhibited a slightly inferior syndio-regularity (Pr = 0.78 vs. 0.82). Finally, catalyst 2b (tBu substituents) produced the highest enrichment of PBPLCH2OtBu (Pr = 0.84), again reminiscent of that of PBPLCH2OiPr (Pr = 0.85). Hence, the general reactivity trend of the 2a–d catalysts to yield syndio-enriched PBPLCH2OtBu is the same as the one observed for PBPLCH2OiPr, but with minor differences in the PHA enrichment in the case of catalysts 2a,c.
O) = ca. 169.77, δ m(CO) = ca. 169.70 ppm; δ r(CH) = ca. 62.18, δ m(CH) = ca. 62.11 ppm; δ r(CH2) = ca. 35.97, δ m(CH2) = ca. 36.06 ppm). While 2d (Cl substituents) afforded almost the same enrichment of PBPLCH2OtBu as that of PBPLCH2OiPr (Pr = ca. 0.70), catalyst 2c (Me substituents) contributed to a slightly higher syndiotacticity (Pr = 0.75 vs. 0.71), while catalyst 2a (cumyl substituents) exhibited a slightly inferior syndio-regularity (Pr = 0.78 vs. 0.82). Finally, catalyst 2b (tBu substituents) produced the highest enrichment of PBPLCH2OtBu (Pr = 0.84), again reminiscent of that of PBPLCH2OiPr (Pr = 0.85). Hence, the general reactivity trend of the 2a–d catalysts to yield syndio-enriched PBPLCH2OtBu is the same as the one observed for PBPLCH2OiPr, but with minor differences in the PHA enrichment in the case of catalysts 2a,c.
 | ||
| Fig. 3 Zoomed regions of the 13C{1H} NMR spectra (125 MHz, CDCl3, 23 °C) of PBPLCH2OtBu prepared by ROP of rac-BPLCH2OtBu, except for the top spectrum of enantiopure (S)-BPLCH2OtBu (Table 2, entry 12), mediated by the 2a, 2b, 2c, or 2d/iPrOH (1:1) catalytic systems (Table 2, entries 2, 4, 6 and 11, respectively). | ||
ROP of rac-BPLCH2OTBDMS
Representative results of the investigation of the ROP of rac-BPLCH2OTBDMS mediated by the 2a–d/iPrOH catalytic systems are summarized in Table 3. With both 2c–d systems (Me, Cl substituents), incomplete low monomer conversions were obtained for rac-BPLCH2OTBDMS loadings of 30–60 equiv. after 2–3 days (entries 1–3), exhibiting very low TOF2c–d of ca. 0.1–0.2 h−1. The 2a–b systems (tBu and cumyl substituents) led to complete or almost complete conversions of 30–500 equiv. of rac-BPLCH2OTBDMS after 1–8 h (entries 4–9), with higher TOF2a–b > 100 h−1. Hence, the 2a–d/iPrOH catalytic systems featured a regular trend toward the ROP of rac-BPLCH2OTBDMS, yet with an overall lower activity as compared to the ROP of rac-BPLCH2OiPr and rac-BPLCH2OtBu.| Entry | Cat. | [BPLCH2OTBDMS]0/[2]0/[iPrOH]0 | Timeb (h) | BPLCH2OTBDMS Conv. c (%) |
M
n,theo
(g mol−1) |
M
n,NMR
(g mol−1) |
M
n,SECf (g mol−1) |
Đ
Mf |
P
rMg |
|---|---|---|---|---|---|---|---|---|---|
| a Reactions performed with [BPLCH2OTBDMS]0 = 1.0 M in toluene at room temperature. b Reaction times were not necessarily optimized. c Conversion of BPLCH2OTBDMS as determined by 1H NMR analysis of the crude reaction mixture. d Molar mass calculated according to Mn,theo = ([BPLCH2OTBDMS]0/[2]0 × conv.BPL(CH2OTBDMS) × MBPL(CH2OTBDMS)) + MiPrOH with MBPL(CH2OTBDMS) = 216 g mol−1 and MiPrOH = 60 g mol−1. e Molar mass determined by 1H NMR analysis of the isolated polymer, from the resonances of the terminal OiPr group. f Number-average molar mass (uncorrected values) and dispersity (Mw/Mn) determined by SEC analysis in THF at 30 °C vs. polystyrene standards. g P r is the probability of racemic linkages between BPLCH2OTBDMS units as determined by 13C{1H} NMR analysis of the isolated PBPLCH2OTBDMSs. h ROP of enantiopure (S)-BPLCH2OTBDMS. i Not determined. | |||||||||
| 1 | 2d | 30:1:1 |
48 | 16 | 1800 | 2500 | 1000 | 1.07 | 0.76 |
| 2 | 2c | 30:1:1 |
8 | 30 | 2000 | 1600 | 2500 | 1.14 | n.d.i |
| 3 | 2c | 60:1:1 |
72 | 25 | 3300 | 3750 | 3000 | 1.12 | 0.77 |
| 4 | 2b | 60:1:1 |
4 | 96 | 12500 |
13500 |
9000 | 1.13 | 0.83 |
| 5 | 2b | 120:1:1 |
8 | 95 | 24700 |
23400 |
19200 |
1.12 | 0.84 |
| 6 | 2b | 250:1:1 |
5 | 100 | 47100 |
37600 |
24300 |
1.07 | 0.81 |
| 7 | 2b | 500:1:1 |
5 | 100 | 94300 |
86900 |
30000 |
1.06 | 0.81 |
| 8 | 2a | 30:1:1 |
1 | 98 | 6400 | 7600 | 8000 | 1.11 | 0.87 |
| 9 | 2a | 60:1:1 |
4 | 99 | 12900 |
12350 |
10000 |
1.10 | 0.87 |
| 10h | 2a | 50:1:1 |
4 | 99 | 10300 |
9100 | 8400 | 1.15 | <0.05 |
The Mn and dispersity data summarized in Table 3, the linear variation of the Mn,NMR and Mn,SEC molar mass values of PBPLCH2OTBDMS as a function of the monomer loading/conversion and the NMR spectra (see the ESI Fig. S12–S16†), all testify a similar well-controlled polymerization of rac-BPLCH2OTBDMS to the 2a–d/iPrOH catalytic systems, as that observed for rac-BPLCH2OiPr and rac-BPLCH2OtBu, affording well-defined α-isopropoxycarbonyl, ω-hydroxy end-capped PHAs. Also, similar to PBPLCH2OiPr and PBPLCH2OtBu discussed above, all the PBPLCH2OTBDMS systems revealed to be syndio-enriched (Pr = 0.76–0.87; Fig. 4) (δ r(CO) = ca. 169.61, δ m(CO) = ca. 169.52 ppm; δ r(CH) = ca. 71.16, δ m(CH) = ca. 71.08 ppm; δ r(CH2O) = ca. 63.50, δ m(CH2O) = ca. 63.35 ppm). Obviously, the stereochemistry of the ROP of rac-BPLCH2OTBDMS is controlled by steric components only, where the ascending catalyst selectivity trend is as follows: 2d (Cl substituents); Pr = 0.76 < 2c (Me substituents); Pr = 0.77 < 2b (tBu substituents); Pr = 0.84 < 2a (cumyl substituents); Pr = 0.87.
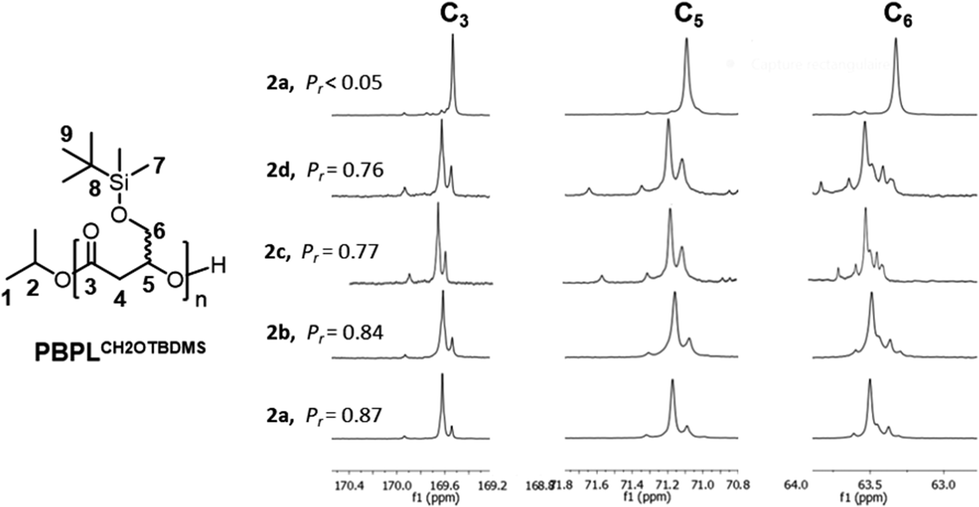 | ||
| Fig. 4 Zoomed regions of the 13C{1H} NMR spectra (125 MHz, CDCl3, 23 °C) of PBPLCH2OTBDMS prepared by ROP of rac-BPLCH2OTBDMS, except for the top spectrum of enantiopure (S)-BPLCH2OTBDMS (Table 3, entry 10), mediated by the 2a, 2b, 2c, or 2d/iPrOH (1:1) catalytic systems (Table 3, entries 9, 5, 3 and 1, respectively); *stands for residual monomer resonances. | ||
Kinetics of the ROP of rac-BPLCH2OiPr/OtBu/OTBDMS
Monitoring of NMR-scale polymerizations of rac-BPLCH2OiPr/OtBu/OTBDMS performed with 2a–d/iPrOH confirmed the kinetic trends assessed from the batch experiments (Tables 1–3). Linear semi-logarithmic plots established that all reactions were first-order in the monomer, with apparent rate constants kapp > 55 min−1 for BPLCH2OiPr/OtBu/2a–b (complete conversion was observed after only 5 min under these conditions; see Table 1, entry 9 and Table 2, entry 11); kapp = 1.143 ± 0.072 min−1 for BPLCH2OTBDMS/2a; 0.797 ± 0.031 min−1 for BPLCH2OTBDMS/2b; 0.323 ± 0.033 min−1 for BPLCH2OtBu/2c; 0.265 ± 0.032 min−1 for BPLCH2OiPr/2c; 0.046 ± 0.041 min−1 for BPLCH2OtBu/2d; 0.0088 ± 0.0021 min−1 for BPLCH2OiPr/2d; 0.0037 ± 0.0033 min−1 for BPLCH2OTBDMS/2c (Fig. 5). Overall, the major trend for the ability of the monomers to ring-open polymerize was thus BPLCH2OtBu ≥ BPLCH2OiPr ≫ BPLCH2OTBDMS, while the catalysts’ activity thus generally followed the order 2a–b ≫ 2c ≫ 2d, as previously observed for the ROP of various BPLFGs β-lactones (FG = CH2OAll, CH2OBn, CH2OMe, CH2OPh, CH2CH2OBn, and CH2SPh) promoted by these catalytic systems (vide supra).7,10 | ||
| Fig. 5 Semi-logarithmic first-order plots for the ROP of rac-BPLFGs (FG = CH2OiPr, CH2OtBu, and CH2OTBDMS) mediated by 2a–d/iPrOH (20 °C, toluene; [BPLFG]0/{[2a–c]0/[iPrOH]0} = 60–75:1:1 and [BPLFG]0/{[2d]0/[iPrOH]0} = 25–30:1:1): 2a (Table 1, entry 9; Table 2, entry 11; Table 3, entry 9); 2b (Table 1, entry 6; Table 2, entry 6; Table 3, entry 4); 2c (Table 1, entry 2; Table 2, entry 4; Table 3, entry 3) and 2d (Table 1, entry 1; Table 2, entry 1); plots from 2a–b all overlap due to similar higher activity of these catalysts regardless of the monomer functionality, and are represented as ▲. The slow kinetics of the ROP of rac-BPLOTBDMS with 2d (Table 3, entry 1) is not shown. | ||
The thermal characteristics of PBPLCH2ORs synthesized by ROP of rac-BPLCH2OR (R = iPr, tBu, and TBDMS) were enhanced by the 2a–d catalytic systems.
The thermal signature of the new functional PHAs synthesized in this work was briefly investigated by differential scanning calorimetry (DSC, Fig. S17–S20†). The glass transition temperature (Tg) values of syndio-enriched PBPLCH2OiPr/CH2OtBu/CH2OTBDMS slightly changed from one to another PHA, and ranged from −18 to +9 °C (Table 4). When compared to the corresponding values gathered for PBPLCH2OMe/CH2OAllyl/CH2OBn (Tg ranging from −38 to 0 °C, Table 4), these values appear to grossly increase with the steric hindrance imparted by the alkoxy/silyloxy side-functionality which decreases the motion of the macromolecules. Also, among the different syndio-enriched polymers herein prepared, only PBPLCH2OTBDMS featured a semi-crystalline behavior with Tm values of 118–119 °C (Fig. S19 and S20†).
| rac-BPLFGs |
|
|
|
|
|
|
|---|---|---|---|---|---|---|
| Cat 2 (R′ = R′′) | rac-BPLCH2OMe [7] | rac-BPLCH2OAll [7] | rac-BPLCH2OBn [7] | rac-BPLCH2OiPr (this work) | rac-BPLCH2OtBu (this work) | rac-BPLCH2OTBDMS (this work) |
| a Table 1, entry 8. b Table 2, entry 5. c Table 3, entry 6; similar values (Tg = 8 °C, Tm = 118 °C) were recorded for the sample in Table 3, entry 7. d Not determined. | ||||||
| Crowded (cumyl, tBu) ( 2a–b ) | Syndiotactic | Syndiotactic | Syndiotactic | Syndiotactic | Syndiotactic | Syndiotactic |
| P r = 0.78–0.81 | P r = 0.81–0.84 | P r = 0.85–0.90 | P r = 0.82–0.86 | P r = 0.78–0.84 | P r = 0.81–0.87 | |
| T g = −12 °C | T g = −38 °C | T g = 0 °C | T g = −18 °Ca | T g = −6 °Cb | T g = 9 °Cc | |
| T m = 116 °C | T m = 85 °C | no Tm obsv. | no Tm obsv. a | no Tm obsv. b | T m = 119 °C c | |
| Aliphatic non-crowded (Me; 2c) | Atactic | Atactic | Atactic | Syndiotactic | Syndiotactic | Syndiotactic |
| P r = 0.49 | P r = 0.49 | P r = 0.50 | P r = 0.71–0.72 | P r = 0.74–0.75 | P r = 0.77 | |
| T g = −18 °C | T g = −40 °C | T g = −6 °C | T g, Tm = n.d.d | T g, Tm = n.d.d | T g, Tm = n.d.d | |
| Halogenated non-crowded (Cl, 2d) | Isotactic | Isotactic | Isotactic | Syndiotactic | Syndiotactic | Syndiotactic |
| P r = 0.10 | P r = 0.09 | P r = 0.10 | P r = 0.70 | P r = 0.70–0.71 | P r = 0.76 | |
| T g = −18 °C | T g = −39 °C | T g = 0 °C | T g, Tm = n.d.d | T g, Tm = n.d.d | T g, Tm = n.d.d | |





Conclusions
Table 4 summarizes the stereoselectivity outcome of the ROP of rac-BPLCH2OMe/CH2OAll/CH2OBn/CH2OiPr/CH2OtBu/CH2OTBDMS as a function of yttrium catalytic systems, differentiating the latter ones according to the presence of highly sterically crowded substituents (i.e., tBu, cumyl; 2a–b), simple aliphatic non-crowded substituents (Me, 2c) and halogeno non-crowded substituents (Cl, 2d). Highly syndio-enriched PBPLCH2OiPr/OtBu/OTBDMS were obtained from 2a–b, alike PBPLCH2OMe/OAll/OBn (Pr = 0.78–0.87 vs. 0.78–0.90; respectively). However, substitution of one or two hydrogen atoms in the alkoxide “outer” methylene group of rac-BPLCH2OMe/OAll/OBn by one or two methyl groups – as in rac-BPLCH2OiPr and rac-BPLCH2OtBu – or with rac-BPLCH2OTBDMS resulted in: (i) changing the stereoregularity of the polymer from atactic to syndio-enriched polymers with catalyst 2c (Pr = 0.49/0.50 vs. 0.71–0.77, respectively), and (ii) switching from isotactic to syndio-enriched polymers with catalyst 2d (Pr = 0.09–0.10 vs. 0.76–0.71, respectively). Obviously, these observations evidence that the stereocontrol in the ROP of racemic 4-alkoxymethylene-β-propiolactones is driven, systematically by steric considerations, but in a few specific cases by electronic ones as well. For rac-BPLCH2OiPr, rac-BPLCH2OtBu and rac-BPLCH2OTBDMS, apparently due to the large steric constraints induced by the alkoxy(silyloxy) side-functionality, all reactions lead to the formation of syndio-enriched polymers, regardless of the catalyst – a crowded one or a non-crowded one – used. This is what is expected from a ‘regular’ chain-end stereocontrol mechanism, in which minimization of steric repulsions in the transition state favors the enchainment of monomer units alternately with opposite absolute configurations (and hence the formation of syndiotactic/heterotactic polymers).1 Only the specific combination of a BPLFG monomer containing two methylene hydrogens apart from the central oxygen on the methylene alkoxy side-functionality (i.e., FG = CH2OMe, CH2OAllyl, and CH2OBn) with a catalyst bearing chloro-substituents (2a) produced isotactic PHAs; this is assumed to arise from attractive interactions between the ligand chloro substituents and the hydrogen atoms on the alkoxy (methoxy, allyloxy, and benzyloxy) side chain of the ring-opened monomer/growing polymer chain.7 On the other hand, a catalyst with highly sterically crowding substituents on the ligand platform is necessary to recover syndio-enriched PHAs from the latter BPLCH2OMe,CH2OAll,CH2OBn monomers, which show no major steric bulkiness on the side chain alkoxymethylene moiety. Thus, at this stage of our investigations, in a ROP mediated by a typically isoselective yttrium catalyst, bulkiness of the –OR methylene-alkoxy/silyloxy moiety of BPLCH2OR monomers is not sufficient to impart isoselectivity, while the presence of two methylene groups adjacent to the oxygen within these BPLCH2OMe,CH2OAllyl,CH2OBn still appears mandatory to access desirable synthetic isotactic PHAs that mimic their natural analogues. Ongoing work by our group aims at examining further the contribution of the two methylene groups, apart from the oxygen in the side-functionality, in the stereoselective ROP of functional β-lactones towards the synthesis of isotactic functional PHAs.Author contributions
CRediT: Rama M. Shakaroun: investigation (lead) and writing – original draft (supporting); Ali Dhaini: investigation (supporting) and writing – review & editing (supporting); Romain Ligny: investigation (supporting); Ali Alaaeddine: supervision (supporting); Sophie Guillaume: conceptualization (lead), supervision (lead), writing – original draft (supporting), and writing – review & editing (lead); Jean-François Carpentier: conceptualization (lead), supervision (lead), writing – original draft (lead), and writing – review & editing (lead).Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research was financially supported in part by the University of Rennes (Ph.D. grants to R. S., A. D. and R. L.), the Lebanese University (Ph.D. grants to R. S. and A. D.) and the Région Bretagne (Ph.D. ARED grant to R. L.). We are grateful to CRMPO and UAR ScanMAT, especially to Philippe Jéhan, Elsa Caytan, and Marielle Blot for MS, NMR and chiral HPLC chromatography analyses, respectively.References
- For leading reviews on stereoselective metal-catalyzed ROP of lactones and related derivatives, see: (a) O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS PubMed; (b) P. Dubois, O. Coulembier and J.-M. Raquez, Handbook of Ring-Opening Polymerization, Wiley, Weinheim, 2009 CrossRef; (c) C. M. Thomas, Chem. Soc. Rev., 2010, 39, 165–173 RSC; (d) M. J. Stanford and A. P. Dove, Chem. Soc. Rev., 2010, 39, 486–494 RSC; (e) P. J. Dijkstra, H. Du and J. Feijen, Polym. Chem., 2011, 2, 520–527 RSC; (f) A. Buchard, C. M. Bakewell, J. Weiner and C. K. Williams, Top. Organomet. Chem., 2012, 39, 175–224 CrossRef CAS PubMed; (g) S. M. Guillaume, E. Kirillov, Y. Sarazin and J.-F. Carpentier, Chem. – Eur. J., 2015, 21, 7988–8003 CrossRef CAS PubMed; (h) J. C. Worch, H. Prydderch, S. Jimaja, P. Bexis, M. L. Becker and A. P. Dove, Nat. Rev. Chem., 2019, 3, 514–535 CrossRef CAS; (i) M. J.-L. Tschan, R. M. Gauvin and C. M. Thomas, Chem. Soc. Rev., 2021, 50, 13587–13608 RSC; (j) L. Al-Shok, D. M. Haddleton and F. Adams, Progress in Catalytic Ring-Opening Polymerization of Biobased Lactones, in Advances in Polymer Science, 2022, Springer Search PubMed; (k) A. H. Westlie, E. C. Quinn, C. R. Parker and E. Y.-X. Chen, Prog. Polym. Sci., 2022, 134, 101608 CrossRef CAS.
- For recent examples of achiral organocatalysts for highly isoselective ROP of racemic lactide, see: (a) B. Koca, D. Akgul and V. Aviyente, Eur. Polym. J., 2019, 121, 109291 CrossRef CAS and references cited therein; (b) Y. Liu, J. Zhang, X. Kou, S. Liu and Z. Li, ACS Macro Lett., 2022, 11, 1183–1189 CrossRef CAS PubMed; (c) F. Ren, X. Li, J. Xian, X. Han, L. Cao, X. Pan and J. Wu, J. Polym. Sci., 2022, 60, 2847–2854 CrossRef CAS.
- For selected examples of metal-based catalysts bearing non-chiral ligands for isoselective ROP of rac-LA, see: (a) N. Nomura, R. Ishii, Y. Yamamoto and T. Kondo, Chem. – Eur. J., 2007, 13, 4433–4451 CrossRef CAS PubMed; (b) P. McKeown, M. G. Davidson, G. Kociok-Köhn and M. D. Jones, Chem. Commun., 2016, 52, 10431–10434 RSC; (c) D. Myers, A. J. P. White, C. M. Forsyth, M. Bown and C. K. Williams, Angew. Chem., Int. Ed., 2017, 56, 5277–5282 CrossRef CAS PubMed; (d) Z. Mou, B. Liu, M. Wang, H. Xie, P. Li, L. Li, S. Lia and D. Cui, Chem. Commun., 2014, 50, 11411–11414 RSC; (e) Z. Zhuo, C. Zhang, Y. Luo, Y. Wang, Y. Yao, D. Yuan and D. Cui, Chem. Commun., 2018, 54, 11998–12001 RSC; (f) C. Kan, J. Hu, Y. Huang, H. Wang and H. Ma, Macromolecules, 2017, 50, 7911–7919 CrossRef CAS.
- (a) X. Tang and E. Y.-X. Chen, Nat. Commun., 2018, 9, 2345–2356 CrossRef PubMed; (b) E. Y.-X. Chen and X. Tang, US Pat. Appl, 2019, 0211144 Search PubMed; (c) X. Tang, A. H. Westlie, E. M. Watson and E. Y.-X. Chen, Science, 2019, 366, 754–758 CrossRef CAS PubMed; (d) Z. Zhang, C. Shi, M. Scoti, X. Tang and E. Y.-X. Chen, J. Am. Chem. Soc., 2022, 144, 20016–20024 CrossRef CAS PubMed.
- (a) For a review on stereoselective ROP of β-lactones, see: J.-F. Carpentier, Macromol. Rapid Commun., 2010, 31, 1696–1705 CrossRef CAS PubMed. β-Butyrolactone (BL, aka BPLMe) is the most common chiral β-lactone; few catalyst systems have shown isoselectivity towards rac-BL with Pm values (i.e., the probability of meso/isotactic enchainment) in the range 0.65–0.85; see: (b) Z. Zhuo, C. Zhang, Y. Luo, Y. Wang, Y. Yao, D. Yuan and D. Cui, Chem. Commun., 2018, 54, 11998–12001 RSC; (c) N. Ajellal, G. Durieux, L. Delevoye, G. Tricot, C. Dujardin, C. M. Thomas and R. M. Gauvin, Chem. Commun., 2010, 46, 1032–1034 RSC; (d) M. Zintl, F. Molnar, T. Urban, V. Bernhart, P. Preishuber-Pflügl and B. Rieger, Angew. Chem., Int. Ed., 2008, 47, 3458–3460 ( Angew. Chem. , 2008 , 120 , 3508–3510 ) CrossRef CAS PubMed; (e) R. Reichardt, S. Vagin, R. Reithmeier, A. K. Ott and B. Rieger, Macromolecules, 2010, 43, 9311–9317 CrossRef CAS; (f) S. Vagin, M. Winnacker, A. Kronast, P. T. Altenbuchner, P. Deglmann, C. Sinkel, R. Loos and B. Rieger, ChemCatChem, 2015, 7, 3963–3971 CrossRef CAS.
- (a) A. Amgoune, C. M. Thomas, S. Ilinca, T. Roisnel and J.-F. Carpentier, Angew. Chem., Int. Ed., 2006, 45, 2782–2784 CrossRef CAS PubMed; Angew. Chem. 2006 118 2848–2850 Search PubMed. For recent accounts, see: (b) J.-F. Carpentier, Organometallics, 2015, 34, 4175–4189 CrossRef CAS; (c) H. Li, R. M. Shakaroun, S. M. Guillaume and J.-F. Carpentier, Chem. – Eur. J., 2020, 26, 128–138 CrossRef CAS PubMed.
- (a) R. Ligny, M. M. Hänninen, S. M. Guillaume and J.-F. Carpentier, Angew. Chem., Int. Ed., 2017, 56, 10388–10393 ( Angew. Chem. , 2017 , 129 , 10524–10529 ) CrossRef CAS PubMed; (b) R. Ligny, M. M. Hänninen, S. M. Guillaume and J.-F. Carpentier, Chem. Commun., 2018, 54, 8024–8031 RSC.
- For a recent example of isoselective ROP of racemic β-thiobutyrolactone by the same type of achiral alkoxy-amino-bisphenolate-yttrium catalysts, see: H. Li, J. Ollivier, S. M. Guillaume and J.-F. Carpentier, Angew. Chem., Int. Ed., 2022, 61, e202202386 CAS.
- Other types of NCIs have also been proposed to play a beneficial role in stereoselective ROP of rac-LA promoted by Zn-β-diketiminate and Al-bis(cathecolato-amino) catalysts; see: (a) E. L. Marshall, V. C. Gibson and H. S. Rzepa, J. Am. Chem. Soc., 2005, 127, 6048–6051 CrossRef CAS PubMed; (b) S. Gesslbauer, R. Savela, Y. Chen, A. J. P. White and C. Romain, ACS Catal., 2019, 9, 7912–7920 CrossRef CAS.
- R. M. Shakaroun, H. Li, P. Jéhan, M. Blot, A. Alaaeddine, J.-F. Carpentier and S. M. Guillaume, Polym. Chem., 2021, 12, 4022–4034 RSC.
- Rac-BPLCH2OiPr and rac-BPLCH2OtBu are new monomers. For the synthesis and organocatalyzed ROP of rac-BP CH2OTBDMS, see: R. M. Shakaroun, H. Li, P. Jéhan, A. Alaaeddine, J.-F. Carpentier and S. M. Guillaume, Polym. Chem., 2020, 11, 2640–2652 RSC.
- (a) J. A. Schmidt, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2005, 127, 11426–11435 CrossRef CAS PubMed; (b) J. W. Kramer and G. W. Coates, Tetrahedron, 2008, 64, 6973–6978 CrossRef CAS PubMed.
- M. Bouyahyi, N. Ajellal, E. Kirillov, C. M. Thomas and J.-F. Carpentier, Chem. – Eur. J., 2011, 17, 1872–1883 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: General conditions, synthesis and characterization of BPLCH2OR monomers, NMR and mass spectra, and DSC traces. See DOI: https://doi.org/10.1039/d2py01573k |
| ‡ Pm is the probability of meso linkages, that is, the enchainment of two monomer units with the same configuration. Pm = 1 − Pr, where Pr is the probability of racemo linkage, that is, the enchainment of two monomer units with the opposite configuration. |
| § Note that, similarly, the ROP of the parent sulfur BPLCH2SPh recovered the corresponding syndiotactic PHA; see ref. 10. |
| This journal is © The Royal Society of Chemistry 2023 |