
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced stability of complex coacervate core micelles following different core-crosslinking strategies†
Riahna
Kembaren
ab,
J. Mieke
Kleijn
a,
Jan Willem
Borst
 b,
Marleen
Kamperman
c and
Anton H.
Hofman
*c
b,
Marleen
Kamperman
c and
Anton H.
Hofman
*c
aPhysical Chemistry and Soft Matter, Wageningen University & Research, Stippeneng 4, 6708 WE Wageningen, The Netherlands
bLaboratory of Biochemistry, Microspectroscopy Research Facility, Wageningen University & Research, Stippeneng 4, 6708 WE Wageningen, The Netherlands
cPolymer Science, Zernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: a.h.hofman@rug.nl
First published on 1st April 2022
Abstract
Complex coacervate core micelles (C3Ms) are formed by mixing aqueous solutions of a charged (bio)macromolecule with an oppositely charged-neutral hydrophilic diblock copolymer. The stability of these structures is dependent on the ionic strength of the solution; above a critical ionic strength, the micelles will completely disintegrate. This instability at high ionic strengths is the main drawback for their application in, e.g., drug delivery systems or protein protection. In addition, the stability of C3Ms composed of weak polyelectrolytes is pH-dependent as well. The aim of this study is to assess the effectiveness of covalent crosslinking of the complex coacervate core to improve the stability of C3Ms. We studied the formation of C3Ms using a quaternized and amine-functionalized cationic-neutral diblock copolymer, poly(2-vinylpyridine)-block-poly(ethylene oxide) (QP2VP-b-PEO), and an anionic homopolymer, poly(acrylic acid) (PAA). Two different core-crosslinking strategies were employed that resulted in crosslinks between both types of polyelectrolyte chains in the core (i.e., between QP2VP and PAA) or in crosslinks between polyelectrolyte chains of the same type only (i.e., QP2VP). For these two strategies we used the crosslinkers 1-ethyl-3-(3′-dimethylaminopropyl)carbodiimide hydrochloride (EDC) and dimethyl-3,3′-dithiopropionimidate dihydrochloride (DTBP), respectively. EDC provides permanent crosslinks, while DTBP crosslinks can be broken by a reducing agent. Dynamic light scattering showed that both approaches significantly improved the stability of C3Ms against salt and pH changes. Furthermore, reduction of the disulphide bridges in the DTBP core-crosslinked micelles largely restored the original salt-stability profile. Therefore, this feature provides an excellent starting point for the application of C3Ms in controlled release formulations.
1 Introduction
Complex coacervate core micelles (C3Ms), also known as block ionomer complexes1 or polyion complex micelles2 are polymeric nanostructures with a complex coacervate core and a neutral hydrophilic shell.3–5 C3M formation is based on self-assembly of ionic-neutral hydrophilic diblock copolymers and oppositely charged macromolecules at concentrations exceeding the critical micelle concentration (CMC).3,6–8 The driving forces for C3M formation are Coulombic attraction between the oppositely charged parts of the polyelectrolyte chains and the entropy gain from the release of the counterions upon complex formation.3,9–11 C3Ms are easy to prepare as it only involves a simple mixing step of aqueous solutions. C3Ms can be used as carrier for charged macromolecules such as proteins, DNA, and RNA.3,12–14 Furthermore, C3Ms are interesting for many other applications, including their use as bionanoreactors or nanoprobes, for the encapsulation of biomolecules, and as drug delivery systems.2,3,15–19Despite their ease of preparation, C3Ms can easily disintegrate, in particular at high ionic strength, which is the main limitation for their practical application.3,14,15,19–21 Increasing the salt concentration decreases the driving forces for C3M formation, resulting in a reduced stability of the C3Ms. Above a certain salt concentration, also known as the critical salt concentration (CSC), C3Ms will completely disintegrate. In addition, in case of weak polyelectrolytes, the pH is an essential parameter for the stability of C3Ms as well.3,15
A strategy to overcome undesired disintegration of C3Ms is by crosslinking the polymers. This can be done reversibly or irreversibly, either in the core or in the corona of the micelles.15,22–25 However, crosslinking the corona/shell may affect the surface characteristics and reduces the hydrophilicity and thus the solubility of the micelles.24 With the core-crosslinking strategy, on the other hand, we expect that the micelles' surface characteristics will remain the same. Core-crosslinking of C3Ms can be achieved by binding together one or both types of polymers present in the core of C3Ms, either physically or covalently.26–28 Chemical crosslinking of the components in the core will provide a more stable network. Chemical crosslinks can be introduced, for example, by irradiation (photo-polymerization), an enzymatic reaction, or by using “click” chemistry.27 To crosslink C3Ms, the polymers must contain reactive functional groups, such as amines, thiols, carboxylates, hydroxyls, acetoacetyl groups, acetal groups, acrylamide derivates, or carbonyl groups.29,30
The aim of this study is to determine the effectiveness of covalently crosslinking the core of C3Ms to improve their stability against salt addition and pH changes. C3Ms were prepared using a cationic-neutral diblock copolymer, poly(2-vinylpyridine)128-b-poly(ethylene oxide)477 (P2VP128-b-PEO477), and an anionic homopolymer, poly(acrylic acid)118 (PAA118). Beforehand, the P2VP-containing diblock copolymer was functionalized with primary amine groups via quaternization. Since primary amines are known to participate in quaternization reactions themselves, protection chemistry was required during the quaternization procedure. Several amine protecting groups are convenient to use like 9-fluorenylmethyl carbamate (FMOC), t-butyl carbamate (BOC), and phthalimides31–33 We have chosen N-(2-bromoethyl)phthalimide because of its stability under the conditions required for the quaternization reaction (including heating to 150 °C). After deprotection, the amine can be used for core-crosslinking by adding a bifunctional crosslinker to the C3M solution.15,24 In this study, we used two types of crosslinkers, 1-ethyl-3-(3′-dimethylaminopropyl)carbodiimide hydrochloride (EDC) and dimethyl 3,3′-dithiopropionimidate dihydrochloride (DTBP). EDC forms irreversible crosslinks between amine and carboxylic groups, while DTBP connects two amine groups. Furthermore, since DTBP crosslinks contain disulphide bridges, these crosslinks can be broken again by addition of a reducing agent. We have investigated these two types of crosslinkers to crosslink the core of C3Ms (Scheme 1). Next, we have compared these different core-crosslinking strategies, i.e., network formation between only one or between both types of polymers in the core, and permanent versus reversible crosslinking. Using dynamic light scattering (DLS), we investigated the formation of C3Ms, their size and stability as a function of ionic strength and pH before and after crosslinking, and in case of DTBP also after breaking the crosslinks.
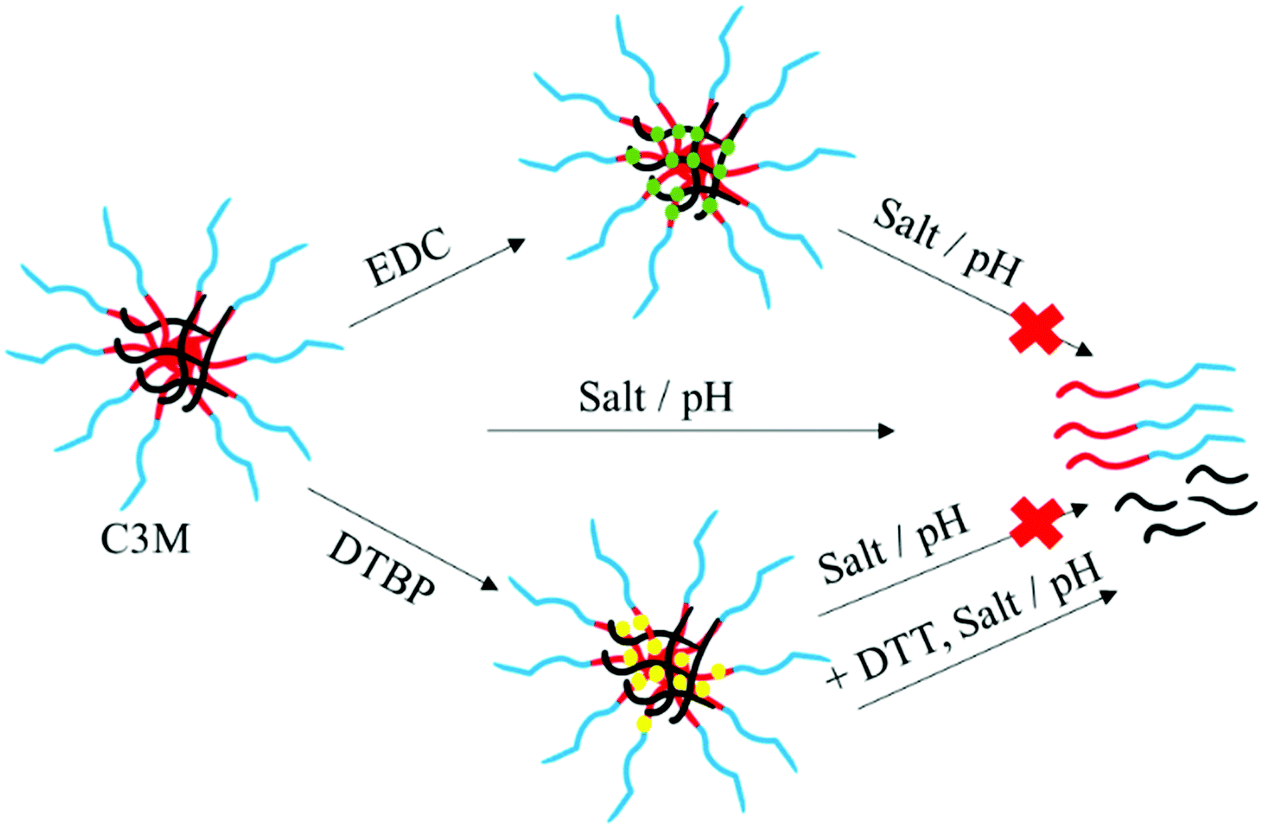 | ||
| Scheme 1 Schematic overview of core-crosslinking strategies of C3Ms using the crosslinkers EDC and DTBP. The use of EDC results in permanent crosslinks, whereas DTBP provides crosslinks that can be broken by reducing agents like 1,4-dithiothreitol (DTT). Red-blue: charged-neutral diblock copolymer. Black: oppositely charged homopolymer. Green and yellow: crosslinks formed by EDC and DTBP, respectively. | ||
2 Experimental section
2.1 Materials
The diblock copolymer poly(2-vinylpyridine)128-block-poly(ethylene oxide)477 (P2VP128-b-PEO477) (Mn = 34.5 kg mol−1, Đ = 1.1) and homopolymer poly(acrylic acid)118 (PAA118) (Mn = 8.5 kg mol−1, Đ = 1.07) were obtained from Polymer Source Inc. The crosslinkers 1-ethyl-3-(3′-dimethylaminopropyl)carbodiimide hydrochloride (EDC) and dimethyl 3,3′-dithiopropionimidate dihydrochloride (DTBP) were purchased from Thermo Fisher Scientific. The quaternization reagent N-(2-bromoethyl)phthalimide and the hydrazine hydrate solution (78–82% in H2O) were obtained from Sigma-Aldrich. 1,4-Dithiothreitol (DTT) was purchased from Carl Roth.2.2 Polymer quaternization
N-(2-Bromoethyl) phthalimide (5 eq. with respect to 2VP, 1.18 g, 4.64 mmol) and diblock copolymer poly(2-vinylpyridine)128-block-poly(ethylene oxide)477 (0.25 g, 0.93 mmol of 2VP units) were mixed and subsequently heated at 150 °C for 2 hours in a small vial with stirring bar. No additional solvents were added; the molten quaternization agent simultaneously acts as a solvent (Tm = 82 °C). After cooling to room temperature, about 5 mL of dichloromethane was added to dissolve the solidified product and then the solution was precipitated in diethyl ether. The precipitated polymer was filtered and washed five times with diethyl ether. Finally, the polymer was dried in a vacuum oven, resulting in an off-white powder.2.3 Deprotection of the quaternized diblock copolymer
The polymer powder (0.3 g) that was obtained after the quaternization reaction was dissolved in 8 mL of ethanol and heated at 80 °C with reflux under an N2 atmosphere. Subsequently, hydrazine hydrate solution (78–82% in H2O) (10 eq. with respect to 2VP) was added for deprotection of the quaternized diblock copolymer. The mixture was kept at 80 °C for 18 hours. After that, the mixture was cooled to 4 °C and filtered to remove phthalhydrazide. The filtrate was collected, and the volume of filtrate was reduced to about 3 mL by using a rotary evaporator. The solution was then dialyzed (MWCO 3.5 kg mol−1) for three days against water to remove excess hydrazine hydrate and the side product phthalhydrazide.34 After lyophilization the polymer product was obtained as a white powder and analyzed by using 1H-NMR (using a Bruker Avance III 400 MHz NMR spectrometer), FTIR (using a Bruker Tensor 27 IR spectrometer), and a ninhydrin assay. For the ninhydrin assay, the polymer (0.5 mg) was dissolved in 50 μL of water, then 200 μL of ninhydrin solution (2% in ethanol) was added. The solution was heated at 90 °C for 3 min, then cooled to room temperature. An overview of the quaternization and deprotection reactions is presented in Scheme 2. | ||
| Scheme 2 Synthetic pathway for the preparation of amine-functionalized diblock copolymer, P2VP128-b-PEO477, via quaternization and deprotection. | ||
2.4 Formation of C3Ms
PAA and quaternized diblock copolymer solutions were prepared separately in a 10 mM sodium phosphate buffer at pH 7.4. All solutions were filtered through 0.2 μm polyethersulfone membrane syringe filters. To determine the preferred micellar composition (PMC), i.e., the optimal polymer composition for C3M formation, different concentrations of poly(acrylic acid) from the stock solution of 60 μM were added to a constant concentration of quaternized diblock copolymer solution (5 μM) with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume ratio (volume total of 1 mL). The mixed solutions were left to equilibrate at room temperature overnight before measurement. The extent of C3M formation was determined by using DLS. After establishment of the PMC, all C3M formulations were made at the PMC and several dilution series with 10 mM sodium phosphate buffer at pH 7.4 were prepared to determine the critical micelle concentration (CMC).
:1 volume ratio (volume total of 1 mL). The mixed solutions were left to equilibrate at room temperature overnight before measurement. The extent of C3M formation was determined by using DLS. After establishment of the PMC, all C3M formulations were made at the PMC and several dilution series with 10 mM sodium phosphate buffer at pH 7.4 were prepared to determine the critical micelle concentration (CMC).
2.5 Core-crosslinking of C3Ms
Core-crosslinking of the C3Ms (at the PMC) was achieved by using two different kinds of crosslinking agents. After mixing of the quaternized diblock copolymer with PAA to form C3Ms (total volume of 5 mL), either EDC (2.0 eq. to amine) or DTBP (3.9 eq. to amine) crosslinker was added to the micellar solutions. The crosslinking reaction was performed in 10 mM buffer sodium phosphate pH 7.4 at room temperature for at least 3 hours. Unreacted crosslinking agent is expected to degrade eventually due to the hydrolysis of linker.2.6 C3M stability
The stability of C3Ms was investigated by salt and acid–base titrations while monitoring the scattering intensity of the micelles by using DLS. For salt stability observation, a 4.0 M NaCl solution was titrated into the C3M solution. For assessment of the pH stability, 0.1 M NaOH and 0.1 M HCl solutions were titrated into the C3M solutions. To cleave the crosslinks originating from DTBP, a final DTT concentration of 50 mM was added to DTBP core-crosslinked C3Ms and incubated for 30 minutes. Subsequently, the solution was titrated with 4 M NaCl and the extent of micelle disintegration was monitored by using DLS.2.7 Dynamic light scattering (DLS)
Light scattering measurements were performed on an ALV-LSE 41/CGS-8F goniometer system equipped with a DPSS laser (λ = 660 nm, 200 mW).6,35 The PMC, mean hydrodynamic radius of the micelles (Rh), and polydispersity index (PDI) were measured at a fixed 90° angle. The shape of the C3Ms was resolved by using multi-angle DLS at angles ranging from 50° to 120° in steps of 10°. Intensity correlation functions were recorded for 10 seconds and averaged over 8 runs per angle. Hydrodynamic radii and polydispersity indices were acquired through a second order cumulant analysis.3 Results and discussion
3.1 Functionalization of the diblock copolymer for crosslinking purposes
Quaternization of P2VP using N-(2-bromoethyl)phthalimide aims to simultaneously introduce positive charges and primary amine functional groups onto the P2VP-b-PEO diblock copolymer.36–39 Primary amines are interesting, as they allow a large variety of click reactions for crosslinking. We used a phthalimide-protected primary amine group in order to prevent self-cyclization or polymerization during the quaternization reaction, which can be easily deprotected afterwards by hydrazinolysis using hydrazine hydrate.33,40 In this study, we tested the homopolymer P2VP first before using the diblock copolymer P2VP128-b-PEO477. The homopolymer functionalization was successful with a degree of quaternization (DQ) of 92% (Fig. S1 and S2, ESI†).Fig. 1A shows the 1H-NMR spectrum of the P2VP128-b-PEO477 copolymer before quaternization. After quaternization with N-(2-bromoethyl)phthalimide, the signals from P2VP's aromatic ring are clearly shifted (6–8.5 ppm) and an intense peak appeared at 7.78 ppm from the phthalimide aromatic rings (Fig. 1B; protons [j] and [k]). Moreover, a new broad signal showed up around 3.5-5.5 ppm originating from the two CH2 groups of ethyl phthalimide (protons [h] and [i]). This result indicates that the polymer was successfully quaternized and a degree of quaternization of about 85% was calculated from the ratio of the integral area of the aromatic rings and the integral area of P2VP's backbone (Fig. S3 and S4, ESI†). After treatment of the quaternized diblock copolymer with hydrazine hydrate, the disappearance of the peak at 7.78 ppm originating from protons [j] and [k] indicates full deprotection of the primary amine groups (Fig. 1C).36,40,41
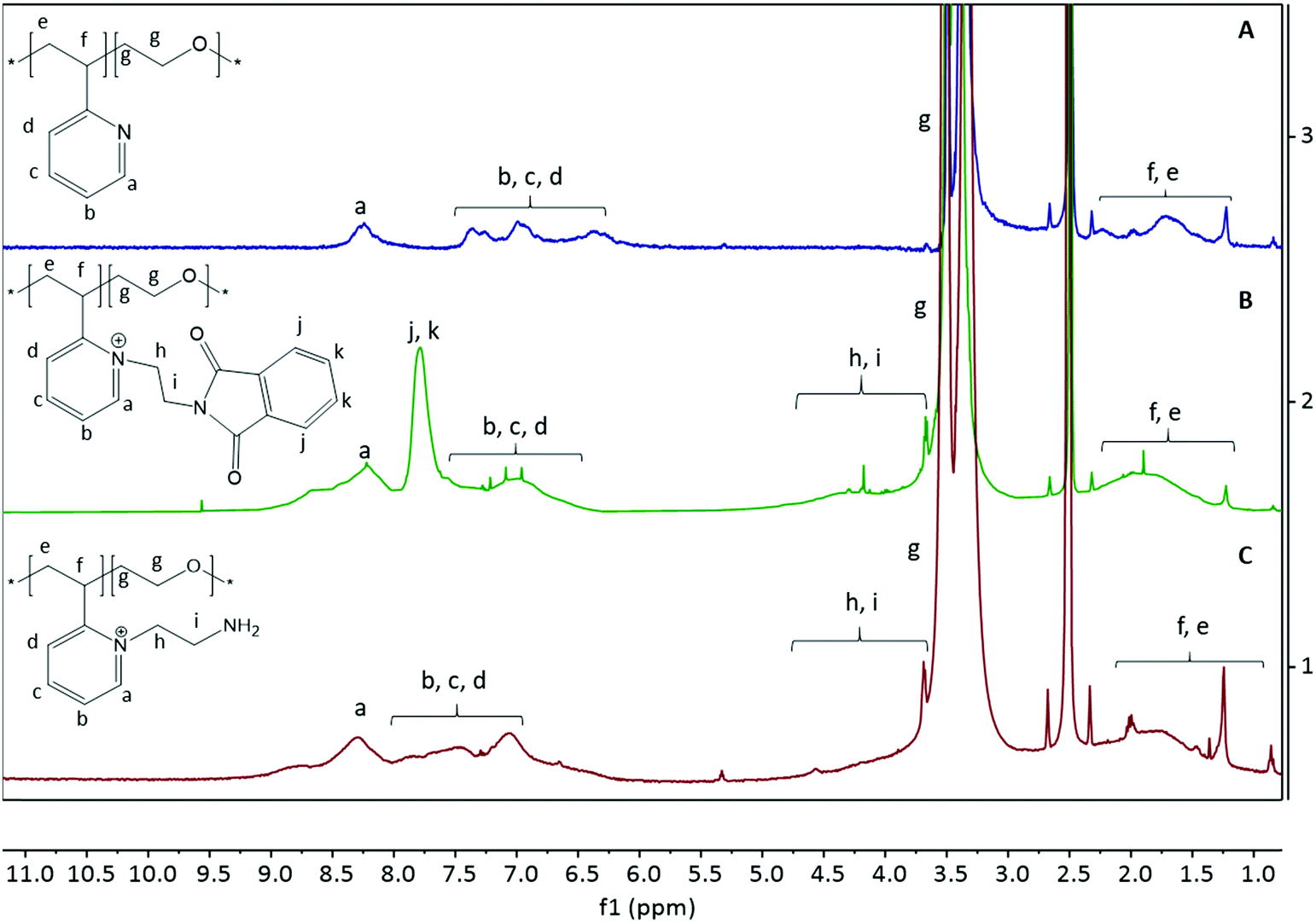 | ||
| Fig. 1 1H-NMR spectra of the functionalized diblock copolymer P2VP128-b-PEO477. (A) Diblock copolymer before quaternization, (B) diblock copolymer after quaternization using N-(2-bromoethyl)phthalimide, (C) quaternized diblock copolymer after deprotection using hydrazine hydrate. Spectra A, B, and C were recorded in DMSO-d6. | ||
Next, we conducted Fourier-transform infrared spectroscopy (FTIR) to further characterize the functionalized diblock copolymer. Fig. 2 demonstrates FTIR spectra of quaternized P2VP128-b-PEO477. After quaternization of P2VP128-b-PEO477, the spectrum of the diblock copolymer shows new bands at 1774 cm−1 and 1710 cm−1 originating from the carbonyls (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching) of the phthalimide groups (Fig. 2B). These changes confirm that the copolymer was successfully quaternized with N-(2-bromoethyl)phthalimide. After hydrazinolysis and dialysis (Fig. 2C), the removal of the phthalimide groups led to the disappearance of the stretching modes of the carbonyl group (CO) in the 1774–1710 cm−1 region. This result proves that the deprotection reaction was successful and that the phthalhydrazide by-product was completely removed.42–44 Furthermore, a new band appeared at 1641 cm−1 which can be assigned to the bending vibration of the NH group, as well as an increase in the intensity of the bands in the 3250–3400 cm−1 region that can be assigned to the stretching vibration of –NH2. This confirms the successful formation of primary amine groups.
O stretching) of the phthalimide groups (Fig. 2B). These changes confirm that the copolymer was successfully quaternized with N-(2-bromoethyl)phthalimide. After hydrazinolysis and dialysis (Fig. 2C), the removal of the phthalimide groups led to the disappearance of the stretching modes of the carbonyl group (CO) in the 1774–1710 cm−1 region. This result proves that the deprotection reaction was successful and that the phthalhydrazide by-product was completely removed.42–44 Furthermore, a new band appeared at 1641 cm−1 which can be assigned to the bending vibration of the NH group, as well as an increase in the intensity of the bands in the 3250–3400 cm−1 region that can be assigned to the stretching vibration of –NH2. This confirms the successful formation of primary amine groups.
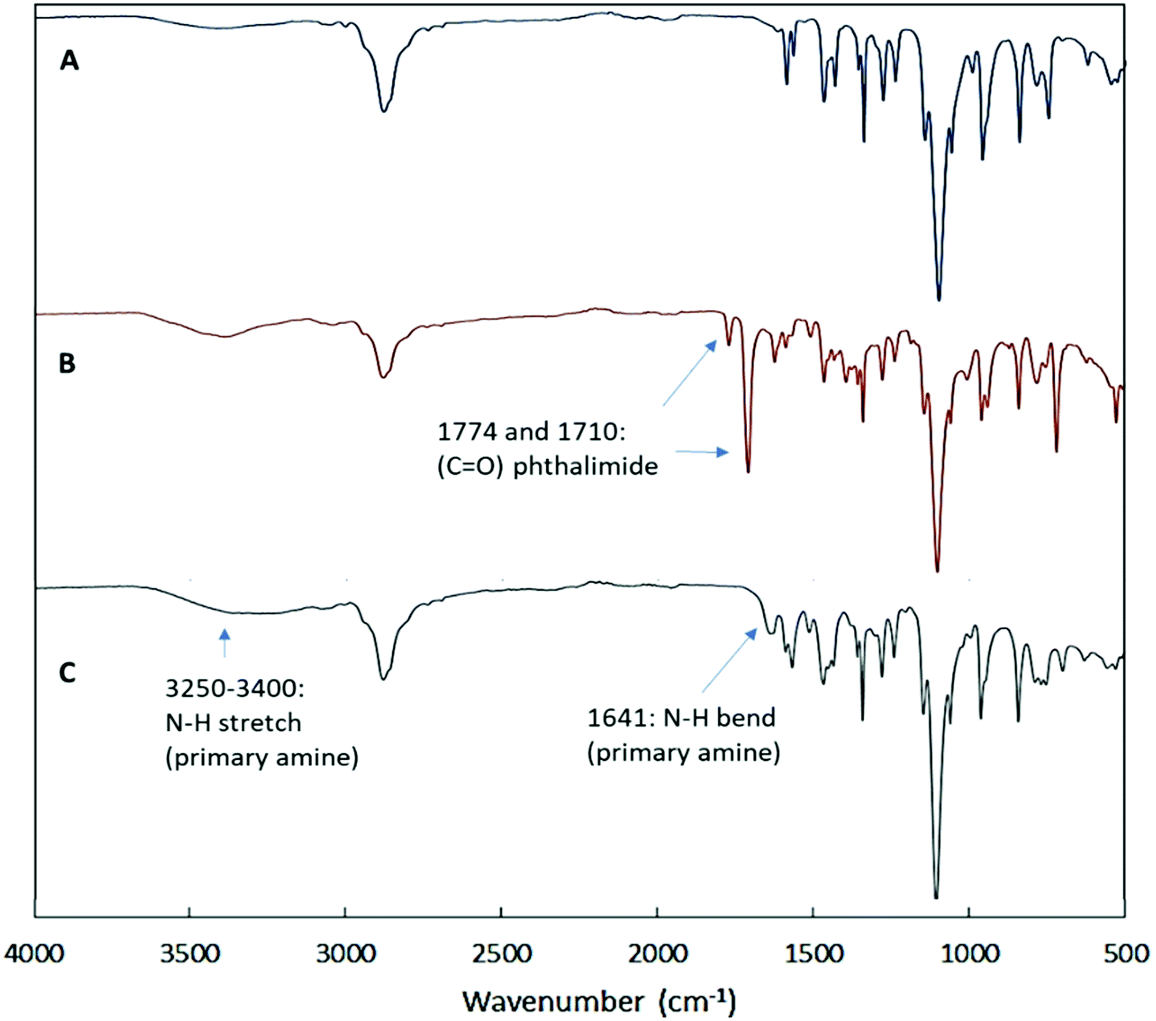 | ||
| Fig. 2 FTIR spectra of the functionalized diblock copolymer P2VP128-b-PEO477. (A) Diblock copolymer before quaternization, (B) diblock copolymer after quaternization using N-(2-bromoethyl)phthalimide, (C) quaternized diblock copolymer after deprotection using hydrazine hydrate. | ||
To further support the presence of accessible primary amines in the deprotected diblock copolymer, we have conducted a ninhydrin assay. This assay is a colorimetric method that enables qualitative and quantitative determination of amino groups.45–47 The presence of primary amines will give a dark purple product known as “Ruhemann's purple”. Fig. 3 shows that the diblock copolymer solutions before quaternization and deprotection only give a pale-yellow colour, identical to the control sample, indicating that there are no primary amines in these polymer samples present. This is in large contrast to the deprotected diblock copolymer, for which the solution turned deep purple upon addition of ninhydrin as a direct result of ninhydrin-amine complex formation. Taken together, we have generated diblock copolymers that have accessible primary amines suitable for crosslinking validated by different approaches like 1H-NMR and FTIR.
 | ||
| Fig. 3 Ninhydrin test (using ninhydrin 2%) of the functionalized diblock copolymer (2 mg mL−1). (A) Blank/negative control, (B) diblock copolymer before quaternization, (C) diblock copolymer after quaternization, (D) diblock copolymer after being deprotected. | ||
3.2 C3M formation between the functionalized diblock copolymer and homopolymer
After successful quaternization of the diblock copolymer, this copolymer was mixed with homopolymer to form C3Ms. DLS measurements were performed to observe the formation of these C3Ms. Different aliquots of a solution of the anionic homopolymer (PAA118) were added to solutions with a constant concentration of cationic-neutral hydrophilic diblock copolymer, quaternized P2VP128-b-PEO477, to determine the preferred micellar composition (PMC). The PMC is identified as the mixing composition where the scattering intensity reaches its maximum. After equilibrating the mixtures overnight, the light scattering intensity (I), the hydrodynamic radius of the micelles (Rh), and the polydispersity index (PDI) were determined as a function of the PAA concentration.Fig. 4 shows the DLS results for mixtures with different concentrations of PAA and a constant concentration of quaternized P2VP128-b-PEO477 (final polymer concentration of 2.50 μM). The light scattering intensity (Fig. 4A) increases rapidly with increasing PAA concentration, indicating formation of C3Ms. At a concentration of 1.81 μM PAA118 the light scattering intensity reached a maximum, implying that this mixing composition contains the highest number and most well-defined micelles. Thus, this point is considered to be the PMC.
 | ||
| Fig. 4 DLS results for mixtures of different concentrations of PAA and a constant concentration of quaternized P2VP128-b-PEO477 (final polymer concentration of 2.50 μM). (A) Absolute scattering intensity (I), (B) hydrodynamic radius (Rh), and (C) polydispersity index (PDI). | ||
At the pH used (pH 7.4), the primary amines of the quaternized diblock copolymer are protonated (pKa of ethyleneamine = 10.4)48,49 and bear a positive charge, thereby contribute to the total number of positive charges on this polymer; it amounts to 218 elementary charges per molecule (DQ × n(2VP) × 2 = 0.85 × 128 × 2 = 218). At this pH the carboxylic groups of PAA are all deprotonated, so the number of negative charges on PAA is 118. Thus, at the PMC, the ratio between the total concentration of negative charges on the PAA and the total concentration of positive charges on the diblock ([−]/[+]) is 0.39 ((1.81 × 118)/(2.5 × 218) = 0.39). Clearly, the PMC of these C3Ms is far from charge stoichiometry ([−]/[+] = 1), indicating that interactions other than electrostatic play a role in micelle formation for this polymer couple. These are probably hydrophobic interactions, since the diblock copolymer still contains hydrophobic pyridine units due to incomplete quaternization.3,12,15
At the PMC, the C3Ms have a hydrodynamic radius of 25.1 ± 1.0 nm (Fig. 4B), and a minimum value for the PDI of 0.1 ± 0.03 is found (Fig. 4C), indicating a narrow size distribution of the C3Ms.50 Addition of PAA beyond the PMC leads to excess charge and results in reduction of light scattering intensity because of disintegration of micelles into negatively charged soluble complexes.7,8,20,51
Besides the ratio between the charged homopolymers and diblock copolymers, micelle formation also depends on their absolute concentration: micelles are only formed above a certain concentration, the CMC.1 Diluting a 2.50 μM C3M stock solution prepared at the PMC with 10 mM sodium phosphate buffer (pH 7.4) initially leads to a linear decrease of the scattering intensity as observed by DLS (Fig. S5, ESI†), reflecting the decrease in number of micelles.52,53 Although the intensity becomes small compared to the background at low polymer concentrations, from this dilution series it is safe to assume the CMC being equal or possibly even lower than 0.59 μM, the concentration at which C3Ms can still be identified. For comparison, the typical range of CMC values reported for polymeric micelles is in the order of 10−6 to 10−7 M.3,52–54 The CMC of low molecular weight surfactant micelles is in the order of 10−3 to 10−4 M. This makes polymeric micelles less prone to dissociation at low concentrations compared to surfactant-based micelles.55
3.3 Core-crosslinking of C3Ms using two different strategies
The aim of our present study is to enhance the stability of the micelles for which we have explored different crosslinking strategies. Here, we used two types of crosslinking agents that allow crosslinking of the core of C3Ms: EDC and DTBP. After 3 hours, the crosslinking reaction was expected to stop due to the short half-life of these linkers in aqueous solution.56–58,71 EDC facilitates formation of a permanent amide bond between the primary amine groups of the quaternized P2VP-b-PEO diblock copolymer and the carboxylic acid groups of PAA in the core of C3Ms. This amide bond formation is irreversible (Scheme 3). Efficient amide formation by the EDC linker can be achieved at a pH in the range of 4.5–7.5.59–61 | ||
| Scheme 3 Crosslink formation between a primary amine group of the quaternized diblock copolymer P2VP-b-PEO and a carboxylate group of the homopolymer PAA using EDC. | ||
On the other hand, DTBP has imido ester groups that can only react with amine-containing polymers to form an amidine bond, and works best in a pH range of 7 to 10. DTBP has an internal disulphide group that can be cleaved using a reducing agent.62–65 The addition of DTBP to a solution of C3Ms results in crosslinking of the core of the micelles by amidine bond formation between amine groups of two P2VP units of the quaternized diblock copolymer (Scheme 4). Thus, if DTBP is used as crosslinker, PAA is not incorporated in the crosslinked network. Furthermore, the imido ester groups of DTBP maintain the overall positive charge on the diblock copolymer after reacting with primary amines66 ensuring that the negatively charged PAA remains in the core. The addition of reducing agents such as 1,4-dithiothreitol (DTT) and tris(2-carboxyethyl)phosphine (TCEP) results in dissociation of the crosslinks. DLS demonstrated that the hydrodynamic radius of EDC and DTBP core-crosslinked micelles was 23.9 ± 0.9 nm and 24.3 ± 1.4 nm, respectively. These sizes are similar to the size of the non-crosslinked micelles. Multi-angle DLS revealed that their shape remained spherical (Fig. 5), which can be deduced from the three overlapping linear relationships between the decay rate Γ and wave vector q for each type of micelle67,68 which are unaffected by the crosslinking agents.
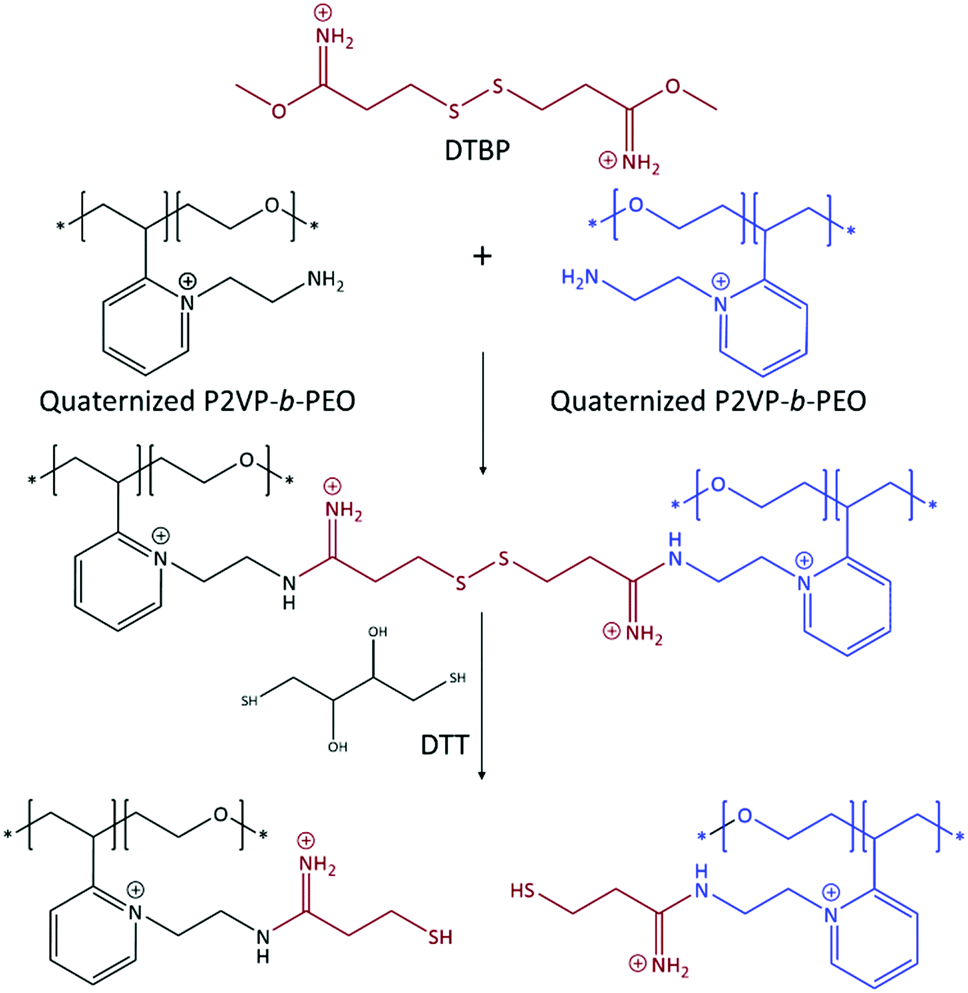 | ||
| Scheme 4 Crosslink formation between primary amine groups of the diblock copolymer P2VP-b-PEO using DTBP. The crosslinks can be cleaved using reducing agents such as DTT. | ||
 | ||
| Fig. 5 Multi-angle DLS results for all C3M formulations. The decay rate Γ as a function of the wave vector q was obtained from the DLS correlation curves by a first (blue), second (orange), and third (grey) order cumulant fit. (A) C3Ms without crosslinks, (B) C3Ms core-crosslinked with EDC, (C) C3Ms core-crosslinked with DTBP. | ||
3.4 Stability of C3Ms
The stability of the micelles, prepared at the PMC, was studied using a salt titration. In Fig. 6, the normalized scattering intensity, Rh and PDI for four different C3Ms samples (C3M control, C3M crosslinked by EDC, C3M crosslinked by DTBP, and C3M crosslinked by DTBP and treated with DTT) are plotted against the salt concentration, up to 1.0 M of NaCl. All samples showed a decrease in scattering intensity with increasing salt concentration. However, for non-crosslinked C3Ms, the scattering intensity decreases significantly faster than for core-crosslinked C3Ms. At 1.0 M NaCl, the scattering intensity of non-crosslinked C3Ms is practically zero, indicating total disintegration of the micelles. However, core-crosslinked micelles turned out to be much more resistant to salt addition. Both EDC and DTBP core-crosslinked C3Ms demonstrate a more gradual decrease in scattering intensity, which seems to reach a plateau at the highest NaCl concentrations applied. At 1.0 M NaCl a large fraction of the micelles was still present. The salt stability of EDC core-crosslinked micelles seems to be higher than that of the DTBP core-crosslinked ones, which can be explained from the fact that in EDC core-crosslinked micelles the two types of polymer chains are bonded together in the core. For EDC crosslinked micelles the scattering intensity at 1.0 M NaCl was about 77% of its initial value. This may be caused by the loss of some polymer chains that where not properly connected via crosslinks. For DTBP core-crosslinked micelles the scattering intensity at 1.0 M salt is about 55% of its value before NaCl addition. Since the PAA chains are not covalently attached to the crosslinked P2VP chains, they can diffuse out from the micelles via reptation69,70 at high salt conditions when all charges are screened. However, possibly a part of the PAA chains remains entangled in the cross-linked network formed by the P2VP chains and cannot dissociate from the complexes, even at very high salt concentrations.15,66 | ||
| Fig. 6 Stability measurements of C3Ms as a function of salt concentration using DLS. C3Ms without crosslinks (control) (blue), C3Ms core-crosslinked with EDC (red), C3Ms core-crosslinked with DTBP (orange), C3Ms core-crosslinked with DTBP after addition of 50 mM DTT (green). (A) Normalized scattering intensity, (B) hydrodynamic radius (Rh), (C) polydispersity index (PDI). | ||
An additional feature of DTBP crosslinking is the presence of a disulphide bridge, offering the possibility to cleave the network by simply adding a reducing agent. We treated DTBP core-crosslinked C3Ms with the reducing agent DTT and observed a gradual disintegration of C3Ms upon addition of salt (see Fig. 6A). After core-crosslinking using DTBP and subsequently reducing the disulphide bridges, the original salt-stability profile of the non-crosslinked C3Ms was largely restored.
Fig. 6B presents the hydrodynamic radii of different C3Ms as a function of NaCl concentration, showing that these are rather constant in all cases. It should be noted that for low scattering intensities the error in Rh is relatively large. DTT-treated crosslinked micelles are larger than non-crosslinked micelles, presumably due to the (cleaved) DTBP moieties present in the cores. These micelles seem to have a much stronger tendency to swell than other C3Ms.15,54
The PDI of non-crosslinked C3Ms and DTT-treated crosslinked micelles increased with increasing salt concentration and reached above 0.4 at 1.0 M NaCl, pointing to highly polydisperse samples due to disintegration of the micelles (Fig. 6C). However, the PDI of crosslinked C3Ms only slightly increased with increasing salt concentration, and reached PDI values of 0.2 at most at 1.0 M salt, which indicates moderately polydisperse samples with the micelles still being intact.50
We also investigated the effect of pH on micelle stability since the pH determines the degree of dissociation of carboxylic groups of poly(acrylic acid) and amine functional groups of the quaternized diblock copolymer. In Fig. 7 and 8, the pH-dependence of the scattering intensity, Rh, and PDI are plotted for the different micellar systems: non-crosslinked C3Ms, EDC core-crosslinked C3Ms, and DTBP core-crosslinked C3Ms. When the pH decreases by the addition of 0.1 M HCl, the negative charge on PAA becomes lower (pKa of PAA = 4.5). At pH 3 the charge of the PAA functional groups is practically zero (dissociation degree α = 0.03). For the non-crosslinked micelles, we observed a decrease in scattering intensity with decreasing pH and especially below pH 4 the intensity decreased sharply (Fig. 7A). Obviously, in this case the reduction of negative charge on PAA results in disruption of the C3Ms. On the other hand, core-crosslinked C3Ms are stable over the full pH range covered by the HCl titration, as evidenced by a constant high scattering intensity. For crosslinking using EDC, the core-crosslinked C3Ms cannot fall apart because a network between PAA and diblock copolymer is established. Surprisingly, for the crosslinking using DTBP, which only connects the QP2VP chains, the PAA apparently also remains trapped inside the core. At low pH, PAA adopts a globular conformation (but not collapsed for PAA with molar mass less than 16.5 kg mol−1).59,72 This globular structure is most likely the cause that PAA cannot diffuse out of the QP2VP network in the core of micelles.59,72
 | ||
| Fig. 7 Stability measurements as a function of the pH (with addition of 0.1 M HCl) as observed by using DLS. Blue (C3Ms without crosslinks – control), red (C3Ms core-crosslinked with EDC), orange (C3Ms core-crosslinked with DTBP. (A) Normalized scattering intensity, (B) hydrodynamic radius (Rh), (C) polydispersity index (PDI). | ||
 | ||
| Fig. 8 Stability measurements as a function of the pH (with addition of 0.1 M NaOH) as observed by using DLS. Blue (C3Ms without crosslinks – control), red (C3Ms core-crosslinked with EDC), orange (C3Ms core-crosslinked with DTBP. (A) Normalized scattering intensity, (B) hydrodynamic radius (Rh), (C) polydispersity index. | ||
The size of the non-crosslinked micelles decreased at a pH close to the pKa of PAA, suggesting that some PAA and also diblock copolymer leave the micelles before they fully disintegrate (Fig. 7B). The PDIs of these non-crosslinked micelles increased upon addition of acid (Fig. 7C) and measured values above 0.4 at a pH below the pKa of PAA, an indication for highly polydisperse samples.50 However, for the core-crosslinked C3Ms, both EDC and DTBP, the size of the micelles and the PDI are rather constant upon the addition of acid.
When the pH is increased by addition of 0.1 M NaOH, the total number of positive charges on the diblock copolymer decreases because the deprotected amino groups become deprotonated (pKa of ethyleneamine = 10.4).48,49 Above pH 11, these groups are barely charged anymore. However, as the quaternization reaction leads to permanent (pH-independent) charges on the pyridine rings, a high pH does not lead to such a dramatic decrease in scattering intensity of the non-crosslinked C3Ms compared to the addition of acid. In other words, non-crosslinked C3Ms do not entirely fall apart since the positive charges of the quaternized pyridine ring are still present at a high pH (Fig. 8A). However, we still observe a lower C3M stability than for the core-crosslinked C3Ms.
The size of the non-crosslinked micelles slightly increased at a pH above the pKa of ethyleneamine, and is accompanied by an increase of the PDI (Fig. 8B and C). This suggests a slight swelling of the micelles and possibly a structural rearrangement and some loss of PAA due to the deprotonation of amino groups. However, with the addition of base, the Rh and PDI of the core-crosslinked C3Ms, both EDC and DTBP, are rather constant showing that the micelles stay intact and unchanged.
In summary, crosslinks between the two components in the core (EDC) result in a slightly higher stability against pH changes compared to crosslinks between only one component (DTBP). Again, this is likely a result of EDC being able to keep both components of the C3Ms together in the core, while with DTBP, PAA may diffuse out as it is not included in the network. We note that the difference between the two crosslinking strategies is less pronounced for stability against pH changes than for the stability against salt addition. In the latter case all charges on both polymers gradually become effectively screened, while changing the pH has no effect on the permanent positive charges of the diblock copolymer and only limited effect (at low pH) on the charges of the PAA homopolymer.
4 Conclusions
In this study, we modified a P2VP-b-PEO diblock copolymer via quaternization that simultaneously resulted in permanent positive charges on the pyridine rings and, after deprotection, in primary amine functional groups. C3Ms were prepared by mixing aqueous solutions of the anionic homopolymer PAA and the quaternized diblock copolymer. DLS measurements confirmed the formation of C3Ms. The PMC of the C3Ms was not found at the charge balance between negative charges on PAA and positive charges on the quaternized P2VP-b-PEO but at a lower ratio ([−]/[+] = 0.39). This points to the role of other types of interactions besides electrostatic attraction in the complex formation, such as hydrophobic interactions. Covalent core-crosslinking of the micelles resulted in a significant improvement of the stability of C3Ms against salt addition and pH changes. Using the crosslinker EDC, permanent crosslinks between PAA and quaternized P2VP in the micellar core are formed; the crosslinker DTBP provides cleavable (reversible) bonds between P2VP chains only. Core-crosslinking using EDC provides a slightly higher stability compared to core-crosslinking using DTBP because it keeps these two components of the C3Ms together in the micelle core. In case of DTBP-crosslinked micelles, PAA can diffuse out of the core due to charge screening when the salt concentration is increased, although some of the polymer chains may remain trapped in the crosslinked network. An advantage of the second approach is the ability to cleave the internal disulphide bridges via addition of a reducing agent (DTT), thereby largely restoring the original disintegration profile of C3Ms as a function of salt concentration. This type of reversible crosslinkers offers great opportunities for controlled release of functional ingredients using C3Ms.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the VLAG Graduate School, Wageningen University & Research. We thank Mr Remco Fokkink, Prof. Willem van Berkel and Prof. Jasper van der Gucht for their valuable suggestions and discussions.Notes and references
- T. K. Bronich, A. V. Kabanov and V. A. Kabanov, Macromolecules, 1997, 9297, 3519–3525 CrossRef
.
- A. Harada and K. Kataoka, Macromolecules, 1995, 28, 5294–5299 CrossRef CAS
- I. K. Voets, A. de Keizer and M. A. Cohen Stuart, Adv. Colloid Interface Sci., 2009, 147–148, 300–318 CrossRef CAS PubMed
- M. A. Cohen Stuart, N. A. M. Besseling and R. G. Fokkink, Langmuir, 1998, 14, 6846–6849 CrossRef
- A. Harada and K. Kataoka, Polym. J., 2018, 50, 95–100 CrossRef CAS
- C. Dähling, J. E. Houston, A. Radulescu, M. Drechsler, M. Brugnoni, H. Mori, D. V. Pergushov and F. A. Plamper, ACS Macro Lett., 2018, 7, 341–346 CrossRef
- C. C. M. Sproncken, J. R. Magana and I. K. Voets, ACS Macro Lett., 2021, 10, 167–179 CrossRef CAS PubMed
- J. R. Magana, C. C. M. Sproncken and I. K. Voets, Polymers, 2020, 12, 1953–1990 CrossRef CAS PubMed
- A. E. Marras, M. Ting, K. C. Stevens and M. V. Tirrell, J. Phys. Chem. B, 2021, 125, 7076–7089 CrossRef CAS PubMed
- A. E. Marras, T. R. Campagna, J. R. Vieregg and M. V. Tirrell, Macromolecules, 2021, 54, 6585–6594 CrossRef CAS
- M. Amann, J. S. Diget, J. Lyngsø, J. S. Pedersen, T. Narayanan and R. Lund, Macromolecules, 2019, 52, 8227–8237 CrossRef CAS
- A. Nolles, A. H. Westphal, J. A. De Hoop, R. G. Fokkink, J. M. Kleijn, W. J. H. Van Berkel and J. W. Borst, Biomacromolecules, 2015, 16, 1542–1549 CrossRef CAS PubMed
- N. P. Agarwal, M. Matthies, F. N. Gür, K. Osada and T. L. Schmidt, Angew. Chem., Int. Ed., 2017, 56, 5460–5464 CrossRef CAS PubMed
- S. Lindhoud, R. De Vries, R. Schweins, M. A. Cohen Stuart and W. Norde, Soft Matter, 2009, 5, 242–250 RSC
- N. Bourouina, M. A. Cohen Stuart and J. Mieke Kleijn, Soft Matter, 2014, 10, 320–331 RSC
- S. Lindhoud, R. de Vries, W. Norde and M. A. C. Stuart, Biomacromolecules, 2007, 8, 2219–2227 CrossRef CAS PubMed
- C. E. Mills, A. Obermeyer, X. Dong, J. Walker and B. D. Olsen, Langmuir, 2016, 32, 13367–13376 CrossRef CAS PubMed
- M. Jaturanpinyo, A. Harada, X. Yuan and K. Kataoka, Bioconjugate Chem., 2004, 15, 344–348 CrossRef CAS PubMed
- W. C. Blocher McTigue and S. L. Perry, Small, 2020, 16, 1–17 CrossRef PubMed
- A. Nolles, E. Hooiveld, A. H. Westphal, W. J. H. Van Berkel, J. M. Kleijn and J. W. Borst, Langmuir, 2018, 34, 12083–12092 CrossRef CAS PubMed
- S. Gao, A. Holkar and S. Srivastava, Polymers, 2019, 11, 1097–1131 CrossRef CAS PubMed
- Y. Shao, W. Huang, C. Shi, S. T. Atkinson and J. Luo, Ther. Delivery, 2012, 3, 1409–1427 CrossRef CAS PubMed
- X. Ma, J. Liu, L. Lei, H. Yang and Z. Lei, J. Appl. Polym. Sci., 2019, 136, 1–8 Search PubMed
- M. Talelli, M. Barz, C. J. F. Rijcken, F. Kiessling, W. E. Hennink and T. Lammers, Nano Today, 2015, 10, 93–117 CrossRef CAS PubMed
- M. S. Shim and Y. J. Kwon, Biomaterials, 2010, 31, 3404–3413 CrossRef CAS PubMed
- G. S. Krishnakumar, S. Sampath, S. Muthusamy and M. A. John, Mater. Sci. Eng., C, 2019, 96, 941–954 CrossRef CAS PubMed
- W. E. Hennink and C. F. van Nostrum, Adv. Drug Delivery Rev., 2012, 64, 223–236 CrossRef
- C. F. Van Nostrum, Soft Matter, 2011, 7, 3246–3259 RSC
- G. Tillet, B. Boutevin and B. Ameduri, Prog. Polym. Sci., 2011, 36, 191–217 CrossRef CAS
- O. B. Ayyub, M. B. Ibrahim and P. Kofinas, Polymer, 2014, 55, 6227–6231 CrossRef CAS
- G. Mantovani, F. Lecolley, L. Tao, D. M. Haddleton, J. Clerx, J. J. L. M. Cornelissen and K. Velonia, J. Am. Chem. Soc., 2005, 127, 2966–2973 CrossRef CAS PubMed
- M. Ju, F. Gong, S. Cheng and Y. Gao, Int. J. Polym. Sci., 2011, 2011, 1–8 CrossRef
- P. D. McMaster, E. W. Byrnes, A. J. Block and P. A. Tenthorey, J. Med. Chem., 1981, 24, 53–58 CrossRef CAS PubMed
- J. Nielsen and P. H. Rasmussen, Tetrahedron Lett., 1996, 37, 3351–3354 CrossRef CAS
- R. Kembaren, R. Fokkink, A. H. Westphal, M. Kamperman, J. M. Kleijn and J. W. Borst, Langmuir, 2020, 36, 8494–8502 CrossRef CAS PubMed
- A. A. Dehghani-Firouzabadi and S. Firouzmandi, J. Braz. Chem. Soc., 2017, 28, 768–774 CAS
- A. V. Malkov, A. J. P. Stewart-Liddon, F. Teplý, L. Kobr, K. W. Muir, D. Haigh and P. Kočovský, Tetrahedron, 2008, 64, 4011–4025 CrossRef CAS
- G. Chamoulaud and D. Bélanger, Langmuir, 2004, 20, 4989–4995 CrossRef CAS PubMed
- K. Sadman, Q. Wang, Y. Chen, B. Keshavarz, Z. Jiang and K. R. Shull, Macromolecules, 2017, 50, 9417–9426 CrossRef CAS
- R. Nosov, P. Padnya, D. Shurpik and I. Stoikov, Molecules, 2018, 23, 1–11 CrossRef PubMed
- M. I. Attia, A. A. El-Emam, A. A. Al-Turkistani, A. L. Kansoh and N. R. El-Brollosy, Molecules, 2014, 19, 279–290 CrossRef PubMed
- S. Akrami, B. Karami and M. Farahi, RSC Adv., 2017, 7, 34315–34320 RSC
- F. Luan, L. Wei, J. Zhang, W. Tan, Y. Chen, F. Dong, Q. Li and Z. Guo, Molecules, 2018, 23, 516–529 CrossRef PubMed
- M. R. Hill, S. Mukherjee, P. J. Costanzo and B. S. Sumerlin, Polym. Chem., 2012, 3, 1758–1762 RSC
- E. W. Yemm, E. C. Cocking and R. E. Ricketts, Analyst, 1955, 80, 209–214 RSC
- S. W. Sun, Y. C. Lin, Y. M. Weng and M. J. Chen, J. Food Compos. Anal., 2006, 19, 112–117 CrossRef CAS
- M. Friedman, J. Agric. Food Chem., 2004, 52, 385–406 CrossRef CAS PubMed
- R. Vijisha and K. Muraleedharan, Int. J. Greenh. Gas Control, 2017, 58, 62–70 CrossRef
- I. Juranić, Croat. Chem. Acta, 2014, 87, 343–347 CrossRef
- S. Bhattacharjee, J. Controlled Release, 2016, 235, 337–351 CrossRef CAS PubMed
- S. Lindhoud, W. Norde and M. A. C. Stuart, Langmuir, 2010, 26, 9802–9808 CrossRef CAS PubMed
- Ö. Topel, B. A. Çakir, L. Budama and N. Hoda, J. Mol. Liq., 2013, 177, 40–43 CrossRef
- M. S. Santos, F. W. Tavares and E. C. Biscaia, Braz. J. Chem. Eng., 2016, 33, 515–523 CrossRef CAS
- H. M. van der Kooij, E. Spruijt, I. K. Voets, R. Fokkink, M. A. Cohen Stuart and J. van der Gucht, Langmuir, 2012, 28, 14180–14191 CrossRef CAS PubMed
- C. J. F. Rijcken, O. Soga, W. E. Hennink and C. F. van Nostrum, J. Controlled Release, 2007, 120, 131–148 CrossRef CAS PubMed
- M. A. Gilles, A. Q. Hudson and C. L. Borders, Anal. Biochem., 1990, 184, 244–248 CrossRef CAS PubMed
- S. B. Douglas, T. Browne and H. Kent, Biochem. Biophys. Res. Commun., 1975, 67, 126–132 CrossRef
- M. J. Hunter and M. L. Ludwig, J. Am. Chem. Soc., 1962, 84, 3491–3504 CrossRef CAS
- S. Keleştemur, M. Altunbek and M. Culha, Appl. Surf. Sci., 2017, 403, 455–463 CrossRef
- K. A. Totaro, X. Liao, K. Bhattacharya, J. I. Finneman, J. B. Sperry, M. A. Massa, J. Thorn, S. V. Ho and B. L. Pentelute, Bioconjugate Chem., 2016, 27, 994–1004 CrossRef CAS PubMed
- B. I. Eroglu, Y. B. Kilinc and Z. Mustafaeva, Turk. J. Biochem., 2011, 36, 222–229 CAS
- H. Liu, F. Zhao, B. Koo, Y. Luan, L. Zhong, K. Yun and Y. Shin, Sens. Actuators, B, 2019, 288, 225–231 CrossRef CAS
- X. Xu, A. E. Smith and C. L. McCormick, Aust. J. Chem., 2009, 62, 1520–1527 CrossRef CAS
- A. R. Durney, S. Kawaguchi, G. Pennamon and H. Mukaibo, Mater. Lett., 2014, 133, 171–174 CrossRef CAS
- O. Koniev and A. Wagner, Chem. Soc. Rev., 2015, 44, 5495–5551 RSC
- D. Oupickí, R. C. Carlisle and L. W. Seymour, Gene Ther., 2001, 8, 713–724 CrossRef PubMed
- A. Harada and K. Kataoka, Science, 1999, 283, 65–67 CrossRef CAS PubMed
- A. Harada and K. Kataoka, Langmuir, 1999, 15, 4208–4212 CrossRef CAS
- M. E. Cates, Macromolecules, 1987, 20, 2289–2296 CrossRef CAS
- J. Klein, Nature, 1978, 271, 143–145 CrossRef CAS
-
G. T. Hermanson, Bioconjugate Techniques, Academic Press, Boston, 3rd edn, 2013 Search PubMed
- T. Swift, L. Swanson, M. Geoghegan and S. Rimmer, Soft Matter, 2016, 12, 2542–2549 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2sm00088a |
| This journal is © The Royal Society of Chemistry 2022 |