
Iron encapsulated nitrogen and sulfur co-doped few layer graphene as a non-precious ORR catalyst for PEMFC application†
B. P. Vinayan*a,
Thomas Diemantb,
R. Jürgen Behmab and
S. Ramaprabhu*c
aHelmholtz Institute Ulm for Electrochemical Energy Storage (HIU), Helmholtzstr. 11, 89081 Ulm, Germany. E-mail: vinayan.parambath@kit.edu; Tel: +49 7315034218
bInstitute of Surface Chemistry and Catalysis, Ulm University, Albert-Einstein-Allee 47, 89081 Ulm, Germany
cAlternative Energy and Nanotechnology Laboratory (AENL), Nano Functional Materials Technology Centre (NFMTC), Department of Physics, Indian Institute of Technology Madras, Chennai, Tamil Nadu 600036, India. E-mail: ramp@iitm.ac.in; Fax: +91-44-22570509/22574852; Tel: +91-44-22574862
First published on 30th July 2015
Abstract
A novel strategy was followed to prepare an iron nanoparticle encapsulated nitrogen and sulfur co-doped few layer graphene (Fe-NSG) non precious electrocatalyst. For this purpose, initially graphite oxide was coated with the polyelectrolyte poly-(sodium 4-styrenesulfonate), followed by the nitrogen-containing polymer polyaniline. An iron precursor was added to this suspension and heated to 300 °C in a hydrogen atmosphere. The final heating of this nanocomposite at 900 °C in a N2 atmosphere and further acid leaching gave a non-precious Fe-NSG catalyst. X-ray photoelectron spectroscopy (XPS) data of the Fe-NSG catalyst illustrates the presence of a large amount of pyridinic and graphitic nitrogen species within the catalyst along with sulfur species. Half-cell and full cell electrochemical measurements prove the four electron transfer pathway of the oxygen reduction reaction with a high current density in an acidic environment. The special confined morphology of Fe nanoparticles within the graphene layers suppresses the agglomeration and dissolution of particles and gives long term durability. The present study illustrates a non-precious electrocatalyst for proton exchange membrane fuel cells with promising performance and stability.
1. Introduction
The sluggish kinetics of the oxygen reduction reaction (ORR) at the cathode of proton exchange membrane fuel cells (PEMFC) necessitate a high loading of platinum (Pt) or Pt alloy electrocatalysts at present.1 The extensive implementation of fuel cell technologies for stationary and vehicular applications, however, demands the replacement of high cost and less abundant Pt with low cost and natural abundant electrocatalysts. In this regard, recent studies show the use of various non-precious metals, metal alloys, metal oxides and nitrogen doped carbon as alternative catalysts for the ORR.2,3 However, the majority of these non-precious metal catalysts experience severe dissolution and large agglomeration under vigorous PEMFC operational conditions and subsequent catalyst performance degradation.4 Recent reports suggest that the trapping of non-precious metals, especially iron (Fe), within nitrogen doped carbon materials could give a high ORR activity due to the creation of metal–Nx active centres.5–7 Zhang et al. studied a series of nitrogen containing carbon supports, co-doped with various transition metals and observed that the ORR activity of catalysts follows the order of Fe > Co > Zn > Mn > metal-free ≫ Cu ≫ Ni.8 The advantages of Fe based alternative catalysts include low cost, excellent electrocatalytic activity, improved durability as compared to other NPC catalysts, and an environmentally benign character.9,10Furthermore, it was demonstrated that the amount of Fe metal loading within the nitrogen doped carbon supports also influences the ORR activity and stability of the non-precious electro catalysts (NPC), the optimum Fe loading was determined to be at around 5 wt%.11 The general procedure to prepare these NPC is the combined heating of precursors which contain nitrogen, carbon and Fe or Co transition metals between the temperatures of 500 and 800 °C.12 The ORR activity of the NPC materials is mainly correlated with the morphology of carbon supports, nature of nitrogen and transition-metal precursors used for synthesis and heat treatment temperature etc. Nitrogen doped graphene support materials are very promising in this regard due to their numerous favourable properties such as large surface area, more electrochemical active sites and very good activity towards ORR, excellent electrical conductivity and high mechanical and chemical stability.13–15 Among the different possibilities to achieve N doping of graphene support materials, mixing with various nitrogen containing polymers such as polyaniline (PANI) or polypyrrole (PPy) and their subsequent pyrolysis at high temperatures can provide large amount of pyridinic, pyrrolic and graphitic nitrogen functionalities within the graphene support, which lead to a high ORR activity.7,16 Furthermore, the presence of surfactants/polyelectrolytes can help to achieve the desired uniform polymer coating over the surface of graphene layers during the polymerization of nitrogen containing monomers.17–24 Similar to N doping, substitution of carbon atoms with heteroatoms like boron, phosphorus, or sulfur within the graphene lattice is also reported to significantly improve the ORR activity.25,26 Furthermore, recent reports also show an enhanced electrocatalytic activity for carbon materials by co-doping of different heteroatoms, such as boron/nitrogen,27 nitrogen/oxygen,28 nitrogen/sulfur,29 and nitrogen/phosphorus,30 etc., within the graphene lattice rather than increasing only the doping level of a single heteroatom.
In this paper, we describe the preparation of an iron nanoparticles (Fe NPs) loaded nitrogen and sulfur co-doped graphene material by a new approach and the investigation of its ORR activity and durability in acidic media by half cell and full cell measurements.
2. Experimental details
2.1 Synthesis of materials
Graphite oxide (GO) was prepared from graphite (Sigma Aldrich, particle size < 45 μm) using Hummer's method.31 GO was coated with the polyelectrolyte poly-(sodium 4-styrenesulfonate) (PSS, Mw ∼ 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 30 wt% in H2O), which is explained elsewhere.32 Furthermore, 2.0 mL of aniline monomer were uniformly mixed with 0.4 g of PSS coated GO in 0.5 M HCl solution (300 mL). The suspension was kept below 10 °C during addition of the oxidant (ammonium peroxydisulfate, APS) and FeCl3 metal precursor.33 Here, FeCl3 is the precursor for the Fe loading in the final sample and it also can help in the polymerization of aniline as oxidizing agent along with APS. 24 h of subsequent stirring of the final suspension allow the complete polymerization of aniline (polyaniline, PANI) and uniform mixing of metal precursor. The final suspension was vacuum-dried using a rotary evaporator. Afterwards, the Fe precursor containing PANI–PSS coated GO sample was spread evenly in a quartz boat and kept inside a CVD furnace. The sample was then flushed with an argon gas flow for 10 minutes. Subsequently hydrogen (H2) gas was allowed to flow and the CVD temperature raised to 300 °C. After half an hour, H2 gas supply was switched off and the temperature increased to 900 °C for 2 h in nitrogen gas flow. The heat-treated sample was then pre-leached in 0.5 M H2SO4 at 60 °C for 6 hours to eliminate unstable and inactive species from the catalyst, and thoroughly washed in de-ionized water and dried. Pristine few layer graphene (FLG) was prepared by hydrogen exfoliation of pristine GO at 300 °C.
000, 30 wt% in H2O), which is explained elsewhere.32 Furthermore, 2.0 mL of aniline monomer were uniformly mixed with 0.4 g of PSS coated GO in 0.5 M HCl solution (300 mL). The suspension was kept below 10 °C during addition of the oxidant (ammonium peroxydisulfate, APS) and FeCl3 metal precursor.33 Here, FeCl3 is the precursor for the Fe loading in the final sample and it also can help in the polymerization of aniline as oxidizing agent along with APS. 24 h of subsequent stirring of the final suspension allow the complete polymerization of aniline (polyaniline, PANI) and uniform mixing of metal precursor. The final suspension was vacuum-dried using a rotary evaporator. Afterwards, the Fe precursor containing PANI–PSS coated GO sample was spread evenly in a quartz boat and kept inside a CVD furnace. The sample was then flushed with an argon gas flow for 10 minutes. Subsequently hydrogen (H2) gas was allowed to flow and the CVD temperature raised to 300 °C. After half an hour, H2 gas supply was switched off and the temperature increased to 900 °C for 2 h in nitrogen gas flow. The heat-treated sample was then pre-leached in 0.5 M H2SO4 at 60 °C for 6 hours to eliminate unstable and inactive species from the catalyst, and thoroughly washed in de-ionized water and dried. Pristine few layer graphene (FLG) was prepared by hydrogen exfoliation of pristine GO at 300 °C.
2.2 Characterization
Powder X-ray diffraction (XRD) of samples was recorded with a step size of 0.016° within the 2θ range from 5–90° using a PANalytical X'Pert Pro X-ray diffractometer equipped with Ni-filtered Cu Kα radiation as the X-ray source (40 kV and 30 mA). Thermo-gravimetric analysis (TGA) of the sample was carried out (NETZSCH instruments) from room temperature to 900 °C in an air atmosphere with a heating rate of 20 °C min−1. The vibrational properties of samples were analyzed using a Raman spectrometer (Witec Alpha 300) with a 532 nm wavelength laser as excitation source. The morphology of the samples was studied by field emission scanning electron microscopy (FESEM, FEI QUANTA 3D) and transmission electron microscopy (TEM, Tecnai G2 20 S-TWIN). Energy dispersive X-ray (EDS) spectral analysis of samples was taken by FESEM equipped with Li-doped silicon as X-ray detector. The elemental composition of the sample surfaces was determined by X-ray photoelectron spectroscopy (XPS) measurements using monochromatized Al Kα (1486.6 eV) radiation (PHI 5800 Multi-Technique ESCA System, Physical Electronics). The measurements were done with a detection angle of 45°, using pass energies at the analyser of 93.9 and 29.35 eV for survey and detail spectra, respectively. For binding energy calibration the C (1s) peak was set to 284.5 eV. Before the XPS measurements, the surfaces were sputtered for 3 minutes (5 kV, 1 μA) to remove the topmost surface layer.2.3 Electrochemical measurements
Cyclic voltammetry (CV) and rotating disk electrode (RDE) measurements were carried out in a conventional three-electrode electrochemical cell at room temperature (25 °C) using AUTOLAB PGSTAT 100 and PINE instruments. An Ag/AgCl electrode (3.0 M KCl) was used as a reference electrode. A graphite rod was used (instead of Pt) as counter electrode for the non-precious catalyst to elude any potential Pt contamination to the Fe-SNG non-precious catalyst. Meanwhile, a Pt wire was used as counter electrode for Pt based catalysts. The catalyst ink for half-cell measurements was prepared by dispersing 5 mg of electrocatalyst and Nafion (5 wt%) in 1 mL of isopropanol solution with 30 min sonication. 10 μL of catalyst ink was dropped onto a 3 mm diameter glassy carbon electrode. All the scanning for CV and RDE were carried out at a scan rate of 20 mV s−1 in oxygen or nitrogen saturated 0.1 M HClO4 solution. During RDE measurements, the electrode rotation rate was varied from 400 to 2000 rpm.The performance of a PEMFC single cell with Fe-NSG as cathode catalyst was studied by constructing a membrane electrode assembly (MEA). Each MEA consists of two electrodes (anode and cathode) and a polymer electrolyte membrane in between. The electrodes include three different layers, the backing layer, gas diffusion layer, and catalyst layer. A teflonized carbon fabric (Torray, USA) was used as the gas diffusion layer. The catalyst ink was prepared by dispersing appropriate amount of electrocatalysts in the solution of de-ionized water and 2-propanol with 5 wt% Nafion solution (DuPont, USA). The catalyst inks were uniformly coated over the gas diffusion layers with the cathode catalyst (Fe-NSG) loading of 4 mg cm−2 and anode catalyst (E-Tek Pt/C, 40 wt% Pt loading) loading of 0.25 mgPt cm−2. The MEA was assembled by sandwiching a pre-treated Nafion 212 CS membrane between anode and cathode by hot-pressing at 130 °C and 70 bar for 4 minutes. The effective electrode area was 11.56 cm2. The MEA was fixed between two graphite plates, which had a provision for serpentine type gas flow. The PEMFC measurements were carried out using a Teledyne fuel cell test station at two different temperatures of 50 and 80 °C, in 90% relative humidity with an oxygen back pressure of 20 psi.
3. Results and discussion
3.1 Structural and morphological studies
The schematic of the synthesis procedure of Fe-NSG electrocatalyst is given in Fig. 1. In the first step, graphite oxide (GO) was soft functionalized by the anionic polyelectrolyte poly(sodium 4-styrenesulfonate) (PSS) to enhance the dispersion of GO within the water medium. This procedure gives a thin coating of sulfonate functional groups (SO3−) containing polyelectrolyte on the surface of GO. The PSS functionalized GO (PSS-GO) and aniline were mixed together in HCl medium. Subsequently, the oxidative polymerizing agent (APS) and the iron precursor FeCl3 were added to this suspension. Here, PSS can act as a sulfur dopant of graphene with APS. In addition, a thin coating of anionic polyelectrolyte PSS over GO can help to achieve a uniform coating of PANI over GO due to the electrostatic attraction between SO3− groups of PSS and N+ groups of polymer.32 Heating of the Fe precursor containing PANI coated PSS-GO to 300 °C in H2 gas atmosphere leads to the complete reduction of GO and iron precursor within the composite. Subsequent increase of the temperature of the nanocomposite to 900 °C in N2 atmosphere decomposes the nitrogen rich polymer PANI and the sulfur containing PSS, which results in Fe loaded nitrogen and sulfur co-doped graphene sheets. The final sample was then pre-leached in 0.5 M H2SO4 at 60 °C for 6 hours to remove unstable and inactive species from the catalyst. | ||
| Fig. 1 Schematic diagram of the synthesis procedure adopted for Fe-NSG non-precious electrocatalyst. | ||
The as-synthesized samples were characterized by various techniques. X-ray diffractograms of (a) graphite (Gr), (b) GO, (c) FLG, and (d) Fe-NSG are illustrated in Fig. 2. A shift in the (002) peak of graphite from 26.73° to 10.54° has been observed upon its oxidation. This corresponds to an increase in the d-spacing of GO from 0.34 to 0.84 nm because of the intercalation of hydroxyl, carbonyl and carboxyl functional groups in between the graphene layers.34 After hydrogen reduction at 300 °C, FLG shows a broad (002) peak ranging from 14° to 30° with a d-spacing of 0.37 nm.18 This suggests an amorphous nature of the stacked layers of graphene and removal of a large portion of the oxygen functional groups.35 At the same time, Fe-NSG gives a more sharp (002) graphitic peak as a result of heating at higher temperatures. The characteristic Fe (110) peak at 42.8° is merged with the (101) graphitic peak at 43°.9,11 Fig. 3a shows the thermo gravimetric (TG) and derivative TG (DTG) data of Fe-NSG electrocatalyst in air. The decomposition of the organic part of the material starts at around 400 °C in air atmosphere and this process is completed slightly above 600 °C. TG gives the amount of total Fe loading within the electrocatalyst of approximately ∼4.8 wt%.
 | ||
| Fig. 2 X-ray diffractograms of (a) graphite, (b) graphite oxide, (c) few layer graphene, and (d) Fe-NSG electrocatalyst. | ||
 | ||
| Fig. 3 (a) Thermo gravimetric (TG) and derivative TG (DTG) curve of Fe-NSG electrocatalyst. (b) Raman spectra of FLG and Fe-NSG electrocatalyst. | ||
The doping information within the carbon material has been evaluated from Raman spectroscopy (Fig. 3b). The G band measures the crystallinity of the carbon material, while the D band is sensitive to structural distortion and defects present in the graphene lattice; the 2D band is an overtone of D band.36 Raman spectra of FLG show the existence of a significant number of defects (higher D band) within the graphene lattice. It may be assumed that when nitrogen and sulfur dopants are introduced into this defect rich graphene lattice, they can easily bond with the carbon atoms present at the defect sites. Additionally, some of these N and S functional groups can also attach over the surface of the graphene layers. The intensity ratio between D and G band, (ID/IG) is a direct measure of the defects and crystalline nature of carbon structures.37 The relatively high ID/IG ratio for Fe-NSG catalyst (1.16) as compared to FLG (0.87) is attributed to the N and S doping within the graphene lattice.38 Table 1 gives the G, D and 2D band peak positions, it shows a downward shift for G, D and 2D bands after doping. Similar kind of red shift of peak positions was already reported for graphene samples doped with electron donating species.39,40
| Material | G (cm−1) | D (cm−1) | 2D (cm−1) |
|---|---|---|---|
| FLG | 1596 | 1364 | 2697 |
| Fe-NSG | 1585 | 1349 | 2674 |
XPS measurements were carried out to investigate the elemental composition and oxidation state of the different elements present in Fe-NSG catalyst. The XPS survey spectrum of Fe-NSG (Fig. 4a) shows the characteristic peaks at the binding energy positions 164, 285, 399, 532 and 710 eV, corresponding to S 2p, C 1s, N 1s, O 1s and Fe 2p, respectively. The atomic percentage of C, N, O, S and Fe within the surface layer of the Fe-NSG catalyst is 89.5%, 3.7%, 3.8%, 1.9% and 1.1% respectively (Table 2). The high-resolution N 1s spectrum of Fe-NSG (Fig. 4b) illustrates the presence of three different kinds of nitrogen, pyridinic N (398.1 eV), graphitic N (400.4 eV) and oxidized N (404.0 eV) are found with relative amounts of 28%, 58% and 14%, respectively.41 The S 2p spectrum of Fe-NSG shows two main peaks at 163.2 eV and 164.4 eV corresponding to the S 2p3/2 and S 2p1/2 peak components of S atoms, which were doped in the graphene layer. In the deconvoluted S 2p spectrum, the broad feature at higher binding energy (168.3 eV) indicates the presence of oxidized S species like sulfoxide; the peak couple at lower binding energy (161.4 and 162.6 eV) is in the typical BE range of sulfides and is therefore assigned to iron–sulfur bond formation (FeSx).39,42 The deconvoluted Fe 2p high resolution spectrum gives three peaks in the Fe 2p3/2 range, the first one at 707.5 eV is assigned to metallic Fe, the second and third one at 710.6 eV (main peak) and 715.9 eV (satellite) are due to oxidized Fe (Fe2+/Fe3+). The three Fe 2p1/2 peak components (at 720.3, 723.8, and 729.5 eV) are assigned in the same way.43 The peaks corresponding to oxidized Fe can be attributed to the formation of iron oxide and iron sulfides.11,16
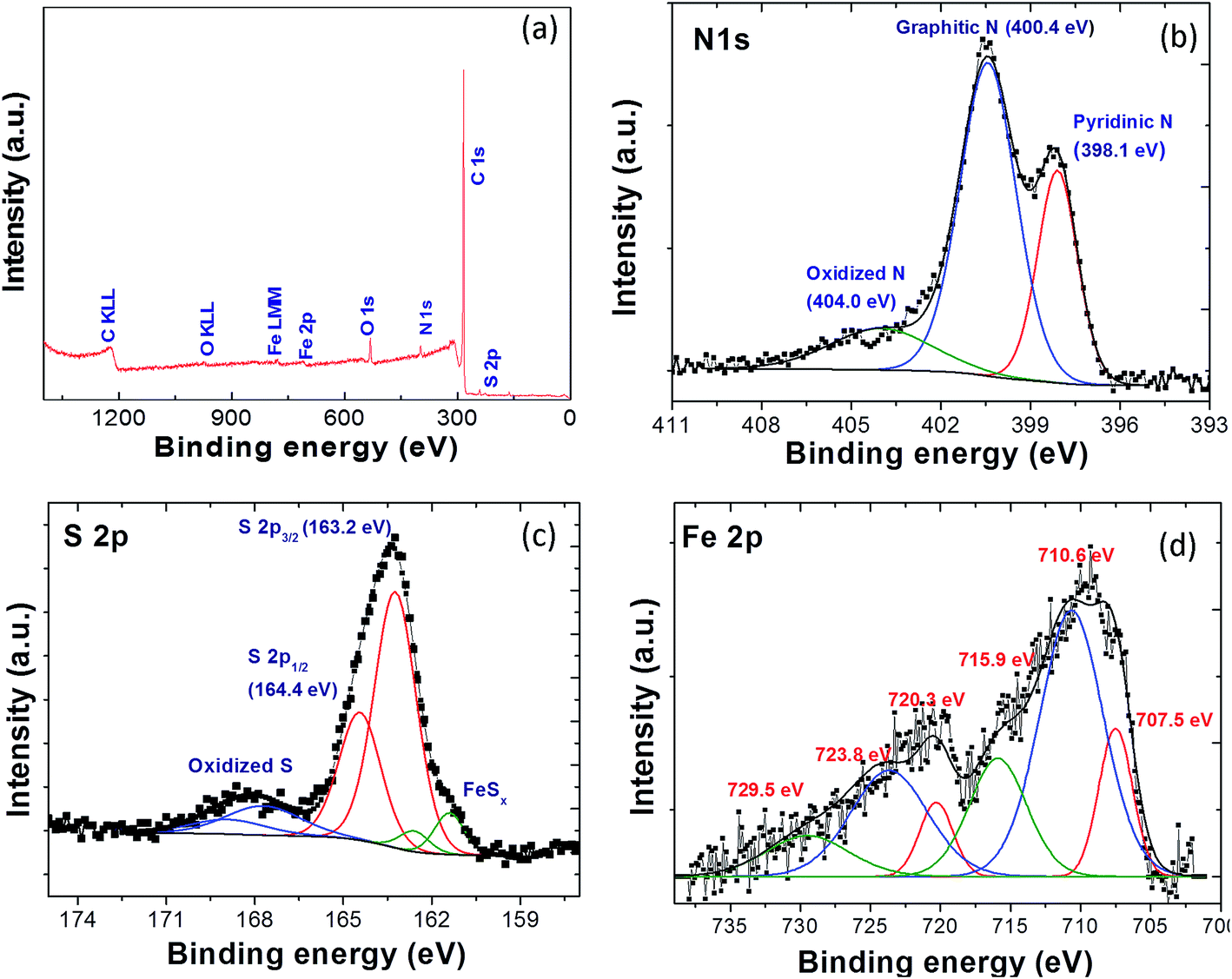 | ||
| Fig. 4 (a) XPS survey spectra of Fe-NSG electrocatalyst. High resolution XPS spectra in the (b) N 1s, (c) S 2p, and (d) Fe 2p binding energy range of the Fe-NSG sample. | ||
| Elements | XPS (%) | EDS (%) |
|---|---|---|
| Carbon | 89.5 | 89.0 |
| Nitrogen | 3.7 | 3.8 |
| Oxygen | 3.8 | 3.5 |
| Sulfur | 1.9 | 2.4 |
| Iron | 1.1 | 1.3 |
The TEM image of FLG (Fig. 5a) shows a thin sheet like morphology of the sample with a small number of wrinkles. The TEM image of the Fe precursor containing PANI coated PSS-GO (Fig. S1: ESI†) reveals a uniform coating of PANI over the PSS-GO surface. TEM and SEM images (Fig. 5b and c) of the Fe-NSG material show a more crumpled structure in which the graphene sheets overlap each other resulting in a porous interconnected network morphology. Finally, the TEM imaging of Fe-NSG (Fig. 5b) also reveals the dispersion of the Fe metal catalyst nanoparticles within the N and S doped carbon host matrix. Furthermore, HRTEM (Fig. S2: ESI†) confirms that the NSG sheets of the Fe-NSG sample contain only a few layers of graphene (≤9 layers). From EDS (Fig. 5d), the atomic percentages of C, N, O, S and Fe present in the catalyst were derived, the results are listed in Table 2. The weight percentage of Fe determined from XPS and EDX is approximately ∼5 wt% and matches with the TGA result. Elemental mapping (Fig. 5e–i) illustrates the homogeneous distribution of C, N, S, Fe and O atoms within the Fe-NSG catalyst.
 | ||
| Fig. 5 Transmission electron micrographs of (a) FLG and (b) Fe-NSG electrocatalyst. (c) Scanning electron micrograph of Fe-NSG electrocatalyst. (d) EDS spectrum of Fe-NSG electrocatalyst. Elemental mapping of Fe-NSG electrocatalyst (e–i) showing the distribution of C, N, O, S and Fe. | ||
3.2 Electrochemical analysis
The electrocatalytic activity of the Fe-NSG was evaluated by half cell and full cell measurements. The half-cell measurements were carried out using a three electrode system with Pt as counter electrode, Ag/AgCl as reference electrode and Fe-NSG coated glassy carbon as working electrode in nitrogen or oxygen saturated 0.1 M HClO4 solution. Fig. 6a shows the cyclic voltammograms (CV) of the bare working electrode, pristine graphene, and Fe-NSG in N2 or O2 saturated electrolyte. The CV diagram of Fe-NSG in O2 saturated electrolyte depicts a clear cathodic peak current at 0.29 V; while in N2 saturated electrolyte the Fe-NSG catalyst gives a smaller cathodic peak current at 0.45 V with major capacitive background current. This suggests a high ORR activity of the Fe-NSG catalyst in O2 environment and the same catalyst retains its ORR activity even after 1000 CV cycles with a cathodic peak current at 0.28 V. To further study the ORR kinetics, rotating disk electrode (RDE) measurements were carried out. Fig. 6b compares the RDE polarization curves of pristine FLG, Fe-NSG in N2 and O2 saturated electrolyte and commercial Pt/C electrode materials at a rotation speed of 1600 rpm and a scan rate of 20 mV s−1. | ||
| Fig. 6 (a) CVs of FLG and Fe-NSG samples in N2- and O2-saturated 0.1 M aqueous HClO4 electrolyte solution at a scan rate of 20 mV s−1. (b) Comparison of RDE data of the FLG, Pt/C and Fe-NSG samples at a rotation speed of 1600 rpm in O2-saturated 0.1 M aqueous HClO4 electrolyte solution. (c) RDE voltammograms recorded for Fe-NSG electrocatalyst in O2-saturated 0.1 M HClO4 electrolyte solution at a scan rate of 20 mV s−1 and different rotation rates; (d) the Koutecky–Levich (KL) plot i−1 vs. ω−1/2 at 0.65 V. | ||
The Fe-NSG catalyst exhibits high ORR activity in O2 saturated electrolyte, with an onset potential up to 0.65 V (vs. Ag/AgCl), the half-wave potential is 110 mV less than that of commercial Pt/C. In N2 saturated electrolyte, the Fe-NSG catalyst delivers only a very small diffusion limited current density as compared to the current in O2 saturated electrolyte which corroborates its high ORR activity. To find out the number of electron transfer during the ORR of Fe-NSG catalyst, RDE measurements have been carried out at different rotation speeds (400 to 2000 rpm) as shown in Fig. 6c. The Koutecky–Levich (KL) equation was used (eqn (1) and (2)) to analyse the data.44
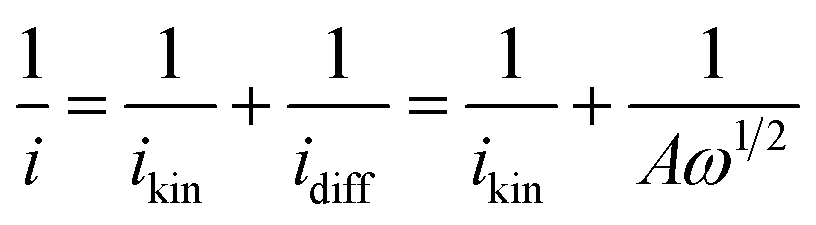 | (1) |
| A = 0.62nFD2/3v−1/6C | (2) |
In eqn (1), i, ikin, idiff are measured, kinetic-limiting and diffusion-limiting current densities, respectively; ω is the angular velocity of the disk (ω = 2πN, N is the linear rotation speed). In eqn (2), n is the number of electrons transferred during the ORR, F is the Faraday constant (96485 C mol−1), D is the diffusion coefficient of O2 in the 0.1 M HClO4 electrolyte (1.93 × 10−5 cm2 s−1), v is the kinetic viscosity of the electrolyte (1.009 × 10−2 cm2 s−1) and C is the bulk concentration of O2 in solution (C = 1.26 × 10−3 mol L−1).43 The electron transfer number during ORR at 0.65 V (vs. Ag/AgCl) for Fe-NSG catalyst has been calculated from the plot between ω−1/2 and i−1 using the KL equation (Fig. 6d) and it was around ∼4. This reveals that the ORR process of Fe-NSG is a one-step four-electron pathway rather than consecutive two-electron pathways.45
Fig. 7a shows the full cell PEMFC test results, where Fe-NSG as the cathode catalyst (4 mg cm−2), Pt/C as the anode catalyst (0.25 mgpt cm−2) and pretreated Nafion 212 polymer as the electrolyte were used in the membrane electrode assembly (MEAs). The polarization curves were taken at two different temperatures (50 °C and 80 °C) with 20 psi back pressure. The maximum power density was reached with 225 mW cm−2 and 118 mW cm−2, at 80 °C and 50 °C, respectively. At 80 °C, the current density values were 885, 386, 161 and 54 mA cm−2 for the voltages of 0.25 V, 0.4 V, 0.5 V and 0.6 V, respectively. To compare the PEMFC full cell performance of Fe-NSG cathode catalysts with commercial Pt/C, we have carried out the full cell measurements for commercial Pt/C (Fig. S3: ESI†). The catalyst loading at the cathode and anode was 0.5 and 0.25 mgPt cm−2, respectively. A current density of 550 mA cm−2 was observed at 0.5 V at 80 °C temperature and 20 psi back pressure, i.e., compared to commercial Pt/C, Fe-NSG cathode catalyst is giving lower PEMFC performance. Meanwhile PEMFC full cell performance of the Fe-NSG cathode catalysts compares well with other non-precious catalysts. Zelenay et al. synthesized a cobalt–polypyrrole–carbon (Co–PPY–C) composite catalyst via a simple chemical method and showed a PEMFC performance of 200 mA cm−2 at 0.5 V and a maximum power density of 140 mW cm−2 at 80 °C temperature and 29.4 psi back pressure.6 Dodelet et al. prepared an iron based non-precious catalyst and reported a PEMFC performance of 300 mA cm−2 at 0.50 V at 80 °C temperature and 45.3 psi back pressure.46 The back pressure applied during the PEMFC measurements of present Fe-NSG electrocatalyst (20 psi) was less as compared to the above reports.
 | ||
| Fig. 7 (a) PEMFC single cell polarization curves of the Fe-NSG cathode electrocatalyst with Pt/C as anode catalyst at two different temperatures (50 °C and 80 °C) with 20 psi back pressure. (b) Stability study of the PEMFC constructed with Fe-NSG cathode electrocatalyst at the temperature of 80 °C and 20 psi back pressure. | ||
The stability studies of MEA constructed with Fe-NSG cathode catalyst (Fig. 7b) were evaluated by running the cell for 25 h at the voltage of 0.4 V and temperature of 80 °C. The PEMFC performance data reveal the stable operation of PEMFC running with Fe-NSG cathode catalyst. The high ORR activity of Fe-NSG electrocatalyst can be attributed to the following reasons. The comparatively higher electronegativity of doped N and S atoms (N: 3.04, S: 2.58) with respect to carbon (C: 2.55) induces more charged sites within the graphene lattice which are favorable for oxygen adsorption and reduction.47 XPS results confirm that Fe-NSG contains a large amount of graphitic and pyridinic type of nitrogen. Previous studies showed that graphitic and pyridinic nitrogen can give a significant contribution to ORR activity.48,49 Another important contribution to ORR activity is coming from the formation of Fe–Nx species, especially Fe–N4.2,5 The large amount of pyridinic nitrogen within Fe-NSG catalyst can coordinate with iron cations and can increase the density of ORR active Fe–Nx centers.50 Presence of an optimum amount of sulfur within the non-precious catalyst can suppress the iron carbide formation, and enhances the formation of Fe–N4 ORR active species.16 The better durability of Fe-NSG electrocatalyst can be ascribed to the unique confined morphology of Fe nanoparticles within the graphene layers which may suppress the agglomeration/dissolution of Fe particles and increase their interfacial contact.
4. Conclusion
In summary, we have developed a facile approach to prepare Fe impregnated, nitrogen and sulfur co-doped graphene based non-precious catalyst and tested its electrochemical performance in acidic electrolyte medium. RDE measurements of the Fe-NSG catalyst confirm a four electron transfer ORR process with high current density. PEMFC full cell measurements of the Fe-NSG catalyst give a maximum power density of 225 mW cm−2 at 80 °C and very good stability at high current density.References
- F. Jaouen, E. Proietti, M. Lefevre, R. Chenitz, J.-P. Dodelet, G. Wu, H. T. Chung, C. M. Johnston and P. Zelenay, Energy Environ. Sci., 2011, 4, 114–130 CAS.
- Z. Chen, D. Higgins, A. Yu, L. Zhang and J. Zhang, Energy Environ. Sci., 2011, 4, 3167–3192 CAS.
- M. Lefèvre, E. Proietti, F. Jaouen and J.-P. Dodelet, Science, 2009, 324, 71–74 CrossRef PubMed.
- C. W. B. Bezerra, L. Zhang, K. Lee, H. Liu, A. L. B. Marques, E. P. Marques, H. Wang and J. Zhang, Electrochim. Acta, 2008, 53, 4937–4951 CrossRef CAS PubMed.
- U. I. Kramm, J. Herranz, N. Larouche, T. M. Arruda, M. Lefevre, F. Jaouen, P. Bogdanoff, S. Fiechter, I. Abs-Wurmbach, S. Mukerjee and J.-P. Dodelet, Phys. Chem. Chem. Phys., 2012, 14, 11673–11688 RSC.
- R. Bashyam and P. Zelenay, Nature, 2006, 443, 63–66 CrossRef CAS PubMed.
- G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443–447 CrossRef CAS PubMed.
- H.-J. Zhang, Q.-Z. Jiang, L. Sun, X. Yuan, Z. Shao and Z.-F. Ma, Int. J. Hydrogen Energy, 2010, 35, 8295–8302 CrossRef CAS PubMed.
- K. R. Reddy, K.-P. Lee, A. I. Gopalan and H.-D. Kang, React. Funct. Polym., 2007, 67, 943–954 CrossRef CAS PubMed.
- K. R. Reddy, K.-P. Lee, A. I. Gopalan, M. S. Kim, A. M. Showkat and Y. C. Nho, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 3355–3364 CrossRef CAS PubMed.
- K. Parvez, S. Yang, Y. Hernandez, A. Winter, A. Turchanin, X. Feng and K. Müllen, ACS Nano, 2012, 6, 9541–9550 CrossRef CAS PubMed.
- C. W. B. Bezerra, L. Zhang, H. Liu, K. Lee, A. L. B. Marques, E. P. Marques, H. Wang and J. Zhang, J. Power Sources, 2007, 173, 891–908 CrossRef CAS PubMed.
- H. Wang, T. Maiyalagan and X. Wang, ACS Catal., 2012, 2, 781–794 CrossRef CAS.
- W. J. Lee, U. N. Maiti, J. M. Lee, J. Lim, T. H. Han and S. O. Kim, Chem. Commun., 2014, 50, 6818–6830 RSC.
- B. P. Vinayan, N. I. Schwarzburger and M. Fichtner, J. Mater. Chem. A, 2015, 3, 6810–6818 CAS.
- M. Ferrandon, A. J. Kropf, D. J. Myers, K. Artyushkova, U. Kramm, P. Bogdanoff, G. Wu, C. M. Johnston and P. Zelenay, J. Phys. Chem. C, 2012, 116, 16001–16013 CAS.
- K. R. Reddy, M. Hassan and V. G. Gomes, Appl. Catal., A, 2015, 489, 1–16 CrossRef CAS PubMed.
- M. Hassan, K. R. Reddy, E. Haque, A. I. Minett and V. G. Gomes, J. Colloid Interface Sci., 2013, 410, 43–51 CrossRef CAS PubMed.
- S. H. Choi, D. H. Kim, A. V. Raghu, K. R. Reddy, H.-I. Lee, K. S. Yoon, H. M. Jeong and B. K. Kim, J. Macromol. Sci., Part B: Phys., 2011, 51, 197–207 CrossRef PubMed.
- K. R. Reddy, H. M. Jeong, Y. Lee and A. V. Raghu, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1477–1484 CrossRef CAS PubMed.
- Y. Lee, S. Kim, H.-I. Lee, H. Jeong, A. Raghu, K. Reddy and B. Kim, Macromol. Res., 2011, 19, 66–71 CrossRef CAS.
- J.-H. Park, A. Choudhury, B. L. Farmer, T. D. Dang and S.-Y. Park, Polymer, 2012, 53, 3937–3945 CrossRef CAS PubMed.
- C.-H. Wu, W.-Y. Chiu and T.-M. Don, Polymer, 2012, 53, 1086–1092 CrossRef CAS PubMed.
- S. J. Han, H.-I. Lee, H. M. Jeong, B. K. Kim, A. V. Raghu and K. R. Reddy, J. Macromol. Sci., Part B: Phys., 2014, 53, 1193–1204 CrossRef CAS PubMed.
- M. del Cueto, P. Ocón and J. M. L. Poyato, J. Phys. Chem. C, 2015, 119, 2004–2009 CAS.
- Y. Zhang, M. Chu, L. Yang, W. Deng, Y. Tan, M. Ma and Q. Xie, Chem. Commun., 2014, 50, 6382–6385 RSC.
- L. Wang, P. Yu, L. Zhao, C. Tian, D. Zhao, W. Zhou, J. Yin, R. Wang and H. Fu, Sci. Rep., 2014, 4, 5184 CAS.
- N. Karthikeyan, B. P. Vinayan, M. Rajesh, K. Balaji, A. K. Subramani and S. Ramaprabhu, Fuel Cells, 2015, 15, 278–287 CrossRef CAS PubMed.
- X. Wang, J. Wang, D. Wang, S. Dou, Z. Ma, J. Wu, L. Tao, A. Shen, C. Ouyang, Q. Liu and S. Wang, Chem. Commun., 2014, 50, 4839–4842 RSC.
- D. Yu, Y. Xue and L. Dai, J. Phys. Chem. Lett., 2012, 3, 2863–2870 CrossRef CAS.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- B. P. Vinayan, R. Nagar, N. Rajalakshmi and S. Ramaprabhu, Adv. Funct. Mater., 2012, 22, 3519–3526 CrossRef CAS PubMed.
- K. R. Reddy, B. C. Sin, K. S. Ryu, J.-C. Kim, H. Chung and Y. Lee, Synth. Met., 2009, 159, 595–603 CrossRef CAS PubMed.
- B. P. Vinayan, R. Nagar, K. Sethupathi and S. Ramaprabhu, J. Phys. Chem. C, 2011, 115, 15679–15685 Search PubMed.
- B. P. Vinayan, R. Nagar and S. Ramaprabhu, J. Mater. Chem., 2012, 22, 25325 RSC.
- M. S. Dresselhaus, G. Dresselhaus and M. Hofmann, Philos. Trans. R. Soc., A, 2008, 366, 231–236 CrossRef CAS PubMed.
- B. P. Vinayan, R. Nagar and S. Ramaprabhu, J. Mater. Chem. A, 2013, 1, 11192–11199 CAS.
- A. C. Ferrari, Solid State Commun., 2007, 143, 47–57 CrossRef CAS PubMed.
- J.-E. Park, Y. J. Jang, Y. J. Kim, M.-S. Song, S. Yoon, D. H. Kim and S.-J. Kim, Phys. Chem. Chem. Phys., 2014, 16, 103–109 RSC.
- H. Liu, Y. Liu and D. Zhu, J. Mater. Chem., 2011, 21, 3335–3345 RSC.
- B. P. Vinayan and S. Ramaprabhu, Nanoscale, 2013, 5, 5109–5118 RSC.
- R. S. C. Smart, W. M. Skinner and A. R. Gerson, Surf. Interface Anal., 1999, 28, 101–105 CrossRef CAS.
- H. Peng, Z. Mo, S. Liao, H. Liang, L. Yang, F. Luo, H. Song, Y. Zhong and B. Zhang, Sci. Rep., 2013, 3, 1765 Search PubMed.
- Z. Lin, G. H. Waller, Y. Liu, M. Liu and C.-P. Wong, Nano Energy, 2013, 2, 241–248 CrossRef CAS PubMed.
- C. Zhang, R. Hao, H. Liao and Y. Hou, Nano Energy, 2013, 2, 88–97 CrossRef CAS PubMed.
- C. Médard, M. Lefèvre, J. P. Dodelet, F. Jaouen and G. Lindbergh, Electrochim. Acta, 2006, 51, 3202–3213 CrossRef PubMed.
- B. P. Vinayan, K. Sethupathi and S. Ramaprabhu, Int. J. Hydrogen Energy, 2013, 38, 2240–2250 CrossRef CAS PubMed.
- H. Jin, H. Zhang, H. Zhong and J. Zhang, Energy Environ. Sci., 2011, 4, 3389–3394 CAS.
- D. W. Boukhvalov and Y.-W. Son, Nanoscale, 2012, 4, 417–420 RSC.
- R. Kothandaraman, V. Nallathambi, K. Artyushkova and S. C. Barton, Appl. Catal., B, 2009, 92, 209–216 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra09030j |
| This journal is © The Royal Society of Chemistry 2015 |